

Abstract
A new small molecule that targets the vacuolar H+-ATPase activates autophagy, inhibits mTORC1 signaling, and displays potential for clearing toxic protein aggregates involved in neurodegenerative diseases.
Macroautophagy (hereafter referred to as autophagy) is a multistage and complex self-eating process that recycles building blocks from intracellular macromolecules1. Pharmacological activation of autophagy is a promising therapeutic avenue for neurodegenerative diseases such as Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD). These diseases are characterized by the buildup of protein aggregates in nerve cells, resulting in cell death and loss of brain function2. The mammalian target of rapamycin (mTOR) complex 1 (mTORC1) signaling pathway regulates autophagy, and mTORC1 inhibitors can activate autophagy3,4. However, current mTORC1 inhibitors have several drawbacks limiting their therapeutic application as autophagy inducers. In this issue of Nature Chemical Biology, Chung and Shin et al. identify a novel covalent ligand called EN6 as an autophagy activator and mTORC1 inhibitor5.
mTORC1 promotes anabolic processes such as protein translation and represses catabolic pathways like autophagy3,4. mTOR is a conserved Ser/Thr protein kinase that complexes with other components (Raptor and mLST8) to form mTORC1. Under nutrient-rich conditions mTORC1 is activated and inhibits autophagy, whereas under nutrient-limiting conditions mTORC1 is inhibited promoting autophagy. mTORC1 represses autophagy by phosphorylating and inhibiting the autophagy-initiating kinase ULK1 or key components and regulators of the Beclin1–VPS34 complex downstream of ULK1 (refs.3,4; Fig. 1). Additionally, mTORC1 transcriptionally regulates autophagy through phosphorylation and cytoplasmic retention of transcriptional factor TFEB. Thus, inhibition of mTORC1 is critical in effectively enhancing autophagic flux.
Fig. 1 |. EN6 promotes autophagy by enhancing vacuolar H+-ATPase (v-ATPase) activity and inhibiting mTORC1 signaling.
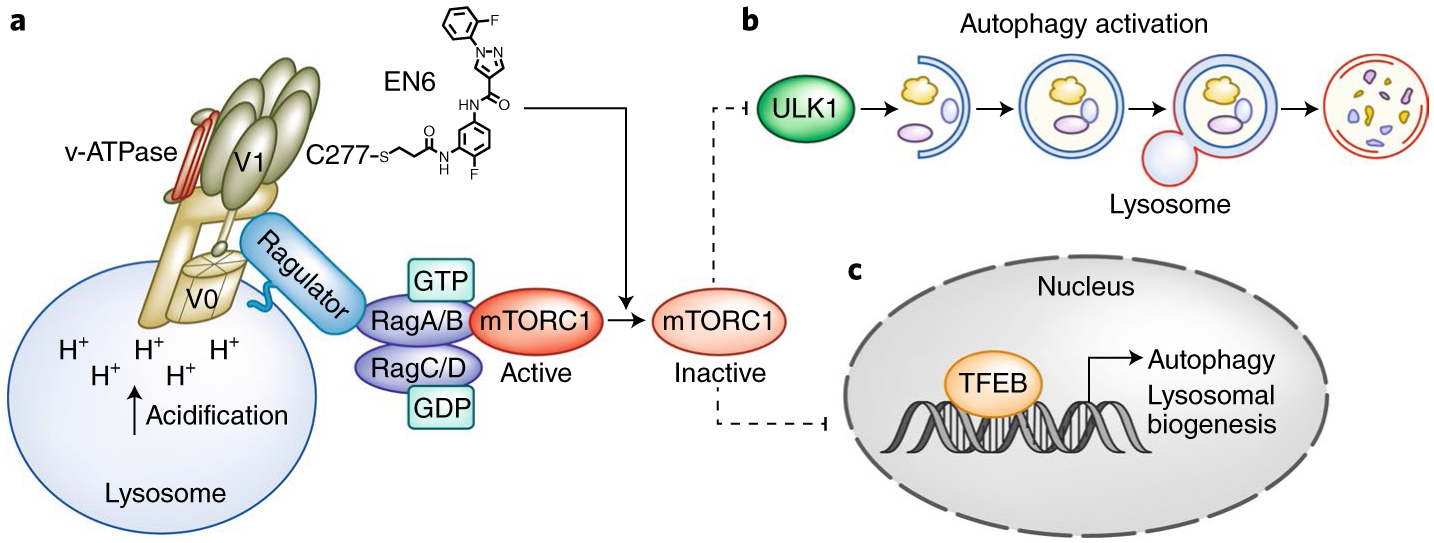
a, EN6 (chemical structure shown) targets Cys277 (C277) of the v-ATPase subunit ATP6V1A (within the V1 domain of the v-ATPase complex). The v-ATPase consists of a V1 domain, which is crucial for v-ATPase ATP hydrolysis, and a V0 domain, which pumps H+ into the lysosome, acidifying it. EN6 modification of v-ATPase enhances its catalytic activity and acidifies lysosomes, which is required for the last stage of autophagy to degrade cargo such as toxic protein aggregates. Moreover, this modification causes a decrease in mTORC1 lysosomal localization and activation. Under nutrient-sufficient conditions, mTORC1 is recruited to the lysosomes and activated by the small GTPase Rheb (not illustrated). The v-ATPase-Ragulator-Rag GTPase complex plays a critical role in regulating mTORC1 lysosomal translocation. In mammals there are four Rag genes: RagA, RagB, RagC, and RagD. RagA/B forms a heterodimer with RagC/D, and the active Rag complex (RagA/B GTPbound and RagC/D GDP-bound) recruits mTORC1 to the lysosome. Inactivation of mTORC1 leads to the dephosphorylation and activation (denoted by dashed line, illustrating release of inhibition) of two downstream substrates ULK1 (b) and TFEB (c). b, Unphosphorylated ULK1 promotes the initiation of autophagy. Autophagy is a multistage process including phagophore initiation, elongation, autophagosome maturation, fusion of autophagosomes with lysosomes (forming autolysosomes), and degradation of autophagic targets. c, Unphosphorylated TFEB is localized in the nucleus and activates transcription of genes promoting autophagy and lysosomal biogenesis.
When nutrients are sufficient, mTORC1 is recruited to the lysosomal surface and subsequently activated by the small G protein Rheb (Fig. 1a)3,4. Mechanistically, mTORC1 is tethered to the lysosomal surface by the Rag GTPases via the Ragulator complex. The vacuolar H+-adenosine triphosphatase ATPase (v-ATPase) was identified as a critical mediator for mTORC1 activation and directly interacts with the Rag GTPase Ragulator on the lysosomal surface6. The v-ATPase pumps protons into the lysosomal lumen to maintain an acidic environment that is ideal for proteolytic enzymes to degrade proteins. Once mTORC1 is activated, it phosphorylates its downstream substrates, ULK1 (Fig. 1b) and TFEB (Fig. 1c), regulating autophagy activation.
First-generation mTORC1 inhibitors, like rapamycin and analogs (rapalogues), allosterically inhibit mTORC1. These mTORC1 inhibitors can block the phosphorylation of some downstream substrates, but not the autophagy-associated mTORC1 substrates ULK1 and TFEB3,4. Second-generation mTORC1 inhibitors are ATP-competitive compounds that inhibit mTORC1 kinase activity and the phosphorylation of all mTORC1 substrates. However, these compounds have unfavorable side effects such as pancreatic β-cell toxicity and death, and they strongly suppress the AKT signaling pathway, which is critical for neuronal cells7. Therefore, the identification of new therapeutic targets that can potently inhibit mTORC1 and activate autophagy will perhaps be beneficial in treating neurodegenerative diseases.
To identify novel compounds and targets for autophagy activation, Chung and Shin et al.5 performed a covalent ligand screen in a cell-based autophagic flux assay and identified the small molecule EN6. Subsequent chemoproteomics profiling identified Cys277 of ATP6V1A, the catalytic subunit of v-ATPase, as the direct target of EN6 (ref.5). The authors show that EN6 disrupts nutrient-induced mTORC1 lysosomal localization and activation (Fig. 1a) and potently inhibits the phosphorylation of mTORC1 downstream targets, including ULK1 and TFEB. In turn, ULK1 is able to promote autophagy activation (Fig. 1b) and TFEB is nuclear localized, upregulating the expression of genes involved in lysosomal biogenesis and autophagy (Fig. 1c). Moreover, the authors demonstrate that EN6 activates the v-ATPase and increases lysosomal acidification. Importantly, in a physiological setting in which enhancing autophagy is beneficial, EN6 can increase the cellular clearance of induced protein aggregates of Tar-binding protein 43 (TDP43). Toxic TDP43 protein aggregates drive the pathogenesis of ALS. Furthermore, intraperitoneally injected mice with EN6 inhibited mTORC1 and induced autophagy in skeletal muscle and heart tissues. Previous studies have shown that inhibition of mTORC1 signaling plays a role in cardiac and skeletal muscle in a variety of diseases7.
Genetic variations of the v-ATPase have also been implicated in human diseases. Although most of them cause loss of function, mutations in the v-ATPase components were recently identified in follicular lymphoma and appear to activate autophagic flux and mTORC1 (refs.8,9). EN6 also enhances v-ATPase activity, which potentially could provide more clues to the mechanistic details of v-ATPase function. The results of Chung and Shin et al.5 show that EN6 decouples the v-ATPase from the Rag GTPases, resulting in mTORC1 inhibition. However, EN6 did not dramatically disrupt the interaction between v-ATPase and the Ragulator complex under nutrient-rich conditions. Thus, how the modification of EN6 on Cys277 specifically regulates mTORC1 and autophagy warrants further investigation, especially from a structural perspective.
This work illustrates how a covalent ligand screen followed by chemoproteomics profiling provides a powerful tool to discover new small molecules and targets for human disease. It also reveals novel insights into the molecular regulation of nutrient-induced mTORC1 activation in respect to the autophagy signaling axis. Thus, EN6 may be useful as an mTORC1 inhibitor and autophagy activator. There has been a great interest in upregulating autophagy for neurodegenerative disorders2. By contrast, inhibition of autophagy has been tested in the clinic for the treatment of cancer. However, given the critical role of autophagy in optimal immune function, activating autophagy might also benefit cancer patients by potentiating immunogenic chemotherapy or radiation therapy10. Thus, the discovery of this novel mTORC1 inhibitor and autophagy activator has potential clinical implications in a wide range of diseases. The efficacy and toxicity needs to be further tested in more relevant preclinical models, such as mouse models of neurodegenerative disease and cancer models.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Choi AM, Ryter SW & Levi ne BN Engl. J. Med 368, 651–662 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Harris H & Rubinsztein DC Nat. Rev. Neurol 8, 108–117 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Jewell JL, Russell RC & Guan KL Nat. Rev. Mol. Cell Biol 14, 133–139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence RE & Zoncu R Nat. Cell Biol 21, 133–142 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Chung CY et al. Nat. Chem. Biol 10.1038/s41589-019-0308-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoncu R et al. Science 334, 678–683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saxton RA & Sabatini DM Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okosun J et al. Nat. Genet 48, 183–188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang F et al. J. Clin. Invest 130, 1626–1640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC & Kroemer G Nat. Rev. Clin. Oncol 14, 247–258 (2017). [DOI] [PubMed] [Google Scholar]