

Abstract
The 3′-one-third of the severe acute respiratory syndrome coronavirus (SARS-CoV) genome contains genes for four essential structural proteins and eight virus-specific genes. The expression of this genomic information of SARS-CoV involves synthesis of a nested set of subgenomic RNAs (sgRNAs). In this study, we showed that the translational levels of 10 SARS-CoV sgRNAs including the two low-abundance sgRNAs 2-1 and 3-1 varied considerably in translation reporter assays. We also demonstrated that the initiator AUG codon of sgRNA-8 was silent and the repressive control was most likely positioned in the upstream untranslated region (UTR) of sgRNA-8. The initiator AUG codons of most sgRNAs are in poor Kozak contexts and the translation of truncated proteins from downstream AUG codons by leaky scanning was common in our experimental settings. No significant correlation was found between complexity of 5′-UTR and the sequence context of AUG codon with the level of translation of SARS-CoV sgRNAs. These results will be helpful for further studies to reveal the biological functions and translation regulatory mechanisms of sgRNAs in the coronavirus life cycle and pathogenesis.
Keywords: SARS coronavirus, Subgenomic RNA, Translational control, Group-specific genes, Leaky scanning
Introduction
Coronaviruses are the largest RNA viruses that are enveloped and contain a single-stranded, positive-sense RNA genome ranging from 27 to 31.5 kb in length. The genome of coronaviruses is polycistronic and possesses a 5′-cap structure and a 3′-poly (A) tail [1]. At the 5′-end, the two large open reading frames (ORFs) (1a and 1b) comprise about two-thirds of the entire coronaviruses genome, which encode the viral replicase and are translated directly from the genomic RNA [2]. Besides four essential structural proteins spike (S), envelope (E), membrane (M), and nucleocapsid (N), the 3′-one-third of the genome comprises variable number of group-specific genes, which are expressed through a set of nested 3′-coterminal subgenomic RNAs (sgRNAs) (Fig. 1a). A key feature of these sgRNAs is that their 5′- and 3′-terminal sequences are identical to those of the genome. This nested set structure results from a fusion of the sequence representing the genomic 5′-end (leader sequence) and sequences representing different 3′-regions of the genome, the so-called mRNA bodies (body sequences). Though the 5′-end of genome greatly affects coronavirus discontinuous transcription to produce sgRNAs [3], the regulatory mechanism of coronavirus gene expression is not well understood.
Fig. 1.
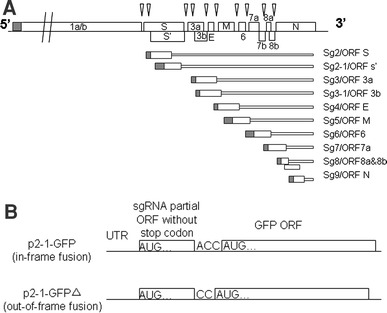
Schematic diagram of the SARS-CoV genome and the sgRNAs. a SARS-CoV genome, the sgRNA and their ORFs. The small grey boxes represent the 5′-UTR of the genomic and sgRNAs, the white boxes represent the ORFs analyzed in study. The SARS-CoV structural proteins (S, E, M, and N) and accessory proteins 3a, 3b, 6, 7a and 7b could be detected in infected cells or SARS patient samples. b Construction of GFP fused protein. The open reading frame of subgenomic RNA 2-1 was fused in-frame and out-of-frame at 5′-end of GFP gene in pEGFP-N1 vector. Translation from predicted initiator AUG codon will result in accumulation of GFP-fusion protein. Leaky translation from downstream GFP AUG codon will result in synthesis of wild-type GFP
The 3′-proximal one-third of severe acute respiratory syndrome coronavirus (SARS-CoV) genome includes eight virus-specific genes: 3a and 3b genes (located between the S and E genes), 6, 7a, 7b, 8a, and 8b genes (located between the E and N genes), as well as 9b and 9c gene (located within the N gene) [4]. In our previous work, we identified 10 sgRNAs from SARS-CoV-infected cells and showed that the transcription of sgRNAs was in a discontinuous manner at the stage of negative strand synthesis [5]. As all the sgRNAs contain a common leader of about 72 nucleotides (nt), it is still not clear how expressions of the 3′-proximal genes are controlled at the translational level. Revelation of the translational control mechanism will help to explain the roles of the group-specific genes and their encoded accessory proteins in viral life cycle and pathogenesis.
In this study, we showed that nine SARS-CoV sgRNAs could be expressed in the reporter system at different levels and the 5′-upstream untranslated regions (UTRs) of individual sgRNAs controlled the translational efficiency of their encoded proteins.
Materials and methods
Cells and viral cDNAs
Baby hamster kidney (BHK) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco Invitrogen) supplemented with 10% heat inactivated fetal bovine serum (Gibco Invitrogen), 2 mM l-glutamine, 100 U/ml of penicillin and 100 μg/ml streptomycin (Gibco Invitrogen Corporation). The cDNAs of SARS coronavirus strain WHU (GenBank accession no.AY394850) were prepared as described previously [5, 6].
Plasmid construction
The 5′-end of SARS-CoV sgRNA 2-1 (including the leader sequence and 146 nt of the 5′-end body sequence) was PCR amplified as described [5] and cloned into pEGFP-N1 vector (Clontech) (Table 1). The ORF of sgRNA 2-1 was fused in-frame and out-of-frame with that of the green florescent protein (GFP) gene, respectively, resulting in plasmids p2-1-GFP and p2-1-GFP∆ (Fig. 1b).
Table 1.
Primer sequences and cloning sites used for plasmid constructions
| Name | Sequence | Restriction site | Description | Vector |
|---|---|---|---|---|
| sgRNAF | 5′-GCGCTAGCATATTAGGTTTTTACCTACCCA-3′ | NheI | 5′ primer for all sgRNAs | pEGFP-N1 and pGL3.0 |
| sgRNA2R1 | 5′-CGGATCCTTCTCTGTGGCAGCAAAATAAATA-3′ | BamHI | 3′ primer for sgRNA2 | pEGFP-N1 |
| sgRNA2R2 | 5′-CGCCATGGTCTCTGTGGCAGCAAAATAAATA-3′ | NcoI | pGL3.0 | |
| sgRNA2-1R1 | 5′-CGACCGGTGGCCCATCTTTATTTTTAAA-3′ | AgeI | 3′ primer for sgRNA2-1 | pEGFP-N1 |
| sgRNA2-1R2 | 5′-GCCTCGAGCCCCATCTTTATTTTTAAACACAAA-3′ | XhoI | pGL3.0 | |
| sgRNA3R1 | 5′-CGACCGGTGGCGCCTCCATACCTGCAGCGACAA3′ | AgeI | 3′ primer for sgRNA3 | pEGFP-N1 |
| sgRNA3R2 | 5′-CGCCATGGTCGCCTCCATACCTGCAGCGACAA-3′ | NcoI | pGL3.0 | |
| sgRNA3-1R1 | 5′-CGACCGGTGGTCGGCTCATCATAAATTGGATCCATTG-3′ | AgeI | 3′ primer for sgRNA3-1 | pEGFP-N1 |
| sgRNA3-1R2 | 5′-CGCCATGGTTCGGCTCATCATAAATTGGATCCATTG-3′ | NcoI | pGL3.0 | |
| sgRNA4R1 | 5′-CGACCGGTGGGACCAGAAGATCAGGAACTCCTTCAGAA-3′ | AgeI | 3′ primer for sgRNA4 | pEGFP-N1 |
| sgRNA4R2 | 5′-CGCCATGGTGACCAGAAGATCAGGAACTCCTTCAGAA-3′ | NcoI | pGL3.0 | |
| sgRNA5R1 | 5′-CGACCGGTGGACTTTCCATGAGCGGTCTGGT-3′ | AgeI | 3′ primer for sgRNA5 and sg8/5 | pEGFP-N1 |
| sgRNA5R2 | 5′-CGCCATGGTACTTTCCATGAGCGGTCTGGT-3′ | NcoI | pGL3.0 | |
| sgRNA6R1 | 5′-CGACCGGTGGTGGATAATCTAACTCCATA-3′ | AgeI | 3′ primer for sgRNA6 | pEGFP-N1 |
| sgRNA6R2 | 5′-CGCCATGGTTGGATAATCTAACTCCATA-3′ | NcoI | pGL3.0 | |
| sgRNA7R1 | 5′-CGACCGGTGGTTGTTGAACCTCCTCTTGTCTGATG-3′ | AgeI | 3′ primer for sgRNA7 | pEGFP-N1 |
| sgRNA7R2 | 5′-CGCCATGGTTTGTTGAACCTCCTCTTGTCTGATG-3′ | NcoI | pGL3.0 | |
| sgRNA8R1 | 5′-CGACCGGTGGATTTGTTCGTTTATTTAAA-3′ | AgeI | 3′ primer for sgRNA8 and sg5/8 | pEGFP-N1 |
| sgRNA8R2 | 5′-CGCCATGGTATTTGTTCGTTTATTTAAA-3′ | NcoI | pGL3.0 | |
| sgRNA8aR | 5′-CGACCGGTGGGATGCACAGCGCTGTACT-3′ | AgeI | 3′ primer for sgRNA8a | pEGFP-N1 |
| sgRNA9R1 | 5′-CGACCGGTGGACTATTGGTGTTGATTGGA-3′ | AgeI | 3′ primer for sgRNA9 | pEGFP-N1 |
| sgRNA9R2 | 5′-CGCCATGGTACTATTGGTGTTGATTGGA-3′ | NcoI | pGL3.0 |
In another set of experiments, the 5′-ends (including the leader sequence and 200–400 nt of the 5′-end of body sequence) of all 10 sgRNAs were amplified by RT-PCR and cloned into pEGFP-N1 vector, with their open reading frames fused in-frame with GFP gene (p2/S, p2-1, p3/3a, p3-1, p4/E, p5/M, p6, p7/7a, p8 and p9/N) (Fig. 1 and Table 1). To circumvent the problem of wild-type GFP expression by leaky scanning, the initiator AUG codon of GFP gene was substituted with GUG by PCR-based mutagenesis, resulting in pGFP* as a negative control.
In parallel experiments, the same 5′-terminal sequences of 10 sgRNAs were cloned into pGL3.0 vector (Promega) by fusing the viral ORF in-frame with luciferase gene to quantitatively measure the sgRNAs translational level. The sequence and position of primers used for plasmid constructions were shown in Table 1.
Transfection and western blot analysis
Baby hamster kidney (BHK) cells were grown to 70–80% confluence on a 35-mm2 plate and transfected with the DNA plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The protein expression was measured by fluorescence microscopy and western blots after 36 h post-transfection. Briefly, transfected BHK cells were lysed with 2× SDS loading buffer and separated on a 12% SDS-polyacrylamide gel. Proteins were transferred onto polyvinylidene difluoride membranes (Bio-Rad). The membranes were blocked overnight with 5% non-fat milk in PBS and incubated with the monoclonal anti-GFP antibody (1:10,000, Clontech). After washing with PBST (PBS with 0.05% Tween-20) for three times, the membranes were incubated with 0.2 ng/ml of horseradish peroxidase-labeled secondary antibody (Lab Vision, USA) for 2 h. Immune complexes were visualized using the LumiGLOTM chemilumiscent substrate kit (Kirkegaard and Perry Lab, Maryland USA).
Firefly luciferase activity assay
In the firefly luciferase reporter gene assays, BHK cells were plated in 24-well plates at 1 × 105 cells per well, and transfected with recombinant sgRNA-luciferase fusion plasmids as described above at 2 μg per well. To assess the expression level of sgRNAs, firefly luciferase activity was quantified using a Steady-Glo Luciferase Assay System Kit (Promega) at different time points post-infection. The empty pGL3.0 transfected cells were used as a positive control while the mock-transfected cells were used as negative control. All the values were expressed as a mean of three independent experiments.
Results and discussion
Translatability of the low-abundance sgRNA 2-1
To determine whether the low-abundance sgRNA 2-1 discovered in the previous study [5] is a functional message RNA, the 5′-proximal 220 nt of the sgRNA 2-1 was fused with the GFP gene both in-frame and out-of-frame. Recombinant plasmids were transfected into BHK cells and the expression of GFP was assessed by fluorescence microscopy (Fig. 2a). The GFP out-of-frame construct (p2-1-GFP∆) was used as a control to monitor any possible leaky scanning-mediated expression of the reporter gene. Empty pEGFP-N1 vector was used as positive control to assess the transfection efficiency.
Fig. 2.
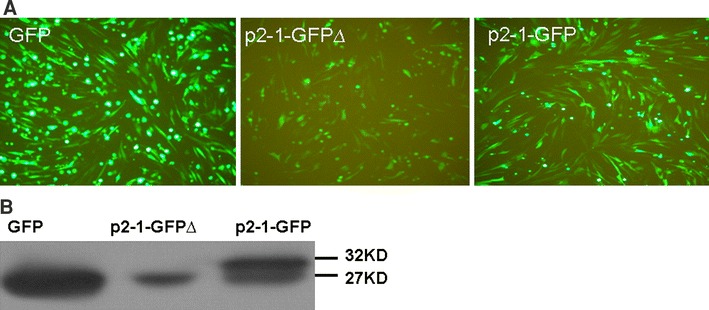
Expression of sgRNA 2-1 in BHK cells. a Fluorescence analysis of the translatability of sgRNA 2-1. The ORF 2b of sgRNA 2-1 was fused in-frame (p2-1-GFP) and out-of-frame (p2-1-GFPΔ) with GFP ORF. After it was transfected into BHK cells, the expression of GFP was assessed by fluorescence microscopy. b Western blot analysis of fusion proteins under control of 5′-sequence of sgRNA 2-1. GFP: pEGFP-N1 as control, which expresses wild-type GFP; p2-1-GFP: in-frame fusion; p2-1-GFPΔ: out-of-frame fusion. Proteins were extracted from transfected cells 48 h post-transfection, separated on 12% SDS-PAGE, and the resolved proteins were transferred to PVDF membrane. The fusion proteins were detected by using anti-GFP monoclonal antibody
As shown in Fig. 2a, relative to p2-1-GFP∆ transfected cells, robust GFP fluorescence was observed in p2-1-GFP and wild-type GFP transfected cells. We also observed the expression of GFP in p2-1-GFP∆ from a downstream AUG codon by leaky scanning. To further confirm the expression of GFP from a downstream start codon, AUG codon usage of the sgRNA 2-1 and the existence of fusion protein in transfected cells, we performed western blot to detect the fusion protein using an anti-GFP antibody. As shown in Fig. 2b, a 32 kDa fusion protein and a relatively less intense 27 kDa band of wild-type GFP were detected in cells transfected with p2-1-GFP, whereas only the 27 kDa band was detected in the cells transfected with p2-1-GFP∆. These data suggest that the authentic AUG codon of ORF 2b in sgRNA2-1 was used for translation, leading to expression of fusion protein, while leaky expression from the AUG of GFP gene also took place.
Scanning ribosome may initiate translation from the weak AUG in sgRNAs at a low frequency or bypass it in favor of the stronger downstream AUG codon of GFP, which is located at only 144 nt downstream from the initiator AUG of ORF2b. Thus, leaky scanning could probably lead to the expression of wild-type GFP from both in-frame and out-of-frame fusion constructs (Fig. 2).
Taken together, we have shown that the sgRNA 2-1 could be a functional mRNA in SARS-CoV-infected cells although it was of low-abundance in the host cells. According to the prediction from the sgRNA 2-1 sequence, expression of ORF 2b in the sgRNA may result in production of a truncated S protein, which is predicted to lack the N-terminal 143 amino acids of the spike protein. Therefore, further studies are required to characterize the biological functions of this novel sgRNA and the encoded proteins in the viral life cycle and pathogenesis.
Varied translational levels of SARS-CoV subgenomic RNAs in translation reporter system
Ten sgRNAs have been identified [5] and the SARS-CoV accessory proteins 3a, 3b, 6,7a, and 7b can be detected in infected cells or SARS patients besides structural proteins [7, 8], while the expression of 8a and 8b is controversial [9–13]. Elucidation of the regulatory mechanism in the translation is important for understanding the pathogenesis of SARS-CoV; however, it is hard to compare the differential translation of sgRNAs because the steady-state level of viral proteins in infected cells reflects the sum of transcription, translation, and the relative stabilities of these transcriptional and translational products. In this study, we adopted the reporter gene system by fusing with partial sgRNA ORF of similar size under the control of the same promoter. This system was supposed to specifically address and compare the translation efficiency of individual sgRNAs by circumventing the problem resulted from different transcription efficiency and protein stability.
We cloned the 5′-ends containing a full leader sequence and the 5′-200–400 nt of the body sequence of all 10 sgRNAs into the pEGFP-N1 vector (Fig. 1). The predicted start codon AUG of each ORF was cloned in-frame with the GFP gene and the start codon of GFP was replaced with GUG. Strong fluorescence was observed in cells transfected with fusion constructs p2/S, p2-1, p3/3a, p5/M, p6, p4/E, and p9/N, whereas relatively weak fluorescence was observed in cells transfected with fusion constructs p3-1, p7/7a, and p8 (Fig. 3a). Expressions of GFP-fusion proteins of expected sizes were detected in cells transfected with plasmids p2-1, p3/3a, p3-1, p4/E, p5/M, p6, p7/7a, and p9/N (Fig. 3b). The major protein band of sgRNA 2-GFP fusion construct (Fig. 3b) was larger than theoretically calculated size (Table 2). This discrepancy could be due to the post-translational modification of protein or not fully denatured protein complex. One minor band below the major band may represent the correct fusion translation product (Fig. 3b). On the other hand, protein bands with smaller sizes were detected in cells transfected with constructs of p3/3a, p4/E, and p5/M, which might result from leaky expression from downstream AUG codons, premature termination or degradation product by cellular proteinases (Fig. 3b).
Fig. 3.
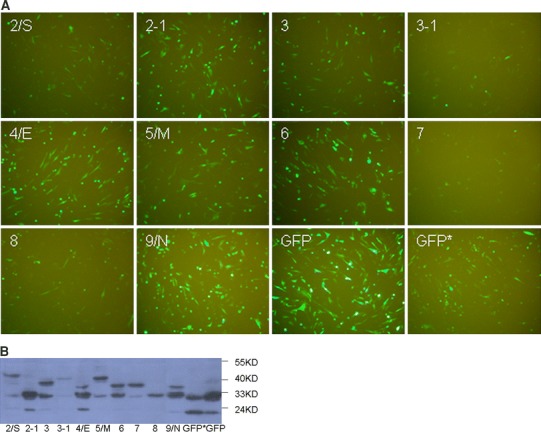
Expression of SARS-CoV sgRNAs in GFP-fusion in BHK cells. a Fluorescence analysis of the expression of 10 SARS-CoV sgRNAs. Florescence micrographs of individual fusion constructs are marked according to the name of subgenomic RNA. Modified GFP with mutated initiator codon (AUG to GUG) is named as GFP*, which indicated that GUG or closely located downstream AUG could be used as translation start codon. b Western blot analysis of GFP-fusion protein. Names of the individual sgRNA are marked at bottom and molecular weight (kDa) is marked on the right side of image. Modified GFP with mutated initiator codon (AUG to GUG) is indicated by an asterisk (*)
Table 2.
Kozak context, length, G + C% and ΔG of sgRNA 5′-UTR and expected size of fusion protein in the reporter assays
| sgRNA | Kozak contexta | ORF Lengthb (nt) | Fusion proteinc (kDa) | Length of 5′ UTR (nt) | % G + C of 5′-UTR | ΔGd (kcal/mol) |
|---|---|---|---|---|---|---|
| 2 | gaaCgAaCAUGuuu | 279 | 38.20 | 72 | 42 | −14.6 |
| 2-1 | CuaaacCCAUGGgu | 147 | 33.10 | 121 | 40 | −15.8 |
| 3 | aCGaacuuAUGGau | 309 | 39.30 | 74 | 39 | −15.8 |
| 3-1 | auuaCuuuAUGGug | 378 | 42.00 | 86 | 38 | −15.8 |
| 4 | aCGaacuuAUGuac | 267 | 37.70 | 74 | 39 | −15.8 |
| 5 | ugcuuAuCAUGGca | 405 | 43.00 | 116 | 34 | −19.3 |
| 6 | gacaacagAUGuuu | 219 | 35.90 | 227 | 42 | −39.8 |
| 7 | aAaCgAaCAUGaaa | 285 | 38.40 | 72 | 42 | −14.6 |
| 8 | uaaaCcuCAUGugc | 273 | 38.00 | 155 | 39 | −30.9 |
| 9 | aaauuAaaAUGucu | 237 | 36.60 | 80 | 36 | −17.1 |
| 8/5 | ugcuuAuCAUGugc | 273 | 38.00 | 116 | 34 | – |
| 5/8 | uaaaCcuCAUGGca | 360 | 41.0 | 155 | 39 | – |
| 8a | aaaCgAaCAUGaaa | 84 | 30.70 | 72 | 42 | – |
aThe AUG codons are represented in bold characters and the bases in uppercase letters indicate the nucleotides that match with consensus Kozak sequence
bLength of sgRNAs ORF fused with GFP sequence
cTheoretical molecular weight of GFP-fusion proteins
dFree energy was calculated for the major loops in the predicted secondary structure of the 5′-UTR
Although the initiator AUG of GFP was replaced with GUG in the fusion constructs, strong fluorescence was still observed with pGFP*, indicating that GUG may serve as a non-canonical translation start codon (Fig. 3a). This result was further confirmed by western blot analysis in cells transfected with pEGFP-N1 and pGFP* (Fig. 3b). It may be due to the flanking primary sequence that closely matches to the consensus motif GCCACCAUGG, which is the optimal context for initiation of eukaryotic mRNAs translation [14, 15]. It is known that GUG can function as an efficient start codon in mammalian cells [16].
When sgRNAs expression levels shown in Fig. 3 are compared, fluorescence intensities represent total expression of GFP in transfected cells, including GFP-fusion protein and GFP expression by leaky scanning from downstream start codon. For example cells transfected with p6 showed stronger fluorescence signal than p7/7a (Fig. 3a), however, western blot result indicated comparable level of fusion protein in p7/7a and p6 transfected cells (Fig. 3b). These results suggest that more GFP was translated from downstream start codon in p6 transfected cells as compared to p7/7a transfected cells. Therefore, the western blot analysis provided more specific information on translation initiation efficiency from either the first AUG codon in sgRNAs or downstream AUGs by leaky scanning.
In order to confirm the above results, we cloned the 5′-ends of all 10 sgRNAs into the pGL3.0 vector to fuse in-frame with luciferase gene for sensitive and quantitative measurement of the varied sgRNAs translation. The luciferase activity expressed from sgRNA 2-1 (sg2-1), sg3, sg5/M, sg6, sg7/7a to sg9/N was 24–491 fold higher than that from sg8 at 18, 24, and 36 h post-transfections, respectively (Fig. 4). These results are consistent with the observations in the GFP-fusion assay system.
Fig. 4.
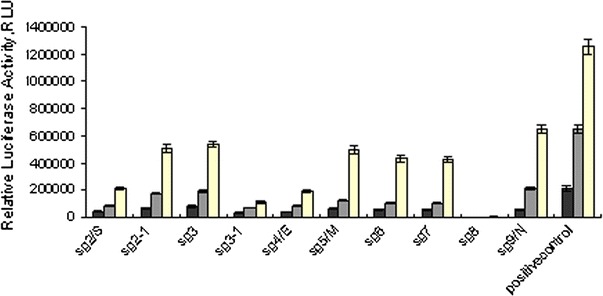
Expression of SARS-CoV sgRNAs in luciferase-fusion in BHK cells. The cells were harvested 18, 24, and 36 h after transfection and luciferase activities were measured. The pGL3.0 was used as a positive control
RNA viruses employ various mechanisms to regulate their gene expression at the translational level. Leaky scanning allows the translation of multiple ORFs from a common mRNA substrate, and such leaky scanning has already been reported for viral RNA translation [17, 18]. For coronaviruses, it has been reported that the SARS-CoV ORF7b and the infectious bronchitis virus (IBV) ORF3b are translated by leaky ribosomal scanning [19, 20]. Our data showed that leaky scanning, which leads to translation from downstream AUG codon, may be common for coronavirus RNAs. Messenger RNAs in which the first AUG codon lacks the preferred nucleotide at both of the key positions (−3, +4) in the Kozak context have the special property of initiating translation at the first and downstream AUG codons, thereby producing two or more proteins from one mRNA. Further studies are needed to investigate the translation of downstream ORFs as well as the role of truncated proteins (if any such protein exists in SARS-CoV infected cells) expressed from downstream AUG codons by leaky scanning.
The 5′-UTR of sgRNA8 could be a cis-acting suppressor element
In most human isolates of SARS-CoV, the sgRNA 8 contains two ORFs, ORF8a and ORF8b. The SARS-CoV WHU strain has a deletion of two nucleotides corresponding to the nucleotides 27,808 and 27,809 in ORF 8a of SARS-CoV Tor2 and Urbani [4, 21]. This 2-nt deletion leads to a shifted ORF 8a of only 24 amino acids instead of 39 amino acids.
Although SARS-CoV 8b gene product could be expressed in vivo when cloned directly behind a promoter [11–13]; the expression of 8a and 8b in SARS-CoV-infected cells is still controversial [9, 10, 13]. As shown above, we were unable to detect the protein expression of sgRNA 8 with the 5′ viral leader sequence, which corroborated with a recent report on ORF8 expression [13]. Cells transfected with p8 displayed significant fluorescence (Figs. 3a, 5a), but the expression of fusion protein could not be detected in western blot (Fig. 5b). To investigate a possible role of the sgRNA8 5′-UTR in translation, the 5′-UTR of sgRNA 8 was replaced by the 5′-UTR of sgRNA 5 to create the plasmid p8/5 because the initiator AUG codon of sgRNA 5 was shown to be functional (Fig. 3b), and the length and the secondary structure of both 5′-UTRs were predicted to be similar to sgRNA 8 5′-UTR. Interestingly, the replacement of the 5′-UTR resulted in the translation of fusion protein from initiator AUG codon of ORF 8b. As expected, when the 5′-UTR of sgRNA 5 was replaced by the 5′-UTR of sgRNA 8 to create the plasmid p5/8, the expression of p5/8 could not be detected (Fig. 5b). We speculated that a small ORF (8a), which is present in the 5′-UTR of sgRNA 8, might play a role in translational suppression from downstream initiator AUG codon of ORF 8b. To study any possible role of upstream ORF in translation suppression, the GFP was fused with ORF 8a to create the plasmid p8a but no fusion protein was detected in cells transfected with this recombinant plasmid (Fig. 5b). This shows that translational suppression from the initiator AUG codon of ORF 8b was not the result of the expression of 8a but could be due to other cis-acting elements present in the 5′-UTR region. Taken together, these data indicate that the 5′-UTR may act as a suppression regulatory element that led to the inhibition of expression of both ORF 8a and 8b in sgRNA 8.
Fig. 5.
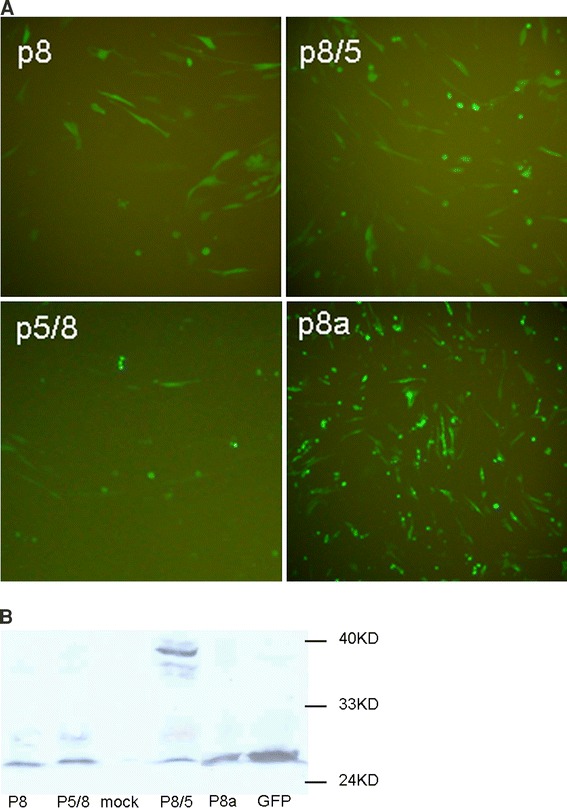
Expression of sgRNA 8 in BHK cells. a Translatability of sgRNA 8 by fluorescence analysis. b Western blot analysis of fusion proteins in cells transfected with different fusion constructs of sgRNA 8. The ORF 8a and 8b were fused in-frame with GFP open reading frame in pEGFP-N1 vector. p8: sgRNA8 ORF8b fused with GFP; p8/5: the 5′-UTR of sgRNA 8 was replaced with that of sgRNA 5; p5/8: the 5′-UTR of sgRNA 5 with that of sgRNA 8; p8a: ORF 8a fused with GFP; GFP: pEGFP-N1 as control; mock: non-transfected cells
The conservation of Kozak context alone has no correlation with the translation efficiency of SARS-CoV sgRNAs
As the expression levels of SARS-CoV sgRNAs were significantly different, we determined whether the sequence context around the start codon AUG (Kozak sequence) plays an important role in the translation of sgRNAs. The optimal context for initiation of translation in vertebrate mRNAs is ACCAUGG [14, 15]. In this consensus motif, two nucleotides at the highly conserved positions (a G residue following the AUG codon (position +4) and a purine, preferably A, three nucleotides upstream AUG condon (position −3)) exert the strongest effect. Sequence analysis revealed that AUG codons of sgRNAs 2-1, 5/M and 8 are in better Kozak context (Table 2). They have only one nucleotide mismatch with the consensus sequence motif ACCAUGG. A pyrimidine (C) is present at position −3 in sgRNA 2-1, whereas a mismatch at position −2 and −1 is present in sgRNA 5 and 8 respectively (Table 2). However, sgRNA 8 has a low-translation efficiency as shown above. The sequence surrounding the AUG initiator codon of sgRNAs 2 and 7 has two nucleotides mismatch with consensus Kozak sequence, and notably they are lacking a guanine (G) at position +4. The sgRNAs 3, 3-1, 4, 6 and 9 possess poor Kozak sequence context for translation initiation (Table 2) but most could be translated efficiently (Fig. 3b). Taken together, no significant correlation was found between the Kozak context around AUG initiator codon of sgRNAs and the translational level of the fusion proteins.
The length of 5′-UTR, the G + C content, and the secondary structure near the 5′-end of an mRNA can drastically affect the translational level of mRNAs [22–25]. We next analyzed whether the properties of the 5′-UTR could influence the translation efficiency of sgRNAs. All SARS-CoV sgRNAs contain the same leader sequence but the 5′-UTR lengths are variable, ranging from 72 nt to 265 nt (Table 2). We calculated the G + C contents of different sgRNA 5′-UTRs and analysed the secondary structures and the free energy (ΔG) of the major loops of sgRNA 5′-UTRs (Table 2). Surprisingly, no significant correlation was found between the length, the G + C content, the secondary structure of 5′-UTR, and the translational level of reporter gene (Table 2).
In summary, the current work addressed the difference of SARS-CoV sgRNA translation efficiency, but it would not correlate with the actual steady-state levels of SARS-CoV proteins in infected cells because the latter is also influenced by the abundance of sgRNA resulted from different transcription levels and regulation as well as the different stability of individual viral proteins. Therefore, further studies are required to determine the relationship between the level of transcription, translation, and relative abundance of protein in cells which were infected by SARS-CoV. At the translational step, our data showed that translation from the downstream initiator codon by leaky scanning was common to SARS-CoV sgRNAs and this could lead to synthesis of truncated viral protein products (if the downstream AUG is in the same reading frame) or altered proteins, which may act as decoys to fool immune system and favor viral replication.
Acknowledgments
This study was supported by China Natural Science Foundation (grant #30670450), the Ph.D. Programs Foundation of Ministry of Education of China (grant No. 20060486023) and China “973” basic research program (grant #2006CB504300).
Footnotes
Yaling Yang and Snawar Hussain contributed equally to this work.
References
- 1.L. Enjuanes, W. Spaan, E. Snijder, D. Cavanagh, in Nidovirales Virus taxonomy: Classification And Nomenclature of Viruses, ed. by C.M.F.M.H.V. van Regenmortel, D.H.L. Bishop, E.B. Carsten, M.K. Estes, S.M. Lemon, D.J. McGeoch, J. Maniloff, M.A. Mayo, C.R. Pringle, R.B. Wickner (Academic Press, San Diego, 2000), pp. 827–834
- 2.Thiel V, Ivanov KA, Putics A, Hertzig T, Schelle B, Bayer S, Weissbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 3.Sawicki SG, Sawicki DL. Adv. Exp. Med. Biol. 1998;440:215–219. doi: 10.1007/978-1-4615-5331-1_26. [DOI] [PubMed] [Google Scholar]
- 4.Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 5.Hussain S, Pan J, Chen Y, Yang Y, Xu J, Peng Y, Wu Y, Li Z, Zhu Y, Tien P, Guo D. J. Virol. 2005;79:5288–5295. doi: 10.1128/JVI.79.9.5288-5295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu Y, Liu M, Zhao W, Zhang J, Zhang X, Wang K, Gu C, Wu K, Li Y, Zheng C, Xiao G, Yan H, Guo D, Tien P, Wu J. Virus Genes. 2005;30:93–102. doi: 10.1007/s11262-004-4586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A. von Brunn, C. Teepe, J.C. Simpson, R. Pepperkok, C.C. Friedel, R. Zimmer, R. Roberts, R. Baric, J. Haas, Plos One 23, 2(5) e459 (2007) [DOI] [PMC free article] [PubMed]
- 8.Narayanan K, Huang C, Makino S. Virus Res. 2008;133:113–121. doi: 10.1016/j.virusres.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keng CT, Choi YW, Welkers MR, Chan DZ, Shen S, Gee LS, Hong W, Tan Y. J. Virol. 2006;354:132–142. doi: 10.1016/j.virol.2006.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CY, Ping YH, Lee HC, Chen KH, Lee YM, Chan YJ, Lien TC, Jap TS, Lin CH, Kao LS, Chen YA. J. Infect. Dis. 2007;196:405–415. doi: 10.1086/519166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee TM, Wong HH, Tay FPL, Fang S, Keng C-T, Tan YJ, Liu DX. FEBS J. 2007;274:4211–4222. doi: 10.1111/j.1742-4658.2007.05947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law PY, Liu YM, Geng H, Kwan KH, Waye MM, Ho YY. FEBS Lett. 2006;580:3643–3648. doi: 10.1016/j.febslet.2006.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oostra M, de Haan CAM, Rottier PJM. J. Virol. 2007;81:13876–13888. doi: 10.1128/JVI.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozak M. J. Mol. Biol. 1987;196:947–950. doi: 10.1016/0022-2836(87)90418-9. [DOI] [PubMed] [Google Scholar]
- 15.Kozak M. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 16.Wegrzyn JL, Drudge TM, Valafar F, Hook V. BMC Bioinformatics. 2008;9:232. doi: 10.1186/1471-2105-9-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pavlakis GN, Felber BK. New Biol. 1990;2:20–31. [PubMed] [Google Scholar]
- 18.Schwartz S, Felber BK, Fenyo EM, Pavlakis GN. J. Virol. 1990;64:5448–5456. doi: 10.1128/jvi.64.11.5448-5456.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaecher SR, Mackenzie JM, Pekosz A. J. Virol. 2007;81:718–731. doi: 10.1128/JVI.01691-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu D.X., Inglis S.C. J. Virol. 1992;66:6143–6154. doi: 10.1128/jvi.66.10.6143-6154.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 22.Fraser SD, Wilkes-Johnston J, Browder LW. Oncogene. 1996;12:1223–1230. [PubMed] [Google Scholar]
- 23.Kozak M. Mol. Cell. Biol. 1989;9:5134–5142. doi: 10.1128/MCB.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozak M. Proc. Natl Acad. Sci. USA. 1986;83:2850–2854. doi: 10.1073/pnas.83.9.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viglianti GA, Rubinstein EP, Graves KL. J. Virol. 1992;66:4824–4833. doi: 10.1128/jvi.66.8.4824-4833.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]