

Abstract
Aminopeptidase N (APN) inhibitors have been reported to be effective in treating of life threatening diseases including cancer. Validated ligand- and structure-based pharmacophore mapping approaches were combined with Bayesian modeling and recursive partitioning to identify structural and physicochemical requirements for highly active APN inhibitors. Based on the assumption that ligand- and structure-based pharmacophore models are complementary, the efficacy of ‘multiple pharmacophore screening’ for filtering true positive virtual hits was investigated. These multiple pharmacophore screening methods were utilized to search novel virtual hits for APN inhibition. The number of hits was refined and reduced by recursive partitioning, drug-likeliness, pharmacokinetic property prediction, and comparative molecular-docking studies. Four compounds were proposed as the potential virtual hits for APN enzyme inhibition.
Electronic supplementary material
The online version of this article (doi:10.1007/s11030-013-9422-5) contains supplementary material, which is available to authorized users.
Keywords: Aminopeptidase N, Pharmacophore mapping, Bayesian modeling, Recursive partitioning, Molecular docking, Virtual hits
Introduction
Aminopeptidase N (APN, EC 3.4.11.2) is a zinc-dependent membrane type II endopeptidase. It belongs to the M1 family of MA clan (metallo-peptidase superfamily) of peptidases. It contains 967 amino acids and has a relative molecular mass (M of
of  99,000 [1]. APN is mainly involved in ATP-dependent downstream processing during cytosolic protein degradation. It enables the utilization of amino acids as nutrients [2]. This enzyme is found in a variety of species, such as mammals, insects, plants, and bacteria. Human APN is identical to the human lymphocyte surface cluster differentiation antigen CD13. It is also known as APN/CD13, which is widely distributed in mammals. It is expressed on the surface of different cells, such as epithelial cells of the intestine and kidney, synaptic membranes in the central nervous system, fibroblasts, endothelial cells, epithelial cells, sebocytes, and keratinocytes [1]. Deregulation of APN/CD13 expression is observed in many diseases, such as cancer, chronic pain [3], and various forms of joint effusions, rheumatoid arthritis [4], multiple sclerosis [5], systemic sclerosis, and systemic lupus erythematosus [6]. APN plays a crucial role in tumor invasion, metastasis, and angiogenesis [7] in cancer. The over-expression of APN was reported in breast, colon, gastric, ovarian, pancreatic, renal, and thyroid cancer [7]. Therefore, it is used as an important clinical marker in cancer [1]. APN also functions as a receptor for corona virus like gastroenteritis virus and human corona virus 229E [2]. Inhibition of APN/CD13 may be useful for treatment of such diseases [1]. Bestatin was first identified as APN inhibitor, isolated from Streptomyces olivoreticuli [8], and reported to improve immunological functions by enhancing the chemotaxis of T lymphocytes. Although several small-molecule APN/CD13 inhibitors have been synthesized and evaluated biologically so far, it is still challenging to develop potent inhibitor with higher binding potential and efficacy. Recently, the X-ray crystal structure of human APN (hAPN) complex with bestatin [9] was reported, and it provided the opportunity to understand the mechanism of action of different APN inhibitors by structure-based analysis. In this study, an attempt was made to highlight the structural and physicochemical requirements of APN inhibitors. We also tried to find novel scaffolds as virtual hits to design potent drug-like APN inhibitors. Both ligand- and structure-based pharmacophore mapping studies were performed to identify the 3D structural requirements for potent APN inhibitory activity. Two different QSAR approaches, i.e., Bayesian modeling and recursive partitioning (RP) techniques, were adopted to understand the physicochemical and 2D structural requirements for better APN inhibitory activity. Simultaneously, ‘multiple pharmacophore-based virtual screening’ was employed for the selection of virtual hits from the diverse chemical databases. The best RP model was utilized to select virtual hits from the ligands obtained from ‘multiple pharmacophore-based virtual screening’. Comparative molecular docking studies with three different docking tools were performed to search the most potential drug-like virtual hits as well as to predict possible interactions between proposed virtual hits and the catalytic residues at the binding site of APN enzyme.
99,000 [1]. APN is mainly involved in ATP-dependent downstream processing during cytosolic protein degradation. It enables the utilization of amino acids as nutrients [2]. This enzyme is found in a variety of species, such as mammals, insects, plants, and bacteria. Human APN is identical to the human lymphocyte surface cluster differentiation antigen CD13. It is also known as APN/CD13, which is widely distributed in mammals. It is expressed on the surface of different cells, such as epithelial cells of the intestine and kidney, synaptic membranes in the central nervous system, fibroblasts, endothelial cells, epithelial cells, sebocytes, and keratinocytes [1]. Deregulation of APN/CD13 expression is observed in many diseases, such as cancer, chronic pain [3], and various forms of joint effusions, rheumatoid arthritis [4], multiple sclerosis [5], systemic sclerosis, and systemic lupus erythematosus [6]. APN plays a crucial role in tumor invasion, metastasis, and angiogenesis [7] in cancer. The over-expression of APN was reported in breast, colon, gastric, ovarian, pancreatic, renal, and thyroid cancer [7]. Therefore, it is used as an important clinical marker in cancer [1]. APN also functions as a receptor for corona virus like gastroenteritis virus and human corona virus 229E [2]. Inhibition of APN/CD13 may be useful for treatment of such diseases [1]. Bestatin was first identified as APN inhibitor, isolated from Streptomyces olivoreticuli [8], and reported to improve immunological functions by enhancing the chemotaxis of T lymphocytes. Although several small-molecule APN/CD13 inhibitors have been synthesized and evaluated biologically so far, it is still challenging to develop potent inhibitor with higher binding potential and efficacy. Recently, the X-ray crystal structure of human APN (hAPN) complex with bestatin [9] was reported, and it provided the opportunity to understand the mechanism of action of different APN inhibitors by structure-based analysis. In this study, an attempt was made to highlight the structural and physicochemical requirements of APN inhibitors. We also tried to find novel scaffolds as virtual hits to design potent drug-like APN inhibitors. Both ligand- and structure-based pharmacophore mapping studies were performed to identify the 3D structural requirements for potent APN inhibitory activity. Two different QSAR approaches, i.e., Bayesian modeling and recursive partitioning (RP) techniques, were adopted to understand the physicochemical and 2D structural requirements for better APN inhibitory activity. Simultaneously, ‘multiple pharmacophore-based virtual screening’ was employed for the selection of virtual hits from the diverse chemical databases. The best RP model was utilized to select virtual hits from the ligands obtained from ‘multiple pharmacophore-based virtual screening’. Comparative molecular docking studies with three different docking tools were performed to search the most potential drug-like virtual hits as well as to predict possible interactions between proposed virtual hits and the catalytic residues at the binding site of APN enzyme.
Materials and methods
Data set
175 APN inhibitors were collected from the literature [10–19]. 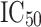 values of all these compounds were determined by the same group of authors using l-leucine-
values of all these compounds were determined by the same group of authors using l-leucine- -nitroanilide as the substrate and microsomal aminopeptidase N from porcine kidney microsomes as the enzyme. This data set (internal data set) was used for the development as well as preliminary validation of pharmacophore models. The molecules of the data set have wide range of inhibitory activity (
-nitroanilide as the substrate and microsomal aminopeptidase N from porcine kidney microsomes as the enzyme. This data set (internal data set) was used for the development as well as preliminary validation of pharmacophore models. The molecules of the data set have wide range of inhibitory activity (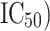 , from 1.8 to 9108.6
, from 1.8 to 9108.6 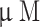 . Moreover, a second data set (external data set) containing 175 compounds was collected from the binding database (BD) [20]. Unlike internal dataset, the biological activities of these compounds were determined by various assay methods by different group of authors, and APN inhibitory activity of these molecules was expressed either in
. Moreover, a second data set (external data set) containing 175 compounds was collected from the binding database (BD) [20]. Unlike internal dataset, the biological activities of these compounds were determined by various assay methods by different group of authors, and APN inhibitory activity of these molecules was expressed either in 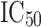 or in
or in 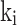 (the dissociation constant for inhibitor binding). From the BD compounds, 101 compounds were arbitrarily chosen as ‘active’ APN inhibitors (reported
(the dissociation constant for inhibitor binding). From the BD compounds, 101 compounds were arbitrarily chosen as ‘active’ APN inhibitors (reported 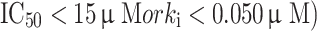 and 74 compounds were arbitrarily selected as ‘inactive’ APN inhibitors (reported
and 74 compounds were arbitrarily selected as ‘inactive’ APN inhibitors (reported 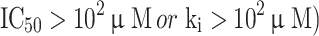 . Comparing with the internal data set, the external compounds contain more structural and biological activity variations. The purpose of using an external data set was to assess the overall performance of pharmacophore models and to develop Bayesian and RP models. The detailed information of internal and external data set compounds is provided in the supporting information (Tables S1 and S2, respectively).
. Comparing with the internal data set, the external compounds contain more structural and biological activity variations. The purpose of using an external data set was to assess the overall performance of pharmacophore models and to develop Bayesian and RP models. The detailed information of internal and external data set compounds is provided in the supporting information (Tables S1 and S2, respectively).
Pharmacophore model development
One ‘ligand-based pharmacophore’ and two ‘structure-based pharmacophore’ models were developed in this study. The first ‘structure-based pharmacophore’ was generated from ligand-receptor interaction, where both receptor and co-crystallized ligand were utilized for the model generation. The second ‘structure-based pharmacophore’ was built only from the receptor structure using the information on the binding site interaction. Each pharmacophore model was independently generated and validated using different protocols in Accelrys Discovery Studio 3.0 (DS) [21].
Ligand-based 3D-QSAR pharmacophore modeling (model 1)
3D QSAR pharmacophore model was developed with the Hyporefine module in DS [21]. In this method, 25 structurally diverse compounds with a logarithmic biological activity, range of approximately 3.7, were selected from internal dataset with the help of Find diverse molecules protocol in DS. Fingerprint feature FCFP_4 (fingerprints of maximum diameter 4) and the biological activity value (IC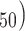 were used as the ‘property’ for the selection of these diverse molecules. These 25 compounds were subsequently used as the training set for the generation of ligand-based pharmacophore hypotheses.
were used as the ‘property’ for the selection of these diverse molecules. These 25 compounds were subsequently used as the training set for the generation of ligand-based pharmacophore hypotheses.
Pharmacophore model generation
All compounds were initially minimized with the Smart minimizer protocol of DS which applies steepest descent algorithm, followed by conjugate gradient algorithm. Prior to pharmacophore generation, the Feature mapping tool was used to predict the favorable features for highly active compounds. It was also found that the addition of ‘exclusion volumes (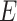 )’ improves the overall statistical quality of the model. Moreover, uncertainty value was fixed to 1.5. A diverse set of conformations of these compounds was generated using the FAST conformer generation method, where maximum number of conformers and the energy range were chosen as 255 and 20 kcal/mol, respectively.
)’ improves the overall statistical quality of the model. Moreover, uncertainty value was fixed to 1.5. A diverse set of conformations of these compounds was generated using the FAST conformer generation method, where maximum number of conformers and the energy range were chosen as 255 and 20 kcal/mol, respectively.
Validation of model 1
The developed 3D pharmacophore hypotheses were validated by three different methods, (1) cost analyses, (2) Fischer randomization test and (3) test set prediction [22]. In the cost analysis method, three cost values—fixed, total, and null cost of the generated hypotheses were analyzed. The fixed or ideal cost is the cost of simple possible hypothesis that fits all the data in a perfect manner. The null cost or no correlation cost is the cost of the hypothesis without any feature. The value of the total cost of the generated hypothesis should lie between the fixed cost and the null cost. It should be close to that of the fixed cost value, but away from the null cost value. The correlation coefficient, root mean square deviation (RMSD) and configuration cost are the other parameters considered in the cost analysis. The configuration cost (determines the complexity of the hypothesis) should be less than 17 and the RMSD value (explains the quality of the correlation between the experimental and estimated values) should be less than 1.5 for a standard pharmacophore hypothesis [23].
Fischer randomization test was used to ensure whether a strong correlation exists between the chemical structures and the biological activities of the training set compounds. The test was performed at 95 % confidence level, where the biological activities of the training set data were scrambled using the same features and parameters used in development of original hypothesis. If any randomization run produces hypothesis with better correlation coefficient and/or less total cost, the original hypothesis may be considered to be developed by chance. The third validation method, i.e. the test set prediction was used to ensure that the pharmacophore hypotheses were well predictive on a set of diverse compounds other than the training set compounds. The test set containing 150 compounds was subjected to Ligand pharmacophore mapping protocol in DS [21] to map these compounds in the generated hypothesis, and to obtain the estimated activity value for each compound. The higher correlation coefficient between the experimental and the estimated activity of the test set indicates better external predictability of the generated hypothesis.
Structure-based pharmacophore mapping I: ligand–receptor interaction (model 2)
In most of the cases, microsomal APN from porcine kidney is used to assay compounds against APN. Porcine APN has a high sequence identity with hAPN (80 % overall, 100 % at the active site, and 94 % in the peptide-binding region) [24]. In this study, the X-ray crystal structure of hAPN was used for all structure-based analyses (PDB ID: 4FYR) [9]. The Receptor–ligand pharmacophore generation protocol in DS allowed extracting information from a ligand–protein interaction to generate a number of hypotheses.
Protein structure preparation
The receptor structure was prepared by the Prepare protein method in DS. All water molecules, except some selected molecules that play important roles in ligand–receptor binding were removed. Subsequently, hydrogen atoms were added to the protein, and CHARMM force-field was assigned to the protein atoms. The binding site of cocrystallized bestatin was defined as the receptor active site, and receptor-bound bestatin was defined as the ligand for hypotheses generation.
Hypotheses generation
Default chemical features, such as hydrogen-bond acceptor (A), hydrogen bond donor (D), hydrophobic (Y), positive ionizable (P), negative ionizable (N), and ring aromatic (RA) were considered for hypotheses generation. The excluded volume (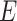 ) was added based on locations of atoms in the protein. During hypotheses generation, the following default parameters were chosen, minimum feature 3; maximum charge distance 8.0; maximum hydrogen-bond distance 4.0; maximum hydrophobic distance 5.5; maximum exclusion volume distance 5.0 and minimum interfeature distance 2.0.
) was added based on locations of atoms in the protein. During hypotheses generation, the following default parameters were chosen, minimum feature 3; maximum charge distance 8.0; maximum hydrogen-bond distance 4.0; maximum hydrophobic distance 5.5; maximum exclusion volume distance 5.0 and minimum interfeature distance 2.0.
Validation of pharmacophore hypotheses
Two validation procedures were implemented: (1) test set validation, and (2) selectivity score-based ranking approach. To assess the ability of the generated hypotheses to discriminate between active and inactive ligands, 25 most active compounds (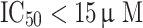 ) and 25 least active compounds (
) and 25 least active compounds (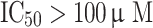 ) of the internal data set were selected to construct ‘active’ and ‘inactive’ sets. These were, subsequently, allowed to map the generated pharmacophore queries. The following parameters were chosen for screening—conformation generation: FAST, maximum conformations: 255, energy threshold: 20 kcal/mol and maximum omitted feature: 0.
) of the internal data set were selected to construct ‘active’ and ‘inactive’ sets. These were, subsequently, allowed to map the generated pharmacophore queries. The following parameters were chosen for screening—conformation generation: FAST, maximum conformations: 255, energy threshold: 20 kcal/mol and maximum omitted feature: 0.
Goodness of Hit score (GH score) was chosen as the critical criteria for the selection of best pharmacophore [25]. The GH score of 0.6–0.7 indicates a good pharmacophore model. Apart from this, sensitivity (Se), specificity (Sp), and accuracy (Acc) [26] were considered as other important parameters. The details of GH, Se, Sp, and Acc are provided in supporting information (Text S1).
In selectivity score determination, a genetic functional algorithm (GFA) model for the selectivity of a pharmacophore was built from a training set of 3285 pharmacophore models, using the total number of features in the model and the feature–feature distance bin values as descriptors. This set was used to find the CapsDiverse database in DS. The logarithmic values of virtual hits were used as the targets. Higher value of selectivity score is indicative of better model.
Structure-based pharmacophore mapping II: LUDI interaction (model 3)
Hypotheses generation
In this method, a LUDI [27] interaction map was created in the receptor (PDB ID: 4FYR) [9] active site sphere, comprising the catalytic residues within 9Å distance from the bound ligand (bestatin) using Interaction generation protocol in DS. This map consisted of all possible hydrogen-bond acceptor (A), hydrogen-bond donor (D), and hydrophobic (Y) features. Initially, the Edit and cluster pharmacophore tool in DS was utilized to edit redundant and pharmacophore features with no catalytic importance. From the generated pharmacophore query, some features were carefully selected on the basis of information reported on ligand–receptor interaction [9].
Validation of pharmacophore
The same ‘Active’ and ‘Inactive’ data sets (used in model 2) were used for the Model 3 validation. The Screen library protocol in DS was utilized for the selection of optimized pharmacophore query. During this analysis, one feature was fixed as ‘required feature’ that is required to be selected by all input test molecules during screening. The selected parameters for this screening method were—minimum and maximum features: 4 and 5, respectively, maximum subset of pharmacophore: 100, conformation search method: FAST and minimum interfeature distance: 2.0.
QSAR analysis
To understand the structural and physicochemical factors responsible for potent APN inhibitory activity, two different QSAR modeling techniques, namely Bayesian modeling [28] and recursive partitioning (RP) modeling [29], were performed. These methods rely only on the activity profile of these compounds (active or inactive) and not on the numerical experimental data. Therefore, compounds assayed by various methods, and biological activities expressed by different units could be used for QSAR modeling. The external dataset, containing 101 active and 74 inactive compounds, was used for these two studies.
Generation of the training and the test sets
The external data set was divided into the training and the test sets (80 and 20 % of the data set, respectively). The selections of the training and the test sets were based on fingerprints of maximum diameter 4 (FCFP_4). To ensure the applicability domain the test compounds [30], the whole data set was subjected to principal component analysis (PCA). In this process, the biological activity (IC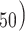 of these molecules along with some molecular properties (AlogP, MW, nHBD, nHBA, nRB, nR, nArR, nFr, and MPSA) were selected. The number of components and minimum variance explained were fixed at 3 and 0.75 respectively. In the PCA score plot, each test compound should remain close to at least one of the training set compounds for an unbiased selection of the training-test sets.
of these molecules along with some molecular properties (AlogP, MW, nHBD, nHBA, nRB, nR, nArR, nFr, and MPSA) were selected. The number of components and minimum variance explained were fixed at 3 and 0.75 respectively. In the PCA score plot, each test compound should remain close to at least one of the training set compounds for an unbiased selection of the training-test sets.
Recursive partitioning (RP) QSAR study
The create recursive partitioning model protocol in DS was used to build the classification model. RP tree consists of internal nodes and leafs where each internal node corresponds to a test on one descriptor, and each leaf is assigned a classification label. The model was built by minimizing the Gini index [31] to divide these compounds into branches. Detailed protocol of RP is provided in supporting information (Text S2).
Bayesian modeling
2D descriptors including function class fingerprints of maximum diameter 6 (FCFP_6), AlogP, MW, nRB, nR, nArR, nHBD, nHBA, and MFPSA were used for the Bayesian model generation using the Create Bayesian model protocol in DS. The model predictability was tested on the test set compounds.
Screening of database
The developed pharmacophore models were used as 3D query in database screening to find out novel and diverse virtual hits suitable for further development. Chemical databases, such as NCI (2,60,071 compounds), Specs (2,53,000 compounds), Maybridge (59,632), and Asinex (36,032 compounds) were screened in this study. The retrieved compounds with higher fit values were further filtered for drug likeliness. At first, these compounds were tested against the Lipinski rules [32] (Molecular weight 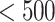 , hydrogen-bond donor
, hydrogen-bond donor 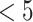 , hydrogen-bond acceptor
, hydrogen-bond acceptor 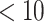 , log
, log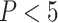 ) and Veber rules [33] (rotatable bond
) and Veber rules [33] (rotatable bond 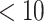 , polar surface area
, polar surface area 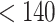 Å
Å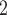 and sum of hydrogen-bond donor and acceptor
and sum of hydrogen-bond donor and acceptor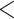 12) for drug-likeliness properties. The filtered drug-like compounds were tested for drug-like absorption, distribution, metabolism, and excretion (ADME) properties calculated by ADMET module in DS [21]. Compounds that successfully passed these filtering methods were treated as the test set compounds, and the activities were predicted through the best developed RP model to identify molecules that possess 2D structural and physicochemical features essential for APN inhibition.
12) for drug-likeliness properties. The filtered drug-like compounds were tested for drug-like absorption, distribution, metabolism, and excretion (ADME) properties calculated by ADMET module in DS [21]. Compounds that successfully passed these filtering methods were treated as the test set compounds, and the activities were predicted through the best developed RP model to identify molecules that possess 2D structural and physicochemical features essential for APN inhibition.
Molecular docking study
The screened drug-like virtual hits were analyzed thoroughly for ligand–receptor-binding affinity by three molecular docking tools: LibDock/DS [34], GOLD [35, 36], and GLIDE [37].
Protein selection
The X-ray structure of APN complexed with bestatin (PDB ID: 4FYR [9]) was used for docking studies. The binding site of the cocrystallized bestatin was defined as the receptor active site. In each docking method, self-docking was carried out initially. The docking trial was considered successful if self-docking could reproduce the cocrystallized conformation/pose of the ligand to a considerable extent. After getting satisfactory result from self-docking, other ligands were cross-docked similarly. Compounds showing consistent as well as comparable docking scores in three different tools (LibDock, GOLD, and GLIDE) were proposed as the final virtual hits.
LibDock/DS docking
In LibDock, the protein structure was prepared by removing bound water molecules, protonating the protein structure (protein dielectric constant 10, pH for protonation 7.4), and applying CHARMm force-field for minimization. LibDock/DS docks ligands into a binding site guided by polar and apolar interaction sites, which are known as hotspots. A binding site sphere radius of 9 Å surrounding the center of cocrystallized ligand (bestatin) was generated. The ligand conformations were generated using FAST conformation generation method that considered the maximum number of conformations to be 255, utilizing a 20 kcal/mol limit for conformational analysis. The other default parameters were kept unchanged during docking study.
GOLD docking
The water molecules in the protein were removed, and the hydrogen atoms were added using GOLD protein preparation. The binding site was defined with a 6 Å radius around the bound inhibitor. Docking calculations were performed using Goldscore fitness function. The ten top scoring conformations of each ligand were considered at the end of the process.
GLIDE docking
In GLIDE, bound water molecules in the protein were deleted, and formal charges of the ligand as well as protein atoms were adjusted. The restrained minimization of hydrogen of the protein was done using protein preparation wizard and OPLS2005 force field. Molecules to be docked were prepared using ligand preparation step. Conformations were generated for each input ligand after generating tautomers, removing defective structures and optimizing ligand geometries. Epik was used to generate all possible states of the ligand in a pH range of 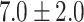 . Docking simulations were performed using GLIDE with OPLS-AA force field. A grid of 10Å
. Docking simulations were performed using GLIDE with OPLS-AA force field. A grid of 10Å  10Å
10Å  10 Å box centered on the centroid of the bound ligand was defined as the binding region. Ligand docking was performed utilizing the prepared protein and ligands as inputs. Extra precision (XP) docking was used for this purpose. Conjugate gradient method (100 steps) was used for energy minimization of poses, which had passed through the selection of initial poses scoring phase. Top ten poses were generated for each ligand.
10 Å box centered on the centroid of the bound ligand was defined as the binding region. Ligand docking was performed utilizing the prepared protein and ligands as inputs. Extra precision (XP) docking was used for this purpose. Conjugate gradient method (100 steps) was used for energy minimization of poses, which had passed through the selection of initial poses scoring phase. Top ten poses were generated for each ligand.
Results and discussion
Ligand-based 3D QSAR pharmacophore mapping
Twenty-five structurally diverse APN inhibitors (P1–P25), selected from the internal data set, were considered as the training set compounds for developing pharmacophore hypotheses by the Hyporefine method. The training set compounds are listed in Fig. 1.
Fig. 1.
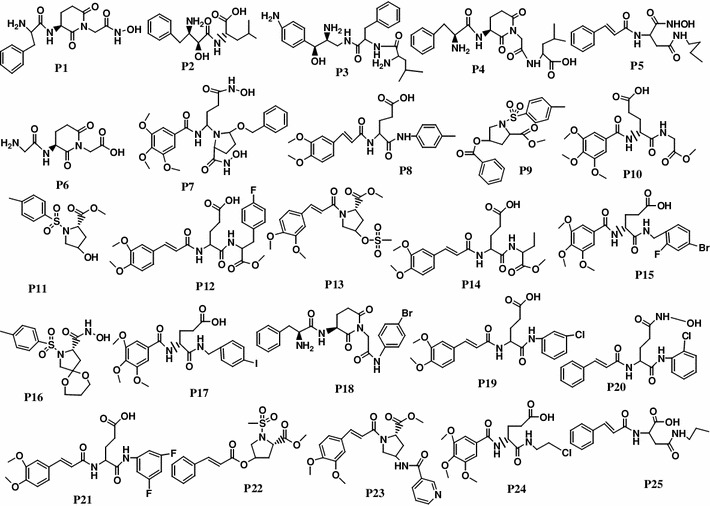
Structures of training set molecules (P1–P25) used for 3D QSAR pharmacophore generation
The standard APN inhibitor, bestatin (P2) was included as a member of the training set. Based on Feature mapping of DS, hydrogen-bond acceptor (A), hydrogen-bond donor (D), ring aromatic (RA), hydrophobic (Y), and positive ionizable (P) features were included in pharmacophore hypothesis generation. Hyporefine generated ten pharmacophore hypotheses with fixed and null cost values of 74.43 and 275.83, respectively. Among ten hypotheses, the first hypothesis (Model 1) was selected as the best pharmacophore on the basis of the cost values, correlation coefficient and RMSD value. Model 1 showed the lowest total cost (91.24) with a cost difference (null cost–total cost) of 184.58, indicating that the model and data are correlated by more than 90 %. The correlation value of this hypothesis was 0.961, which suggests the model is capable of predicting the activity of the training set compounds. In addition, lower RMSD value (1.15) of the hypothesis further supports the predictive ability of the model. The configuration cost of the model was found as 13.89, which was much lower than the cutoff value of 17. The Model 1 consisted of one each A, D, P, and RA feature along with five exclusion volumes ( ) (Fig. 2). The representative result of Model 1 is shown in supporting information (Table S3).
) (Fig. 2). The representative result of Model 1 is shown in supporting information (Table S3).
Fig. 2.
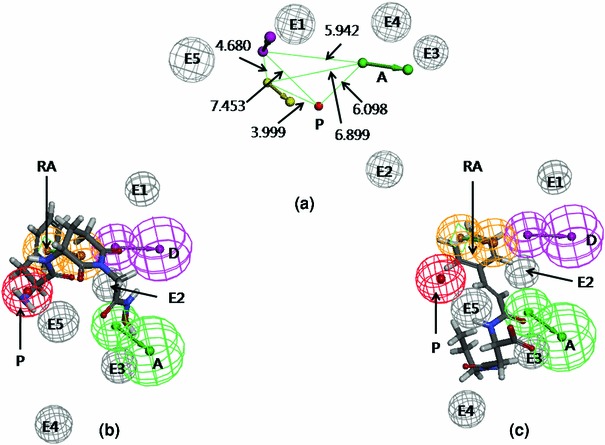
a 3D-spatial arrangement and inter-feature distance constraints between the chemical features (Model 1). b Mapping of the most active compound (P1) in the training set on Model 1. c Mapping of the least active compound (P25) in the training set on Model 1
Apart from the cost analyses, the merit of hypothesis was justified by its ability to predict the activity of individual compounds within the set. For this purpose, the training set compounds were approximately classified into three different categories: active ( and low active/inactive (
and low active/inactive ( ). All the compounds in the training set were accurately predicted with low error values between the experimental and estimated
). All the compounds in the training set were accurately predicted with low error values between the experimental and estimated 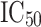 .
.
Test set prediction
The test set contained 150 compounds (supporting information, Table S1) with diverse structures and a wide range of biological activity. It was used to evaluate the best ligand-based hypothesis, i.e. Model 1. The relation between the experimental and the estimated activity values of the test set compounds showed a correlation coefficient (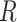 ) of 0.81 and the
) of 0.81 and the 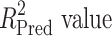 of 0.65. The correlation plots of both training and test set compounds are depicted in supporting information (Fig. S1). The observed and estimated activities of test set compounds are provided in supporting information (Table S4).
of 0.65. The correlation plots of both training and test set compounds are depicted in supporting information (Fig. S1). The observed and estimated activities of test set compounds are provided in supporting information (Table S4).
Fischer randomization test
The Fischer randomization test at 95 % confidence (supporting information, Table S5) showed that 4 out of 19 randomized runs could produce correlation (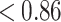 ) less than the original hypothesis. Moreover, the total costs of the randomized runs were much higher than the original hypothesis. It justified that the Model 1 was not generated by chance.
) less than the original hypothesis. Moreover, the total costs of the randomized runs were much higher than the original hypothesis. It justified that the Model 1 was not generated by chance.
Figure 2a represents the arrangement of the pharmacophoric features along with their inter-feature distance constraints. Figure 2b, c shows the mapping of the most (P1) and the least active (P25) compounds of the training set on Model 1, respectively. The hydroxyl group in the hydroxamic acid of P1 mapped upon the A feature, while the secondary amino (protonated) and the phenyl group of P1 mapped over P and RA features, respectively. The phenyl and carbonyl groups of the compound P25 mapped upon the RA and A features, respectively. The fit values of P1 and P25 were 5.61 and 1.95, respectively. It is observed that most of the highly active compounds in the training set contained positive ionizable primary amine group. Although some active (e.g. P5, of fit value 4.47) and moderately active (e.g., P8, fit value 4.29) compounds lacked this feature, showed considerable APN inhibitory activity. These compounds contained amide group that mapped over the donor feature. Therefore, it may be assumed that some polar groups, in addition to positively charged groups, also determine higher binding potential of these ligands.
In the X-ray crystal structure of hAPN complexed with bestatin [9], it was found that apart from catalytic amino acid residues, some bound water molecules (w1176, w1118, w1315, w1382, w1919, and w1445, the atom numbers were assigned from the PDB structure) play significant roles in the receptor binding by forming bridged hydrogen-bonding interactions (both intra and inter) with the ligand. The 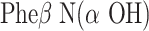 fragment of bestatin is pushed deeply into the S1 pocket of the enzyme, where amino group, carbonyl oxygen atom and
fragment of bestatin is pushed deeply into the S1 pocket of the enzyme, where amino group, carbonyl oxygen atom and  -hydroxyl groups formed water-mediated hydrogen bonds with hAPN. The schematic representation of ligand–receptor interactions [9] is shown in Fig. 3a. The mapped conformation of bestatin (P2) in Model 1 is shown in Fig. 3b. It was found that bestatin could map three out of four features in Model 1. The RA feature of phenyl ring could be compared to the
-hydroxyl groups formed water-mediated hydrogen bonds with hAPN. The schematic representation of ligand–receptor interactions [9] is shown in Fig. 3a. The mapped conformation of bestatin (P2) in Model 1 is shown in Fig. 3b. It was found that bestatin could map three out of four features in Model 1. The RA feature of phenyl ring could be compared to the  interaction with Phe472, whereas P feature of secondary amine (protonated) could be compared with interactions with water molecules, w1176, w1445, and w1382. The hydrogen-bond acceptor feature of the carboxylic group could be compared with interactions with Tyr477 and Glu389. However, other interactions with carbonyl groups and hydroxyl group could not be interpreted from the current model. Because of good predictive quality of Model 1, it could be inferred that mapped interactions are responsible for determination of the biological activity of the internal data set compounds.
interaction with Phe472, whereas P feature of secondary amine (protonated) could be compared with interactions with water molecules, w1176, w1445, and w1382. The hydrogen-bond acceptor feature of the carboxylic group could be compared with interactions with Tyr477 and Glu389. However, other interactions with carbonyl groups and hydroxyl group could not be interpreted from the current model. Because of good predictive quality of Model 1, it could be inferred that mapped interactions are responsible for determination of the biological activity of the internal data set compounds.
Fig. 3.
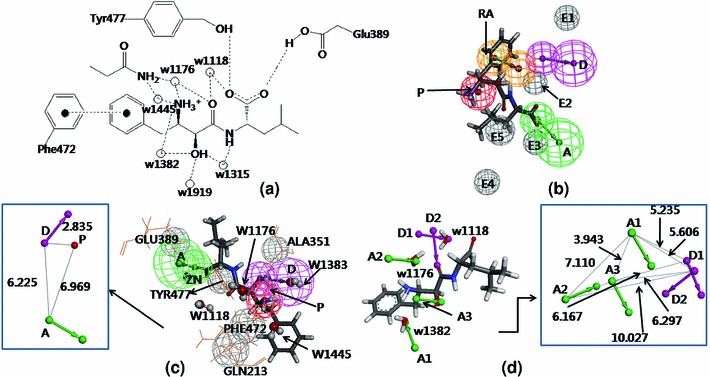
a Interaction of bestatin at the hAPN active site, hydrogen bonds and  interaction are shown in dotted line, water molecules are shown as spheres. b Mapping of bestatin (P2) on Model 1. c Mapping of bestatin on Model 2 (water molecules are shown in ball and stick form) and 3D-spatial arrangement and inter-feature distance constraints between the chemical features of Model 2 (in box). d Mapping of Model 3 with bestatin and catalytic water molecules (presented in stick form), 3D-spatial arrangement and inter-feature distance constraints between the chemical features of Model 3 (in box)
interaction are shown in dotted line, water molecules are shown as spheres. b Mapping of bestatin (P2) on Model 1. c Mapping of bestatin on Model 2 (water molecules are shown in ball and stick form) and 3D-spatial arrangement and inter-feature distance constraints between the chemical features of Model 2 (in box). d Mapping of Model 3 with bestatin and catalytic water molecules (presented in stick form), 3D-spatial arrangement and inter-feature distance constraints between the chemical features of Model 3 (in box)
Structure-based pharmacophore mapping I: ligand–receptor interaction
The receptor–ligand pharmacophore mapping generated hypotheses based on the receptor–ligand interaction with 50 internal data set compounds, comprising 25 actives and 25 inactives. The result is shown in Table 1.
Table 1.
Representative result of structure-based pharmacophore modeling
| Pharm | Features | Selectivity | Act | Inact | TP | TN | FP | FN | Sn | Sp | Acc | GH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RLPharm1 | DHHP | 1.1148 | 25 | 25 | 10 | 25 | 0 | 15 | 0.40 | 1.00 | 0.70 | 0.16 |
| RLPharm2 | AADH | 0.69736 | 25 | 25 | 16 | 3 | 22 | 9 | 0.64 | 0.12 | 0.38 | 0.41 |
| RLPharm3 | AADH | 0.69736 | 25 | 25 | 16 | 3 | 22 | 9 | 0.64 | 0.12 | 0.38 | 0.41 |
| RLPharm4 | AHHP | 0.47716 | 25 | 25 | 8 | 25 | 0 | 17 | 0.32 | 1.00 | 0.66 | 0.10 |
| RLPharm5 | AAHP | 0.47716 | 25 | 25 | 14 | 25 | 0 | 11 | 0.56 | 1.00 | 0.78 | 0.31 |
| RLPharm6 | AHHP | 0.47716 | 25 | 25 | 9 | 25 | 0 | 16 | 0.36 | 1.00 | 0.68 | 0.13 |
| RLPharm7 | DHH | 0.33951 | 25 | 25 | 11 | 4 | 21 | 14 | 0.44 | 0.16 | 0.68 | 0.19 |
| RLPharm8 | DHP | 0.33951 | 25 | 25 | 13 | 25 | 0 | 12 | 0.52 | 1.00 | 0.76 | 0.27 |
| RLPharm9 | ADP | 0.33951 | 25 | 25 | 23 | 25 | 0 | 2 | 0.92 | 1.00 | 0.96 | 0.85 |
| RLPharm10 | DHP | 0.33951 | 25 | 25 | 14 | 25 | 0 | 11 | 0.56 | 1.00 | 0.78 | 0.31 |
Selection and validation of hypotheses
It is evident from Table 1 that the RLPharm9 showed the most acceptable result in terms of Se, Sp, Acc, and GH score. The mapped conformation of bestatin in this pharmacophore (Model 2) is shown in Fig. 3c. The hypothesis contained two features (A and P), which are mapped over the functionalities as in Model 1. The third feature, D was mapped over the hydroxyl group that forms hydrogen-bond interaction with the water molecule w1382. Figure 3 justified that a strong relation exists between ligand- and structure-based pharmacophore modeling. However, both of these models could not explain the hydrogen-bond interaction of the carbonyl group. Figure 3c (box) represents the spatial arrangement of features in Model 2 along with their inter-feature distance constraints.
Structure-based pharmacophore mapping II: LUDI interaction
The Interaction generation protocol generated total 434 features (A, D, and H) at the active site (bestatin-binding site) of the enzyme (PDB ID: 4FYR [9]). These features were clustered and redundant interactions were removed. From the remaining features, some were selected on the basis of ligand–receptor interactions reported in the literature [9]. The final hypothesis (Model 3) comprising five features [three hydrogen-bond acceptors (A1–A3) and two hydrogen-bond donors (D1–D2)] were chosen on the basis of predictability on the internal test set compounds. Figure 3d shows the features mapped with the ligand and catalytic site of receptor molecules. A3 and D2 are directed towards hydroxyl and carbonyl groups of the ligand, respectively. The other features (A1, A2, and D1) correspond to the water molecules (w1382, w1176, and w1118) play important roles in ligand–receptor interaction. A2 was selected as ‘required feature’, as it showed the most consistent result for the test set validation. Noticeably, A2 is mapped over w1382 that interacts with the positive ionizable amino group (Fig. 3a), which was found to be the most important feature from SAR study, and it appeared in both Models 1 and 2.
Validation of model
The same internal test set was used as in Model 2. After screening, 25 compounds out of 50 were retrieved and 22 of these were found to be active. Based on this study, enrichment factors and GH value were calculated [details are provided in supporting information (Table S6)]. The GH value of 0.77 justifies that the developed LUDI-based pharmacophore (Model 3) was considerably selective for active compounds.
QSAR modeling
Generation of training and test sets
Generate training and test data protocol of DS created the training and the test sets with 144 and 31 compounds, respectively. The applicability domains of the sets were analyzed by PCA. In the PCA score plot (Fig. S2 of supporting information), it was found that each test compound remains comparatively close to at least one of the training compound.
Recursive partitioning (RP) QSAR modeling
The RP on 144 training set compounds generated three classification trees (Tree 1, 2, and 3). Tree 1 was selected as the best model (Model 4) on the basis of receiver operating characteristic (ROC) score (0.926) and cross-validated ROC curve (0.807). Moreover, when the model was validated with 31 test set compounds, the ROC score was 0.790. The details statistical analysis of Model 4 is provided in supporting information (Table S7). The schematic representation of the RP-tree 1 model is shown in Fig. 4.
Fig. 4.
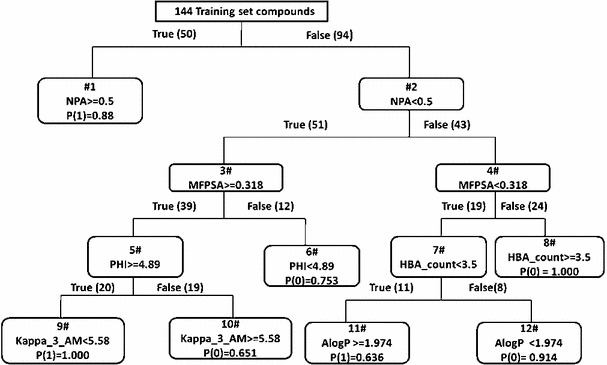
The schematic representation of the RP-tree 1 model
Figure 4 depicts the development of Model 4 with six selected descriptors along with their cut-off values that were used for developing and validating the model. It demonstrates that nPA, MFPSA, Kappa_3_AM, AlogP, PHI, and HBA_count played important roles in distinguishing actives from inactives. The functionality of these descriptors are listed in Table S8 (supporting information). In Fig. 4, the 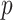 values are for winning class and reflect sample weight. 1 means active and 0 stands for inactive. In this model, the training set contains 87 actives and 57 inactive compounds. Each active sample is assigned a weight of 94 and each inactive sample is assigned a weight of 87, e.g. in node 1, 46 active and 4 inactive compounds are observed. Therefore,
values are for winning class and reflect sample weight. 1 means active and 0 stands for inactive. In this model, the training set contains 87 actives and 57 inactive compounds. Each active sample is assigned a weight of 94 and each inactive sample is assigned a weight of 87, e.g. in node 1, 46 active and 4 inactive compounds are observed. Therefore, 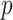 (1) can be calculated by the expression:
(1) can be calculated by the expression: 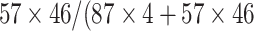 ).
).
In the RP model, NPA (total number of atoms in a molecule with a positive charge) was found to be the main classifier, which may be correlated with the fact that the positive ionizable features, like primary amines play important role in the ligand–receptor interaction. The physicochemical descriptor MFPSA is the ratio of the polar surface area to that the total surface area. It is evident from Fig. 3a that the catalytic site of APN contains amino acids having polar side chains (e.g., glutamic acid, tyrosine, etc.) as well as polar water molecules that participate in the receptor–ligand interactions. Although the model highlights the importance of ionizable groups, these are not the sole determinant for the higher activity of these molecules. Hydrogen-bond acceptor and donor groups also play vital roles in the ligand–receptor interactions. Therefore, the significance of MFPSA as a second major classifier of the model (for the compounds that lacked nPA) reconfirms that the polarity of the molecule has considerable importance for APN inhibition. The number of hydrogen-bond acceptor was also found to be important, and the significance of it was found in the pharmacophore mapping approach. In addition, Alog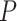 (logarithm of octanol–water partition coefficient determined by the Ghose and Crippen’s method [38]) was found to be important for APN inhibition of compounds that had lower cutoff values of both nPA and MFPSA. The function of hydrophobic descriptor may be easily understood as the binding site of hAPN comprises of hydrophobic pockets and hydrophobic interactions with residues like Phe472 and Ser469 were reported for bestatin. In addition, two topological kappa-shaped index descriptors, PHI and Kappa_3_AM were appeared in the model. The Kappa_3_AM is alpha-modified shape index of order three and PHI is the molecular flexibility index. These topological descriptors appeared in the model for compounds having higher values of MFPSA. Therefore, it may be interpreted that flexibility of the molecule is an important criterion for higher activity.
(logarithm of octanol–water partition coefficient determined by the Ghose and Crippen’s method [38]) was found to be important for APN inhibition of compounds that had lower cutoff values of both nPA and MFPSA. The function of hydrophobic descriptor may be easily understood as the binding site of hAPN comprises of hydrophobic pockets and hydrophobic interactions with residues like Phe472 and Ser469 were reported for bestatin. In addition, two topological kappa-shaped index descriptors, PHI and Kappa_3_AM were appeared in the model. The Kappa_3_AM is alpha-modified shape index of order three and PHI is the molecular flexibility index. These topological descriptors appeared in the model for compounds having higher values of MFPSA. Therefore, it may be interpreted that flexibility of the molecule is an important criterion for higher activity.
Bayesian modeling
Eight interpretable descriptors (AlogP, MW, nHBA, nHBD, nR, nArR, and MFPSA) and one fingerprint feature FCFP_6 (molecular function class fingerprints of maximum diameter 6) were used in developing the Bayesian model with the same training and test sets used in RP modeling. The model was validated using LOO cross-validation and ROC was generated. The ROC curve for the training set is provided in supporting information (Fig. S3). The model developed with a LOO cross-validation statistics of 0.918, and enrichments data (Table S9, supporting information) was obtained. The ‘best split’ value was calculated by picking the split that reduced the sum of the percent misclassified for inhibitors and noninhibitors using the cross-validation score for each compound. The ‘best split’ value obtained in the current model (Model 5) was 0.577, which demarcated highly active compounds from nonpotent inhibitors. Based on this value, a contingency table was obtained, which calculated true positive (TP), true negative (TN), false positive (FP), and false negative (FN). The LOO cross-validated result of training set is provided in supporting information (Fig. S3).
After leaving 20 % out 100 times, the ROC (0.907), concordance (0.903), specificity (0.895), and sensitivity (0.908) were observed in the Model 5. The five-fold cross-validation result is given in the supporting information (Table S10). Based on Bayesian score, top 20 favorable and unfavorable molecular fingerprints for APN inhibitory activity were identified by FCFP_6. All identified good and bad fingerprints are shown in the supporting information (Table S11), and five each selected favorable and unfavorable fingerprints are listed in Fig. 5.
Fig. 5.
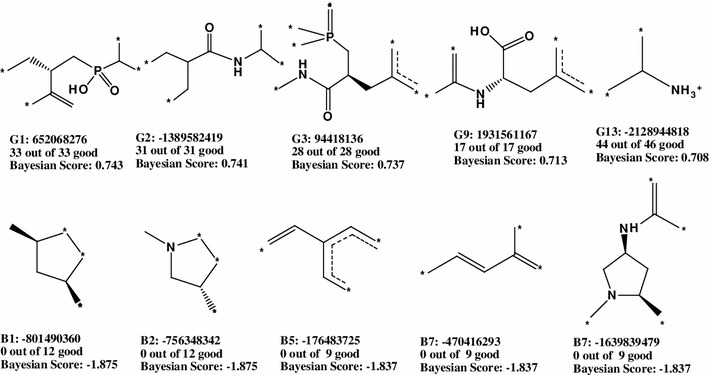
Good (G) and bad (B) molecular fingerprints identified by FCFP_6 descriptor
The retrieved fingerprints of the Bayesian modeling confirmed that features containing more number of polar groups are favorable. From both pharmacophore mapping and RP modeling, it was inferred that polar groups and positive ionizable features play essential role in the enzyme inhibition. The Bayesian modeling reconfirmed that polar moieties, like amide, carboxylic acid, and phosphinyl groups are favorable. The functionality of aromatic moieties may be intriguing because these appeared in both favorable and unfavorable fingerprints. However, unfavorable aromatic rings were either attached to polar residues or have exocyclic conjugation. Both of the functionalities may be related to aromaticity of these rings. It was interesting to find that acyclic rings appeared as the most unfavorable fingerprints due to lack of features (positive charge, polarity, and ring aromaticity) required for higher activity.
Thirty-one test set compounds were used to validate Model 5 and produced ROC score of 0.941, which indicated high predictive quality of the selected Bayesian model. The results showed concordance of 0.871, specificity of 0.882 and sensitivity of 0.857. The results and ROC plot for test set validation are provided in supporting information (Fig. S3).
Multiple pharmacophore screening and its validation
Pharmacophore models (both ligand- and structure-based) are used for the screening of databases containing large number of diverse molecules to find compounds that could be proposed as potential virtual hits [39–46]. An ideal pharmacophore query is able to capture active molecules selectively from the databases discarding inactive molecules. Therefore, prior to virtual screening the selectivity of the model should be properly assessed. The decoy set validation is the most popular and widely used approach to confirm overall specificity of a model [43–46]. The decoy set is a set of compounds, where some known active compounds are combined with inactives or decoys, and the whole set is screened with the selected pharmacophore query. The selection of inactive compounds for decoy set validation is very crucial for unbiased virtual screening. The selectivity of the pharmacophore model may be represented either by ROC curve [43, 45] or by GH score [44, 46]. The largest public available database of decoys is directory of useful decoys (DUD), which provides decoys for limited number of targets [47]. In the absence of DUD for a particular target, the selection of inactives depends on the users. However, the latter approach may have two major limitations, (a) it often presumes the inactivity of the decoys despite lack of supporting evidence [48] and (b) the decoy selection method may lack transparency. Moreover, Verdonk et al. [49] earlier demonstrated that some 1D molecular properties may affect the results of docking-based virtual screening, and their proposal may be extrapolated to pharmacophore-based virtual screening as well [43]. According to Verdonk et al., it is not sufficient to use a random library (subsets of public databases) for performance assessments, but it is essential to build up a focused library that reflects the physicochemical properties of the active set. Verdonk principle may be utilized for the selection of decoys [49]. In the current approach an alternative procedure has been adopted which includes the following steps, (a) selection of an well identified and recognizable decoy set from a public database (i.e. BD), which is considerably less focused than internal dataset used for the pharmacophore model development, (b) validation of the decoy sets by multiple screening with three different pharmacophore models developed in the current study, and (c) determination of activity profile by RP model developed from the external data set. The justification of these steps (a, b and c) is discussed in the supporting information (Text S3).
To substantiate the idea of multiple screening, the external data set was screened with three different pharmacophore queries (Models 1, 2, and 3) as shown in Fig. 6.
Fig. 6.
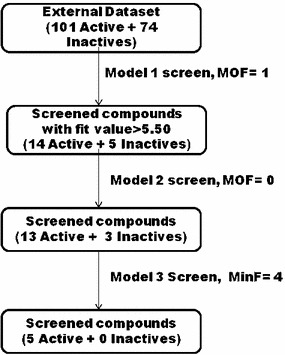
Multiple screening using external data set (MOF maximum omitted feature, MinF minimum number of feature)
The ligand-based pharmacophore model (Model 1) was selected for initial screening as it is able to quantify the estimated activity of the screened ligands. The structure-based models were utilized to refine these ligands filtered through Model 1. A cutoff fit value of 5.5 was set for Model 1, because top most active compounds (including bestatin) in the internal data set showed ‘fit value’ more than 5.50 in mapping on Model 1 (Table 1). In the first screening (Model 1), 26.3 % of the retrieved virtual hits were found to be seeded with known inactive compounds. The percentage of known inactives was gradually lowered in the subsequent screenings (18.75 and 0 % of the retrieved virtual hits screened though model 2 and 3, respectively). Therefore, it may be concluded that the number of inactives may be gradually reduced by ‘multiple pharmacophore screening’, and this approach could be useful for screening of public databases.
Screening of databases
Four databases—NCI, Specs, Maybridge, and Asinex were screened to find out potential virtual hits for APN inhibitory activity. These compounds were screened with three pharmacophore models (Models 1–3) as shown in Fig. 7.
Fig. 7.
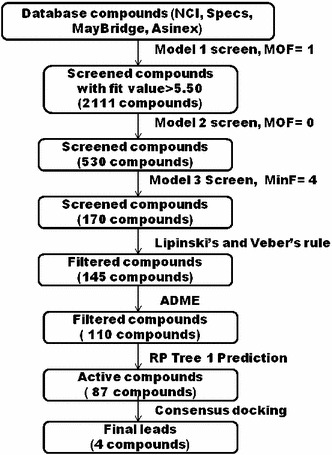
Database screening and filtering protocol (MOF maximum omitted feature, MinF minimum number of feature)
The Model 1 filtered 2,111 compounds with fit values more than 5.50. These compounds were further screened subsequently with Models 2 and 3. The remaining compounds (170 compounds) were subjected to Lipinski and Veber rules for drug likeliness and ADME properties, employing human intestinal absorption, aqueous solubility, and blood–brain barrier penetration. 60 out of 170 compounds were found to violate any one of the Lipinski and/or Veber rules for drug likeliness and ADME properties and thus rejected. The remaining 110 compounds were subjected to the RP prediction. The necessity of applying RP prediction may be substantiated by the work of Verdonk et al. [49], as they pointed out the influence of 1D descriptors in virtual screening. As the RP model was created with the help of descriptors that highlight the overall structural requirements for APN inhibition, application of this model in virtual screening may further help in reducing the number of inactive virtual hits in the database.
The RP model (Model 4, Fig. 5) predicted 87 compounds as active which have physicochemical and structural features similar to the reported highly active APN inhibitors. Finally, 87 compounds were subjected to molecular docking studies to predict binding affinities and explore interactions.
Comparative molecular docking study
In each docking method, the bound inhibitor bestatin was self-docked at the catalytic site of the receptor (PDB ID: 4FYR) [9] by three different docking tools: LibDock, GOLD, and GLIDE. The best conformer (supporting information, Fig. S4) of bestatin was observed in LibDock method with the lowest self-docking RMSD value of 0.38.
The 87 filtered drug-like compounds were docked by three docking tools, and the docking scores were compared with bestatin. It was found that only four compounds showed consistent docking results for all three molecular docking tools, and were proposed as the potential virtual hits for APN inhibition. The dockscores, predicted activities and ADME profiles of these four compounds are shown in Table 2.
Table 2.
Docking scores, ADME property, and estimated activity of virtual hits
| Ligand | Libdock score | Glide XP | Gold score | Model 1 | Solubilty | BBB | AlogP | PSA | |
|---|---|---|---|---|---|---|---|---|---|
| Fit value |
 ) ) |
(ADME) | (ADME) | (ADME) | (ADME) | ||||
| Bestatin | 138.15 |
 8.80 8.80 |
82.69 | 5.59 |

|

|

|

|

|
| MFCD00830868 | 153.99 |
 8.45 8.45 |
78.17 | 5.54 | 2.31 |
 5.16 5.16 |
 0.05 0.05 |
4.42 | 76.71 |
| MFCD00830876 | 146.50 |
 6.94 6.94 |
77.12 | 5.58 | 2.13 |
 3.56 3.56 |
 0.46 0.46 |
3.09 | 76.71 |
| MFCD02680015 | 137.92 |
 6.24 6.24 |
78.72 | 5.60 | 2.03 |
 3.70 3.70 |
 0.67 0.67 |
2.57 | 85.73 |
| MFCD02681563 | 134.18 |
 7.94 7.94 |
72.23 | 5.62 | 1.92 |
 3.43 3.43 |
 0.76 0.76 |
2.85 | 96.54 |
The docking interactions (LibDock/DS) and pharmacophore (Model 1)- mapped conformations of these virtual hits are provided in supporting information (Figs. S5 and S6, respectively). Two of these virtual lead compounds (MFCD00830868, MFCD00830876) contain piperazine residues. The protonated nitrogen atoms of these residues were mapped over the positive ionizable feature, and also interacted with the catalytic amino acid residues. Flippo et al. [50] earlier investigated some quinoline antimalarial agents, which strongly inhibited mammalian APN enzyme (lowest IC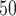 = 0.028
= 0.028 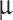 M). These compounds also contained piperazine moiety as the only positive ionizable group. Chelerythrin, a natural product containing quaternary nitrogen, also showed 82 % inhibition of APN [3]. Ligand–receptor interactions of these compounds were not reported earlier. Apart from the positive ionizable features, the proposed virtual hits also contained polar groups and aromatic rings, which were found to be important in APN inhibitory activity. All virtual hits were found to form
M). These compounds also contained piperazine moiety as the only positive ionizable group. Chelerythrin, a natural product containing quaternary nitrogen, also showed 82 % inhibition of APN [3]. Ligand–receptor interactions of these compounds were not reported earlier. Apart from the positive ionizable features, the proposed virtual hits also contained polar groups and aromatic rings, which were found to be important in APN inhibitory activity. All virtual hits were found to form  interaction with the catalytic amino acid residues. The pharmacophore mapped conformations were comparable with the docked conformations (supporting information, Figs. S5 and S6).
interaction with the catalytic amino acid residues. The pharmacophore mapped conformations were comparable with the docked conformations (supporting information, Figs. S5 and S6).
Conclusion
The current chemometric approach was performed to achieve three objectives (1) to highlight structural and physicochemical factors important in APN enzyme inhibition (2) to focus on aspects of ‘multiple pharmacophore screening’ approach in pharmacophore-based virtual screening and (3) to propose new virtual hits for APN inhibition. Ligand- and structure-based pharmacophore mapping, RP and Bayesian modeling were carried out and all models were properly validated to achieve the first objective. Both ligand- and structure-based pharmacophore mapping suggested that four features: (a) positive ionizable, (b) acceptor, (c) donor, and (d) ring aromatic are essential for the receptor–ligand interactions. The 3D QSAR pharmacophore model may be utilized for prediction of bioactivity of the ligands. RP modeling was performed on a diverse set of molecules, and it showed positive ionizable (P) feature may be the most important determinant for APN inhibition, and increased polarity of the molecule has positive contribution for higher biological activity. Besides, hydrophobicity [related to ring aromatic (RA)] was also found to be involved in receptor–ligand interaction. RP modeling presented some cut-off values of important descriptors that demarcate the active inhibitors from that of inactives. These may be utilized for the design of new molecules. In addition, Bayesian model highlighted some favorable and unfavorable fingerprints, which may also be utilized for the novel lead design purpose. The results of pharmacophore mapping, RP and Bayesian modeling were comparable and consistent enough to indicate the success of each modeling approach. To explore the second objective, an external random data set was constructed from reported APN inhibitors available in a public database (BD). It was shown that multiple screening by the ligand- and structure-based models may decrease the number of false positives gradually even from a random decoy set, the use of which is not recommended [49] for screening approaches in order to avoid low enrichment. Moreover, it was also found that the ligand- and structure-based pharmacophore models are complementary [51] because sequential screening by ligand- and structure-based pharmacophore queries gradually reduced the number of inactive virtual hits. The multiple screening followed by the RP prediction further reduced the number of false positive hits. This composite approach of screening also decreased the number of true positives to a considerable extent (Fig. 7). However, the main success of this approach lies in the fact that it is less complicated and maintains transparency in terms of the selection of decoys as well as the validation of the model. The last objective of the work was to search some potential virtual hits of APN inhibitors. Multiple pharmacophore screenings followed by RP prediction were carried out from some public databases. The retrieved virtual hits were tested for drug-likeness and pharmacokinetic properties. The filtered virtual hits were docked at the receptor active site, and four compounds showing consistent results in three different docking tools were ultimately proposed as the most potential virtual hits. All virtual hits were found to contain essential features like positive charge, polarity, and ring aromaticity, and may help to obtain useful APN inhibitors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Authors are thankful to the All India Council for Technical Education (AICTE) New Delhi and the Council of Scientific and Industrial Research (CSIR) New Delhi for providing financial support. One of the authors (AKH) is grateful to Council of Scientific and Industrial Research (CSIR), New Delhi for awarding a Senior Research Fellowship. We are also thankful to the authority of Jadavpur University for providing us the facility required for the work.
Abbreviations
- APN
Aminopeptidase N
- A
Hydrogen-bond acceptor
- BD
Binding database
- D
Hydrogen-bond donor
- MFASA
Molecular fractional polar surface area
- MPSA
Molecular polar surface area
- MW
Molecular weight
-
 HBA
HBA Number of hydrogen-bond acceptor
-
 HBD
HBD Number of hydrogen-bond donor
-
 Fr
Fr Number of fragments
-
 RB
RB Number of rotatable bonds
-
 PA
PA Number of positive atoms
-
 R
R Number of rings
-
 ArR
ArR Number of aromatic rings
- P
Positive ionisable
- R
Ring aromatic
- RP
Recursive partition
- Y
Hydrophobic
References
- 1.Zhang X, Fang H, Zhang J, Yuan Y, Xu W. Recent advances in aminopeptidase N (APN/CD13) inhibitor research. Curr Med Chem. 2011;18:5011–5021. doi: 10.2174/092986711797535155. [DOI] [PubMed] [Google Scholar]
- 2.Ito K, Nakashima Y, Onohara Y, Takeo M, Nakashima K, Matsubara F. Crystal structure of aminopeptidase N (proteobacterial alanyl aminopeptidase) from Escherichia coli and conformational change of methionine 260 involved in substrate recognition. J Biol Chem. 2006;281:33664–33676. doi: 10.1074/jbc.M605203200. [DOI] [PubMed] [Google Scholar]
- 3.Bauvois B, Dauzonne D. Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med Res Rev. 2006;26:88–130. doi: 10.1002/med.20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimizu T, Tani K, Hase K, Ogawa H, Huang L, Shinomiya F, Sone S. CD13/aminopeptidase N-induced lymphocyte involvement in inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46:2330–2338. doi: 10.1002/art.10517. [DOI] [PubMed] [Google Scholar]
- 5.Hafler DA, Hemler ME, Christenson L, Williams JM, Shapiro HM, Strom TB, Strominger JL, Weiner HL. Investigation of in vivo activated T cells in multiple sclerosis and inflammatory central nervous system diseases. Clin Immunol Immunopathol. 1985;37:163–171. doi: 10.1016/0090-1229(85)90147-3. [DOI] [PubMed] [Google Scholar]
- 6.Luan YP, Xu WF. The structure and main functions of aminopeptidase N. Curr Med Chem. 2007;14:639–647. doi: 10.2174/092986707780059571. [DOI] [PubMed] [Google Scholar]
- 7.Wickstrom M, Larsson R, Nygren P, Gullbo J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2011;102:501–508. doi: 10.1111/j.1349-7006.2010.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umezawa H, Aoyagi T, Suda H, Hamada M, Takeushi T. Bestatin an inhibitor of aminopeptidase B produced by actinomycetes. J Antibiotic. 1976;29:97–99. doi: 10.7164/antibiotics.29.97. [DOI] [PubMed] [Google Scholar]
- 9.Wong AHM, Zhou D, Rini JM. The x-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J Biol Chem. 2012;287:36804–36813. doi: 10.1074/jbc.M112.398842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng X-C, Wang Q, Fang H, Tang W, Xu W-F. Design synthesis and preliminary evaluation of novel pyrrolidine derivatives as matrix metalloproteinase inhibitors. Eur J Med Chem. 2008;43:2130–2139. doi: 10.1016/j.ejmech.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 11.Cheng X-C, Wang Q, Fang H, Tang H, Xu W-F. Design synthesis and evaluation of novel sulfonyl pyrrolidine derivatives as matrix metalloproteinase inhibitors. Bioorg Med Chem. 2008;16:5398–5404. doi: 10.1016/j.bmc.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Fang H, Wang X, Xu W. Novel cyclic-imide peptidomimetics as aminopeptidase N inhibitors structure-based design chemistry and activity evaluation II. Eur J Med Chem. 2010;45:1618–1626. doi: 10.1016/j.ejmech.2009.12.071. [DOI] [PubMed] [Google Scholar]
- 13.Jia M, Yang K, Fang H, Xu Y, Sun S, Su L, Xu W. Novel aminopeptidase N (APN/CD13) inhibitors derived from chloramphenicol amine. Bioorg Med Chem. 2011;19:5190–5198. doi: 10.1016/j.bmc.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Fang H, Zhu H, Wang X, Zhang L, Li M, Li Q, Yuan Y, Xu W. Novel aminopeptidase N (APN/CD13) inhibitors derived from 3-phenylalanylyl-N’-substituted-26-piperidinedione. Bioorg Med Chem. 2010;18:5981–5987. doi: 10.1016/j.bmc.2010.06.078. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Shang L, Fang H, Zhu H, Mu J, Wang Q, Wang X, Yuan Y, Xu W. Design synthesis and preliminary studies of the activity of novel derivatives of N-cinnamoyl-l-aspartic acid as inhibitors of aminopeptidase N/CD13. Bioorg Med Chem. 2009;17:7398–7404. doi: 10.1016/j.bmc.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Wang Y, Wu J, Li Y, Wang Q, Xu W. Novel aminopeptidase N inhibitors derived from antineoplaston AS2-5 (part II) Bioorg Med Chem. 2009;17:3061–3071. doi: 10.1016/j.bmc.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Wang J, Zhang L, Xu W. Design, synthesis and preliminary activity evaluation of novel peptidomimetics as aminopeptidase N/CD13 inhibitors. Arch Pharm Chem Life Sci. 2011;344:494–504. doi: 10.1002/ardp.201100109. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Wang J, Li J, Wu J, Li Y, Zhu H, Fan R, Xu W. Novel aminopeptidase N inhibitors derived from antineoplaston AS2-5 (Part I) Bioorg Med Chem. 2009;17:3053–3060. doi: 10.1016/j.bmc.2009.02.063. [DOI] [PubMed] [Google Scholar]
- 19.Cheng X-C, Wang Q, Fang H, Tang W, Xu W-F. Synthesis of new sulfonyl derivatives as matrix metalloproteinase inhibitors. Bioorg Med Chem. 2008;16:7932–7938. doi: 10.1016/j.bmc.2008.07.073. [DOI] [PubMed] [Google Scholar]
- 20.http://www.bindingdb.org
- 21.Accelrys Inc Discovery Studio 3.0 San Diego USA 2011
- 22.Debnath AK. Pharmacophore mapping of a series of 24-diamino-5-deazapteridine inhibitors of Mycobacterium avium complex dihydrofolate reductase. J Med Chem. 2002;45:41–53. doi: 10.1021/jm010360c. [DOI] [PubMed] [Google Scholar]
- 23.Thangapandian S, John S, Sakkiah S, Lee KW. Discovery of potential integrin VLA-4 antagonists using pharmacophore modeling virtual screening and molecular docking studies. Chem Biol Drug Des. 2011;78:289–300. doi: 10.1111/j.1747-0285.2011.01127.x. [DOI] [PubMed] [Google Scholar]
- 24.Chen L, Lin Y-L, Peng G, Li F. Structural basis for multifunctional roles of mammalian aminopeptidase N. PNAS. 2012;109: 17966–17971. doi: 10.1073/pnas.1210123109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osman FG, Douglas RH. Metric for analyzing hit lists and pharmacophores. In: Guner OF, editor. Pharmacophore perception development and use in drug design. 1. San Diego, CA: International University Line La Jolla; 2000. pp. 193–210. [Google Scholar]
- 26.Triballeau N, Acher F, Brabet I, Pin I-J, Bertrand H-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem. 2005;48:2534–2547. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- 27.Bohm HJ. The Computer Program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput-Aided Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]
- 28.Prathipati P, Ma NL, Keller TH. Global Bayesian models for the prioritization of antitubercular agents. J Chem Inf Model. 2008;48:2362–2370. doi: 10.1021/ci800143n. [DOI] [PubMed] [Google Scholar]
- 29.Lu J, Zheng M, Wang Y, Shen Q, Luo X, Jiang H, Chen K. Fragment-based prediction of skin sensitization using recursive partitioning. J Comput Aided Mol Des. 2011;25:885–893. doi: 10.1007/s10822-011-9472-7. [DOI] [PubMed] [Google Scholar]
- 30.Ojha PK, Roy K. Comparative QSARs for antimalarial endochins: importance of descriptor-thinning and noise reduction prior to feature selection. Chem Int Lab Sys. 2011;109:146–161. doi: 10.1016/j.chemolab.2011.08.007. [DOI] [Google Scholar]
- 31.Li S, Fedorowicz A, Singh H, Soderholm SC. Application of the random forest method in studies of local lymph node assay based skin sensitization data. J Chem Inf Model. 2005;45:952–964. doi: 10.1021/ci050049u. [DOI] [PubMed] [Google Scholar]
- 32.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev 23:3–25. doi:10.1016/S0169-409X(96)00423-1 [DOI] [PubMed]
- 33.Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 34.Rao SN, Head MS, Kulkarni A, Lalonde JM. Validation studies of the site-directed docking program LibDock. J Chem Inf Model. 2007;47:2159–2171. doi: 10.1021/ci6004299. [DOI] [PubMed] [Google Scholar]
- 35.http://www.ccdc.cam.ac.uk/products/life_sciences/gold
- 36.Jones G, Willet P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a descriptor of desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/S0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 37.Glide ver 5.5 Schrodinger LLC, New York 2012
- 38.Ghose AK, Crippen G. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure–activity relationships. I. Partition coefficient as a measure of hydrophobicity. J Comp Chem. 1986;7:565–577. doi: 10.1002/jcc.540070419. [DOI] [PubMed] [Google Scholar]
- 39.Thangapandian S, John S, Sakkiah S, Lee KW. Potential virtual lead identification in the discovery of renin inhibitors: application of ligand and structure-based pharmacophore modeling approaches. Eur J Med Chem. 2011;46:2469–2476. doi: 10.1016/j.ejmech.2011.03.035. [DOI] [PubMed] [Google Scholar]
- 40.Saxena S, Chaudhaery SS, Varshney K, Saxena AK. Pharmacophore based virtual screening and docking studies of HSP90 inhibitors. SAR QSAR Env Res. 2010;21:445–462. doi: 10.1080/1062936X.2010.501817. [DOI] [PubMed] [Google Scholar]
- 41.Lu S-H, Wu JW, Liu H-L, Zhao J-H, Liu K-T, Chuang C-K, Lin H-Y, Tsai W-B, Ho Y. The discovery of potential acetylcholinesterase inhibitors: a combination of pharmacophore modeling virtual screening and docking studies. J Biomed Sci. 2011;18:8–13. doi: 10.1186/1423-0127-18-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.John S, Thangapandian S, Sakkiah S, Lee KW. Potent bace-1 inhibitor design using pharmacophore modeling in silico screening and molecular docking studies. BMC Bioinformatics. 2011;12:S1–S28. doi: 10.1186/1471-2105-12-S1-S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
43.Taha MO, Tarairah M, Zalloum H, Abu-Sheikha G. Pharmacophore and QSAR modeling of estrogen receptor
 ligands and subsequent validation and in silico search for new hits. J Mol Graph Model. 2010;28:383–400. doi: 10.1016/j.jmgm.2009.09.005. [DOI] [PubMed] [Google Scholar]
ligands and subsequent validation and in silico search for new hits. J Mol Graph Model. 2010;28:383–400. doi: 10.1016/j.jmgm.2009.09.005. [DOI] [PubMed] [Google Scholar] - 44.Vadivelan S, Sinha BN, Rambabu G, Boppanna K, Jagarlapudi Pharmacophore modeling and virtual screening studies to design some potential histone deacetylase inhibitors as new hits. J Mol Graph Model. 2008;26:935–946. doi: 10.1016/j.jmgm.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 45.Vijayan RSK, Prabu M, Mascarenhas NM, Ghoshal N. Hybrid structure-based virtual screening protocol for the identification of novel BACE1 Inhibitors. J Chem Inf Model. 2009;49:647–657. doi: 10.1021/ci800386v. [DOI] [PubMed] [Google Scholar]
- 46.Sakkiah S, Thangapandian S, John S, Lee KW. Pharmacophore based virtual screening molecular docking studies to design potent heat shock protein 90 inhibitors. Eur J Med Chem. 2011;46:2937–2947. doi: 10.1016/j.ejmech.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 47.Kirchmair J, Markt P, Distino S, Wolber G, Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparison enrichment assessment and decoy selection: what can we learn from earlier mistakes? J Comput Aided Mol Des. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 48.Taha MO, Habash M, Al-Hadidi Z, Al-Bakri A, Younis K, Sisan S. Docking-based comparative intermolecular contacts analysis as new 3-D QSAR concept for validating docking studies and in silico screening: NMT and GP inhibitors as case studies. J Chem Inf Model. 2011;51:647–669. doi: 10.1021/ci100368t. [DOI] [PubMed] [Google Scholar]
- 49.Verdonk ML, Berdini V, Hartshorn MJ, Mooji WTM, Murray CW, Taylor RD, Watson P. Virtual screening using protein–ligand docking: avoiding artificial enrichment. J Chem Inf Comp Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 50.Flippo M, Florent I, Grellier P, Sergheraert C, Deprez-Poulain P. Design, synthesis and antimalarial activity of novel quinoline-based zinc metallo-aminopeptidase inhibitors. Bioorg Med Chem Lett. 2003;13:2659–2662. doi: 10.1016/S0960-894X(03)00550-X. [DOI] [PubMed] [Google Scholar]
- 51.Sanders MPA, McGuire R, Roumen L, Esch IJP, de Vlieg J, Klomp JPG, Graff C. From the protein’s perspective: the benefit and challenges of protein structure-based pharmacophore modeling. Med Chem Commun. 2012;3:28–38. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.