

Abstract
Porcine epidemic diarrhea virus (PEDV) is a pathogen of swine that causes severe diarrhea and dehydration resulting in substantial morbidity and mortality in newborn piglets. Phage display is a technique with wide application, in particular, the identification of key antigen epitopes for the development of therapeutic and diagnostic reagents and vaccines. To identify antigen epitopes with specificity for PEDV, a monoclonal antibody (MAb-5E12) against the immunodominant region of the PEDV Spike protein (S1) was used as the target for biopanning a 12-mer phage display, random peptide library. After multiple rounds of biopanning and stringent washing, three phage-displayed peptides, designated L, W and H, were identified that recognize MAb-5E12. Sequence analysis showed that the one or more of the peptides exhibited partial sequence similarity to the native S1 sequence ‘MQYVYTPTYYML’ (designated peptide M) at position 201–212. In combination with software analysis for the prediction of B cell epitopes, aa 201–212 exhibited characteristics of a linear epitope on the PEDV S1 protein. In contrast to peptide M, a consensus motif ‘PxxY’ was identified on both peptides L and W, and on the S1 protein, but not on peptide H. Peptide M and the MAb-5E12-recognizing peptides L and W significantly inhibited the adsorption of PEDV on the cell surface as monitored through plaque-reduction assays. Furthermore, data from real-time PCR and indirect immunofluorescence assays were consistent with the ability of peptides M, L and W to block viral protein expression and thereby function as antiviral agents for PEDV.
Keywords: Porcine epidemic diarrhea virus (PEDV), S1 protein, Monoclonal antibody, Putative epitope, Phage display technique
Introduction
Porcine epidemic diarrhea virus (PEDV) is a primary pathogen of the genus Alphacoronavirus of Coronaviridae family and Nidovirales order, that causes persistent diarrhea in swine [1]. The virus has been discovered in many countries of Europe, Asia, and South America [2–4], suggesting that it indeed is a cosmopolitan disease among swine producing countries. Although in recent years progress has been made in the development of vaccines to control and prevent disease, outbreaks continue to rise and pose problems to the swine industry and international trade.
PEDV is an enveloped, single-stranded, RNA virus approximately 28 kb in size, and encodes four structural proteins; the spike (S) protein, nucleocapsid (N) protein, membrane (M) protein and small envelope (E) protein [5]. Like other coronavirus, PEDV infects susceptible cells via interactions between the S protein and specific cellular receptors [6, 7]. The S protein is also the dominant viral antigen and induces neutralizing host antibodies and protective immunity [2, 8]. The PEDV S protein was subdivided into the S1 (aa 1–789) and S2 (aa 790–1383) domains. The globular part of S protein is formed by the S1 domain. It contains the major neutralizing and cellular receptor-binding residues, and plays an important role in mediating virus invasion of host cells [8, 9].
Phage display is a technique used to identify putative antigenic epitopes or other macromolecule-binding ligands by probing a random phage library expressing peptide ligands on the phage surface, with specific target molecules i.e., cell-surface receptors, enzymes, or viral or bacterial proteins. Reports have shown that small peptides derived from phage display libraries exhibit excellent prospects as therapeutic agents against these diseases [10, 11]. Using hybridoma technology, we developed a monoclonal antibody to the PEDV S1 protein (designed MAb-5E12), which binds to the recombinant PEDV S1 protein (PEDV rS1) and recognizes the native PEDV [12]. In this study, MAb-5E12 was used as the immobilized target to biopan a 12-mer phage display random peptide library. Ten positive phage clones that specifically bind to MAb-5E12 were identified and constitute three distinct peptide sequences. Upon alignment with the native S1 protein, results indicated high sequence similarity to a region of the PEDV S1 protein. Using bioinformatics to assess B cell epitopes on the PEDV S1 protein indicated that the motif 201MQYVYTPTYYML212 was a putative linear, antigenic epitope. Among the sequences identified, two phage-derived peptides were synthesized and found to inhibit PEDV adsorption in a dose-dependent manner and in so doing exhibited anti-viral activity.
Materials and methods
Cell, virus and reagents
An African green monkey kidney cell line (VeroE6) was cultured in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 8 % fetal bovine serum (FBS, US) at 37 °C, 5 % CO2. PEDV strain CV777 was kept in our laboratory, and propagated in VeroE6 cells in serum-free DMEM containing 2.5 µg/ml of trypsin. A monoclonal antibody 5E12 to the S1 protein of PEDV, and anti-PEDV rabbit polyclonal antibodies were previously generated in our laboratory [12]. A phage display peptide library kit (Ph.D.-12) was purchased from New England BioLabs (Hitchin, Hertfordshire, UK). Fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG, horseradish peroxidase (HRP)-conjugated goat anti-rabbit and anti-mouse IgG were purchased from the Zhongshan Company (Beijing, China).
Biopanning
To obtain high amount of the MAb-5E12, adult female BALB/c mice were injected with 106 hybridoma cells in 0.5 ml of DMEM. The ascites were produced within 1–2 weeks. The ascites were centrifuged at 12,000 rpm for 10 min, and the supernatant was purified by the caprylic acid–ammonium sulfate method as previously described [13]. Biopanning with a 12-mer phage display peptide library was done according to the manufacturer’s instructions with minor modifications [14]. During first to third rounds of panning, the purified MAb-5E12 was incubated with the crude phage library or with eluted phage (1.5 × 1011 pfu). In order to remove non-specific, antibody-binding molecules, in the fourth round of biopanning, an unrelated MAb kept in our laboratory was incubated with the third round-eluted phage. The unbound phage was subjected to a fifth round of panning at a concentration of 1.5 × 1011 pfu. The concentration of MAb-5E12 was reduced with each successive round of biopanning at 20, 15, 10 and 5 µg/well, respectively. The concentration of the unrelated MAb (Negative control, which against a recombinant VP7 protein of Porcine rotavirus, a normal mouse MAb, and has not cross-reactions with MAb-5E12) (round four) was 10 µg/well. Thirteen individual phage clones were picked after the last round of biopanning.
Binding affinity analyzed by direct and competitive ELISA
For direct ELISA, the plates were coated with 2.5 μg/well of MAb-5E12 or unrelated MAb in 0.1 M NaHCO3 (pH 8.6) at 4 °C overnight. The plates were blocked with 1 % bovine serum albumin (BSA) in TBS-0.1 % Tween (50 mM Tris buffered saline containing 0.1 % Tween, TBST) at 37 °C for 1 h, then subsequently incubated with the selected phage clone and wild-type M13 phage (1.5 × 1011 pfu/well). After four rounds of washing in TBST, the M13 polyclonal antibody (dilution 1:1000 in TBST; Abcam) was added, and the mix incubated for 1 h at 37 °C, then washed 4× and incubated with HRP-conjugated anti-rabbit IgG antibody (diluted 1:5000 in TBST). The plates were washed again, and developed using o-phenylenediamine for 10 min at room temperature. The reactions were stopped using 2 M H2SO4 and the OD490 values were determined. Competitive ELISA was performed under similar conditions with the following modifications: individual phage (1.5 × 1011 pfu) were mixed with MAb-5E12 (2.5 μg) prior to adding to the PEDV rS1 protein-coated plates (2.5 μg/well), and then incubated with the HRP-conjugated anti-mouse IgG antibody. The inhibition rate (%) was calculated as follows: {[OD490 MAb-5E12 (no phage) − OD490 MAb-5E12 (with phage)]/OD490 MAb-5E12 (no phage)} × 100.
Sequence alignment of phage-displayed peptides
The DNA from 10 selected phage clones was extracted and purified using iodide buffer (pH 8.0) and ethanol precipitation according to manufacturer’s instructions. The phage dodecapeptide-gIII fusion gene was amplified from the purified DNA template using sense (5′-GTATGGGATTTTGCTAAACAAC) and antisense (5′-CCCTCATAGTTAGCGTAACG) primers. The PCR products were sequenced, and the deduced amino acid sequences were aligned with the PEDV S1 protein (NCBI accession number AF353511.1) using Lasergene DNAStar Megalign™ 9.0.
Prediction of B cell epitopes in the PEDV S1 protein
Bioinformatics predicted antigenic peptides (BPAP) (http://imed.med.ucm.es/Tools/antigenic.pl) was used to assess putative B cell epitopes within the PEDV S1 protein. This system is straightforward and predicts antigenic determinants with 75 % accuracy [15].
Cytotoxicity of synthesized peptides
Peptides were synthesized by the China Peptides Co., Ltd. (Shanghai, China), and the purity confirmed to be >95 %. The cytotoxicity of the peptides was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay [16]. Briefly, cells (2 × 104 cells/well) were seeded onto 96-well plates for 24 h, treated with serially-diluted peptides (100–3.125 µg/ml), and then incubated for an additional 48 h. Afterwards, the plates were supplemented with 10 μl of MTT solution (0.5 mg/ml, Invitrogen, US), and incubated for an additional 4 h. The media was discarded and 100 μl of dimethyl sulfoxide (DMSO) was added to solubilize the formazan crystals. After 15 min, the OD490 was read and cell viability was calculated as follows: (OD490 treatment)/(OD490 control) × 100. The concentration of peptide that inhibited cell proliferation to 50 % was defined as the 50 % cytotoxic concentration (CC50).
Plaque-reduction assay
Anti-PEDV activity of the peptides was evaluated by plaque-reduction assays as described previously [17]. In this test, PEDV was diluted to MOI = 0.05, to which was added dilutions of peptides (3.125, 6.25, 12.5, 25 and 50 µg/ml) not to exceed the CC50. Two different approaches were used to treat the cells: [1] PEDV was first incubated with cells for 1 h and then removed; the cells were washed extensively with phosphate-buffered saline (PBS) and then incubated with peptides at 37 °C for another 1 h; [2] dilutions of peptide were incubated with the cells for 1 h prior to the addition of PEDV; the cells were washed extensively with PBS, then incubated with PEDV for 1 h; and then removed virus and washed with PBS, subsequently supplemented with 1 % methylcellulose in serum-free DMEM at 37 °C for 72 h. All cells were fixed in 4 % formaldehyde for 1 h at room temperature, and then stained with 1 % crystal violet solution for 20 min. The cells were washed several times until the plaques were visible for counting. The inhibition rate was calculated as follows: [1 − (plaques in treated wells/plaques in control wells)] × 100.
Real-time RT-PCR
In 24-well plates, confluent VeroE6 cells (2 × 105 cells/well) were treated with different dilutions of peptide (3.125–50 µg/ml) and PEDV (MOI = 0.05) for 1 h. The medium was replaced with serum-free DMEM and incubated for an additional 24 h. Total RNA was extracted using TriQuick reagent (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) according to the manufacturer’s instructions. cDNA was synthesized using murine leukemia virus reverse transcriptase (GBI Labs/Golden Bridge International, Inc., Mukilteo, WA, USA) with oligo dT (HaiGene Technology, Harbin, China). The cDNA was then subjected to real-time PCR with primer pairs specific for PEDV N and β-actin [17]. PEDV N specific primers were 5′-CACTGGTTGGGCTTTCTATGTC and 5′-TGTTAGTGGGTACAGCGTTGTT, while β-actin primers were 5′-GGCTCAGAGCAAGAGAGGTATCC and 5′-GGTCTCAAACATGATCTGAGTCATCT, respectively. The relative transcription of PEDV N was calculated by the method [18].
Indirect immunofluorescence assay (IFA)
VeroE6 cells were seeded onto 96-well plates at a density of 2 × 104 cells/well and grown for 24 h. An equal volume of peptide (50 µg/ml) and PEDV (MOI = 0.05) was added to each well and incubated at 37 °C for 1 h. PEDV-infected or mock-infected cells were used as controls. The media was replaced with the serum-free media and the incubation continued for 24 h after which the cells were washed 3× with ice-cold PBS, fixed with 4 % formaldehyde for 30 min and then quenched with 0.1 M glycine for 5 min. The cells were again washed 3× in PBS, permeabilized with 0.1 % Triton X-100 for 10 min and then incubated at 37 °C for 1 h with rabbit anti-PEDV polyclonal antibody (1:500 in PBS). After washing, the cells were incubated with FITC-conjugated goat anti-rabbit IgG antibody at 37 °C for 30 min and visualized by fluorescence microscopy (Leica, Wetzlar, Germany).
Results
Phage display mimotope of MAb-5E12
To identify the epitope within PEDV recognized by MAb-5E12, purified MAb-5E12 was used as the immobilized target to biopan a random, 12-mer phage peptide library. After five rounds of biopanning including one round to remove non-specific binding to IgG molecules, 13 individual phage clones (designated ph1-13) were identified by direct ELISA. The results showed that 10 phages specifically recognized MAb-5E12 (OD490 > 1.2), but did not bind to unrelated MAb or BSA (Fig. 1a). Competitive ELISA was used to assess the binding efficiency of these 10 phage clones to MAb-5E12. As shown in Fig. 1b, the phage inhibited binding of MAb-5E12 to the PEDV S1 protein ranging from 34 to 68 % relative to controls (no phage).
Fig. 1.
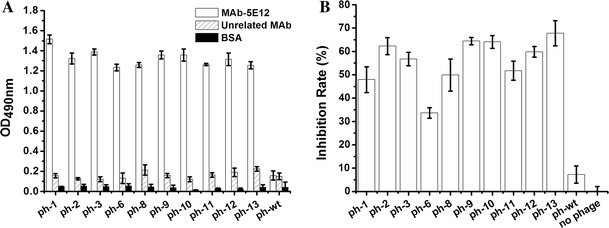
Binding analysis of the phage display peptides to MAb-5E12 by direct and competitive ELISA. a Selected phages named ph-1 to ph-13 and wild-type M13 phage (ph-wt) were incubated with MAb-5E12, an unrelated MAb, and BSA in ELISA plates to assess their binding affinities. The OD490 values are proportional to binding affinities of each phage. b Ten positive individual phage and MAb-5E12 were added to PEDV rS1 protein-coated plates; ph-wt and no phage were used as negative controls. The inhibition rates {[OD490 MAb-5E12 (no phage) − OD490 MAb-5E12 (with phage)]/OD490 MAb-5E12 (no phage)} × 100 of the positive individual phage are shown. The experiment was performed in triplicate
Alignment of the peptide sequences
DNA sequencing revealed that 10 phage clones presented three distinct peptide sequences (Table 1). The deduced amino acid sequences were LMQINPTYYQIM, WSFNPSTYTIAG and HDFVADMYQLAQ, designated peptides L, W and H, respectively. As shown in Fig. 2, multiple sequence alignment revealed that the one or more of the peptides exhibited partial sequence similarity to the native S1 sequence at position 180–220. We found that peptide L has a similarity index of 50 % to the sequence ‘MQYVYTPTYYML’ (designated peptide M) at position of 201–212. Further, both peptides L and W had a consensus motif ‘PxxY’ that is present within the PEDV S1 protein sequence. The data suggest that the motif 201MQYVYTPTYYML212 is the putative epitope of the PEDV S1 protein recognized by MAb-5E12.
Table 1.
Deduced amino acid sequences of phage clones
| Phage clones | Amino acid sequence |
|---|---|
| ph-(2, 3, 8, 9, 10, 11, 12, 13) | LMQINPTYYQIM (L) |
| ph-1 | WSFNPSTYTIAG (W) |
| ph-6 | HDFVADMYQLAQ (H) |
After five rounds of biopanning, 10 individual clones were characterized by DNA sequencing. The deduced amino acid sequences are displayed. Three unique peptide sequences were identified and have been designated as peptides L, W and H (in parenthesis)
Fig. 2.

Alignment of deduced phage display amino acid sequences and the PEDV S1 protein. Peptides L, W and H displayed one or more amino acids with similarity to PEDV S1 protein in aa 180–220 (NCBI accession number AF353511.1, 1–789 aa)
Prediction of B cell epitopes in the PEDV S1 protein
The method of Kolaskar and Tongaonkar has been used to predict B cell epitopes. This same method identified 30 antigenic determinants in the PEDV S1 protein (Table 2) with an average antigenic propensity of 1.0469. Among the 30 antigenic determinants, we found B cell epitopes located in the 198–214 region which encompasses the putative binding site of MAb-5E12. Previous reports [8, 19–22] demonstrated that functional epitopes within the PEDV S protein, include aa 499–638, 636–789, 744–759, 756–771, 748–755, 764–771, and 1368–1374. Studies presented herein are consistent with aa 201–212 representing a new linear epitope on the S1 protein of PEDV.
Table 2.
Prediction of B cell epitopes of PEDV S1 protein
| Number | Start position | Sequence | End position |
|---|---|---|---|
| 1 | 4 | LIYFWLLLPVLPTLSLPQDVTRC | 26 |
| 2 | 37 | SKFNVQAPAVVVLGGY | 52 |
| 3 | 69 | TASGVHGIFLSYI | 81 |
| 4 | 97 | DPSGYQLYLH | 106 |
| 5 | 115 | AIARLRICQF | 124 |
| 6 | 130 | LGPTVND | 136 |
| 7 | 139 | TGRNCLFNKAIPA | 151 |
| 8 | 168 | DRVTVFADKIYHFY | 181 |
| 9 | 198 | SCAMQYVYTPTYYMLNV | 214 |
| 10 | 222 | IYYEPCTANCTGYAANVFA | 240 |
| 11 | 255 | NWFLLSNDSTLLHGKVVSNQPLLVNCLLAIPKIYGLGQ | 292 |
| 12 | 300 | MDGVCNG | 306 |
| 13 | 308 | AVDRAPE | 314 |
| 14 | 322 | DTSVILAEGSIVLHT | 336 |
| 15 | 338 | LGTNLSFVCSN | 348 |
| 16 | 350 | SDPHLAIFAIPLGATEVPYYCFLKVDTYNSTVYKFLAVLPPTVREIVITKYGDVYVNGFGYLHLGLLDAVTI | 421 |
| 17 | 440 | STNFVDALIEVQG | 452 |
| 18 | 455 | IQRILYCDDPVSQLKCSQVAF | 475 |
| 19 | 479 | DGFYPISSRNLLSHEQPISFVTL | 501 |
| 20 | 506 | DHSFVNITVSAAFGG | 520 |
| 21 | 522 | SSANLVAS | 529 |
| 22 | 535 | GFSSFCVDT | 543 |
| 23 | 546 | FTITLFYNV | 554 |
| 24 | 556 | NSYGYVSK | 563 |
| 25 | 565 | QDSNCPFTLQSVNDYLSFSKFCVSTSLLAGACTIDLFGYPAFGSGVKLTSLYFQ | 618 |
| 26 | 639 | SFMTLDVCTKYTI | 651 |
| 27 | 658 | GIITLTNSSILAGVYY | 673 |
| 28 | 677 | SGQLLAFK | 684 |
| 29 | 687 | TSGAVYSVTPCSFSEQAAYVNDDIVGVISS | 716 |
| 30 | 741 | CTEPVLVYSNIGVCKSGSIGYVPSQYGQVKIAP | 773 |
The number 9 is a B cell epitope of PEDV S1 protein which encompasses the putative binding site of MAb-5E12 in a bold type
Anti-PEDV activity of peptides in vitro
To determine whether or not peptides M, L, W and H could inhibit PEDV infection, plaque-reduction, real-time RT-PCR and IFA assays were performed at or below the empirically-determined CC50 values for each peptide. Assays were performed pre-and post-cell treatment with PEDV to assess the mode of action. Plaque assays demonstrated that pre-PEDV infection, none of the peptides had a demonstrable effect on virus production (date not shown). However, peptides M, L and W significantly blocked the adsorption of virus in a dose-dependent manner (Fig. 3a) when cells were treated prior to the addition of PEDV; inhibitions of 72, 68 and 51 % were observed, respectively. This is consistent with inhibition occurring at the cell surface. In contrast to peptide M, we note that the motif ‘PxxY’ is present in both peptides L and W which inhibit the binding of the virus to the cells and is not present in peptide H which showed little or no effect on the binding of PEDV to the cell surface.
Fig. 3.
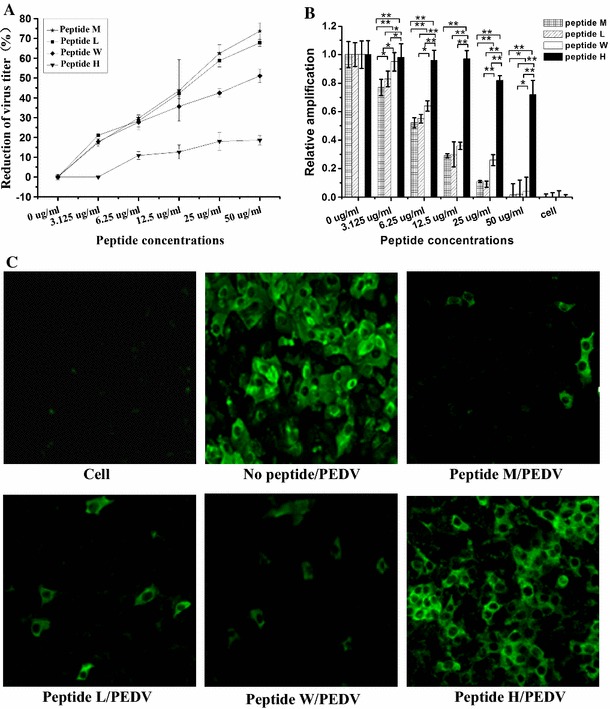
Anti-PEDV activity of peptides M, L, W and H. a Plaque-reduction assay. The reduction in virus titer is presented with changing peptide concentration. Inhibition rates were calculated as follows: [1 − (plaques in treated wells/plaques in control wells)] × 100. b Real-time PCR. Total RNA was extracted and PEDV N and β-actin mRNA were subjected to real-time PCR. Relative amplification ( method)of the PEDV N gene was normalized to β-actin. All data are expressed as mean ± SD. Statistical significance is represenced by *p < 0.05; **p < 0.01 relative to peptides M, L, W or H. c IFA. Results show VeroE6 cells treated with peptides M, L, W or H (50 µg/ml each) prior to PEDV infection. Untreated VeroE6 cells and PEDV-infected VeroE6 cells were used as controls
Secondarily, we examined the effects of these peptides on viral mRNA synthesis by real-time PCR. As shown in Fig. 3b, the level of viral mRNA in cells pretreated with peptides M, L or W decreased with increasing peptide concentration (3.125–50 µg/ml); however, no significant changes were observed in cells pretreated with peptide H or in peptide-untreated controls.
Finally, we used IFA to confirm the inhibitory activity of the peptides on the binding and uptake of PEDV. Using the experimentally-determined CC50 concentrations for each peptide i.e., 50 µg/ml, we observed significantly lower fluorescence intensities in cells treated with peptides M, L or W relative to peptide-untreated controls; the effects of peptide H on IFA intensities were unremarkable (Fig. 3c).
Discussion
Phage display has been successfully used to identify epitopes of different viruses such as foot-and-mouth disease virus (FMDV) [23, 24], classical swine fever virus (CSFV) [25], Japanese encephalitis virus (JEV) [26], Epstein–Barr virus (EBV) [27], porcine reproductive and respiratory syndrome virus (PRRSV) [28] and mouse hepatitis virus (MHV) [29]. Therefore, one purpose of this study was to identify potential antigenic epitopes in PEDV S1 protein using this technique. Antigenic epitopes can be divided into linear epitopes and conformational epitopes. The linear epitopes are a few amino acids adjacent to each other on the primary structure of antigen. The conformational epitopes are discontinuous on the primary structure of antigen, but some amino acid residues near each other on the spatial structure [30]. Here, under stringent panning conditions, we identified the peptide sequence that had the binding ability to MAb-5E12. Three sequences, LMQINPTYYQLM, WSFNPSTYTIAG and HDFVADMYQLAQ, which were identified that displayed on the phages (Table 1). Our findings suggested the motif 201MQYVYTPTYYML212 might be a critical linear epitope in the PEDV S1 protein recognized by MAb-5E12.
PEDV S protein is a glycoprotein located on the surface of the virion and has two subunits S1 and S2. S1 subunit makes up the globular part of S protein, which contains the specific receptor-binding domains (RBD) responsible for adsorption of virions to the receptor of host cells, while S2 subunit is responsible for mediating fusion [6]. So far, limited reports [7, 31, 32] showed that porcine aminopeptidase N (pAPN) may act as a functional receptor of PEDV, but the accurate positions of RBD in PEDV S protein remain unclear. Studies have shown that phage display peptides which recognize pAPN are able to inhibit TGEV and PEDV infection of cells [17, 33]. Therefore, it is a good strategy for inhibition of virus enter into cells by interfering with the attachment of virus on the cell surface. Interestingly, in spite of the MAb 5E12 had no neutralizing activity against PEDV, using plaque-reduction, real-time PCR and indirect immunofluorescence assays, our data showed that peptide M and the MAb-5E12-recognizing peptides L and W significantly inhibited the adsorption of PEDV on the cell surface and thereby function as antiviral agents for PEDV. We assumed that it was possibly the smaller sizes of peptides M, L and W allowing for better recognition and/or binding to cellular receptors.
Currently, there are no effective prophylactic measures against PEDV infection. As such, research efforts continue to search for functional epitopes that can be exploited as candidates for subunit vaccines [34–36]. Here, two peptides derived from phage display libraries were identified with sequence homology to a specific region of the PEDV S1 protein and that affect binding of the PEDV to the cell surface. Inasmuch as peptides L and W interacted with MAb-5E12, these may function both as modulators to enhance host immunity to infection, and as small molecule therapeutic agents against this disease.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (31372438; 31201911; 31340003); Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2013BAD12B04), Harbin Science and Technology Bureau (RC2012XK002003), State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (SKLVEB2012KFKT007); and the Doctoral Scientific Fund Project (20122325110019) of the Ministry of Education, P.R. China.
References
- 1.Pijpers A, van Nieuwstadt AP, Terpstra C, Verheijden JH. Vet. Rec. 1993;132:129–131. doi: 10.1136/vr.132.6.129. [DOI] [PubMed] [Google Scholar]
- 2.Song D, Park B. Virus Genes. 2012;44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang YW, Dickerman AW, Pineyro P, Li L, Fang L, Kiehne R, Opriessnig T, Meng XJ. mBio. 2013;4:e00713–e00730. doi: 10.1128/mBio.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ. J. Vet. Diagn. Invest. 2013;25:649–654. doi: 10.1177/1040638713501675. [DOI] [PubMed] [Google Scholar]
- 5.Egberink HF, Ederveen J, Callebaut P, Horzinek MC. Am. J. Vet. Res. 1988;49:1320–1324. [PubMed] [Google Scholar]
- 6.Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li BX, Ge JW, Li YJ. Virology. 2007;365:166–172. doi: 10.1016/j.virol.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun D, Feng L, Shi H, Chen J, Cui X, Chen H, Liu S, Tong Y, Wang Y, Tong G. Vet. Microbiol. 2008;131:73–81. doi: 10.1016/j.vetmic.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh J, Lee KW, Choi HW, Lee C. Arch. virol. 2014;159:2977–2987. doi: 10.1007/s00705-014-2163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kay BK, Kurakin AV, Hyde-DeRuyscher R. Drug Discov. Today. 1998;3:370–378. doi: 10.1016/S1359-6446(98)01220-3. [DOI] [Google Scholar]
- 11.Levin BR, Bull JJ. Am. Nat. 1996;147:881–898. doi: 10.1086/285884. [DOI] [Google Scholar]
- 12.Cao L, Qin Z, Ge X, Yin X, Xia C, Bu RE, Fang Y, Liu J, Gao Y, Ren X. Monoclon. Antib. Immunodiagn. Immunother. 2013;32:371–374. doi: 10.1089/mab.2013.0045. [DOI] [PubMed] [Google Scholar]
- 13.McKinney MM, Parkinson A. J. Immunol. Methods. 1987;96:271–278. doi: 10.1016/0022-1759(87)90324-3. [DOI] [PubMed] [Google Scholar]
- 14.Zou H, Zarlenga DS, Sestak K, Suo S, Ren X. Antiviral Res. 2013;99:383–390. doi: 10.1016/j.antiviral.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolaskar AS, Tongaonkar PC. FEBS Lett. 1990;276:172–174. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- 16.Dai JP, Chen J, Bei YF, Han BX, Wang S. J. Oral Pathol. Med. 2009;38:276–281. doi: 10.1111/j.1600-0714.2008.00738.x. [DOI] [PubMed] [Google Scholar]
- 17.Meng F, Suo S, Zarlenga DS, Cong Y, Ma X, Zhao Q, Ren X. Virology. 2014;456–457:20–27. doi: 10.1016/j.virol.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Sun DB, Feng L, Shi HY, Chen JF, Liu SW, Chen HY, Wang YF. Acta Virol. 2007;51:149–156. [PubMed] [Google Scholar]
- 20.Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, Laude H, Yang MS, Jang YS. Mol. Cells. 2002;14:295–299. [PubMed] [Google Scholar]
- 21.Cruz DJ, Kim CJ, Shin HJ. Virology. 2006;354:28–34. doi: 10.1016/j.virol.2006.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cruz DJ, Kim CJ, Shin HJ. Virus Res. 2008;132:192–196. doi: 10.1016/j.virusres.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Zhao L, Li W, Zhou G, Yu L. Vet. Microbiol. 2011;148:189–199. doi: 10.1016/j.vetmic.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Yang D, Zhang C, Zhao L, Zhou G, Wang H, Yu L. Virus Res. 2011;155:291–299. doi: 10.1016/j.virusres.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 25.Zhang F, Yu M, Weiland E, Morrissy C, Zhang N, Westbury H, Wang LF. Arch. Virol. 2006;151:37–54. doi: 10.1007/s00705-005-0623-9. [DOI] [PubMed] [Google Scholar]
- 26.Wu SC, Lin CW. Virus Res. 2001;76:59–69. doi: 10.1016/S0168-1702(01)00246-5. [DOI] [PubMed] [Google Scholar]
- 27.Zhang D, Mao Y, Cao Q, Xiong L, Wen J, Chen R, Zhu J. Viruses. 2013;5:1131–1142. doi: 10.3390/v5041131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostrowski M, Galeota JA, Jar AM, Platt KB, Osorio FA, Lopez OJ. J. Virol. 2002;76:4241–4250. doi: 10.1128/JVI.76.9.4241-4250.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu MW, Scott JK, Fournier A, Talbot PJ. Virology. 2000;271:182–196. doi: 10.1006/viro.2000.0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regenmortel MHVV. Biologicals. 2001;29:209–213. doi: 10.1006/biol.2001.0308. [DOI] [PubMed] [Google Scholar]
- 31.Nam E, Lee C. Vet. Microbiol. 2010;144:41–50. doi: 10.1016/j.vetmic.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navari M, Zare M, Javanmardi M, Asadi-Ghalehni M, Modjtahedi H, Rasaee MJ. Immunopharmacol. Immunotoxicol. 2014;36:309–315. doi: 10.3109/08923973.2014.945127. [DOI] [PubMed] [Google Scholar]
- 33.Ren X, Wang M, Yin J, Li G. J. Clin. Microbiol. 2010;48:1875–1881. doi: 10.1128/JCM.01707-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurr W, Shaw M, Herzog RI, Li Y, Sherwin R. PLoS One. 2013;8:e69464. doi: 10.1371/journal.pone.0069464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Y, Zhang Y, Mitchell WJ, Zhang G. J. Immunol. 2012;189:4909–4920. doi: 10.4049/jimmunol.1201622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meola A, Delmastro P, Monaci P, Luzzago A, Nicosia A, Felici F, Cortese R, Galfre G. J. Immunol. 1995;154:3162–3172. [PubMed] [Google Scholar]