

Abstract
Kawasaki disease is an acute, self-limited vasculitis of childhood and has become the leading cause of acquired pediatric heart disease in the USA. Prompt treatment is essential in reducing cardiac-related morbidity and mortality. The underlying etiology remains unknown. The disease itself may be the characteristic manifestation of a common pathway of immune-mediated vascular inflammation in susceptible hosts. The characteristic clinical features of fever for at least 5 days with bilateral nonpurulent conjunctivitis, rash, changes in lips and oral cavity, changes in peripheral extremities, and cervical lymphadenopathy remain the mainstay of diagnosis. Supplementary laboratory criteria can aid in the diagnosis, particularly in cases of incomplete clinical presentation. Diagnosis of Kawasaki disease can be challenging as the clinical presentation can be mistaken for a variety of other pediatric illnesses. Standard of care consists of intravenous immune globulin and aspirin. Corticosteroids, infliximab, and cyclosporine A have been used as adjunct therapy for Kawasaki disease refractory to initial treatment. There is ongoing research into the use of these agents in the initial therapy of Kawasaki disease.
Keywords: Kawasaki disease, Update, Pathogenesis, Etiology, Treatment
Introduction
Kawasaki disease (KD) is an acute self-limited vasculitis which presents with characteristic clinical features first described in 1967 [1]. The high prevalence of coronary artery aneurysm/ectasia and its associated cardiovascular morbidity in untreated children is well characterized [2]. Kawasaki disease is now the leading cause of pediatric acquired heart disease in the USA [3]. Prompt treatment with high-dose intravenous immune globulin (IVIG) reduces the incidence of coronary artery abnormalities from 18 to 4 % [4]. However, significant gaps remain in our understanding of KD. The underlying etiology remains unknown. Epidemiological studies have recently attributed the possible source of the etiologic agent of KD to northern China with subsequent spread to Japan via tropospheric winds [5•].
The diagnosis of KD remains primarily based upon the characteristic clinical features of ≥5 days of fever, bilateral nonpurulent conjunctivitis, rash, cervical lymphadenopathy, and mucocutaneous changes [6••]. However, diagnostic challenges often arise given the significant overlap in clinical features of KD and other pediatric illnesses. Additionally, KD can also present with incomplete clinical features and a number of atypical presentations. Yet, accurate diagnosis is integral in prevention of morbidity and mortality of KD as prompt treatment with IVIG significantly reduces the incidence of long-term cardiac complications.
Epidemiology
The most recent epidemiological data describes a divergence in the incidence of KD between the West (North America, Australia, Europe) and East Asia (Japan, Taiwan, Korea). In the West, the current incidence of KD is 4–25/100,000 in children <5 years. In the last decade, the overall incidence in the West has plateaued following an initial increase. By comparison, the incidence of KD in East Asian countries has continued to increase and currently report an incidence 10–20 times higher than USA and Europe. Japan has the highest incidence of KD in children <5 years of any country (265/100,000), followed by Korea (134/100,000), and Taiwan (83/100,000) [7•].
Ethnic variation in the incidence of KD suggests genetic susceptibility as a significant factor in the higher prevalence of KD in East Asian countries. Within the USA, the incidence of KD ranges from 17.5 to 20.8/100,000 in children <5 years [8]. However, the incidence of KD is significantly higher in Pacific Islanders and African Americans. In particular, Americans of Japanese ancestry in Hawaii exhibit a similar incidence of KD to that of the general population of Japan (>200/100,000 in children <5 years) [9].
Etiology
Many infectious etiologies for KD have been proposed, although a putative agent has not been isolated. The young age of children affected by KD suggests that KD is triggered by an agent to which the population becomes immune to by late childhood [10]. A variety of bacterial and viral pathogens have been isolated from patients with KD. These include Staphylococcus aureus, Streptococcus pyogenes, adenovirus, parvovirus B19, human herpes virus 6, rotavirus, human coronavirus, bocavirus, parainfluenza, Epstein-Barr, dengue, varicella-zoster, measles, and influenza viruses. While intracytoplasmic inclusion bodies found in the bronchial epithelium on autopsy may support a viral etiology [11], the etiologic importance of the concurrent infectious pathogens seems to be limited.
The possibility of a superantigen/bacterial toxin etiology has been postulated due to the similar desquamation found in scarlet fever, toxic shock syndrome, and KD. However, significant supportive data is lacking. Paralysis of the adaptive immune system typically seen in superantigen-mediated illness is not seen in KD. Additionally, no bacterial toxin has been detected in the peripheral blood of KD patients making such a toxin unlikely to be responsible for the diffuse inflammation found in acute KD [11]. A primary autoimmune etiology is unlikely due to the self-limited and nonrecurring nature of KD.
The correlation of the seasonality of KD with tropospheric winds from densely cultivated region of northeastern China to Japan suggests a possible windborne pathogen as the underlying etiology of KD [5•]. Nevertheless, the clinical features of KD may be the result of a common pathway of immune-mediated vascular inflammation after a variety of inciting infections rather than infection with a specific pathogen [12]. The fact that a putative infectious agent has not been isolated despite significant investigation supports this hypothesis.
Pathogenesis
Through exposure to a causative trigger(s), systemic inflammation is induced in the host through an incompletely understood cascade [13]. The inflammation primarily targets the medium-sized muscular arteries, most notably the coronary arteries. The initial infiltrate is primarily neutrophilic in the first 2 weeks. This is followed by infiltration of eosinophils and CD8 T cells >2 weeks after disease onset [10].
Investigation of coronary arteries with light and transmission electron microscopy has identified three distinct vascular processes: necrotizing arteritis (NA), subacute/chronic (SA/C) vasculitis, and luminal myofibroblastic proliferation [14]. The extent and permanence of vascular damage depends upon the scope of involvement of vascular inflammation. Mild inflammation and damage generally preserves the internal and external elastic lamina, which may cause mild dilation but allows the arteries to maintain its normal morphology. As the severity of the inflammation increases, the internal and external elastic lamina is destroyed resulting in progressively larger aneurysms. These aneurysms may be fusiform (from mild SA/C vasculitis) or saccular (from severe SA/C vasculitis or NA). These aneurysms are at risk for rupture or development of thrombosis. Thrombosis may result in ischemia/myocardial infarction or can undergo organization and ultimately recanalization [14]. Coronary artery aneurysms have also been previously described to remodel and regress [15–17].
Diagnosis
There is no single diagnostic test or pathognomonic clinical feature of KD. Therefore, a thorough history and physical examination for the established clinical diagnostic criteria is essential. The classic clinical criteria for the diagnosis of KD require the presence of ≥5 days of fever and ≥4/5 clinical features (see Table 1) [6••]. The characteristic clinical features are typically not present all at the same time, and therefore continuous close observation may be necessary to make the diagnosis.
Table 1.
Diagnosis of Kawasaki disease
| Classic clinical criteria | |
| Fever persisting for at least 5 days | |
| Plus | |
| At least four of the following principle features: | |
| 1.Extremity changes | Erythema of palms/soles, edema of hands/feet, periungual peeling of fingers, toes in weeks 2–3 |
| 2.Rash | Polymorphous exanthema, NOT bullous/vesicular |
| 3.Changes in lips and oral cavity | Erythema, cracked lips, strawberry tongue, diffuse injection of oral and pharyngeal mucosa |
| 4.Conjunctival injection | Bilateral, bulbar sparing limbus, nonpurulent |
| 5.Cervical lymphadenopathy | >1.5 cm in diameter, usually unilateral |
| Supplemental laboratory criteria | |
| Hypoalbuminia | <3.0 mg/dL |
| Alanine aminotransferase (ALT) | Elevation above reference range (varies by laboratory) |
| Anemia for age | Decreased in hemoglobin below reference range for age |
| Leukocytosis | White blood cells >15,000/mm3 |
| Sterile pyuria (urinalysis) | ≥10 WBC/HPF (high-power field) |
| Thrombocytosis after 7 days | Platelets >450,000/mm3 after 7 days |
The fever of KD is high-spiking, peaking >39–40 °C, with a mean duration of 11 days, although it has been reported to last for up to 3–4 weeks in the absence of treatment. Upon initiation of treatment, the fever typically resolves within 1–2 days. Extremity changes initially present as erythema, induration, and swelling of the hands and/or feet in the acute phase of KD. Desquamation of the fingers and toes is typically a late finding, beginning 2–3 weeks after onset of the fever. The polymorphous exanthema of KD may take various forms: maculopapular rash (most common), scarlatiniform rash, erythroderma, erythema-multiforme, or micropustular. Bullous/vesicular rashes have not been described. The rash typically presents within the first 5 days of fever as a generalized eruption involving the trunk, extremities, and perineal region. Early desquamation may occur in regions of perineum and is a strong indicator of KD. Bilateral conjunctival injection in KD affects the bulbar conjunctivae and spares the limbus. The injection is nonpurulent and painless, presenting shortly after onset of fever. Mild anterior uveitis may be present on slit lamp examination. Oral cavity and lip changes in KD present include generalized erythema of the lips and oropharyngeal mucosa, peeling/cracking/fissuring of the lips, and erythema and prominent fungiform papillae on the tongue, commonly referred to as a “strawberry tongue.” Strawberry tongue is not unique to KD as it is also associated with streptococcal scarlet fever. Cervical lymphadenopathy in KD is usually unilateral, classically presents in the anterior cervical triangle, and diagnostic criteria require ≥1 enlarged lymph node which is ≥1.5 cm in diameter. Of the classic clinical features, cervical lymphadenopathy is the least commonly observed.
The diagnosis of incomplete KD is made in patients who do not fulfill the classic clinical criteria (see Fig. 1). Incomplete KD is a separate entity from “atypical” KD, which present with symptoms typically not seen in KD. Incomplete KD should be considered in children with unexplained fever for ≥5 days in addition to the presence of 2–3 principal clinical features of KD. There is a higher prevalence of “incomplete” presentation in younger children. Therefore, incomplete KD should be considered in young children (<12 months old) with unexplained fever for ≥5 days associated with any of the clinical features as KD [6••]. In the evaluation for incomplete KD, the presence of supportive laboratory and echocardiogram findings (see Table 1) is useful in ultimately confirming the diagnosis.
Fig. 1.
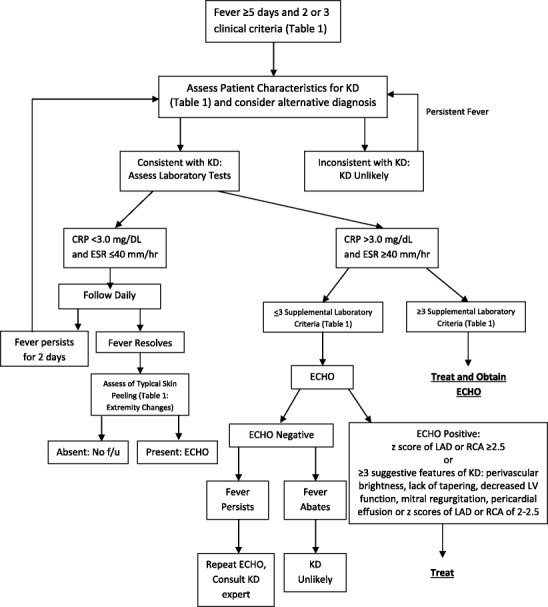
2004 American Heart Association Diagnostic Algorithm for Evaluation of Suspected Incomplete Kawasaki Disease [Adapted & used with permission from Newberger et al. Circulation. 2004;110:2747–2771]
The term “atypical KD” has been used interchangeably with incomplete KD in the past, but should be reserved for cases of KD which present with findings rarely associated with KD. These features include transient unilateral peripheral facial nerve palsy, transient high-frequency sensorineural hearing loss, hepatic enlargement with jaundice, and acalculous gallbladder distention. Testicular swelling, pulmonary nodules, pleural effusions, and hemophagocytic syndrome have also been reported but are less common. Kawasaki shock syndrome, the presentation of KD disease with systolic hypotension or clinical signs of poor perfusion, has also been described as a rare presentation of KD [18•].
The major cardiac sequelae of KD affect the coronary arterial system. The pericardium, myocardium, endocardium, valves, and coronary arteries may all be affected by the diffuse inflammatory process of KD. This may present as pericarditis, myocarditis, valvulitis, and new valvular regurgitation. A gallop rhythm with innocent flow murmur may also be present, commonly in the setting of anemia and fever of acute KD. Poor myocardial function can also result in cardiogenic shock. Arrhythmia, prolonged PR interval, and nonspecific ST and T wave changes may be present on EKG.
Supplementary laboratory criteria supporting the diagnosis of KD are listed in Table 1 and include hypoalbuminemia, anemia for age, elevation of alanine aminotransferase (ALT), thrombocytosis after 7 days, leukocytosis, and sterile pyuria. Additional nonspecific laboratory findings are elevated markers of inflammation (erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP)). Approximately 50 % of patients with KD have WBC >15,000/mm3. Anemia may develop in patients with prolonged duration of active inflammation and is typically present with normal RBC indexes. Acute phase reactant elevation (ESR/CRP) is nearly universally present, although discordant ESR/CRP can be seen in the acute presentation of KD. Thrombocytosis, with platelet counts from 500,000 to >1 million/mm3, is a late feature of KD commonly occurring in the second week of the disease and slowly resolving by weeks 4 to 8 in uncomplicated KD. Elevation of ALT is typically mild to moderate and occur in ≤40 % of cases. Hypoalbuminemia (albumin ≤3.0) is associated with more severe and prolonged disease. Sterile pyuria is present in approximately one third of patients. Sterile pyuria is not found in suprapubic aspiration of urine as underlying urethritis is present in KD.
Diagnostic Challenges
The differential diagnosis of patients presenting with the clinical features of KD is vast. There is considerable overlap in the clinical features of KD with several common pediatric illnesses including but not limited to adenovirus infection, bacterial cervical adenitis, retropharyngeal abscess, systemic onset JRA, and toxic shock syndrome. Incomplete KD often presents the greatest diagnostic challenge as initial presentation may be consistent with any of the previously mentioned clinical illnesses given the limited clinical manifestations. Therefore, it is essential to consider KD in the differential diagnosis of these illnesses.
Adenovirus infection is one of the most common pediatric infections, comprising 7–8 % of all pediatric respiratory illnesses [19]. The presence of adenovirus can be detected through polymerase chain reaction (PCR) testing of the nasopharynx (NP) swab in up to 11 % of healthy children [20]. In patients with KD with subsequent development of coronary artery abnormalities, incidental detection of adenovirus by PCR in NP swab has been described. Therefore, accurate differentiation between the incidental detection of adenovirus in patients with KD and primary adenoviral disease is essential to avoid the subsequent mortality/morbidity associated with undiagnosed KD. Prolonged fever and elevated inflammatory markers are common features of adenovirus infection and KD. The most commonly described clinical feature of KD compared to adenoviral infection reported in a cohort study was conjunctivitis. Features of adenovirus conjunctivitis include unilateral onset, prominence of tearing more frequently than purulence, and follicular hyperplasia. Additionally, adenovirus often presents with pharyngoconjunctival fever, characterized by nonexudative or exudative pharyngitis in addition to conjunctivitis. Extremity changes and unilateral neck swelling were the least common clinical features of KD seen in patients with adenoviral disease [21•].
Node-first presentation of KD (NFKD) is the manifestation of KD with fever and cervical adenopathy prior to the presentation of other clinical features of KD. Compared to typical KD, the patients of NFKD tend to be older and have higher markers of inflammation. Node-first presentation of KD may often be misdiagnosed as bacterial cervical adenitis as fever and cervical adenopathy are characteristic clinical findings in both diseases. Laboratory findings to aid in differentiation of these two entities include lower WBC count, hemoglobin, platelet counts, and higher absolute band count, CRP, ESR, ALT in NFKD. Additionally, radiographic findings in bacterial cervical adenitis favor a single dominant node or conglomerations and suppuration of lymph nodes. Conversely, radiographic findings in NFKD favor multiple discrete nodes [22•].
Retropharyngeal edema is a rare presentation of KD, which due to unfamiliarity with the presentation, is often misdiagnosed as retropharyngeal abscess. Many of these patients have underwent pharyngeal needle aspiration and antibiotic therapy due to misdiagnosis, delaying treatment for KD and increasing the risk of development of coronary artery abnormalities [23, 24]. The diagnosis of KD with retropharyngeal edema should be considered in patients diagnosed with retropharyngeal abscess subsequently found to have sterile culture results from surgical drainage. There were no significant differences in laboratory findings between these two entities. Clinical symptoms classically associated with retropharyngeal abscess such as stridor, neck pain, and dysphagia are less common in the retropharyngeal edema of KD. Radiological findings of ring enhancement and mass effect on neck CT were more commonly found in patients with retropharyngeal abscess [25•].
Systemic onset juvenile idiopathic arthritis (SoJIA) is an autoimmune disease characterized by severe inflammation of multiple organ systems. The clinical features of early stage SoJIA (fever, rash, thrombocytosis, increased markers of inflammation, and arthritis) may resemble KD. Coronary artery dilation can be present in SoJIA, but is less common. Arthritis is a feature that may be present in both KD and SoJIA. It is generally mild and self-limited in KD, as compared to the persistent and severe arthritis of SoJIA which often requires treatment with immunosuppressive agents for resolution [26]. The rash of SoJIA is typically intermittent and coincides with fever spikes as compared to the fixed rash of KD. The presence of mucocutaneous findings suggests KD as this finding is absent in SoJIA. Additionally, thrombocytosis is typically a late finding of KD (occurring in the second week of disease) as compared to the early stages of SoJIA. As IVIG is an adjunctive rather than primary modality of therapy for SoJIA, the diagnosis of SoJIA should be considered in patients with KD which do not respond to multiple IVIG infusions [27].
Toxic shock syndrome (TSS) is a toxin-mediated disease which presents with clinical features of fever, hypotension, rash, and subsequent desquamation of the skin with multi-organ involvement and may be confused with KD. The most common causative organism is S. aureus. The condition has been misdiagnosed as KD due to similar clinical features of rash and fever. The hypotension of TSS can be misattributed to the shock associated with KD. Patients presenting with TSS are on average older (mean age 113 ± 56 months) than patients with KD (37 ± 41 months) [28•]. Other features suggestive of TSS include elevated creatinine and labile blood pressures requiring higher vasopressor support and increased fluid resuscitation. Features suggestive of KD include anemia, thrombocytosis, and cardiac abnormalities [29–31].
Treatment
Routine Management
Intravenous Immune Globulin
The use of intravenous immune globulin (IVIG) in the acute phase of KD to prevent the development of coronary artery abnormalities is well defined [4, 32, 33]. Treatment with IVIG within the first 10 days of illness reduces the risk of development of coronary artery abnormalities to approximately 5 % from 25 % [32]. The mechanism of action remains unknown, but the generalized anti-inflammatory effect of IVIG likely inhibits progression of KD, ultimately preventing the development of coronary artery abnormalities. The use IVIG is recommended for treatment of patients with KD [6••] and has been shown to be cost effective [34].
Dosed at 2 gm/kg in a single infusion, IVIG should be given as initial treatment for KD. Treatment should be instituted within the first 10 days of illness and should still be administered to children after the tenth day of illness should fever persists, or with ongoing systemic inflammation (elevated ESR/CRP) or the presence of coronary artery abnormalities [6••]. Treatment of KD prior to day 5 of illness may be associated with increased need for IVIG retreatment [35]. Live vaccines, particularly measles and varicella, should be deferred for 11 months after IVIG treatment. Children at high risk of measles exposure may be vaccinated earlier, but will require re-immunization 11 months after IVIG treatment. Treatment with IVIG is given concurrently with aspirin (see below).
Aspirin
During the acute phase of KD, aspirin (ASA) is administered at high dosages (80–100 mg/kg/day divided every 6 h). Concurrent administration of high-dose ASA with IVIG during the acute phase of KD seems to provide an additive anti-inflammatory effect. High-dose aspirin alone does not lower the frequency of development of coronary abnormalities despite its anti-inflammatory effect [32]. The duration of high-dose aspirin therapy varies with institution. All institutions require resolution of fever for 48–72 h prior to stopping high-dose ASA therapy. However, some institutions will treat with high-dose ASA until day 14 of illness regardless of fever resolution.
Once high-dose ASA is discontinued, low-dose ASA (3–5 mg/kg given daily) is started. Low-dose ASA is used for its anti-platelet effect to prevent possible complications from coronary artery abnormalities associated with KD. Evaluation for coronary abnormalities should be done at least until 6–8 weeks after the onset of KD. Low-dose ASA is discontinued if there is no evidence of coronary abnormalities and platelet count is normal at that time. Should evidence of coronary abnormalities develop or persist, low-dose ASA is continued indefinitely. Ibuprofen should be avoided while the patient is on ASA as it antagonizes the platelet inhibition of ASA [36].
Children receiving ASA therapy are at risk of development of Reye syndrome, particularly during active infections with varicella or influenza. Reye syndrome has been reported in patients receiving prolonged high-dose ASA therapy for KD [37], although it is unclear whether low-dose ASA increased the risk of Reye syndrome. Children on long-term ASA therapy should receive inactivated influenza vaccine. Physicians should evaluate patients on ASA who are exposed to or develop symptoms of influenza and varicella.
Refractory Kawasaki Disease
Approximately 13 % of patients with KD fail to respond to initial treatment with IVIG [38]. Failure to respond is characterized as persistent fever ≥36–48 h following completion of IVIG infusion. The most likely explanation for persistent fever in these patients is failure of initial IVIG to abort the disease process of KD. However, KD can be misdiagnosed due to its shared clinical characteristics with a wide variety of common pediatric illnesses. Therefore, patients with refractory KD should be assessed for possible misdiagnosis of KD as well (see “Diagnostic Challenges” section).
IVIG Retreatment
Patients with KD who fail to respond to initial IVIG therapy should receive a repeat infusion of IVIG at the same dose (2 gm/kg as a single infusion). Retreatment at lower doses (1 gm/kg) has been associated with increased risk of developing coronary abnormalities [38].
Corticosteroids
Pulse steroid therapy in patients with KD refractory to two IVIG infusions (2 gm/kg infusion followed by 1 gm/kg infusion) has been associated with shorter duration of fever and hospital stay when compared to repeat 1 gm/kg IVIG infusion [39]. The use of corticosteroids in patients with KD is currently reserved for children who have received ≥2 infusions of IVIG on the basis that effects of steroid therapy on coronary artery abnormalities were uncertain at the time of publication of KD treatment guidelines in 2004. The most commonly used steroid regimen is IV methylprednisolone (IVMP) 30 mg/kg daily for 1–3 days [6••].
Further research into the use of corticosteroids in the primary treatment of KD remains ongoing. In 2007, a randomized controlled trial by Newburger et al. showed that the addition of one dose of IVMP therapy to initial IVIG infusion did not reduce the prevalence of coronary artery abnormalities or total length of hospital stay [40]. In Japan, the study on the efficacy of IVIG and steroids in patients with severe presentation of KD (RAISE study), a multicenter, prospective, randomized, open-label, blinded-endpoints trial, showed combination treatment with IVIG and prednisolone had significant advantages over IVIG alone in the prevention of coronary artery abnormalities with rapid defervescence of fever and normalization of inflammatory markers [41•]. Severe KD was determined through a risk score evaluation of five points or higher per Kobayashi risk score scale, which predicts a >30 % probably of nonresponsiveness to initial IVIG treatment (Table 2) [42]. There were significant differences in corticosteroid regimen between the two trials. The median start time of therapy in the RAISE study was 2 days earlier than in the trial by Newburger et al. Additionally, the steroid dosage of the RAISE study was IV prednisone 2 mg/kg divided three times a day over 5 days followed by titration to oral prednisolone dosed at 2 mg/kg/day until resolution of fever and improvement in inflammatory markers. The median duration of steroid therapy in the RAISE study was 21 days. This is a significant increase in total steroid dose as compared to the single dose of 30 mg/kg methylprednisolone in the Newburger study. The earlier initiation of IVIG and corticosteroid therapy with subsequent increased steroid treatment duration was associated with significantly lower rates of coronary artery abnormalities in the RAISE study. This may be a result of early suppression of vasculitis that precedes vascular remodeling due to early initiation of corticosteroid therapy. The addition of corticosteroid therapy to the initial treatment regimen of KD is not the standard of care at this time.
Table 2.
Kobayashi risk score and risk of nonresponse to intravenous immune globulin treatment
| Risk factor | Points |
| Sodium ≤133 mmol/L | 2 |
| Aspartate aminotransferase ≥ 100 IU/L | 2 |
| Days of illness at initial treatment ≤4 | 2 |
| Neutrophil percent ≥80 % | 2 |
| C-reactive protein ≥10 mg/dL | 1 |
| Age ≤12 months | 1 |
| Platelet count ≤30 × 104/mm3 | 1 |
| Risk stratification | Nonresponse rate to intravenous immune globulin treatment |
| Low risk (score 0–3) | 5 % |
| High risk (score >4) | 43 % |
Infliximab
Infliximab (Remicaide), a human monoclonal antibody against tumor necrosis factor (TNF)-α, can be used in the treatment of refractory Kawasaki disease. During the acute phase of KD, TNF-α is markedly elevated and degree of elevation has been correlated to the development of coronary artery abnormalities [43]. The most commonly used regimen is one infliximab infusion dosed at 5 mg/kg.
Two retrospective reviews of patients with refractory KD treated with infliximab showed complete resolution of symptoms in majority of patients with refractory KD following administration of infliximab [44, 45]. Additionally, a recent pilot study randomized 43 patients with KD refractory to initial IVIG infusion to receive either repeat IVIG (2 gm/kg) or infliximab (5 mg/kg). Treatment with infliximab was associated with shortened duration of fever and lengths of hospitalization and possible reduction in the need for retreatment [46•]. The effects of infliximab on the development of coronary artery abnormalities in KD remain unclear. This study did not show reduction in the development of coronary artery abnormalities. However, regression in both the progression of coronary artery dilation/formation [44] and the resolution of dilated coronary arteries [45] following infliximab infusion have been reported. Therefore, infliximab may inhibit progression of coronary artery dilation and injury rather than prevent the formation of coronary artery abnormalities [46•].
In a phase 3 randomized, double-blind placebo-controlled trial, the addition of infliximab did not reduce treatment resistance of initial therapy but did shorten fever duration, time to normalization of inflammatory markers (CRP, ESR), and had a larger decrease in the Z score of the left anterior descending coronary artery [47•]. Currently, there is no indication for the addition of infliximab to the initial treatment regimen of uncomplicated KD.
Cyclosporin A
Cyclosporin A (CsA) is a calcineurin inhibitor, which targets the Ca2+/NFAT signaling pathway. This pathway is associated with immune hyper-reactivity. Unresponsiveness to IVIG therapy and coronary artery abnormalities in KD patients have been associated with single nucleotide polymorphisms of genes (ITPKC, CASP3) which downregulate the Ca2+/NFAT signaling pathway. Therefore, inhibition of the Ca Ca2+/NFAT signaling pathway through CsA may be considered in patients with refractory KD.
In a retrospective review of 19 patients with refractory KD treated with CsA, dosed at 4 mg/kg/day for 14 days, 14/19 patients became afebrile within 5 days. In these patients, significantly lower serum inflammatory cytokines were found [48]. A phase III multicenter, randomized, open-label, blinded-end point trial to evaluate the efficacy and safety of immunoglobulin plus cyclosporin A in patients with severe KD (KAICA trial) is currently ongoing in Japan. The trial aims to compare IVIG with concurrent CsA therapy to IVIG therapy alone in patients with severe KD defined as a Kobayashi risk score scale >5. The primary endpoint is the frequency of coronary artery abnormalities at 12 week follow up [49•]. The dose of CsA in the KAICA trial is 5 mg/kg/day with a target blood concentration level of 60–200 ng/mL for a total of 5 days.
Anticoagulation
Prevention of thrombosis in patients with KD varies with the extent of the coronary artery abnormality present in the patient. Low-dose ASA is appropriate for asymptomatic patients with mild to stable disease. However, as the severity of coronary artery enlargement increase, the combination of ASA with other antiplatelet agents such as clopidogrel is warranted. Additional anticoagulation is necessary with further coronary aneurysm expansion. Frequently, warfarin is added to low-dose aspirin for patients with giant aneurysms. Low molecular weight heparin has also been used in place of warfarin by some physicians.
Early Clinical Trials
The efficacy of other anti-inflammatory agents in the treatment of KD is currently being investigated. Of particular interest is the efficacy of these new agents in the KD complicated by coronary artery abnormalities. The current treatment regimen of IVIG and aspirin is well documented to reduce the incidence of coronary artery abnormalities in KD, but effective agents in the prevention of progression of coronary artery abnormalities are currently under investigation.
Anakinra
The IL-1 inflammatory pathway is upregulated in KD and involved in coronary artery inflammation. Blockade of this pathway in mouse model of KD has been shown to prevent the development of coronary artery abnormalities [50]. Anakinra is an IL-1 receptor antagonist which has be used in rare cases of refractory KD [51, 52].
The ANAKID trial, phase I/IIa trial of anakinra in KD patients with coronary artery abnormalities, is ongoing in the USA. Subjects will receive a 2–6-week course of daily anakinra injections. The study aims to evaluate the safety and tolerability of anakinra to prevent/attenuate coronary artery damage in patients with KD [53•]. The KAWAKINRA trial, a phase IIa multicenter trial of anakinra in KD patients who fail to respond to initial IVIG therapy (persistent fever up to 48 h after infusion), is currently enrolling in Europe (Clinicaltrials.gov NCT02390596).
Atorvastatin
In addition to their well-documented cholesterol lowering effects, statins likely have anti-inflammatory and anti-oxidant effects. Statins have been associated with reduction in inflammatory markers such as CRP [54]. The Cholesterol and Recurrent Events (CARE) trial found a higher risk of recurrent myocardial infarction in patients with normal cholesterol levels who received placebo rather than pravastatin [55]. The anti-inflammatory, anti-oxidant, and endothelial healing properties of statins have been postulated to be beneficial in blocking the progression of coronary artery abnormalities in KD [56•]. Use of pravastatin in a 40-year-old patient with history of KD and giant left coronary artery aneurysm has been shown to have reduction in coronary artery inflammation [57•]. Additionally, treatment of 13 children with KD complicated by medium to giant coronary artery aneurysms with pravastatin resulted in improvement in endothelial function measured by flow-mediated dilation and reduction in CRP [58•]. Currently, there is a phase I/IIa dose escalation study of atorvastatin to determine its safety, pharamacokinetics, and activity in children with acute KD and coronary artery abnormalities (Clinicaltrials.gov NCT01431105).
Conclusion
The diagnosis and initial treatment of KD with IVIG and aspirin remains unchanged from the 2004 American Heart Association (AHA) treatment guidelines. However, diagnostic challenges exist, particularly in the presentation of incomplete KD, due to overlap of clinical features of KD with other common pediatric illnesses. Prompt treatment with IVIG and aspirin remains the mainstay of treatment. Treatment options for refractory KD include repeat IVIG, corticosteroids, TNF-α inhibitors, calcineurin inhibitors, and IL-1 inhibitor. Research on the use of other agents in patients with KD is ongoing, and results may guide future treatment.
Compliance with Ethical Standards
Conflict of Interest
Drs Zhu and Ang have no conflicts of interests to declare.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Footnotes
This article is part of the Topical Collection on Pediatric Infectious Diseases
Contributor Information
Frank H. Zhu, Email: fzhu@dmc.org
Jocelyn Y. Ang, Phone: 313-745-5863, Email: jang@med.wayne.edu
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Kawasaki T, et al. A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan. Pediatrics. 1974;54(3):271–6. [PubMed] [Google Scholar]
- 2.Kato H, et al. Long-term consequences of Kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation. 1996;94(6):1379–85. doi: 10.1161/01.CIR.94.6.1379. [DOI] [PubMed] [Google Scholar]
- 3.Taubert KA, Rowley AH, Shulman ST. Nationwide survey of Kawasaki disease and acute rheumatic fever. J Pediatr. 1991;119(2):279–82. doi: 10.1016/S0022-3476(05)80742-5. [DOI] [PubMed] [Google Scholar]
- 4.Newburger JW, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315(6):341–7. doi: 10.1056/NEJM198608073150601. [DOI] [PubMed] [Google Scholar]
- 5.•.Rodo X, et al. Tropospheric winds from northeastern China carry the etiologic agent of Kawasaki disease from its source to Japan. Proc Natl Acad Sci U S A. 2014;111(22):7952–7. doi: 10.1073/pnas.1400380111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.••.Newburger JW, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114(6):1708–33. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 7.•.Singh S, Vignesh P, Burgner D. The epidemiology of Kawasaki disease: a global update. Arch Dis Child. 2015;100(11):1084–8. doi: 10.1136/archdischild-2014-307536. [DOI] [PubMed] [Google Scholar]
- 8.Uehara R, Belay ED. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J Epidemiol. 2012;22(2):79–85. doi: 10.2188/jea.JE20110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holman RC, et al. Racial/ethnic differences in the incidence of Kawasaki syndrome among children in Hawaii. Hawaii Med J. 2010;69(8):194–7. [PMC free article] [PubMed] [Google Scholar]
- 10.Sundel RP. Kawasaki disease. Rheum Dis Clin North Am. 2015;41(1):63–73. doi: 10.1016/j.rdc.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Rowley AH. Kawasaki disease: novel insights into etiology and genetic susceptibility. Annu Rev Med. 2011;62:69–77. doi: 10.1146/annurev-med-042409-151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi K, Oharaseki T, Yokouchi Y. Update on etio and immunopathogenesis of Kawasaki disease. Curr Opin Rheumatol. 2014;26(1):31–6. doi: 10.1097/BOR.0000000000000010. [DOI] [PubMed] [Google Scholar]
- 13.Yim D, et al. Update on Kawasaki disease: epidemiology, aetiology and pathogenesis. J Paediatr Child Health. 2013;49(9):704–8. doi: 10.1111/jpc.12172. [DOI] [PubMed] [Google Scholar]
- 14.Orenstein JM, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS One. 2012;7(6):e38998. doi: 10.1371/journal.pone.0038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki A, et al. Active remodeling of the coronary arterial lesions in the late phase of Kawasaki disease: immunohistochemical study. Circulation. 2000;101(25):2935–41. doi: 10.1161/01.CIR.101.25.2935. [DOI] [PubMed] [Google Scholar]
- 16.Sasaguri Y, Kato H. Regression of aneurysms in Kawasaki disease: a pathological study. J Pediatr. 1982;100(2):225–31. doi: 10.1016/S0022-3476(82)80639-2. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi M, Mason W, Lewis AB. Regression of coronary aneurysms in patients with Kawasaki syndrome. Circulation. 1987;75(2):387–94. doi: 10.1161/01.CIR.75.2.387. [DOI] [PubMed] [Google Scholar]
- 18.•.Taddio, A., et al., Describing Kawasaki shock syndrome: results from a retrospective study and literature review. Clin Rheumatol, 2016. doi:10.1007/s10067-016-3316-8. This retrospective review compares the main clinical presentation, echocardiogram and laboratory findings as well as treatment options of Kawasaki shock syndrome which is a rare presentation of Kawasaki disease. [DOI] [PubMed]
- 19.Edwards KM, et al. Adenovirus infections in young children. Pediatrics. 1985;76(3):420–4. [PubMed] [Google Scholar]
- 20.Fairchok MP, et al. Epidemiology of viral respiratory tract infections in a prospective cohort of infants and toddlers attending daycare. J Clin Virol. 2010;49(1):16–20. doi: 10.1016/j.jcv.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.•.Song E, et al. Clinical and virologic characteristics may aid distinction of acute adenovirus disease from Kawasaki disease with incidental adenovirus detection. J Pediatr. 2016;170:325–30. doi: 10.1016/j.jpeds.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 22.•.Kanegaye, J.T., et al., Lymph-node-first presentation of Kawasaki disease compared with bacterial cervical adenitis and typical Kawasaki disease. J Pediatr, 2013. 162(6): p. 1259–63, 1263 e1-2. This retrospective review identifies characteristics differentiating the node-first presentation of Kawasaki disease from bacterial cervical lymphadenitis and typical Kawasaki disease to aid the recognization and treatment of this rare presentation of Kawasaki disease. [DOI] [PMC free article] [PubMed]
- 23.Ganesh R, et al. Kawasaki disease mimicking retropharyngeal abscess. Yonsei Med J. 2010;51(5):784–6. doi: 10.3349/ymj.2010.51.5.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hung MC, et al. Kawasaki disease resembling a retropharyngeal abscess—case report and literature review. Int J Cardiol. 2007;115(2):e94–6. doi: 10.1016/j.ijcard.2006.08.095. [DOI] [PubMed] [Google Scholar]
- 25.•.Nomura O, et al. Comparison of patients with Kawasaki disease with retropharyngeal edema and patients with retropharyngeal abscess. Eur J Pediatr. 2014;173(3):381–6. doi: 10.1007/s00431-013-2179-0. [DOI] [PubMed] [Google Scholar]
- 26.Dogra S, et al. Incomplete Kawasaki disease followed by systemic onset juvenile idiopathic arthritis—the diagnostic dilemma. Indian J Pediatr. 2013;80(9):783–5. doi: 10.1007/s12098-012-0893-7. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S, et al. Systemic onset juvenile idiopathic arthritis with macrophage activation syndrome misdiagnosed as Kawasaki disease: case report and literature review. Rheumatol Int. 2013;33(4):1065–9. doi: 10.1007/s00296-010-1650-8. [DOI] [PubMed] [Google Scholar]
- 28.•.Lin YJ, et al. Early differentiation of Kawasaki disease shock syndrome and toxic shock syndrome in a pediatric intensive care unit. Pediatr Infect Dis J. 2015;34(11):1163–7. doi: 10.1097/INF.0000000000000852. [DOI] [PubMed] [Google Scholar]
- 29.Kanegaye JT, et al. Recognition of a Kawasaki disease shock syndrome. Pediatrics. 2009;123(5):e783–9. doi: 10.1542/peds.2008-1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki A, et al. Tricuspid and mitral regurgitation detected by color flow Doppler in the acute phase of Kawasaki disease. Am J Cardiol. 1988;61(4):386–90. doi: 10.1016/0002-9149(88)90950-2. [DOI] [PubMed] [Google Scholar]
- 31.Gamez-Gonzalez LB, et al. Clinical manifestations associated with Kawasaki disease shock syndrome in Mexican children. Eur J Pediatr. 2013;172(3):337–42. doi: 10.1007/s00431-012-1879-1. [DOI] [PubMed] [Google Scholar]
- 32.Durongpisitkul K, et al. The prevention of coronary artery aneurysm in Kawasaki disease: a meta-analysis on the efficacy of aspirin and immunoglobulin treatment. Pediatrics. 1995;96(6):1057–61. [PubMed] [Google Scholar]
- 33.Furusho K, et al. High-dose intravenous gammaglobulin for Kawasaki disease. Lancet. 1984;2(8411):1055–8. doi: 10.1016/S0140-6736(84)91504-6. [DOI] [PubMed] [Google Scholar]
- 34.Klassen TP, Rowe PC, Gafni A. Economic evaluation of intravenous immune globulin therapy for Kawasaki syndrome. J Pediatr. 1993;122(4):538–42. doi: 10.1016/S0022-3476(05)83532-2. [DOI] [PubMed] [Google Scholar]
- 35.Muta H, et al. Early intravenous gamma-globulin treatment for Kawasaki disease: the nationwide surveys in Japan. J Pediatr. 2004;144(4):496–9. doi: 10.1016/j.jpeds.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 36.Catella-Lawson F, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345(25):1809–17. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- 37.Lee JH, Hung HY, Huang FY. Kawasaki disease with Reye syndrome: report of one case. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1992;33(1):67–71. [PubMed] [Google Scholar]
- 38.Burns JC, et al. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J. 1998;17(12):1144–8. doi: 10.1097/00006454-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 39.Hashino K, et al. Re-treatment for immune globulin-resistant Kawasaki disease: a comparative study of additional immune globulin and steroid pulse therapy. Pediatr Int. 2001;43(3):211–7. doi: 10.1046/j.1442-200x.2001.01373.x. [DOI] [PubMed] [Google Scholar]
- 40.Newburger JW, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. N Engl J Med. 2007;356(7):663–75. doi: 10.1056/NEJMoa061235. [DOI] [PubMed] [Google Scholar]
- 41.•.Kobayashi T, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): a randomised, open-label, blinded-endpoints trial. Lancet. 2012;379(9826):1613–20. doi: 10.1016/S0140-6736(11)61930-2. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi T, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113(22):2606–12. doi: 10.1161/CIRCULATIONAHA.105.592865. [DOI] [PubMed] [Google Scholar]
- 43.Matsubara T, Furukawa S, Yabuta K. Serum levels of tumor necrosis factor, interleukin 2 receptor, and interferon-gamma in Kawasaki disease involved coronary-artery lesions. Clin Immunol Immunopathol. 1990;56(1):29–36. doi: 10.1016/0090-1229(90)90166-N. [DOI] [PubMed] [Google Scholar]
- 44.Song MS, et al. Infliximab treatment for refractory kawasaki disease in korean children. Korean Circ J. 2010;40(7):334–8. doi: 10.4070/kcj.2010.40.7.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burns JC, et al. Infliximab treatment for refractory Kawasaki syndrome. J Pediatr. 2005;146(5):662–7. doi: 10.1016/j.jpeds.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 46.•.Youn Y, et al. Infliximab as the first retreatment in patients with Kawasaki disease resistant to initial intravenous immunoglobulin. Pediatr Infect Dis J. 2016;35(4):457–9. doi: 10.1097/INF.0000000000001039. [DOI] [PubMed] [Google Scholar]
- 47.•.Tremoulet AH, et al. Infliximab for intensification of primary therapy for Kawasaki disease: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet. 2014;383(9930):1731–8. doi: 10.1016/S0140-6736(13)62298-9. [DOI] [PubMed] [Google Scholar]
- 48.Hamada H, et al. Inflammatory cytokine profiles during Cyclosporin treatment for immunoglobulin-resistant Kawasaki disease. Cytokine. 2012;60(3):681–5. doi: 10.1016/j.cyto.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 49.•.Aoyagi R, et al. Study protocol for a phase III multicentre, randomised, open-label, blinded-end point trial to evaluate the efficacy and safety of immunoglobulin plus cyclosporin A in patients with severe Kawasaki disease (KAICA Trial) BMJ Open. 2015;5(12):e009562. doi: 10.1136/bmjopen-2015-009562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee Y, et al. Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation. 2012;125(12):1542–50. doi: 10.1161/CIRCULATIONAHA.111.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell AJ, Burns JC. Adjunctive therapies for Kawasaki disease. J Infect. 2016;72(Suppl):S1–5. doi: 10.1016/j.jinf.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 52.Shafferman A, Birmingham JD, Cron RQ. High dose Anakinra for treatment of severe neonatal Kawasaki disease: a case report. Pediatr Rheumatol Online J. 2014;12:26. doi: 10.1186/1546-0096-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.•.Tremoulet AH, et al. Rationale and study design for a phase I/IIa trial of anakinra in children with Kawasaki disease and early coronary artery abnormalities (the ANAKID trial) Contemp Clin Trials. 2016;48:70–75. doi: 10.1016/j.cct.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ridker PM, et al. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100(3):230–5. doi: 10.1161/01.CIR.100.3.230. [DOI] [PubMed] [Google Scholar]
- 55.Ridker PM, et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1998;98(9):839–44. doi: 10.1161/01.CIR.98.9.839. [DOI] [PubMed] [Google Scholar]
- 56.•.Tremoulet AH. The role of statins in inflammatory vasculitides. Autoimmunity. 2015;48(3):177–80. doi: 10.3109/08916934.2015.1027818. [DOI] [PubMed] [Google Scholar]
- 57.•.Suda K, et al. Statin reduces persistent coronary arterial inflammation evaluated by serial (1)(8)fluorodeoxyglucose positron emission tomography imaging long after Kawasaki disease. Int J Cardiol. 2015;179:61–2. doi: 10.1016/j.ijcard.2014.10.057. [DOI] [PubMed] [Google Scholar]
- 58.•.Duan, C., et al., Effect of pravastatin on endothelial dysfunction in children with medium to giant coronary aneurysms due to Kawasaki disease. World J Pediatr, 2014. doi:10.1007/s12519-014-0465-1.This small prospective study documents improvement in endothelial function and reduction in low-grade chronic inflammation in patients with coronary aneurysms due to Kawasaki disease following treatment with pravastatin.