

Abstract
Bats have great potential as reservoirs for emerging viruses such as severe acute respiratory syndrome-coronavirus. In this study, bat coronaviruses (BtCoVs) were detected by RT-PCR from intestinal and fecal specimens of Miniopterus fuliginosus breeding colonies in Wakayama Prefecture caves, where we previously identified bat betaherpesvirus 2. Two primer sets were used for the detection of BtCoV: one was for the RNA-dependent RNA polymerase (RdRp) region and the other was for the spike (S) protein region. Eleven and 73% of intestinal and fecal specimens, respectively, were positive for RdRp region, and 2 and 40% of those were positive for S protein region. Sequencing and phylogenetic analysis showed that the detected BtCoV belonged to the group 1 (alpha) coronaviruses. These data suggest that BtCoV is endemic in M. fuliginosus in Japan.
Keywords: Bats, Coronavirus, Miniopterus fuliginosus, Virus detection
Introduction
An outbreak of severe acute respiratory syndrome (SARS) that was caused by a newly identified SARS coronavirus (SARS-CoV) occurred in China, Singapore, Vietnam, and other countries from 2002 to 2003; 8,098 patients were reported, 774 of whom died [1]. At first, SARS-CoV was considered to be derived from civets and raccoon dogs [2]. However, recent studies have suggested that SARS-CoV is a recombinant of bat-derived coronaviruses [3, 4]. Bat coronaviruses (BtCoVs) have now been identified in China [3, 4] and other countries such as USA [5], Germany [6], Kenya [7], and the Philippines [8]; however, there has been no report of BtCoV detection in Japan. Bats have also great potential as reservoirs for emerging viruses such as Ebola and Nipah [9, 10]. We have recently identified novel viruses from bats, such as bat betaherpesvirus 2 [11] and bat adenovirus 1 [12], in Japan. To confirm the role of bats as host species for viruses, we attempted to detect BtCoV from Miniopterus fuliginosus using intestinal and fecal specimens from specific caves located in the Wakayama prefecture, Japan.
Materials and methods
Bat samples
All bats and fecal specimens were collected in May 2009 and July 2010 from a headrace tunnel and a cave in the Wakayama Prefecture, Japan, with permission from government officials. Those are well-known habitats of M. fuliginosus; the headrace tunnel is a roosting place of male and non-breeding female bats and the cave is a breeding place, where we collected bats and identified a novel virus in a previous study [11, 13]. The following three methods were used to collect intestinal specimens: (1) Captured bats were euthanized using an overdose of anesthetic and small intestines were collected without washing their contents, (2) Forty-five bats were captured with a net and placed in a bag, after which fresh fecal samples were collected, placed in a solution of RNAlater (Ambion, Foster City, CA, USA), and separated into four pools. After fecal samples were collected, the bats were released safely, (3) Three traps were placed under a mass of bats that were hanging from the ceiling of each cave. The next day, fecal samples were collected, placed in RNAlater, and separated into 11 pools.
Extraction of RNA and RT-PCR
Total RNA was first extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and was re-extracted using the QIAampViral RNA Mini Kit (Qiagen, Hilden, Germany). The cDNA was synthesized using M-MLV Reverse Transcriptase (Invitrogen, San Diego, CA, USA) and random hexamers, and two-step reverse transcription PCR was performed using PrimeSTAR Max (Takara Bio, Shiga, Japan) with the default conditions of each master mix, except the annealing temperature, which was 48°C. Two primer sets were used for the detection of BtCoV; primer set for the first PCR: IN-6 primer, 5′-GGTTGGGACTATCCTAAGTGTGA-3′ and IN-7 primer, 5′-CCATCATCAGATAGAATCATCAT-3′ [14]; primer set for the second PCR: IN-6 primer and hemi-nested reverse primer, 5′-ATCAGATAGAATCATCATAGAGA-3′ [15], were used for detection of the sequence in RNA-dependent RNA polymerase (RdRp) region. These primer sets are known to be able to amplify genes of the coronavirus family including BtCoV [5, 8, 14, 16–18]. To amplify the spike (S) region sequence, consensus primers were constructed using the online consensus primer design software (CoCoMo; http://www.geneknot.jp/cocomo/) with seven sequences deposited in GenBank. The constructed primer sets for S protein region were as follows: No. 6, 5′-NSHRYKTATGTHTGYAAYGGHAA-3′ and No. 2 5′-DGAYTGBGAYTTDACACAYTCRTT-3′ were used for the first PCR; No. 1, 5′-HTGTGYBCAGYAYTAYAAYGGYAT-3′ and No. 2 were used for the second PCR. After amplification, amplicons were detected by 1.5% agarose gel electrophoresis.
Phylogenetic analysis
The phylogenetic analysis was performed as follows: obtained sequence data were carefully checked for peaks and 327 bp (RdRp) and 547 bp (S) of each amplicon, except primer sequences were selected to construct phylogenetic trees of nucleotide sequences. Five different sequences were obtained for the RdRp region (accession numbers AB619638–AB619642) and two sequences were obtained for S region (accession numbers AB644273 and AB644274). The same region of other available coronaviral sequences (BtCoV HKU7-1, DQ249226; BtCoV HKU8-1, DQ249228; BtCoV 1A, NC_010437; BtCoV 1B, NC_010436; BtCoV Fujian/773/2005, EF434379; HCoV229E ATCC, NC_002645; HCoVNL63, NC_005831; PEDV CV777, NC_003436; AIBV Beaudette GK, AJ311317; MHV JHM, NC_006852; SARSr-Rh-BatCoV HKU3-7, GQ153542; SARS Frankfurt1, AY291315; civet SARSr-CoV PC4-139, AY613949; and EU420139, BtCoV HKU8) were used to create the phylogenetic trees. The bootstrap test for the phylogenetic tree was constructed using MEGA4 software [19] with the neighbor-joining method with a pairwise deletion option.
Results
As shown in Table 1, PCR products at the expected sizes were obtained from 5/45 (11%) of intestinal and 11/15 (73%) of fecal specimens with the RdRp primer set, and 1/45 of intestinal and 6/15 (40%) of fecal specimens were positive with the S protein primer set. PCR products were gel purified and sequence analysis was performed with specific-sequence primer. The sequences of PCR products showed some similarities to the deposited BtCoV sequences; these sequences were analyzed by a BLASTn search (http://blast.ddbj.nig.ac.jp/top-e.html) and matched to nucleotide sequences of BtCoVs. These suggest that the detected sequences were derived from newly identified BtCoV sequences. Because the PCR product was detected not only in the feces pool but also in the small intestines, these results strongly suggest that M. fuliginosus in Japan is definitely infected by BtCoV.
Table 1.
Detection of BtCoV from bat specimens
| Region | Specimens | RT-PCR | Sequence type | Number | Total |
|---|---|---|---|---|---|
| RdRp | Intestine | 5/45 (11%) | BtCoV M.ful./Japan/01/2009 | 4 | 5/45 (11%) |
| BtCoV M.ful./Japan/02/2009 | 1 | ||||
| Feces from | |||||
| Captured bat | 2/4 (50%) | BtCoV M.ful./Japan/01/2010 | 1 | 11/15 (73%) | |
| BtCoV M.ful./Japan/03/2010 | 1 | ||||
| Trap | 9/11 (82%) | BtCoV M.ful./Japan/01/2010 | 7 | ||
| BtCoV M.ful./Japan/02/2010 | 2 | ||||
| S | Intestine | 1/45 (2%) | BtCoV M.ful./Japan/03/2009 | 1 | 1/45 (2%) |
| Feces from | |||||
| Captured bat | 1/4 (25%) | BtCoV M.ful./Japan/04/2010 | 1 | 6/15 (40%) | |
| Trap | 5/11 (45%) | BtCoV M.ful./Japan/04/2010 | 4 | ||
Obtained sequence data were carefully checked for peaks and 327 bp for the RdRp region and 547 bp for the S protein region of each amplicon, except primer sequences, were selected to construct phylogenetic trees of nucleotide sequences. Five different sequences of BtCoVs were detected in RT-PCR products of the RdRp region and two sequences were detected from RT-PCR products of the S protein region, and they were named in accordance with the rules for nomenclature of influenza virus, that is, BtCoV host species/isolated place/isolation number/isolated year. The viral genomic information was deposited in DDBJ/EMBL/GenBank (accession numbers AB619638–AB619642 and AB644273–AB644274). The phylogenetic trees were constructed along with other available coronaviral sequences deposited in GenBank using the MEGA4 software [19] (Fig. 1a: RdRp region, b: S region). The sequences detected from M. fuliginosus in Japan were classified into group 1 (alpha) coronavirus of BtCoVs and were quite different from SARS-related BtCoVs. In the RdRp region, BtCoVs in Japan are closely related to BtCoV HKU7-1, which was isolated from M. magnater in Hong Kong in 2006 [18], with 95% of nucleotides identified. BtCoV HKU8-1, which was isolated from M. pusillus in Hong Kong in 2006 [18], was the second related strain, but fewer nucleotides were identified (80%) than in HKU7-1. In the S protein region, M.ful./Japan/03/2009 showed 92% identity relative to BtCoV Fujian/773/2005 (GenBank EF434379), which was isolated in the Fujian province in China in 2005 [20]. In contrast, M.ful./Japan/04/2010 showed about 70% similarity with the HKU8 strain and other group 1 BtCoVs (1a and 1b), which were isolated in Hong Kong from 2004 to 2005 [21]. Both of two isolated Japanese sequences belonged to the group 1 (alpha) coronaviruses, but the identity between them was only 64%. Therefore, this suggests that different BtCoV strains were present in the same cave. We could not compare nucleotide sequences of the S gene between Japanese BtCoV and HKU 7-1 because the sequence for HKU 7-1 was not available in GenBank.
Fig. 1.
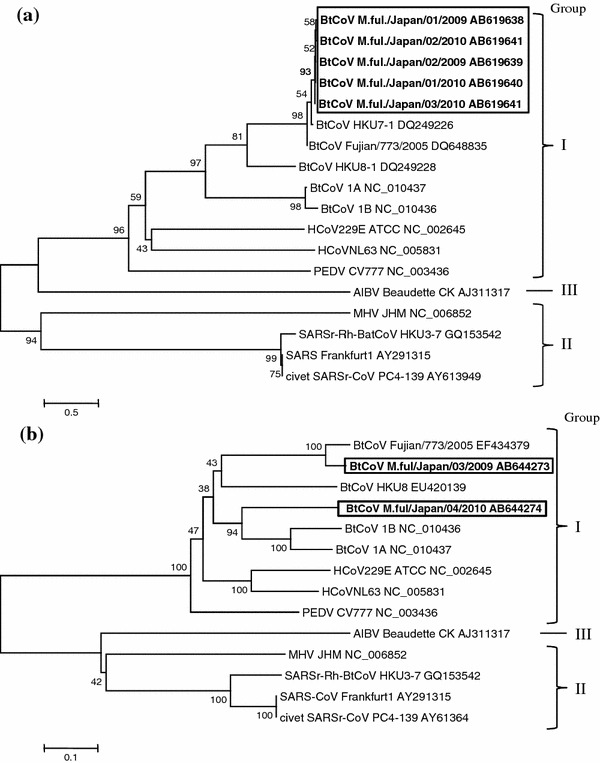
The phylogenetic trees based on the nucleotide sequence of a RdRp and b S regions. The phylogenetic trees were constructed with the MEGA4 software using the bootstrap test command with the neighbor-joining method. The accession numbers of used coronaviral sequences were described in “Materials and methods” section
Discussion
In this study, 45 intestinal specimens and 15 pools of fecal specimens were tested for BtCoV with specific primer sets for RdRp and S regions. The number of PCR-positive pools was high: 5/45 (11%) of intestinal and 11/15 (73%) fecal specimens were positive with the primer set for the RdRp region and 1/45 (2%) of intestinal and 6/15 (40%) fecal specimens were positive with the primer set for the S protein region. The specimens were collected from two places; one was a habitat of M. fuligunosus, which lives in the Kinki region of Japan, and more than 20,000 female bats gather in the cave during the breeding season [13]. This suggests that BtCoV is endemic at a high frequency in M. fuliginosus in the Kinki region of Japan. In addition, BtCoVs in Japan belong to the group 1 (alpha) coronaviruses, and phylogenetic analysis of the RdRp and S protein regions indicated that it is quite different from SARS-related CoV. In particular, S protein of coronavirus is a major determinant of receptor binding and virus–cell membrane fusion [22]. Therefore, the differences in S protein region sequence imply the possibility that Japanese BtCoVs have different infectivity to host species compared to other SARS-related BtCoVs. Moreover, the S protein sequences of M.ful./Japan/03/2009 and M.ful./Japan/04/2010 showed 64% identity between them. This also suggests that different strains of BtCoVs are endemic in the same place, like several different strains of human coronaviruses.
The nucleotide sequences of RdRp of BtCoV in Japan were similar (95%) to those of HKU7-1, which was isolated from M. magnater in Hong Kong, and the nucleotide sequences of S protein of Japanese BtCoVs also showed some similarity to other Hong Kong and Fujian strains. Miniopterus bats are a migratory species and one population has several habitats surrounding its central breeding cave [23]. The distance of bat migration is reported to be several 100 km [13, 24]. This implies the possibility that BtCoV-infected bats have been brought to Japan from Southeast China (or to there from Japan) through unknown routes. In addition, Rodrigues and Palmeirim [24] have reported that female bats nearly always return to their natal colony to give birth, whereas male bats sometimes go to other colonies, suggesting that BtCoV is transmitted between Japan and Hong Kong or Fujian by male bats. Similar cases have been reported for rabies virus. Rabies virus infection is associated with the migratory routes of Nathusius’ pipistrelle (Pipistrellus nathusii) in France. It has also been reported that silver-haired bats (Lasionycteris noctivagans) migrate seasonally from Alaska, across Canada, to Texas, and rabies virus variants have been identified from several locations throughout the geographic range of these bats [25]. Therefore, BtCoV might be transmitted between Hong Kong and Japan as a result of bat migration.
Acknowledgments
We thank Mrs. Momoko Ogata and Miyuki Kawase for their assistance. This study was supported in part by a Grant from the Japan Society for the Promotion of Science, by the Ministry of Health, Labour, and Welfare, Japan.
Footnotes
Kazuya Shirato, Ken Maeda, and Shumpei Tsuda contributed equally to this study.
References
- 1.Peiris JS, Guan Y, Yuen KY. Nat. Med. 2004;10:S88–S97. doi: 10.1038/nm1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 3.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 4.Lau SK, Li KS, Huang Y, Shek CT, Tse H, Wang M, Choi GK, Xu H, Lam CS, Guo R, Chan KH, Zheng BJ, Woo PC, Yuen KY. J. Virol. 2010;84:2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dominguez SR, O’Shea TJ, Oko LM, Holmes KV. Emerg. Infect. Dis. 2007;13:1295–1300. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, Petersen N, Annan A, Grywna K, Muller M, Pfefferle S, Drosten C. Emerg. Infect. Dis. 2008;14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE. Emerg. Infect. Dis. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe S, Masangkay JS, Nagata N, Morikawa S, Mizutani T, Fukushi S, Alviola P, Omatsu T, Ueda N, Iha K, Taniguchi S, Fujii H, Tsuda S, Endoh M, Kato K, Tohya Y, Kyuwa S, Yoshikawa Y, Akashi H. Emerg. Infect. Dis. 2010;16:1217–1223. doi: 10.3201/eid1608.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, bin Adzhar A, White J, Daniels P, Jamaluddin A, Ksiazek T. Emerg. Infect. Dis. 2001;7:439–441. doi: 10.3201/eid0703.010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leroy EM, Kumulungui B, Pourrut X, Rouquet P, Hassanin A, Yaba P, Delicat A, Paweska JT, Gonzalez JP, Swanepoel R. Nature. 2005;438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe S, Maeda K, Suzuki K, Ueda N, Iha K, Taniguchi S, Shimoda H, Kato K, Yoshikawa Y, Morikawa S, Kurane I, Akashi H, Mizutani T. Emerg. Infect. Dis. 2010;16:986–988. doi: 10.3201/eid1608.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeda K, Hondo E, Terakawa J, Kiso Y, Nakaichi N, Endoh D, Sakai K, Morikawa S, Mizutani T. Emerg. Infect. Dis. 2008;14:347–349. doi: 10.3201/eid1402.070932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H, Maeda K, Inoue R, Suzuki K, Sano A, Tsumura M, Hashimoto H, Teranishi T, Okumura K, Abe Y. Bull. Center Nat. Environ. Ed. Nara Univ. Educ. 2005;7:31–37. [Google Scholar]
- 14.Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Lau SK, Woo PC, Suen KY, Yuen KY, Guan Y, Peiris JS. J. Virol. 2005;79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu DK, Poon LL, Chan KH, Chen H, Guan Y, Yuen KY, Peiris JS. J. Gen. Virol. 2006;87:2461–2466. doi: 10.1099/vir.0.82203-0. [DOI] [PubMed] [Google Scholar]
- 16.Chiu SS, Chan KH, Chu KW, Kwan SW, Guan Y, Poon LL, Peiris JS. Clin. Infect. Dis. 2005;40:1721–1729. doi: 10.1086/430301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, Yip BC, Huang Y, Chan KH, Yuen KY. Virology. 2006;351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura K, Dudley J, Nei M, Kumar S. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 20.Vijaykrishna D, Smith GJ, Zhang JX, Peiris JS, Chen H, Guan Y. J. Virol. 2007;81:4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu DK, Peiris JS, Chen H, Guan Y, Poon LL. J. Gen. Virol. 2008;89:1282–1287. doi: 10.1099/vir.0.83605-0. [DOI] [PubMed] [Google Scholar]
- 22.Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramos Pereira MJ, Salgueiro P, Rodrigues L, Coelho MM, Palmeirim JM. J. Hered. 2009;100:533–544. doi: 10.1093/jhered/esp032. [DOI] [PubMed] [Google Scholar]
- 24.Rodrigues L, Palmeirim JM. J. Zool. 2008;274:116–125. doi: 10.1111/j.1469-7998.2007.00361.x. [DOI] [Google Scholar]
- 25.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Clin. Microbiol. Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]