

Lacosamide, an anti-epileptic drug that targets sodium channels, reduces pain in a subset of patients with small fibre neuropathy caused by mutations in Nav1.7 sodium channels. Labau et al. provide biophysical data that may explicate why a subset of Nav1.7 variants differentially respond to treatment with lacosamide.
Keywords: small fibre neuropathy, lacosamide, Nav1.7 mutations, neuropathic pain, electrophysiology
Abstract
Small fibre neuropathy is a common pain disorder, which in many cases fails to respond to treatment with existing medications. Gain-of-function mutations of voltage-gated sodium channel Nav1.7 underlie dorsal root ganglion neuronal hyperexcitability and pain in a subset of patients with small fibre neuropathy. Recent clinical studies have demonstrated that lacosamide, which blocks sodium channels in a use-dependent manner, attenuates pain in some patients with Nav1.7 mutations; however, only a subgroup of these patients responded to the drug. Here, we used voltage-clamp recordings to evaluate the effects of lacosamide on five Nav1.7 variants from patients who were responsive or non-responsive to treatment. We show that, at the clinically achievable concentration of 30 μM, lacosamide acts as a potent sodium channel inhibitor of Nav1.7 variants carried by responsive patients, via a hyperpolarizing shift of voltage-dependence of both fast and slow inactivation and enhancement of use-dependent inhibition. By contrast, the effects of lacosamide on slow inactivation and use-dependence in Nav1.7 variants from non-responsive patients were less robust. Importantly, we found that lacosamide selectively enhances fast inactivation only in variants from responders. Taken together, these findings begin to unravel biophysical underpinnings that contribute to responsiveness to lacosamide in patients with small fibre neuropathy carrying select Nav1.7 variants.
Introduction
Painful peripheral neuropathy represents a major health burden and a globally unmet clinical need. Current first-line therapeutic strategies for the management of chronic pain associated with painful neuropathy, including tricyclic antidepressants, pregabalin, gabapentin or serotonin-noradrenaline reuptake inhibitors, are in many cases reported as unsatisfactory, in part because of adverse effects (Finnerup et al., 2015; Nishikawa and Nomoto, 2017). Furthermore, the degree of pain improvement dramatically varies across the population (Sopacua et al., 2019). More effective pharmacological management of neuropathic pain represents a high priority.
Genetic studies have provided compelling evidence regarding the contribution of altered voltage-gated sodium channel (VGSC) expression and function in neuropathic pain. The Nav1.7 sodium channel has drawn particular interest because it is preferentially expressed in peripheral sensory and sympathetic neurons and has directly been linked to multiple human pain conditions: gain-of-function missense mutations in SCN9A (encoding the Nav1.7 protein) have been reported in patients with inherited erythromelalgia, paroxysmal extreme pain disorder, and some forms of small fibre neuropathy (SFN). Conversely, loss-of-function mutations in Nav1.7 have been identified in individuals with congenital complete insensitivity to pain (Dib-Hajj et al., 2013; Bennett et al., 2019; Dib-Hajj and Waxman, 2019).
SFN is morphologically characterized by injury to the small intraepidermal nerve fibres, specifically the unmyelinated C and thinly myelinated Aδ-fibres, and functionally by dorsal root ganglion (DRG) neuronal and axonal hyperexcitability. Prominent burning pain in the distal extremities, often accompanied with autonomic dysfunction, tend to dominate the clinical picture (Faber et al., 2012; Brouwer et al., 2014; Sopacua et al., 2019). Sodium channel variants have been described in ∼15% of patients with SFN, with a higher frequency in missense mutants of SCN9A (de Greef et al., 2018; Eijkenboom et al., 2019). Many of these variants have been functionally profiled and shown to confer gain-of-function attributes on the channel and render DRG neurons hyperexcitable (Dib-Hajj et al., 2013; Bennett et al., 2019; Dib-Hajj and Waxman, 2019). The gain-of-function attributes of these variants increases confidence in their pathogenicity (Waxman et al., 2014), suggesting that carriers of these variants might benefit from treatments using sodium channel blockers.
Lacosamide, an FDA-approved anti-epileptic drug (AED), has been reported to preferentially enhance slow inactivation of VGSC, including Nav1.7 (Errington et al., 2006, 2008; Niespodziany et al., 2013). However, a recent study has alternatively suggested that lacosamide binds to the fast-inactivated state of Nav1.7, but at a slower rate (Jo and Bean, 2017). There is also evidence that lacosamide may alter the effect of CRMP2 (collapsin-response mediator protein 2) on Nav1.7 channel trafficking (Wilson and Khanna, 2015; Moutal et al., 2017). Furthermore, lacosamide has been found to have a higher affinity for tetrodotoxin-sensitive sodium channels over other ion channels and central receptors (Sheets et al., 2008; Moutal et al., 2017) that are targeted by traditional AEDs (Errington et al., 2006), conferring increased selectivity. A retrospective study of patients with seizures who had switched from other AED drugs to lacosamide, because of deleterious effects from their original AED treatment, has shown successful conversion in many of these patients (Kim et al., 2019), suggesting lacosamide to have improved tolerability. Lacosamide has also been tested in small clinical trials as a potential therapy for painful diabetic neuropathy where the majority of patients reported significant pain improvement with <10% reporting adverse effects (Rauck et al., 2007; Shaibani et al., 2009a, b; Wymer et al., 2009). However, until recently, the clinical effects of lacosamide on patients with Nav1.7-related pain disorders had yet to be investigated.
A recent randomized, placebo-controlled, double-blind, crossover trial (de Greef et al., 2019) assessed the effect of lacosamide treatment in 24 patients with SFN carrying multiple Nav1.7 variants. Fifteen different mutations were identified in this cohort, of which five were carried by several patients. Different subjects in this trial exhibited a spectrum of responses to treatment, with some patients reporting substantial pain relief while others did not show any improvement (Fig. 1). In a different study, lacosamide has been shown to produce pain relief in one patient with SFN (Namer et al., 2019). However, the factors responsible for variable lacosamide responsiveness in the de Greef et al. (2019) study are not known. In the present study, we report the Nav1.7 variants in these patients and assess the pharmacogenomic correlation between the Nav1.7 genetic variants and the response to lacosamide in this series of patients. Using voltage-clamp recordings of HEK293 (human embryonic kidney) cells expressing variants from both responsive and non-responsive patients, we characterized the effect of lacosamide on the biophysical properties of the Nav1.7 channel, at a clinically achievable concentration, and identify for the first time a hyperpolarizing effect on fast inactivation, which reduces the fraction of the channels that are available to open, as a distinguishing property of variants that correlates with lacosamide responsiveness in patients with SFN.
Figure 1.

Clinical response of patients carrying specific Nav1.7 variants. (A) The changes in pain scores in response to either lacosamide (LCM) or placebo are plotted for all subjects carrying Nav1.7 variants assessed in this study. Black and orange colours, adapted from de Greef et al. (2019), represent results from mostly responsive patients, whereas green, red and blue represent the results of mostly unresponsive patients. (B–F) The clinical responses for each variant. (B) The responses from a non-responder carrying variant I228M. (C) The responses from a patient with a large clinical response carrying variant W719C. (D) The responses from the patients carrying the I739V variant (five patients, four responders). (E) The responses from patients carrying the L1267V variant (four patients, three non-responders). (F) The responses from patients carrying the W1538R variant (two patients, both non-responders).
Materials and methods
Patients
Twenty-four participants (18–80 years old) were recruited at the Maastricht University Medical Center+ (UMC+) for the Lacosamide Efficacy-N’-Safety Study (LENSS) (de Greef et al., 2019). Subjects were selected based on diagnosis of pure SFN with no associated conditions (except for diabetes mellitus), with a mutation in the SCN9A gene (Nav1.7 channel) of class III, IV or V (variants of uncertain pathogenicity, likely to be or clearly pathogenic, respectively), using genetic classification criteria, as described previously (Wallis et al., 2013).
The clinical study design and results were published previously (de Greef et al., 2016, 2019). Briefly, the participants received either 200 mg of lacosamide or placebo twice daily for 8 weeks, preceded by a titration period and followed by a tapering period, and a 2-week washout period before receiving the alternative compound. They were asked to rate their pain twice daily using the daily pain intensity numerical rating scale (PI-NRS) ranging from 0 to 10. The patients were considered responsive to treatment when at least a 1-point reduction was reported from the individual baseline pain scores.
Plasmids and HEK293 cell transfection
The construct of the human Nav1.7 wild-type isoform was rendered tetrodotoxin-resistant and fused to an eGFP-2A linker, as previously described (Yang et al., 2016). The construct produces a green fluorescent protein with the Nav1.7 channel as independent proteins from the same transcript, which enables the visual identification of transfected cells based on their fluorescence. Nav1.7-related SFN mutations identified in participants in this study (I739V, I228M, L1267V, W719C and W1538R) were introduced into the constructs using QuikChange® XL site-directed mutagenesis (Stratagene). HEK293 cells, grown under standard culture conditions (5% CO2, 37°C) in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum, were transiently transfected by electroporation with a Nav1.7 variant using Lipojet (SignaGen Laboratories). The transfected cells were resuspended and plated onto coverslips the next day and patch-clamp recordings were performed over the following 48 h.
Electrophysiology
Whole-cell voltage-clamp recordings were obtained at room temperature on isolated HEK293 cells showing green fluorescence. Recordings were alternatively performed on coverslips of HEK293 cells expressing wild-type or a Nav1.7 variant, either treated with vehicle (extracellular solution) or lacosamide. Electrodes were pulled from 1.65-mm outer diameter borosilicate glass micropipettes (WPI) and had a resistance of 0.8–2.0 MΩ when filled with intracellular pipette solution, which contained (in mM): 140 CsF, 10 NaCl, 10 HEPES, 1 EGTA (pH 7.3 with CsOH, adjusted to 320 mOsm with dextrose). The extracellular solution contained (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES (pH 7.3 with NaOH, adjusted to 320 mOsm with dextrose). Voltage-clamp protocols for the biophysical characterization of VGSCs were initiated 5 min after establishing whole-cell configuration on an EPC-10 USB amplifier (HEKA Electronics) and acquired using PatchMaster software (HEKA Electronics) at an acquisition rate of 50 kHz with a low-pass Bessel filter setting of 2.9 kHz. Voltage errors were minimized with 60–90% series resistance compensation, and only cells with <4 mV voltage error after compensation were included for analysis. When appropriate, linear leak currents and capacitance artefacts were corrected by P/6 subtraction.
To examine the current-voltage (I-V) relationship, a series of step depolarizations from −80 mV to +50 mV in 5-mV increments were applied from a holding potential of −120 mV at 5-s interpulse intervals. To evaluate the effect of lacosamide on steady-state fast inactivation, the membrane potential was held at conditioning potentials that varied from −140 mV to 10 mV for 500 ms, and cells were then given a 20-ms test pulse to 0 mV to elicit current from remaining available channels.
For the slow inactivation protocol, the cell was held at conditioning potentials that varied from −130 mV to 10 mV for 30 s; the membrane potential was then pulsed to −120 mV for 100 ms to allow channels not in the slow inactivated state to recover from fast inactivation, and then given a 20-ms test pulse to 0 mV to elicit current from available channels.
To measure the use-dependence of inhibition of lacosamide at 20 Hz stimulation, series of 20-ms pulses were applied at −10 mV. Peak inward currents were measured and normalized to the maximum current amplitude.
In vitro pharmacology
A bi-daily 200 mg lacosamide dose results in a maximum plasma concentration of 30 μM (Cawello, 2015). The patients assessed in this study received lacosamide at this bi-daily dose during the trial (de Greef et al., 2019); thus 30 μM of lacosamide was used for the studies described here.
Lacosamide (Vimpat®, UCB, obtained from the West Haven VA Medical Center pharmacy), with a concentration of 10 mg/ml (39.9 mM) in aqueous saline (pH 4), was diluted in the extracellular bath solution (vehicle; described above) to achieve the final working concentration of 30 µM. Working solutions were prepared fresh daily.
The extracellular bath solution was continuously perfused at a consistent flow rate of 1 ml/min through a 250 μm perfusion pipette, using a pressure-regulated system (AutoMate Scientific). Complete bath exchange was enabled via aspiration from the opposite end of the cell chamber from the perfusion pipette. To control for time-dependent changes in channel properties, independent cell cohorts were exposed to only one test concentration of lacosamide or vehicle at a consistent time after initiation of whole-cell recording. Specifically, the solution was exchanged for 2 min, corresponding to an absolute bath exchange of 2 ml, starting 2 min following breakage of the cell membrane.
Statistical analyses
Whole-cell voltage-clamp data were analysed using Fitmaster (HEKA Electronics), Excel (Microsoft) and Origin (OriginLab Corporation, Microcal Software, Northhampton, MA). Data are expressed as means ± standard error of the mean (SEM). Statistical significance was determined by unpaired Student’s t-test and was reached when P < 0.05.
Data availability
Data that support the findings of this study are available upon reasonable request.
Results
Clinical characterization
Whole-exome sequencing of the patients’ genomes identified 15 different SCN9A mutations, including five recurrent variants (Supplementary Table 1). Patients carrying different Nav1.7 variants displayed a range of responses to lacosamide. We selected variants that were most representative of the range of clinical responsiveness for biophysical analysis. Specifically, we selected two variants from responsive patients (≥1 point on the PI-NRS scale) and three variants from non-responsive patients (≤1 point on the PI-NRS scale), based on the pain scores plot in the clinical trial study (de Greef et al., 2019), shown in Fig. 1.
We considered the W719C (2157G>C; p.Trp719Cys) and the I739V (c.2215A>G; p.Ile739Val) variants to be associated with responders. W719C showed the largest response to lacosamide compared to the other variants, with a 3.65-point decrease from the baseline average score. I739V, which has been functionally profiled in detail previously (Fu et al., 2012; Han et al., 2012), was found in five different patients. While four I739V carriers displayed decreases (1.1 to 2.8 points) in their pain scores, one carrier did not exhibit any significant improvement while treated with lacosamide (Fig. 1D).
By contrast, I228M (c.684C>G; p.Ile228Met), L1267V (c.3799C>G; p.Leu1267Val) and W1538R (c.4612T>C; p.Trp1538Arg) variants were associated with non-responders. L1267V was selected because it was carried by four patients. Three did not respond to lacosamide, while one carrier of this variant responded to both lacosamide and placebo; there was a 3.0-point reduction in the baseline pain score following the lacosamide phase compared to the placebo phase (Fig. 1E). I228M is a well-studied variant with a clearly defined pathogenicity (Estacion et al., 2011; Persson et al., 2013) (Fig. 1B), and W1538R, previously characterized as a gain-of-function variant altering cellular excitability (Cregg et al., 2013), was identified in two carriers with compound mutations in Nav1.7, both of whom were non-responders (Fig. 1F). The two patients carrying the W1538R variation were both found to also carry a M932L/V991L (c.2794A>C and c.2971G>T; p.Met932Leu/Val991Leu) double mutation, with one of them also carrying I739V in addition to the M932L/V991L mutation. Notably, patients who only carry M932L/V991L or I739V variants alone have been responsive to lacosamide treatment (Supplementary Table 1). The pain scores are presented in Fig. 1.
The locations of the variants within the Nav1.7 backbone are shown in Fig. 2. Both responder variants are located in the second domain (DII): I739V is located within the first transmembrane segment of the voltage-sensing domain (VSD; DII/S1) (Faber et al., 2012), while W719C is found 20 amino acids upstream, at the end of the first intracellular loop linking DI/S6 to DII/S1 (Linker 1). The non-responder variants are spread across the three other domains within the VSD (S1–S4) of the channel: I228M is located in DI/S4 (Faber et al., 2012), L1267V in DIII/S3 (Brouwer et al., 2014) and W1538R in DIV/S2 (Cregg et al., 2013; Kapetis et al., 2017).
Figure 2.
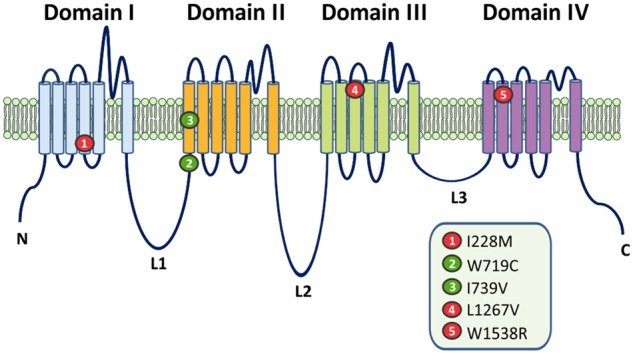
Topology of the human Nav1.7 sodium channel, with the location of I228M, W719C, I739V, L1267V and W1538R. All but one mutation (W719C) are located in the VSDs of the channel. The two lacosamide-responsive mutations are shown in green. Both are located within the vicinity of Domain II: W719C is located at the end of Linker 1 and I739V in VSD II. Non-responsive mutations are indicated in red. All three variants (I228M, L1267V and W1538R) map to the VSD in Domains I, III and IV, respectively.
Lacosamide fails to alter the voltage-dependence of activation in responsive and non-responsive Nav1.7 variants
Representative Nav1.7 sodium currents recorded after perfusion with 30 μM lacosamide or vehicle control are shown in Fig. 3. Consistent with previous data (Errington et al., 2006, 2008), the voltage-dependence of activation, represented as the Boltzmann fit of normalized conductance, was unaffected by the clinically achievable concentration of 30 µM lacosamide in both the mutant and wild-type cells (Fig. 3).
Figure 3.

Lacosamide effects on voltage-dependence of activation for each Nav1.7 variant. (A) Sodium current traces elicited by the activation protocol from different cells exposed to either lacosamide (LCM) or vehicle control. Sodium inward currents were elicited from a holding potential of −120 mV to various depolarizing steps ranging from −80 to +40 mV in 5 mV increments. (B) Normalized current-voltage relationship obtained by measuring the peak inward current elicited as a function of the stimulus voltage. (C) Voltage-dependence of activation for cells treated with lacosamide or vehicle control. The peak inward currents were transformed to normalized conductance from current-voltage plot.
Lacosamide evokes a hyperpolarizing shift in slow inactivation in most of the Nav1.7 variants
Lacosamide has previously been reported to have large effects on sodium channel slow inactivation (Errington et al., 2006, 2008; Niespodziany et al., 2013). To assess the potential impact of the variants on the lacosamide-mediated shift in slow inactivation, we compared the normalized slow inactivation curves from each of the variants examined in the presence of 30 μM lacosamide or vehicle control, as shown in Fig. 4. As expected, lacosamide shifted the voltage-dependence of slow inactivation of the wild-type channel to more hyperpolarized potentials (i.e. enhancing slow inactivation, thus reducing the number of Nav1.7 channels that are available to open at physiological membrane potentials; Fig. 4A). The half-inactivation voltage (V1/2) of slow-inactivation Boltzmann fits from each variant are shown in Table 1. The variants from patients who were scored as responders showed substantially enhanced slow inactivation in response to lacosamide: W719C demonstrated a −20 mV hyperpolarized shift (Fig. 4C, P = 0.02), while I739V channels showed a −12.2 mV shift to more hyperpolarized potentials (Fig. 4D, P < 0.01). The shifts in voltage-dependence of slow inactivation for the non-responsive variants (L1267V, W1538R, and I228M) were −9.3 mV, +1.4 mV, and −12.1 mV, respectively (Fig. 4B, E and F). Except for W1538R, lacosamide induced a significant hyperpolarizing shift in voltage-dependence of slow inactivation in all variants.
Figure 4.

Lacosamide effects on slow inactivation for each Nav1.7 variant. The averaged voltage-dependence of slow inactivation curves in the presence (red) or absence (black) of lacosamide are shown for (A) wild-type (WT), (B) I228M, (C) W719C, (D) I739V, (E) L1267V and (F) W1538R. The average V1/2s calculated from of the Boltzmann fits are listed in Table 1.
Table 1.
Half inactivation voltage (V1/2) and use-dependence of inhibition for the Nav1.7 mutations to lacosamide
| Variant | Slow inactivation (V1/2) | Fast inactivation (V1/2) | Use-dependence | |||
|---|---|---|---|---|---|---|
| Vehicle | Lacosamide | Vehicle | Lacosamide | Vehicle | Lacosamide | |
| Wild-type | −70.6 ± 1.4 (n = 16) | −79.5 ± 2.1** (n = 9) | −86.8 ± 1.4 (n = 19) | −88.0 ± 1.9 (n = 8) | 0.82 ± 0.02 (n = 20) | 0.75 ± 0.02* (n = 9) |
| Responders | ||||||
| W719C | −56.9 ± 4.4 (n = 8) | −77.1 ± 5.3* (n = 4) | −78.7 ± 0.8 (n = 11) | −88.4 ± 1.3** (n = 5) | 0.96 ± 0.01 (n = 10) | 0.87 ± 0.04** (n = 4) |
| I739V | −67.1 ± 1.6 (n = 12) | −79.3 ± 2.1** (n = 5) | −85.2 ± 1.0 (n = 11) | −99.8 ± 1.1** (n = 6) | 0.82 ± 0.01 (n = 16) | 0.73 ± 0.03** (n = 5) |
| Non-responders | ||||||
| L1267V | −74.4 ± 2.1 (n = 9) | −83.7 ± 2.7* (n = 8) | −82.5 ± 2.4 (n = 7) | −87.1 ± 1.6 (n = 11) | 0.89 ± 0.02 (n = 12) | 0.87 ± 0.02 (n = 10) |
| W1538R | −69.4 ± 4.0 (n = 10) | −68.0 ± 2.8 (n = 8) | −79.5 ± 3.0 (n = 10) | −73.9 ± 1.2 (n = 8) | 0.91 ± 0.02 (n = 11) | 0.93 ± 0.01 (n = 8) |
| I228M | −66.0 ± 2.9 (n = 7) | −78.1 ± 2.5** (n = 7) | −87.1 ± 1.6 (n = 11) | −88.9 ± 2.8 (n = 6) | 0.82 ± 0.03 (n = 6) | 0.75 ± 0.01* (n = 7) |
Lacosamide significantly enhanced slow inactivation in wild-type and all mutant channels, except for W1538R, while fast inactivation was only affected by lacosamide in W719C and I739V channels. Use-dependence of inhibition at 20 Hz was changed in the two responder variants as well as in wild-type and I228M. Data are presented as mean ± SEM. Significant values are represented by *P < 0.05, **P < 0.01.
Lacosamide enhances fast inactivation selectively in responsive variants
While recent data suggest the potential of lacosamide to affect steady state fast inactivation in Nav1.7 wild-type channels (Jo and Bean, 2017), most previous reports have shown little to no effects of lacosamide on fast inactivation. Nonetheless, other compounds that bind to the local anaesthetic site, such as carbamazepine, phenytoin, or lamotrigine, have been shown to enhance fast inactivation (Kuo and Lu, 1997; Mantegazza et al., 2010). We compared the effect of lacosamide on fast inactivation for the five variants and found that lacosamide significantly enhanced fast inactivation exclusively in variants carried by patients who were scored as responsive, while having no significant effect on wild-type or on variants from patients who scored as non-responsive (Fig. 5). I739V displayed a large −14.6 mV hyperpolarizing shift (Fig. 5D, P < 0.01), and W719C channels exhibited a − 9.7 mV shift (Fig. 5C, P < 0.01). The V1/2 of the fast-inactivation Boltzmann fits from each variant are documented in Table 1.
Figure 5.

Lacosamide effects on steady state fast inactivation for each Nav1.7 variant. The averaged fast inactivation curves in the presence or absence of lacosamide are shown for (A) wild-type (WT), (B) I228M, (C) W719C, (D) I739V, (E) L1267V and (F) W1538R. The average V1/2s calculated from of the Boltzmann fits are listed in Table 1.
Lacosamide produces a variable effect on use-dependent block in non-responders
We also examined the development of use-dependent block at 20 Hz for each of the variants upon exposure to 30 μM lacosamide or vehicle control (Fig. 6). Lacosamide enhanced use-dependent inhibition in wild-type channels (Fig. 6A) and in variants from patients who were scored as responders (Fig. 6C and D). Interestingly, lacosamide did not increase use-dependent blockade in two of the variants identified in non-responsive patients (L1267V and W1538R; Fig. 6E and F), while it did in the non-responsive I228M variant (Fig. 6B and Table 1). The degree of use-dependent block from each Nav1.7 mutation is shown in Table 1.
Figure 6.

Lacosamide effects on use-dependent block for each Nav1.7 variant. The averaged use-dependent block to a train of 20 pulses at 20 Hz in the presence or absence of lacosamide are shown for (A) wild-type (WT), (B) I228M, (C) W719C, (D) I739V, (E) L1267V and (F) W1538R. The averages of the normalized responses are shown in Table 1.
Discussion
Pharmacotherapy for the treatment of pain in SFN is limited, with many patients reporting inadequate pain relief from currently available medications, including sodium channel blockers. However, while mutations in peripheral sodium channels have been linked to pathological pain, there have only been a few studies documenting treatment outcomes in patients with specific Nav1.7 mutations. We have recently described pharmacogenomically-guided targeting of rare but drug-responsive Nav1.7 mutations with carbamazepine in patients with inherited erythromelalgia (Yang et al., 2012, 2018; Geha et al., 2016; Han et al., 2018). However, thus far there has not been a pharmacogenomic study of patients with more common pain disorders who carry Nav1.7 channel mutations. In this study, we build on the findings of the LENSS trial (de Greef et al., 2019), which investigated the efficacy of lacosamide for the treatment of patients with Nav1.7-related SFN, and provide biophysical data that may explicate why a subset of Nav1.7 variants differentially responded to lacosamide treatment.
Lacosamide has been shown to operate via different mechanisms of action compared to classical sodium channel blockers. Using standard recording protocols, AEDs have been found to enhance both fast and slow inactivation, while lacosamide has been reported to selectively enhance voltage-dependence of slow inactivation. According to this model, lacosamide has been proposed to either bind to a different site, or at significantly slower binding rates than other anticonvulsants (Errington et al., 2008; Sheets et al., 2008). However, using a different recording protocol and a concentration of 100 μM, lacosamide has been reported to effectively enhance steady state fast inactivation when exposed for long depolarizations, possibly due to binding to the fast inactivated state but at a very slow rate (Jo and Bean, 2017). In the present study, we observed that, at clinically achievable concentration of 30 μM, lacosamide does not affect fast inactivation in Nav1.7 wild-type channels when standard recording protocols were used (Fig. 5A). Lacosamide failure to alter fast inactivation in wild-type channels in our study is likely due to the different lacosamide concentration and recording conditions, compared to those used by Jo and Bean (2017).
Lacosamide binding activity is known to share several properties with well-established sodium channel inhibitors. For instance, lacosamide has been found to compete with carbamazepine and lidocaine for channel inhibition in vitro, suggesting that they likely bind at the same site, but with different pharmacodynamics (Jo and Bean, 2017). Classical sodium channel blockers have been shown to share the same binding site located in the pore-forming region of the channel, within S6 of DI, DIII and DIV (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2001, 2002; Catterall and Swanson, 2015). Genetic alterations at these sites have been shown to fully abolish both voltage- and use-dependent block (Fozzard et al., 2011), highlighting the importance of maintaining the integrity of these regions for potent inhibition. Topological mapping shows that none of the five variants described in this study map to the pore domain itself, but instead they map to the VSD (Fig. 2). Therefore, the variants might not directly interfere with the drug binding site in the ion conduction pathway.
While lacosamide did not affect fast inactivation in wild-type channels (Fig. 5A) or any of the three non-responder variants (Fig. 5B, E and F), it caused a significant and substantial hyperpolarizing shift in the two variants identified in responsive patients (W719C and I739V; Fig. 5C and D). These differences in response to lacosamide may result from a novel emerging mechanism of action, conferred by these specific variants. This type of phenomenon has previously been reported in inherited erythromelalgia, where multiple examples of Nav1.7 variants underlying inherited erythromelalgia pathogenicity have been shown to be treatment-responsive to carbamazepine in a disorder in which most patients are treatment-resistant. Three inherited erythromelalgia-causing variants mapping to DI (I234T, S241T and V400M) have been shown to respond to carbamazepine via a depolarizing (corrective) shift in activation (Fischer et al., 2009; Meijer et al., 2014; Geha et al., 2016). Importantly, none of the mutations described above are located near the carbamazepine/local anaesthetic binding site. These findings, together with the present results, indicate that mutations outside the pore domain can alter the response of the mutant channel to drugs traditionally considered to be pore-binding, and suggest that functional testing of these variants in vitro may predict treatment responsiveness.
While fast inactivation seems to be the parameter that discriminates the responders from the non-responders, effects of lacosamide on slow inactivation and use-dependence might also be required for complete and sustained pain relief. In agreement with the known effects of lacosamide on slow inactivation, the wild-type channel as well as all but one mutant channel (W1538R) underwent a significant hyperpolarizing shift in the voltage-dependence of slow inactivation (Fig. 4). Interestingly, the W1538R variant was also found to be the strongest non-responder, with one carrier reporting better pain improvement from placebo than from treatment itself (Fig. 1). This variant specifically has been mapped to the VSD of DIV, and is part of the binding site of the new class of aryl sulfonamide Nav1.7 blockers, such as PF-05089771, which targets both the fast and slow inactivated states of Nav1.7 with high selectivity (McCormack et al., 2013; Focken et al., 2016; Theile et al., 2016). This observation raises the possibility of a potential common mechanism of action between the two drugs and suggests that a disruption at this site might alter lacosamide function.
Whole-exome sequencing (WES) of the two patients carrying the W1538R variant identified additional Nav1.7 mutations in both of these subjects. Both individuals carried the M932L/V991L variant, which has been associated with DRG neuron hyperexcitability (Faber et al., 2012), and one of these subjects also carried the I739V mutation (which also has been shown to produce DRG neuron hyperexcitability) (Faber et al., 2012; Han et al., 2012). WES analysis as carried out in this study does not allow the identification of the distribution of these mutations between the two Nav1.7 alleles, which requires analysis of DRG-specific RNA sequences from these patients. Such analysis is beyond the scope of this study because it requires access to sensory tissues from these patients or the development of induced pluripotent stem cells that can then be differentiated into nociceptors in vitro. Nonetheless, as other subjects in this cohort carrying either the I739V alone or the M932L/V991L compound variants responded to lacosamide (Supplementary Table 1), and fast inactivation of I739V mutant channels was hyperpolarized by exposure to lacosamide (Fig. 5), the W1538R mutation potentially acts as a dominant contributor to the poor efficacy of lacosamide in these subjects carrying the compound genotypes. Future clinical studies on patients carrying only the W1538R mutation will be needed to provide a more definitive conclusion that carriers of this mutation will not respond to treatment with lacosamide.
Interestingly, one of the patients carrying solely the I739V variant was unresponsive to lacosamide, while the four other patients were previously reported to respond well to lacosamide (Fig. 1) (de Greef et al., 2019). Furthermore, in the current study we demonstrated lacosamide to significantly shift all three gating parameters of I739V channels (Table 1). Thus, the cause for the lack of response of one carrier with the I739V mutation is not channel intrinsic, but is likely to be affected by additional genetic, epigenetic or environmental factors. Notably, the non-responder carrier had a relatively low level of pain severity, which may have limited the detection of lacosamide efficacy.
Many factors can impact on an individual’s response to a drug. While focusing on the interaction between drug and target molecule, our data suggest that in vitro pharmacological and biophysical analysis of Nav1.7 mutations may predict the likelihood of specific carriers to be responsive to treatment, which could be important in the future as an approach to personalized treatment based on the patient’s genetic background. Building on the first clinical trial of lacosamide for treatment of patients with SFN and carriers of Nav1.7 variants (LENSS) (de Greef et al., 2019), the present results suggest that, at least for strong positive responders and non-responders, pharmacogenomic analysis in vitro may correlate with clinical responsiveness. The relatively small number of variants that were studied here, two responders and three non-responders, necessitates caution in generalizing these data to suggest that SFN patients who carry Nav1.7 variants that enhance fast inactivation upon exposure to lacosamide will necessarily be responsive to treatment. If supported by studies in larger numbers of patients tested with lacosamide and if extended to other potential medications, the pharmacogenomic approach described in this paper might contribute to the development of selective, individualized pain treatment strategies.
Supplementary Material
Acknowledgements
We thank Fadia Dib-Hajj and Daniel Sosniak for excellent technical assistance.
Funding
This work was supported by Center Grant B9253-C from the U.S. Department of Veterans Affairs Rehabilitation Research and Development Service. This project also received funding from the Molecule-to-Man Pain Network, a European Commission Multi-Center Collaborative Projects through the European Union’s Horizon 2020 research and innovation program under grant agreement No. 721841. The clinical study was supported by a grant from the Prinses Beatrix Spierfonds. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
Competing interests
The authors report no competing interests.
Glossary
Abbreviations
- DRG =
dorsal root ganglion
- SFN =
small fibre neuropathy
- VSD =
voltage-sensing domain
References
- Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev 2019; 99: 1079–151. [DOI] [PubMed] [Google Scholar]
- Brouwer BA, Merkies IS, Gerrits MM, Waxman SG, Hoeijmakers JG, Faber CG. Painful neuropathies: the emerging role of sodium channelopathies. J Peripher Nerv Syst 2014; 19: 53–65. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Swanson TM. Structural basis for pharmacology of voltage-gated sodium and calcium channels. Mol Pharmacol 2015; 88: 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawello W. Clinical pharmacokinetic and pharmacodynamic profile of lacosamide. Clin Pharmacokinet 2015; 54: 901–14. [DOI] [PubMed] [Google Scholar]
- Cregg R, Laguda B, Werdehausen R, Cox JJ, Linley JE, Ramirez JD, et al. Novel mutations mapping to the fourth sodium channel domain of Nav1.7 result in variable clinical manifestations of primary erythromelalgia. Neuromol Med 2013; 15: 265–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Greef BTA, Hoeijmakers JGJ, Geerts M, Oakes M, Church TJE, Waxman SG, et al. Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: a randomized controlled trial. Brain 2019; 142: 263–75. [DOI] [PubMed] [Google Scholar]
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies I. Associated conditions in small fiber neuropathy-a large cohort study and review of the literature. Eur J Neurol 2018; 25: 348–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Greef BT, Merkies IS, Geerts M, Faber CG, Hoeijmakers JG. Efficacy, safety, and tolerability of lacosamide in patients with gain-of-function Nav1.7 mutation-related small fiber neuropathy: study protocol of a randomized controlled trial-the LENSS study. Trials 2016; 17: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Waxman SG. Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci 2019; 42: 87–106. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 2013; 14: 49–62. [DOI] [PubMed] [Google Scholar]
- Eijkenboom I, Sopacua M, Hoeijmakers JGJ, de Greef BTA, Lindsey P, Almomani R, et al. Yield of peripheral sodium channels gene screening in pure small fibre neuropathy. J Neurol Neurosurg Psychiatry 2019; 90: 342–52. [DOI] [PubMed] [Google Scholar]
- Errington AC, Coyne L, Stohr T, Selve N, Lees G. Seeking a mechanism of action for the novel anticonvulsant lacosamide. Neuropharmacology 2006; 50: 1016–29. [DOI] [PubMed] [Google Scholar]
- Errington AC, Stohr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol 2008; 73: 157–69. [DOI] [PubMed] [Google Scholar]
- Estacion M, Han C, Choi JS, Hoeijmakers JG, Lauria G, Drenth JP, et al. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain 2011; 7: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, et al. Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012; 71: 26–39. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015; 14: 162–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer TZ, Gilmore ES, Estacion M, Eastman E, Taylor S, Melanson M, et al. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol 2009; 65: 733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focken T, Liu S, Chahal N, Dauphinais M, Grimwood ME, Chowdhury S, et al. Discovery of aryl sulfonamides as isoform-selective inhibitors of NaV1.7 with efficacy in rodent pain models. ACS Med Chem Lett 2016; 7: 277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozzard HA, Sheets MF, Hanck DA. The sodium channel as a target for local anesthetic drugs. Front Pharmacol 2011; 2: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Wang SJ, Zhou GD, Liu W, Cao Y, Zhang WJ. Residual undifferentiated cells during differentiation of induced pluripotent stem cells in vitro and in vivo. Stem Cells Dev 2012; 21: 521–9. [DOI] [PubMed] [Google Scholar]
- Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV, et al. Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol 2016; 73: 659–67. [DOI] [PubMed] [Google Scholar]
- Han C, Hoeijmakers JG, Ahn HS, Zhao P, Shah P, Lauria G, et al. Nav1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 2012; 78: 1635–43. [DOI] [PubMed] [Google Scholar]
- Han C, Hoeijmakers JG, Liu S, Gerrits MM, Te Morsche RH, Lauria G, et al. Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain 2012; 135: 2613–28. [DOI] [PubMed] [Google Scholar]
- Han C, Themistocleous AC, Estacion M, Dib-Hajj FB, Blesneac I, Macala L, et al. The novel activity of carbamazepine as an activation modulator extends from NaV1.7 mutations to the NaV1.8-S242T mutant channel from a patient with painful diabetic neuropathy. Mol Pharmacol 2018; 94: 1256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Bean BP. Lacosamide inhibition of Nav1.7 voltage-gated sodium channels: slow binding to Fast-inactivated states. Mol Pharmacol 2017; 91: 277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetis D, Sassone J, Yang Y, Galbardi B, Xenakis MN, Westra RL, et al. Network topology of NaV1.7 mutations in sodium channel-related painful disorders. BMC Syst Biol 2017; 11: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Kim HK, Bae EK. Switching from traditional sodium channel blockers to lacosamide in patients with epilepsy. Seizure 2019; 65: 172–5. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Lu L. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br J Pharmacol 1997; 121: 1231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M. Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol 2010; 9: 413–24. [DOI] [PubMed] [Google Scholar]
- McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci U S A 2013; 110: E2724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer IA, Vanasse M, Nizard S, Robitaille Y, Rossignol E. An atypical case of SCN9A mutation presenting with global motor delay and a severe pain disorder. Muscle Nerve 2014; 49: 134–8. [DOI] [PubMed] [Google Scholar]
- Moutal A, Yang X, Li W, Gilbraith KB, Luo S, Cai S, et al. CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide. Pain 2017; 158: 2301–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namer B, Schmidt D, Eberhardt E, Maroni M, Dorfmeister E, Kleggetveit IP, et al. Pain relief in a neuropathy patient by lacosamide: proof of principle of clinical translation from patient-specific iPS cell-derived nociceptors. EBioMedicine 2019; 39: 401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niespodziany I, Leclere N, Vandenplas C, Foerch P, Wolff C. Comparative study of lacosamide and classical sodium channel blocking antiepileptic drugs on sodium channel slow inactivation. J Neurosci Res 2013; 91: 436–43. [DOI] [PubMed] [Google Scholar]
- Nishikawa N, Nomoto M. Management of neuropathic pain. J Gen Fam Med 2017; 18: 56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AK, Liu S, Faber CG, Merkies IS, Black JA, Waxman SG. Neuropathy-associated Nav1.7 variant I228M impairs integrity of dorsal root ganglion neuron axons. Ann Neurol 2013; 73: 140–5. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994; 265: 1724–8. [DOI] [PubMed] [Google Scholar]
- Rauck RL, Shaibani A, Biton V, Simpson J, Koch B. Lacosamide in painful diabetic peripheral neuropathy: a phase 2 double-blind placebo-controlled study. Clin J Pain 2007; 23: 150–8. [DOI] [PubMed] [Google Scholar]
- Shaibani A, Biton V, Rauck R, Koch B, Simpson J. Long-term oral lacosamide in painful diabetic neuropathy: a two-year open-label extension trial. Eur J Pain 2009a; 13: 458–63. [DOI] [PubMed] [Google Scholar]
- Shaibani A, Fares S, Selam JL, Arslanian A, Simpson J, Sen D, et al. Lacosamide in painful diabetic neuropathy: an 18-week double-blind placebo-controlled trial. J Pain 2009b; 10: 818–28. [DOI] [PubMed] [Google Scholar]
- Sheets PL, Heers C, Stoehr T, Cummins TR. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3-methoxypropanamide], lidocaine, and carbamazepine. J Pharmacol Exp Ther 2008; 326: 89–99. [DOI] [PubMed] [Google Scholar]
- Sopacua M, Hoeijmakers JGJ, Merkies ISJ, Lauria G, Waxman SG, Faber CG. Small-fiber neuropathy: expanding the clinical pain universe. J Peripher Nerv Syst 2019; 24: 19–33. [DOI] [PubMed] [Google Scholar]
- Theile JW, Fuller MD, Chapman ML. The selective Nav1.7 inhibitor. Mol Pharmacol 2016; 90: 540–8. [DOI] [PubMed] [Google Scholar]
- Wallis Y, Payne S, McAnulty C, Bodmer D, Sistermans E, Robertson K, et al. Practical guidelines for the evaluation of pathogenicity and the reporting of sequence variants in clinical molecular genetics. Assoc Clin Genom Sci (ACGS) 2013. Available from https://www.acgs.uk.com/media/10791/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf (15 September 2019, date last accessed). [Google Scholar]
- Waxman SG, Merkies ISJ, Gerrits MM, Dib-Hajj SD, Lauria G, Cox JJ, et al. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol 2014; 13: 1152–60. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Khanna R. Specific binding of lacosamide to collapsin response mediator protein 2 (CRMP2) and direct impairment of its canonical function: implications for the therapeutic potential of lacosamide. Mol Neurobiol 2015; 51: 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymer JP, Simpson J, Sen D, Bongardt S, Lacosamide S. Efficacy and safety of lacosamide in diabetic neuropathic pain: an 18-week double-blind placebo-controlled trial of fixed-dose regimens. Clin J Pain 2009; 25: 376–85. [DOI] [PubMed] [Google Scholar]
- Yang Y, Adi T, Effraim PR, Chen L, Dib-Hajj SD, Waxman SG. Reverse pharmacogenomics: carbamazepine normalizes activation and attenuates thermal hyperexcitability of sensory neurons due to Nav 1.7 mutation I234T. Br J Pharmacol 2018; 175: 2261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Dib-Hajj SD, Zhang J, Zhang Y, Tyrrell L, Estacion M, et al. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun 2012; 3: 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Huang J, Mis MA, Estacion M, Macala L, Shah P, et al. Nav1.7-A1632G mutation from a family with inherited erythromelalgia: enhanced firing of dorsal root ganglia neurons evoked by thermal stimuli. J Neurosci 2016; 36: 7511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall WA. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J Biol Chem 2001; 276: 20–7. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug bloc. J Biol Chem 2002; 277: 35393–401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study are available upon reasonable request.