

Abstract
Newcastle disease virus (NDV) and infectious bronchitis virus (IBV) are two poultry pathogens affecting the respiratory tract of chickens, and cause major economic losses in the industry. Rapid detection of these viruses is crucial to inform implementation of appropriate control measures. The objective of our study is developing a simple, rapid and field applicable recombinase polymerase amplification (RPA)–nucleic acid lateral flow (NALF) immunoassay for detection of NDV and IBV. Isothermal amplification of the matrix protein (M) gene of NDV and the nucleoprotein (N) gene of IBV was implemented via recombinase polymerase amplification at 38 °C for 40 min and 20 min, respectively using modified labeled primers. NALF device used in this study utilizes antibodies for detection of labeled RPA amplicons. The results revealed that RPA-NALF immunoassays can detect both viruses after 40 min at 38 °C and only NDV after 20 min. The limit of detection (LOD) was 10 genomic copies/RPA reaction. The assays results on clinical samples collected from diseased chicken farms demonstrated a good performance in comparison with quantitative real time reverse transcription polymerase chain reaction (qRT-PCR). The assays established in this study can facilitate rapid, on-site molecular diagnosis of suspected cases of ND and IB viral infections as the results can be detected by the naked eye without the need for measuring fluorescence. Furthermore, the NALF device could be adapted to detect other infectious agents.
Keywords: Infectious bronchitis virus, Newcastle disease virus, Nucleic acid lateral flow immunoassay, Recombinase polymerase amplification
Introduction
Newcastle disease virus (NDV) and infectious bronchitis virus (IBV) are two poultry pathogens causing major economic losses for the poultry industry. These respiratory viruses can co-circulate in Egyptian chicken farms. Newcastle disease (ND) is a contagious disease causing a high mortality rate (70–100%) in avian species. NDV is a negative sense, single-stranded, monopartite RNA virus of the Avian Paramyxovirus type 1 (APMV-1). NDV strains have been categorized according to virulence as lentogenic, mesogenic, and velogenic strains. Velogenic strains are the most virulent, causing respiratory tract lesions as well as sever hemorrhagic lesions in the digestive tract (viscerotropic), and/or neurological signs (neurotropic). The lentogenic strains have low virulence and are common among domestic poultry [1, 2].
Infectious bronchitis (IB) is an infectious viral disease affecting poultry and induced by a positive-sense single-stranded RNA coronavirus. The disease characterized clinically by respiratory distress and sharp drop in egg production and quality in layers. There have been reports of IBV strains associated with nephritis [3–5].
Rapid identification of these viruses will facilitate control measures implementation and consequently, declines the economic losses. Therefore, portable, accurate, inexpensive, simple, and rapid, on site test is needed for diagnosis on the spot of infection.
Currently, field detection of these viruses depends on clinical manifestations and post-mortem examination. Confirmative laboratory diagnosis can be carried out by virus isolation, haemagglutination inhibition, enzyme-linked immunosorbent assay (ELISA), immunofluorescence techniques and neutralization test [1, 2, 4–7]. Also, reverse transcription-PCR (RT-PCR) and quantitative real time RT-PCR (qRT-PCR) have been reported for molecular detection of these viruses [8–13]. However, the lack of adequate laboratory resources, skilled staff requirement and the time consuming nature of the above mentioned diagnostic methods constitute obstacles to the rapid detection under field condition.
Recombinase polymerase amplification (RPA) is a single-tube isothermal amplification and detection methodology for DNA and has equal sensitivity to real time-polymerase chain reaction. In contrast to PCR, it uses only one constant temperature. The amplification step depends first on a recombinase (UvsX and co-factor UvsY from T4 Enterobacteria phage) binding to specific primers followed by scanning the DNA for the complementary sequence. Once invasion of the strand is maintained by the single strand binding protein (T4 gp32), a primer extension is ensued via a strand displacing DNA polymerase (Staphylococcus aureus) [14–18]. Lateral flow immunoassays are currently widely used due to their simplicity, low cost, good specificity, rapid result, electricity-free operation and long shelf life, making them suitable for on-site pathogen detection particularly in developing countries [19]. RPA combined with lateral flow dipstick (LFD) detection of amplicons has been previously reported for H9 subtype of avian influenza virus detection in Central China and proved to be appropriate for field application [20].
The aim of our study was developing a field applicable, simple, specific and rapid molecular diagnostic method for ND and IB viruses using recombinase polymerase amplification, combined with nucleic acid lateral flow immunoassays for results visualization.
Materials and methods
Template preparation
Synthesized templates of NDV matrix protein (M) gene and IBV nucleoprotein (N) gene were designed according to previously published sequences in GenBank with Accession number AY562988 for NDV and FJ589733 for IBV. The templates were synthesized by Integrated DNA Technologies (IDT), Leuven, Belgium. They were received in lyophilized form (250 ng), centrifuged for 3–5 s at 3000×g to ensure the lyophilized material at the bottom of the tube, then resuspended in TE buffer to reach a final concentration of 10 ng/μl (7 × 1010 gene copies/μl) before incubated at 50 °C for 20 min and centrifuged. The prepared templates are stored at − 20 °C till use.
Oligonucleotide primers
The used specific primers can amplify a specific fragment of M gene of NDV (125 bp) and N gene of IBV (131 bp), as described previously [10, 21]. For the NDV assay, M gene forward primer labeled with DIG while reverse primer labeled with Biotin. For IBV assay, N gene forward primer labeled with FAM and reverse primer conjugated with Biotin. The labeled oligonucleotide primers were obtained from Integrated DNA Technologies (IDT), Leuven, Belgium. These primers were received in lyophilized form and TE buffer was used in resuspension of the primers to obtain a final concentration of 100 μM/μl. Details of modified primers used for the identification of NDV and IBV summarized in Table 1. The primers used in our study were also verified for their cross reactivity with other avian respiratory viruses using NCBI database BLAST search (http://www.ncbi.nlm.nih.gov) and there was no cross reactivity observed with avian influenza viruses (H5N1, H5N8 and H9N2), Infectious laryngeotracheitis virus (ILT) and avian reovirus.
Table 1.
Primers used for the identification of NDV and IBV nucleic acids templates
| Virus | Primers sequences and tags | Target gene | References |
|---|---|---|---|
| Newcastle disease virus (NDV) |
M + 4100 (5′ DIG-AGTGATGTGCTCGGACCTTC-3′) M-4220 (5′ Biotin-CCTGAGGAGAGGCATTTGCTA-3′) |
M gene | [10] |
| Infectious bronchitis virus (IBV) |
AIBV-fr (5′ FAM-ATGCTCAACCTTGTCCCTAGCA-3′) AIBV-as (5′ Biotin-TCAAACTGCGGATCATCACGT-3′) |
N gene | [21] |
NDV and IBV RPA amplification conditions
RPA assays were carried out via the twistAmp® Basic kit (TwistDx Limited, Cambridge, UK). Briefly, 2.4 μl of each primer, 29.5 μl of rehydration buffer and 11.2 μl of nuclease free water were added to the lyophilized pellet in the RPA-tubes strip, followed by adding 2.5 μl of 280 mM Mg acetate in each lid. Then, 1 μl of each nucleic acid template was added. The RPA-tubes strip was centrifuged before placed into Axxin T16 isothermal instrument at 38 °C for period of 40 min. Also, the amplification reaction was examined after incubation period of 20 min. A negative control was included in each run. All experiments were performed three times.
Nucleic acid lateral flow (NALF) detection
For visual detection of labeled RPA amplicons, PCRD nucleic acid detector (Abingdon Health Ltd, National Innovation Campus, Sand Hutton, York, UK) was used. This device utilizes antibodies against primer-tags (DIG, FAM, Biotin) and has been designed for lateral flow detection of amplified products (Fig. 1). The device comprises a sample well, conjugate pad, nitrocellulose membrane and absorbent pad, which are collected together in a plastic container. NALF detection was performed separately for the NDV and IBV assays, using separate devices for each assay. Following RPA reaction, 5 μl of the amplification product was transferred into a 0.5 ml tube and mixed thoroughly with 70 μl of the PCRD extraction buffer and added to the sample well of a PCRD test cassette.
Fig. 1.

Schematic representation of PCRD nucleic acid detector design. At the conjugate pad, the carbon Biotin-conjugated antibodies bind to biotin on one end of the amplicons, and flows towards the test-lines, L1 and L2. L1 is lined with anti-DIG monoclonal antibodies to capture DIG/Biotin labeled amplicons for NDV assay. L2 is lined with anti-FAM monoclonal antibodies to capture FAM/Biotin labeled amplicons for IBV assay. The excess carbon Biotin-conjugated antibodies are captured by anti-mouse antibodies at the control line (C)
Upon sample application, the carbon Biotin-conjugated antibodies were rehydrated and reacted with the tag/Biotin-labeled amplicons in the sample. The first line (L1) consisted of anti-DIG monoclonal antibodies for capturing DIG/Biotin labeled amplicons for the NDV assay. The second line (L2) contains anti-FAM monoclonal antibodies to capture FAM/Biotin labeled amplicons for IBV assay. The excess carbons dots covered with Biotin antibodies flowed pass the two test-lines and were fixed at the control line (C) to ensure that a PCRD test cassette operating correctly. The cassette was kept in a horizontal position, and the results were recorded after 5 min.
The limit of detection (LOD)
To detect the minimum number of genome copies of NDV and IBV that can be reliably detected by RPA-NALF assays. Tenfold serial dilutions of the gene templates were carried out as follows; 10 ng (107 genome copies), 1 ng (106 genome copies), 0.1 ng (105 genome copies), 0.01 ng (104 genome copies), 0.001 ng (103 genome copies), 0.0001 ng (102 genome copies) and 0.00001 ng (10 genome copies). Amplified RPA amplicons were detected using PCRD nucleic acid detector as described above.
Clinical performance of NDV and IBV RT-RPA-NALF immunoassays
Thirty-six oropharyngeal swabs were collected from 12 Egyptian chicken flocks in Dakahlia Governorate (three samples from each flock) with respiratory manifestations (nasal discharge, conjunctivitis, rales and gasping). A total of 12 pools were created. RNA extracts of the pooled clinical samples were tested for NDV and IBV via RT-RPA-NALF using twistAmp® Basic RT kit (Twist Dx, UK) according to the kit manual and results visualization was done via PCRD test cassettes. Testing of pooled samples in parallel using qRT-PCR was carried out [12, 21]. Negative and positive controls were used to assess false positive results.
Results
RPA amplification and detection by NALF
A 75 μl suspension composed of 5 μl of RPA amplicons after incubation for period of 40 min at 38 °C mixed thoroughly with 70 μl of the PCRD extraction buffer was applied to the sample well of PCRD nucleic acid detector. After about 5 min, black lines at test line 1 (L1) and control line (C) in case of NDV assay and two black lines at test line 2 (L2) and control line (C) in case of IBV assay were observed. Negative control included in each run showed only black line at the control line (C) (Fig. 2). NDV RPA assay showed no cross detection with IBV, and vice versa.
Fig. 2.
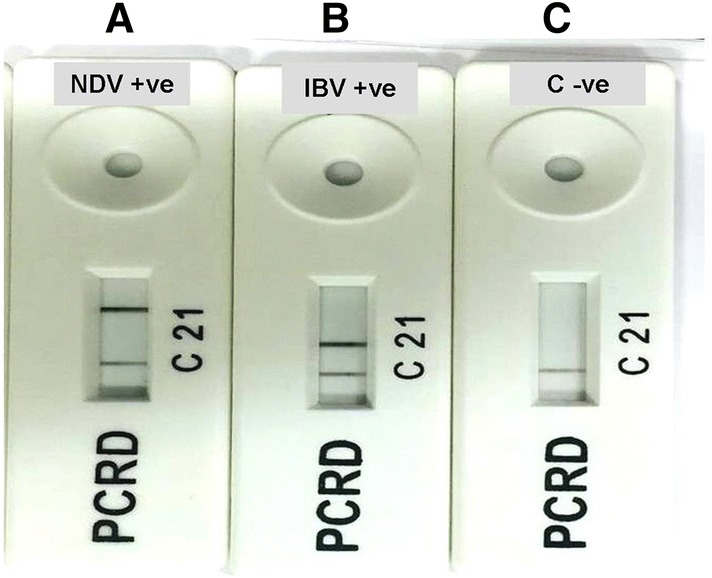
Results showing identification of RPA amplicons after incubation for period of 40 min at 38 °C using PCRD nucleic acid detector: a NDV assay showed positive result; b IBV assay showed positive result; c negative control with negative result
Recombinase polymerase amplification reaction incubation at 38 °C for period of 20 min, followed by detecting the amplified amplicons using PCRD nucleic acid detector revealed in case of NDV, black lines at test line 1 (L1) and control line (C), while in the case of IBV assay and negative control revealed only black line at control line (C) (Fig. 3). The results indicated the LOD to be 10 genome copies (0.00001 ng) per RPA reaction using PCRD nucleic acid detector for both NDV and IBV assays (Fig. 4).
Fig. 3.
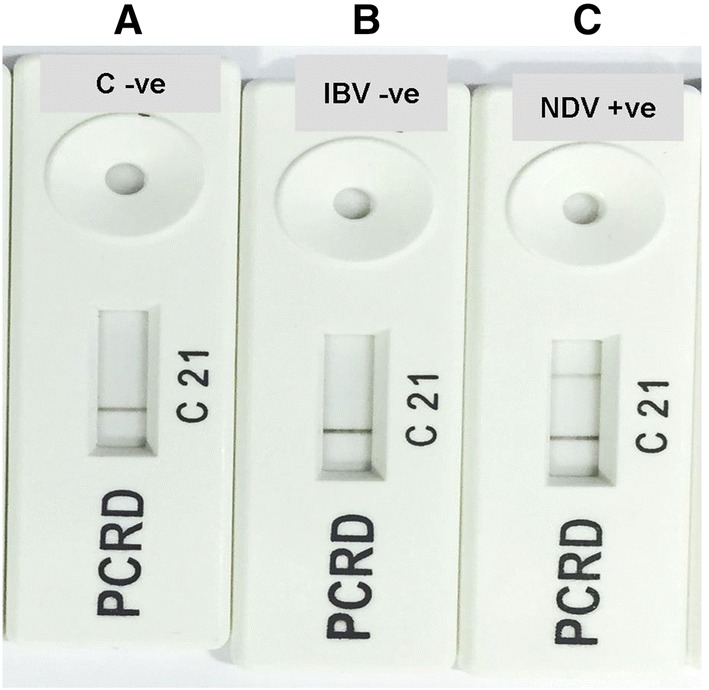
Results showing identification of RPA amplicons after incubation for period of 20 min at 38 °C using PCRD nucleic acid detector: a Negative control; b IBV assay showed negative result; c NDV assay showed positive result
Fig. 4.

The limit of detection (LOD) for both NDV (a) and IBV (b) assays, the NALF results present the LOD as 10 genome copies (0.00001 ng) per RPA reaction for both assays
Assays performance on clinical samples
RT-RPA-NALF immunoassays and qRT-PCR were used to screen 12 pooled clinical samples for NDV and IBV (Fig. 5). NDV positive rate detection by qRT-PCR and RT-RPA-NALF was 66.7% (8/12) and 75% (8/12), respectively and 91.7% agreement was observed for the two methods with a kappa correlation of 0.792. IBV nucleic acid detected in 25% (3/12) and 16.7% (2/12) of the samples by qRT-PCR and RT-RPA-NALF, respectively, and the observed agreement was 91.7% with a kappa correlation of 0.750. All IBV positive samples showed mixed infection with NDV.
Fig. 5.

Performance of RT-RPA-NALF assays on clinical samples for IBV and NDV detection: a Negative control; b positive controls (103 genome copies per reaction) for IBV (left) and NDV (right) assays; c clinical samples showed negative (left) and positive (right) results for NDV; d clinical samples showed negative (left) and positive (right) results for IBV
Discussion
NDV and IBV can cause great economic losses for poultry industry in form of weight loss, decreased egg production and misshaped eggs, immunosuppression as well as vaccination and treatment costs [22–24]. Also, NDV has a minor zoonotic importance and can cause very mild and self-limiting conjunctivitis in human [25].
Rapid and specific diagnosis of ND and IB diseases followed by application of control measures is required for monitoring and limiting the spread of NDV and IBV among chickens. Therefore, the objective of our study was developing simple, field applicable and rapid RPA-NALF immunoassays for NDV and IBV detection.
Herein, we used primers designed previously to identify M gene of NDV and N gene of IBV as these genes are highly conserved and suitable as targets for diagnosis of these diseases [10, 21].
The developed RPA assays for NDV and IBV were highly sensitive (10 genome copies detected/reaction) and rapid (less than 1 h). In case of NDV assay, the total time for detection could be 20 min only.
Several real-time RT-PCRs based tests have been reported for sensitive detection of NDV and IBV [10, 12, 13, 26]. However, they required more than 1 h for test result. Further, to prevent workplace or reagents contamination with amplified products, the master mix preparation, adding of the nucleic acids sample to the master mix and the amplification reaction must be carried out in separate clean benches. Moreover, reagents of real-time PCR must be kept at − 20 °C. Therefore, real-time PCR is unsuitable for pathogen detection on spot of infection.
Recombinase polymerase amplification reactions could be performed by simple hand held heating devices instead of using thermal cyclers as in case of conventional and real time PCR. Also, RPA reagents are cold chain independent making it more suitable rapid on site diagnosis [27, 28].
Recently, reverse transcription loop-mediated isothermal amplification (RT-LAMP) assays were described for molecular detection of NDV and IBV [29, 30]. In contrast to RPA, LAMP reactions use six primers, which are difficult to design in viruses showing high mutation rate like NDV and IBV.
Lateral flow immunoassays (LFIAs) are currently used for pathogen detection depending on antigen or antibody detection under field condition because they are inexpensive, rapid, simple and disposable but with some difficulties to get results with high specificity and sensitivity. Different microfluidic systems were designed for nucleic acid amplification and detection on-chip [31, 32]. However, most of these microfluidic systems use a syringe pump to operate, as well as additional imaging instrumentation to acquire test results. Additionally, NALF can improve the sensitivity and specificity of LFIAs through signaling amplification [33]. The combination of RPA with a lateral flow assay device gives substantial advantage over PCR-based assays especially in laboratories with limited resources.
The limit of detection is one of the factors that determine the sensitivity of any molecular test. The results reveal a LOD of 10 genome copies per RPA reaction for both NDV and IBV assays after incubation for 40 min at 38 °C.
Typically, commercial chickens are vaccinated during their production cycle to prevent NDV and IBV infections. Live vaccines can persist in the farm for long period during the production cycle. Thus, such assays (targeting a conservative region of the viruses genome) could be useful as a preliminary screening test (positive/negative result). We evaluated our assays performance using 12 pooled clinical samples. The results of RT-RPA-NALF revealed good performance in comparison with qRT-PCR in both sensitivity and specificity. Further studies should be directed toward testing the clinical sensitivity using a larger number of clinical specimens.
Further studies are crucial to improve the assays to be able to distinguish field virulent virus strains from vaccine strains which needed to choose appropriate control measures. Also, a simple and field applicable method to extract pathogens’ nucleic acids will greatly facilitate using RPA-NALF assays for on-site diagnosis [34].
In addition to the promising results of our current study, the developed assay could be further improved while the current RPA-NALF test detects NDV and IBV in two separate assays, both assays could be combined in a single test. Accordingly, designing primers and validated RPA conditions capable of identifying mixed infection simultaneously using single assay are crucial.
In conclusion, RPA-NALF tests demonstrated in this study can facilitate rapid, on-site molecular diagnosis of suspected cases of ND and IB diseases as the results can be detected by the naked eye without the need for measuring fluorescence. Furthermore, the NALF device could be adapted to detect other infectious agents.
Acknowledgements
The authors thank Dr. Michael G. Mauk (University of Pennsylvania, Philadelphia, USA) for conducting the English language reviewing. Mohamed El-Tholoth is the recipient of Daniel Turnberg UK/Middle East Travel fellowship funded by Wellcome Trust in UK.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All chickens’ procedures including sampling were done in accordance with the ethical standards established by Mansoura University, Egypt.
Informed consent
Oral consent from chicken farms owners were obtained in compliance with the national ethical regulations.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Alexander DJ. Newcastle disease and other avian paramyxoviruses. Rev Sci Tech. 2000;19:443–462. doi: 10.20506/rst.19.2.1231. [DOI] [PubMed] [Google Scholar]
- 2.Alexander DJ, Manvell RJ, Parsons G. Newcastle disease virus (strain Herts 33/56) in tissues and organs of chickens infected experimentally. Avian Pathol. 2006;35:99–101. doi: 10.1080/03079450600597444. [DOI] [PubMed] [Google Scholar]
- 3.Cavanagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34:439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- 4.Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- 5.Jackwood MW, de Wit S. Infectious bronchitis. In: Swayne DE, Glisson JR, McDougald LR, Nolan LK, Suarez DL, Nair V, editors. Diseases of poultry. 13. Ames: Wiley-Blackwell Publishing; 2013. pp. 139–160. [Google Scholar]
- 6.National Research Council . Methods for examining poultry biologics and for identifying and quantifying avian pathogens. Newcastle disease. Washington, D. C: National Academy of Science; 1971. p. 66. [Google Scholar]
- 7.Cooke JKA, Brown AJ, Bracewell CD. Comparison of haemagglutination inhibition test and serum neutralization test in tracheal organ cultures for typing infectious bronchitis virus strains. Avian Pathol. 1987;16:505–511. doi: 10.1080/03079458708436399. [DOI] [PubMed] [Google Scholar]
- 8.Kant A, Koch G, Van Roozelaar DJ, et al. Differentiation of virulent and nonvirulent strains of Newcastle disease virus within 24 hours by polymerase chain reaction. Avian Pathol. 1997;26:837–849. doi: 10.1080/03079459708419257. [DOI] [PubMed] [Google Scholar]
- 9.Gohm DS, Thur B, Hofmann MA. Detection of Newcastle disease virus in organs and faeces of experimentally infected chickens using RT-PCR. Avian Pathol. 2000;29:143–152. doi: 10.1080/03079450094171. [DOI] [PubMed] [Google Scholar]
- 10.Wise MG, Suarez DL, Seal BS, et al. Development of a real-time reverse-transcription PCR for detection of Newcastle disease virus RNA in clinical samples. J Clin Microbiol. 2004;42:329–933. doi: 10.1128/JCM.42.1.329-338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathivanan K, Kumanan K, Nainar AM. Characterization of Newcastle disease virus isolated from apparently normal guinea fowl (Numida melagridis) Vet Res Commun. 2004;28:171–177. doi: 10.1023/B:VERC.0000012116.60726.9b. [DOI] [PubMed] [Google Scholar]
- 12.Callison SA, Hilt DA, Boynton TO, et al. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J Virol Methods. 2006;138:60–65. doi: 10.1016/j.jviromet.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fellahi S, Harrak ME, Kuhn JH, et al. Comparison of SYBR green I real-time RT-PCR with conventional agarose gel-based RT-PCR for the diagnosis of infectious bronchitis virus infection in chickens in Morocco. BMC Res Notes. 2016;9:231. doi: 10.1186/s13104-016-2037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piepenburg O, Williams CH, Stemple DL, et al. DNA detection using recombination proteins. PLoS Biol. 2006;4:e204. doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Euler M, Wang Y, Nentwich O, et al. Recombinase polymerase amplification assay for rapid detection of Rift Valley fever virus. J Clin Virol. 2012;54:308–312. doi: 10.1016/j.jcv.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Euler M, Wang Y, Otto P, et al. Recombinase polymerase amplification assay for rapid detection of Francisella tularensis. J Clin Microbiol. 2012;50:2234–2238. doi: 10.1128/JCM.06504-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chow WH, McCloskey C, Tong Y, et al. Application of isothermal helicase-dependent amplification with a disposable detection device in a simple sensitive stool test for toxigenic Clostridium difficile. J Mol Diagn. 2008;10:452–454. doi: 10.2353/jmoldx.2008.080008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5:795–800. doi: 10.1038/sj.embor.7400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.St John A, Price CP. Existing and emerging technologies for point-of-care testing. Clin Biochem Rev. 2014;35:155–167. [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Yang PP, Zhang YH, et al. Development of a reverse transcription recombinase polymerase amplification combined with lateral-flow dipstick assay for avian influenza H9N2 HA gene detection. Transbound Emerg Dis. 2019;66:546–551. doi: 10.1111/tbed.13063. [DOI] [PubMed] [Google Scholar]
- 21.Meir R, Maharat O, Farnushi Y, et al. Development of a real-time TaqMan® RT-PCR assay for the detection of infectious bronchitis virus in chickens, and comparison of RT-PCR and virus isolation. J Virol Methods. 2010;163:190–194. doi: 10.1016/j.jviromet.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexander DJ. Newcastle disease, other paramyxoviruses and pneumovirus infections. In: Saif YM, Barnes HJ, Glisson JR, Fadly AM, McDougald LR, Swayne DE, editors. Diseases of poultry. 11. Ames: Iowa State University Press; 2003. pp. 63–99. [Google Scholar]
- 23.Cavanagh D, Naqi SA. Infectious bronchitis. In: Calnek BW, Barnes HJ, Beard CW, Reid WM, Yoder HW, editors. Diseases of poultry. 10. Ames: Iowa State University Press; 1997. pp. 511–526. [Google Scholar]
- 24.Dimitrov KM, Ramey AM, Qiu X, et al. Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus) Infect Genet Evol. 2016;39:22–34. doi: 10.1016/j.meegid.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 25.Chang PW. Newcastle disease. In: Beran GW, editor. CRC handbook series in zoonoses section B: viral zoonoses. CRC: Baton Raton; 1981. pp. 261–274. [Google Scholar]
- 26.Farkas T, Szekely E´, Belak S, et al. Real-time PCR-based pathotyping of Newcastle disease virus by use of TaqMan minor groove binder probes. J Clin Microbiol. 2009;47:2114–2123. doi: 10.1128/JCM.01652-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abd El Wahed A, Weidmann M, Hufert FT. Diagnostics-in-a-Suitcase: development of a portable and rapid assay for the detection of the emerging avian influenza A (H7N9) virus. J Clin Virol. 2015;69:16–21. doi: 10.1016/j.jcv.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faye O, Faye O, Soropogui B, et al. Development and deployment of a rapid recombinase polymerase amplification Ebola virus detection assay in Guinea in 2015. Eurosurveillance. 2015;20:30053. doi: 10.2807/1560-7917.ES.2015.20.44.30053. [DOI] [PubMed] [Google Scholar]
- 29.Pham HM, Nakajima C, Ohashi K, et al. Loop-mediated isothermal amplification for rapid detection of Newcastle disease virus. J Clin Microbiol. 2005;43:1646–1650. doi: 10.1128/JCM.43.4.1646-1650.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen HT, Zhang J, Ma YP, et al. Reverse transcription loop-mediated isothermal amplification for the rapid detection of infectious bronchitis virus in infected chicken tissues. Mol Cell Probes. 2010;24:104–106. doi: 10.1016/j.mcp.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimov IK, Garcia-Cordero JL, O’Grady J, et al. Integrated microfluidic tmRNA purification and real-time NASBA device for molecular diagnostics. Lab Chip. 2008;8:2071–2078. doi: 10.1039/b812515e. [DOI] [PubMed] [Google Scholar]
- 32.Lee SH, Kim SW, Kang JY, et al. A polymer lab-on-a-chip for reverse transcription (RT)-PCR based point-of-care clinical diagnostics. Lab Chip. 2008;8:2121–2127. doi: 10.1039/b811131f. [DOI] [PubMed] [Google Scholar]
- 33.Mark D, Haeberle S, Roth G, et al. Microfluidic lab-on-a-chip platforms: requirements, characteristics and applications. Chem Soc Rev. 2010;39:1153–1182. doi: 10.1039/b820557b. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Macdonald J, Von Stetten F. Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst. 2019;144:31–67. doi: 10.1039/C8AN01621F. [DOI] [PubMed] [Google Scholar]