

Abstract
An antikinetoplastid pharmacomodulation study at position 3 of the recently described 3-bromo-8-nitroquinolin-2(1H)-one hit molecule was conducted. 24 derivatives were synthesized using the Suzuki-Miyaura cross-coupling reaction and evaluated in vitro toward both L. infantum axenic amastigotes and T. brucei brucei trypomastigotes. The results show that the introduction of a para-carboxyphenyl group at position 3 of the scaffold leads to a selective antitrypanosomal hit molecule (21) with a lower reduction potential (-0.56 V) than the initial hit (-0.45 V). Compound 21 displays a micromolar antitrypanosomal activity (IC50 = 1.5 μM) and a low cytotoxicity on the human HepG2 cell line (CC50 = 120 μM), reaching higher selectivity index (IS = 80) than the reference drug eflornithine. Contrary to the results previously obtained in this series, hit compound 21 is not active toward L. infantum and is not efficiently bioactivated by T. brucei brucei type I nitroreductase, which suggests the existence of an additional mechanism of action.
Keywords: Trypanosomatids, 8-nitroquinolin-2(1H)-one series, Antikinetoplastid pharmacomodulation, Parasitic nitroreductases, Pallado-catalyzed cross-coupling reactions
Introduction
Kinetoplastids are flagellated protozoan parasites responsible for lethal neglected tropical diseases such as human African trypanosomiasis (HAT) and visceral leishmaniasis (VL). They are characterised by the presence of a circular DNA called kinetoplast, adjacent to the flagellar basal body. Trypanosoma parasites are transmitted by the bite of an infected sandfly and are the causative agents of HAT, also known as “sleeping sickness”. There are many species of Trypanosoma, but only T. brucei gambiense and T. brucei rhodesiense are responsible for HAT.[1] During the infection, metacylic trypomastigotes enter in the blood circulation via the bite of the “tse tse fly” and disseminate in the whole organism where they differentiate into bloodstream trypomastigotes: this is the hemolymphatic stage. After several weeks or months including symptoms such as headaches, anaemia and hepatosplenomegaly, trypomastigotes cross the blood-brain barrier (BBB) and cause damages to the central nervous system, leading to sleeping disorders, behavioural disorders, seizure, coma and finally death: this is the meningoencephalic stage.[2] Leishmania is another protozoan parasite, at the origin of leishmaniasis and transmitted by the bite of an infected sandfly. L. donovani and L. infantum are the two major species causing the most severe form of the disease: visceral leishmaniasis (VL).[3] Briefly, metacyclic promastigotes penetrate into the skin during the blood meal of an infected sandly. They are internalized by mononuclear phagocytic cells such as macrophages where they differentiate into amastigotes. Parasites multiply in these cells until their destruction and disseminate in many organs such as liver and spleen, leading to death.[4]
HAT and VL represent more than 1 billion people at risk and are responsible for more than 55.000 new cases and 25.000 deaths each year.[5,6] These numbers are surely underestimated because of the difficulty to access to some rural areas and the unspecific symptoms in the early stages of the diseases. Currently, there are very few efficient and safe drugs available on the market against these neglected tropical diseases. Pentamidine and suramin are used for the treatment of the first stage of HAT but these molecules are highly toxic and need hospitalisation for the IV administration.[7] Melarsoprol, an arsenic containing-drug and a combination of eflornithine and nifurtimox are suitable for the second stage of HAT (Fig. 1).[8]. The same observation can be made with VL for which only Amphotericin B, miltefosine, antimonial derivatives, pentamidine and paromomycin are available. Among these drugs, miltefosine is the only orally available drug. These drugs are either expensive (liposomal amphotericin B), present severe side effects (nephrotoxicity of amphotericin B, teratogenicity of miltefosine…) or show an increasing lack of efficacy due to the emergence of resistant parasites (antimony derivatives and miltefosine).[9] This global context calls for the discovery of new antikinetoplastid molecules.
Figure 1.

Structure, indication and route of administration of the antitrypanosomal drugs on the market.
Unfortunately, today, there are only two new chemical entities in clinical trials against HAT and none against VL (Fig 2.).[10] Acoziborole is an orally-active benzoxaborole in phase IIb/III of clinical trials, this molecule is active against both stages of HAT.[11] Fexinidazole, a 5-nitroimidazole, was recently in phase II of clinical trials against VL but showed a lack of efficacy whereas it succeded to a phase IIIb study against HAT.[12,13] This molecule is rapidly metabolised in vivo into two metabolites (a sulfoxide and a sulfone derivative) which are still active against the Trypanosoma parasites. Fexinidazole is selectively bioactivated by type I parasitic nitroreductases (NTR) leading to cytotoxic electrophilic metabolites, through a successive 2 electron reduction, such as nitroso and hydroxylamine derivatives.[14] There are two NTR identified in Leishmania (NTR1 and NTR2)[15,16] and only one in Trypanosoma.[17] These nitroreductases are absent from mammalian cells. Consequently, substrates of these enzymes can afford selective antikinetoplastid candidates. Unfortunately, no X-ray structure of these parasitic NTRs is available, which restricts the use of most of the classical rational medicinal chemistry approaches, such as docking, to design new substrates of these enzymes.
Figure 2.

Molecular structure of drug candidates acoziborole and fexinidazole (with its active metabolites).
Our research team has been working on the synthesis of new antikinetoplastid molecules for several years. Starting from a chemical study about 2-substituted nitroquinoline derivatives with antiparasitic potential,[18] we identified a new antileishmanial hit: the 8-nitroquinolin-2(1H)-one.[19] Pharmacomodulation studies at position 4 of this scaffold were then realized.[20,21] Recently, we described a comprehensive study of this pharmacophore via an electrochemistry-guided work and the development of a computational model, able to predict the redox potentials of each molecule in the series.[22] Thus, a new antikinetoplastid hit molecule was identified (Fig. 3). This molecule was not genotoxic in the comet assay and was selectively bioactivated by type 1 NTRs of L. donovani and T. brucei brucei.[22]
Figure 3.

Structure and biological profile of the previously identified antikinetoplastid hit 3-bromo-8-nitroquinolin-2(1H)-one.
Here, we present a pharmacomodulation study at position 3 of the scaffold, using the Suzuki-Miyaura or Sonogashira cross-coupling reaction. To explore SARs, twenty six molecules were synthesized and evaluated on both L. infantum axenic amastigotes and T. brucei brucei trypomastigotes. All molecules were also assessed for their cytotoxicity on the HepG2 human cell line.
Results and Discussion
Chemistry
In a first time, 3-bromo-8-nitroquinolin-2(1H)-one was prepared in 3 steps, as presented in Scheme 1. [22] The nitration of the 2-chloroquinoline mainly led to the 2-chloro-8-nitroquinoline intermediate which was transformed in a second step into the corresponding lactam, according to a previously reported procedure.[23] The 8-nitroquinolin-2(1H)-one was finally selectively halogenated at position 3 by refluxing in HBr (48% aqueous solution) in the presence of sodium bromate, as reported by O’Brien and co-workers.[24]
Scheme 1.

Synthesis of the initial antikinetoplastid hit. a) H2SO4, HNO3, rt, 2 h; b) CH3CN, HClO4, 100°C, 72 h; c) NaBrO3, HBr (48% in H2O), 100°C, 5 h.
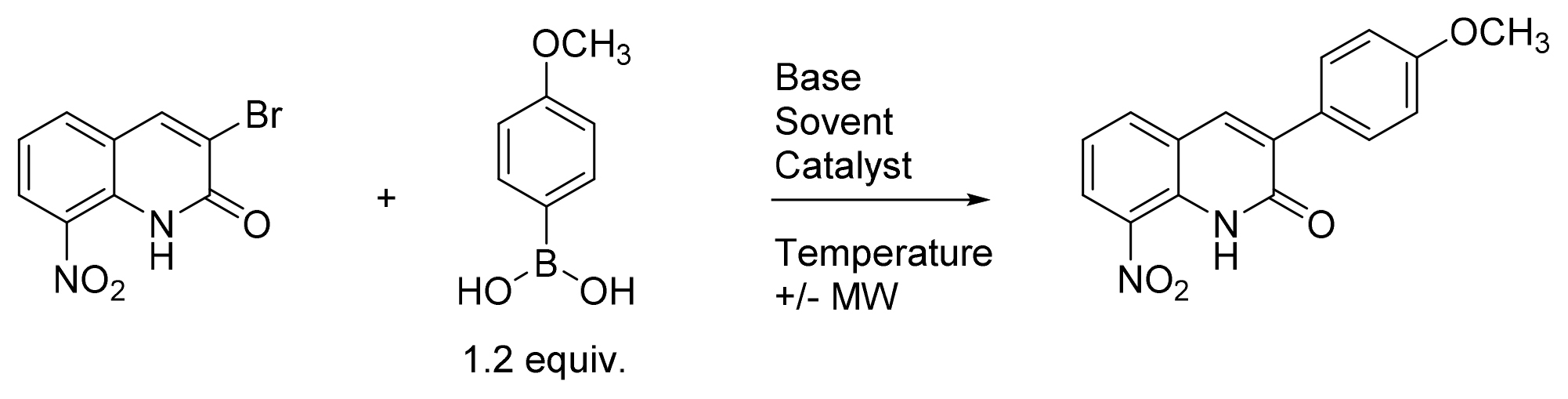
Then, the microwave-assisted Suzuki-Miyaura cross coupling reaction between 3-bromo-8-nitroquinolin-2(1H)-one and p-methoxyphenylboronic acid was studied, looking for optimal conditions. (Table 1). Several parameters were then investigated such as the nature of the solvent, base, and Pd-catalyst or the reaction temperature.
Table 1. Optimization of the Suzuki-Miyaura cross-coupling reaction between 3-bromo-8-nitroquinolin-2(1H)-one and p-methoxyphenylboronic acid.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base, 3 equiv. | Catalyst | Temperature (°C) | Time (h) | Yield (%)[a] |
| 1 | DMF | Na2CO3 | Pd(OAc)2, 0.1 equiv. | 150 | 1 | 43 |
| 2 | DMSO | Na2CO3 | Pd(OAc)2, 0.1 equiv. | 150 | 1 | - [b] |
| 3 | Toluene | Na2CO3 | Pd(OAc)2, 0.1 equiv. | 110 | 8 | - [c] |
| 4 | Dioxane | Na2CO3 | Pd(OAc), 0.1 equiv. | 100 | 8 | - [c] |
| 5 | THF | Na2CO3 | Pd(OAc)2, 0.1 equiv. | 66 | 8 | - [c] |
| 6 | DME | Na2CO3 | Pd(OAc)2, 0.1 equiv. | 85 | 8 | - [c] |
| 7 | DMF/H2O (8/2) | Na2CO3 | Pd(OAc)2, 0.1 equiv. | 100 | 1 | 46 |
| 8 | DMF/H2O (8/2) | K2CO3 | Pd(OAc)2, 0.1 equiv. | 100 | 1 | - [b] |
| 9 | DMF/H2O (8/2) | Cs2CO3 | Pd(OAc)2, 0.1 equiv. | 100 | 1 | - [b] |
| 10 | DMF | Na2CO3 | Pd(PPh3)4, 0.1 equiv. | 150 | 1 | 26 |
| 11 | DME | Na2CO3 | Pd(PPh3)4, 0.1 equiv. | 85 | 3 | 52 |
| 12 | DME | K2CO3 | Pd(PPh3)4, 0.1 equiv. | 85 | 4 | 84 |
| 13 | DME | K2CO3 | Pd(PPh2)Cl2, 0.1 equiv. | 85 | 8 | - [c] |
| 14 | DME | K2CO3 | Pd(dppf)Cl2, 0.1 equiv. | 85 | 8 | - [c] |
| 15 | DME | K2CO3 | Pd(OAc)2, 0.1 equiv. | 85 | 8 | - [c] |
| 16 | DME | Cs2CO3 | Pd(PPh3)4, 0.1 equiv. | 85 | 2 | 88 |
| 17 | DME | CsF | Pd(PPh3)4, 0.1 equiv. | 85 | 2 | 88 |
| 18 | DME | Cs2CO3 | Pd(PPh3)4, 0.05 equiv. | 85 | 4 | 56 |
The yield was calculated after purification by chromatography on silica gel with adapted eluent
Degradation of the substrate was observed on TLC
Only partial conversion of the substrate was observed on TLC.
The first Suzuki-Miyaura reaction conditions (entry 1) were inspired from a previously described protocol to introduce aryl moiety at position 3 of the quinolinone ring.[25] This reaction was realized in DMF under microwave (MW) heating, using 3 equiv. of Na2CO3 as a base, 0.1 equiv. of Pd(OAc)2 as a catalyst and 1.2 equiv of p-methoxyphenylboronic acid in a sealed tube, to afford the desired compound in 43 % yield. The first parameter studied was the nature of the solvent (entries 1-7). By using toluene, dioxane, THF, DME or DMSO, partial conversion or degradation of the substrate was observed whereas the use of a DMF/H2O mixture led to a 46% yield. Then, two others bases were studied (entries 8-9) but led to the degradation of the substrate within 1 h. In DMF, the replacement of Pd(OAc)2 by Pd(PPh3)4 decreased the yield of the reaction from 43% to 26% (entry 10). Then, adapting another previously described protocol using DME and Pd(PPh3)4 (entry 11), a slightly better yield was obtained.[26] This result was then improved by replacing Na2CO3 by K2CO3 which afford a yield of 84%. The nature of the catalyst was then studied (entries 13-15) but none of these assays led to an improvement in the reaction yield and only a few conversion was observed. Finally, the best results were obtained by using Cs2CO3 or CsF as a base (entries 16-17), which afford an efficient procedure of a Suzuki-Miyaura cross coupling reaction on this substrate. In the entry 18, the decrease of the amount of catalyst to 0.05 equiv. led to a lower reaction yield (56%) in comparison to entry 16, which was chosen as the best reaction conditions.
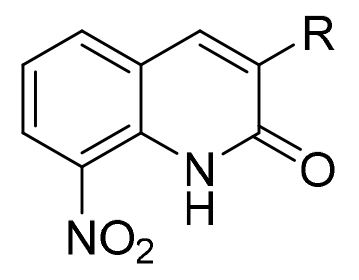
Then, the procedure was extended to 19 other arylboronic acids, to afford new derivatives bearing a phenyl, thiophene, furane or pyridine moiety at position 3 of the scaffold (Scheme 2). The reactions yields were generally superior to 65% (for 14 derivatives) but were lower for 4-hydroxyphenylboronic acid and 4-aminophenylboronic acid with respectively 37% and 41% yields.
Scheme 2.

General procedure for the Suzuki-Miyaura cross coupling reaction between 3-bromo-8-nitroquinolin-2(1H)-one and various arylboronic acids.
Three additional molecules (21-23) were synthesized by the saponification of compounds 18-20 into the carboxylic acid derivatives, using an excess of sodium hydroxide in an ethanol/water mixture (Scheme 3).
Scheme 3.

Preparation of compounds 21-23.
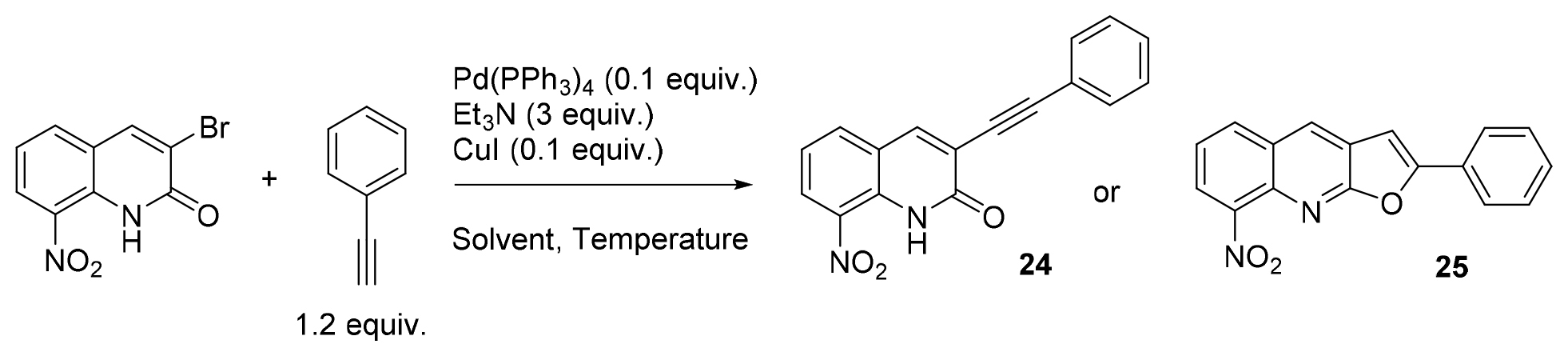
In parallel, in a view to enlarge the chemical diversity at position 3 of the scaffold and introduce alkynyl moieties, a Sonogashira cross-coupling reaction was set up. The initial conditions were adapted from a previously described protocol. [27] As presented in Table 2, after a brief optimization of the reaction between 3-bromo-8-nitroquinolinone and phenylacetylene, DME was chosen as solvent, and the reaction was carried out at 15°C (entry 6). Interestingly, it was noted that temperature had a strong influence on the reaction. At 0°C in DME, the reaction did not take place whereas it worked at 15 and 25°C, affording awaited compound 24. Surprisingly, it led to the unexpected 8-nitro-2-phenylfuro[2,3-b]quinoline 25 when heating at 40°C in DME or DMF. Compound 25 was also obtained when trying to recrystallize 24 from acetonitrile, underlining the instability of this product. Such a consecutive Sonogashira coupling and cyclization reaction, leading to furo[2,3-b]pyridine derivatives, was already reported in the literature [28,29]
Table 2. Optimization of the Sonogashira cross-coupling reaction on 3-bromo-8-nitroquinolin-2(1H)-one substrate.
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Yield 24 (%)[a] | Yield 25 (%)[a] | Time (h) |
| 1 | DMF | 25°C | -[b] | -[b] | 48 |
| 2 | THF | 25°C | -[b] | -[b] | 48 |
| 3 | Et3N | 25°C | -[b] | -[b] | 48 |
| 4 | DME | 25°C | 56 | 0 | 0.25 |
| 5 | DME | 0 | -[b] | -[b] | 48 |
| 6 | DME | 15 | 72 | 0 | 1.5 |
| 7 | DME | 40 | 0 | 51 | 0.5 |
| 8 | DMF | 40 | 0 | 56 | 36 |
The yield was calculated after purification by chromatography on silica gel with adapted eluent;
Only partial conversion of the substrate was observed on TLC.
The optimization of both Suzuki-Miyaura and Sonogashira cross-coupling reactions led to the synthesis of 25 new molecules with an aryl or an alkynyl group at position 3 of the scaffold. Appart molecule 24, which was considered too unstable, all these molecules were then evaluated in vitro to determine their antikinetoplastid potential. Compound 25 could not be evaluated because of a very poor aqueous solubility.
Compound evaluation
Initially, the cytotoxicity of these molecules was assessed in vitro on the HepG2 human cell line and the corresponding cytotoxic concentration 50% (CC50) were compared to the one of the reference drug doxorubicin (Table 3). The biological results showed that compounds 1-11 were not soluble enough in aqueous medium and could not be tested. The water-solubility was improved with compounds 10, 12-17 and 21-23, bearing either a pyridine-3-yl or a phenyl moiety at position 3 of the scaffold, this latter being substituted by a hydrophilic group such as hydroxymethyl and aldehyde or an ionized carboxylic group. These molecules displayed a low cytotoxicity on HepG2 human cell line with CC50 > 25 μM, the compound bearing an aldehyde group in para position of the phenyl ring (15) was the most cytotoxic of this series with a CC50 = 30 μM.
Table 3. In vitro antileishmanial, antitrypanosomal and cytotoxic activities of the synthesized compounds 1-25 and reference standards.
 | |||||
|---|---|---|---|---|---|
| Compound | R (Yield %) | IC50
L. infantum axenic amastigotes (μM) |
IC50
T. brucei brucei trypomastigotes (μM) |
CC50 HepG2 (μM) | Antitrypanosomal selectivity Index[g] |
| 1 | Phenyl (90) | >12[a] | 4.7 ± 2.7 | >12[a] | >2 |
| 2 | 4-OCH3-phenyl (88) | >6[a] | - | >6[a] | - |
| 3 | 4-OH-phenyl (37) | >3[a] | - | >3[a] | - |
| 4 | 4-NH2-phenyl (41) | >3[a] | - | >3[a] | - |
| 5 | 4-Cl-phenyl (73) | >3[a] | - | >3[a] | - |
| 6 | 4-F-phenyl (70) | >6[a] | - | >6[a] | - |
| 7 | 4-CF3-phenyl (60) | >6[a] | - | >6[a] | - |
| 8 | 3-thienyl (92) | >6[a] | - | >6[a] | - |
| 9 | 2-furyl (65) | >3[a] | - | >3[a] | - |
| 10 | 3-pyridyl (62) | >50[a] | 2.8 ± 0.8 | >25[a] | >9 |
| 11 | 4-pyridyl (71) | NS[b] | - | NS[b] | - |
| 12 | 4-CH2OH-phenyl (77) | >25[a] | 1.9 ± 0.3 | >25[a] | >13 |
| 13 | 3-CH2OH-phenyl (87) | 29.3 ± 4.2 | 1.5 ± 0.3 | >25[a] | >17 |
| 14 | 2-CH2OH-phenyl (72) | 22 ± 2.0 | 7.2 ± 0.6 | >100[c] | >14 |
| 15 | 4-CHO-phenyl (51) | 35 ± 1.7 | 0.5 ± 0.1 | 30 ± 3.7 | 60 |
| 16 | 3-CHO-phenyl (55) | 10.2 ± 0.6 | 5.6 ± 0.4 | >100[c] | >18 |
| 17 | 2-CHO-phenyl (65) | 9.8 ± 1.2 | 7.5 ± 0.4 | >50[a] | >7 |
| 18 | 4-COOCH3-phenyl (72) | >12.5[a] | - | >12.5[a] | - |
| 19 | 3-COOCH3-phenyl (71) | NS[b] | - | NS[b] | - |
| 20 | 2-COOCH3-phenyl (71) | NS[b] | - | NS[b] | - |
| 21 | 4-COOH-phenyl (65) | >100[c] | 1.5 ± 0.2 | 120 ± 7 | 80 |
| 22 | 3- COOH-phenyl (60) | >100[c] | 7.5 ± 1.0 | >100[c] | >13 |
| 23 | 2- COOH-phenyl (66) | >100[c] | >50[a] | >100[c] | - |
| Initial Hit[22] | Br | 7.1 ± 1.5 | 1.9 ± 0.44 | 92 ± 13.0 | 48 |
| 8-Nitroquinolinone[22] | H | 15.5 ± 0.5 | 23.4 ± 5.7 | 164 ± 28 | 7 |
| Doxorubicin[d] | - | - | 0.2 ± 0.02 | - | |
| Amphotericin B[e] | 0.06 ± 0.001 | - | 7.0 ± 0.25 | - | |
| Miltefosine[e] | 0.8 ± 0.2 | - | 84.5 ± 8.8 | - | |
| Fexinidazole[e] [f] | 3.3 ± 0.7 | 0.4 ± 0.18 | >100[c] | >250 | |
| Suramin[f] | - | 0.03 ± 0.009 | >100[c] | >3333 | |
| Eflornithine[f] | - | 15.8 ± 2.1 | >100[c] | >6 | |
The product could not be tested at higher concentrations due to a poor solubility in aqueous medium
The product was not soluble at any tested concentration
The IC50 or CC50 value was not reached at the highest tested concentration
Doxorubicin was used as a cytotoxic reference drug
Amphotericin B, miltefosine and fexinidazole were used as antileishmanial reference drugs or drug candidate
Fexinidazole, suramin and eflornithine were used as antitrypanosomal reference drugs or drug candidate
Antitrypanosomal selectivity index was calculated according to the following formula : CC50 HepG2 / IC50 T. brucei brucei
Then, all synthesized compounds were tested in vitro against Leishmania infantum axenic amastigotes. Their inhibitory concentration 50% (IC50) were determined and compared to the ones of two antileishmanial reference drugs (amphotericine B and miltefosine) and to the drug candidate fexinidazole.
Regarding antileishmanial activity, apart for aldehyde-containing compounds 16 and 17, the tested series appeared either poorly active (IC50 = 22 – 35 μM) or even totally inactive (IC50 > 100 μM for carboxylic group containing derivatives) toward L. infantum, in comparison with the reference drugs. Thus, introducing an aryl moiety at position 3 of the pharmacophore did not seem to favour antileishmanial activity.
In a second time, only the compounds with appropriate aqueous solubility were tested in vitro against T. brucei brucei trypomastigotes and compared to reference antitrypanosomal drugs (suramin and eflornithine) and the drug candidate fexinidazole. All tested molecules displayed good antitrypanosomal activity (0.5 μM ≤ IC50 ≤ 7.5 μM), better than the one of eflornithine (IC50 = 15.8 μM) and approaching the one of fexinidazole (IC50 = 0.4 μM). Interestingly, compounds substituted in ortho position of the phenyl ring (14, 17, 23) displayed higher IC50 values than their analogues substituted in meta and para positions. Compounds with a para-substituted phenyl ring generally showed lower IC50 than their meta-substituted analogues, 15 being 11 times more potent than 16, and 21 5 times more potent than 22. Compounds 15 and 21 appeared as the most promising antitrypanosomal molecules in this series with respective IC50 values of 0.5 and 1.5 μM. Compound 21 appears as a new antitrypanosomal hit, with an activity against T. brucei brucei close to the one of the parent compound (IC50 = 1,9 μM) but with a better cytotoxicity profile, leading to a better selectivity index (80 versus 48 for the initial hit). By comparison with reference drugs, compound 21 appears less active than suramin but more active than eflornithine. Regarding fexinidazole, another nitroheterocycle, compound 21 presents the same cytotoxic profile with CC50 = 120 μM and is three times less active than this drug candidate. Indeed, unlike fexinidazole and 3-bromo-8-nitroquinolin-2(1H)-one, compound 21 displayed a selective antitrypanosomal activity, being inactive (IC50 > 100 μM) against L. infantum. This is a first element indicating that molecule 21 shows a specific antiparasitic profile in the studied series.
Finally, the O-methylated analogue 26 of hit compound 21 was synthesized in two steps: O-methylation of 18, using methyl iodide in DMF under inert atmosphere, followed by a saponification with the same procedure as described for compounds 21-23 (Scheme 4).It was then tested in vitro against both L. infantum and T. brucei brucei (Fig. 4). It was as active against T. brucei brucei as 21 (IC50 = 2.2 μM). Molecule 26 was also active against L. infantum (IC50 = 12.8 μM) whereas 21 was not (IC50 > 100 μM). These results are surprising, considering that the hydrogen bond between the lactam function and the nitro group appeared mandatory for providing antileishmanial activity in 8-nitroquinolin-2(1H)-one series by increasing the reduction potential value [22]. This is a second element indicating that the introduction of an aryl group at position 3 of the 8-nitroquinolin-2(1H)-one scaffold could lead to a new antikinetoplasid mechanism of action, in comparison with 3-bromo-8-nitroquinolin-2(1H)-one.
Scheme 4.

Preparation of compound 26.
Figure 4.

In vitro antiparasitic activities and cytotoxicity of compound 26.
To assess if the antitrypanosomal nitroheterocycle 21 was bioactivated by the NTR of T. brucei brucei, its IC50 value was measured on both a wild type T. brucei brucei trypomastigotes strain and a NTR-overexpressing one (Table 4). The results were compared with the ones obtained for the initial hit.[22] This latter is clearly bioactivated by the trypanosomal NTR, being 4.5 more active against the strain overexpressing the NTR than on the wild type one, whereas 21 is only 1.3 time more active on the strain overexpressing the NTR. This result is a third element that suggests that 21, which is less intensively bioactivated by the trypanosomal NTR than the initial hit, could present another parasitic target. This assay also showed that compound 21 presents the same level of activity toward T. brucei brucei than the drug nifurtimox, used as a bioactivation control.
Table 4. Study of the bioactivation of 21 by trypanosomal nitroreductase NTR.
| Compound | IC50
T. brucei brucei trypomastigotes wild type strain (μM) |
IC50
T. brucei brucei trypomastigotes NTR over-expressing strain (μM). |
Fold change |
|---|---|---|---|
| 21 | 5.4 +/- 0.12 | 4.2 +/- 0.2 | 1.3 |
| Initial Hit | 17.7 +/- 1.0 | 3.9 +/- 0.1 | 4.5 |
| Nifurtimox | 1.9 +/- 0.05 | 0.6 +/- 0.05 | 3.1 |
In parallel, an electrochemistry study was carried out by measuring in DMSO the reduction potentials of five 8-nitroquinolin-2(1H)-one derivatives bearing an aryl group at position 3, using cyclic voltammetry (Table 5). For all compounds, a reversible single-electron reduction was observed (formation of an anion radical). The redox potentials of the new compounds bearing an aryl group at position 3 ranged between -0.53 V and – 0.59 V, higher than for the initial hit (-0.45 V) but quite close to the one of 8-nitroquinolin-2(1H)-one (-0.54 V). As previously noted in the studied series [22], the O-methylation of compound 21 leading to compound 24, is responsible for an important decrease in the redox potential value from -0.56 V to -0.93 V. This shift is mainly due to the removal of the intramolecular hydrogen bond between the lactam function and the nitro group. It can be concluded that the introduction of a phenyl ring at the position 3 of the scaffold has no significant impact on the redox potential of the studied series but that it allows to reach novel antitrypanosomal molecules that display lower reduction potential than the initial hit with the same level of efficacy.
Table 5. Effects of the substitution of the phenyl ring on reduction potentials in Suzuki-Miyaura series.
| Compound | Structure | E° (V)[a] |
|---|---|---|
| 1 | 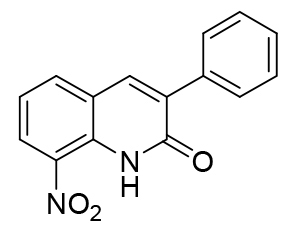 |
-0.59 |
| 7 | 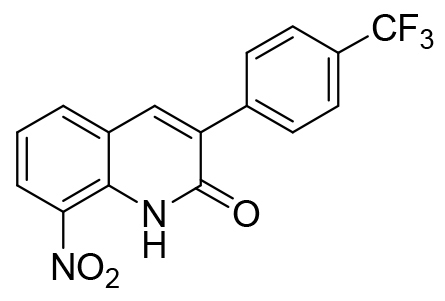 |
-0.53 |
| 13 | 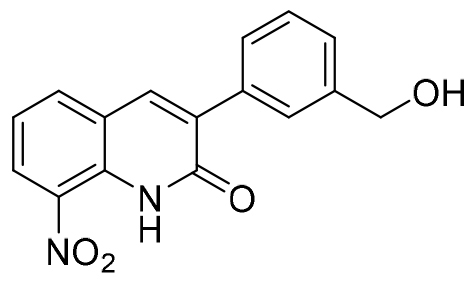 |
- 0.53 |
| 21 | 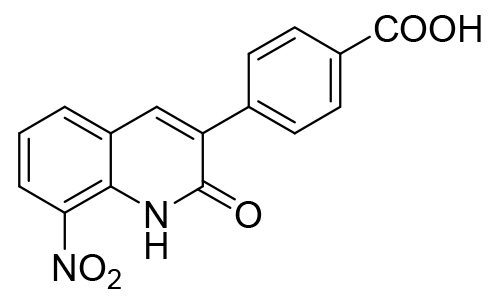 |
-0.56 |
| 26 | 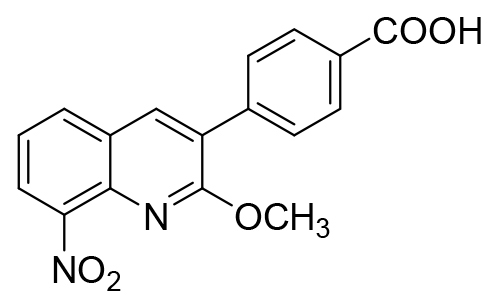 |
- 0.93 |
| Initial Hit | 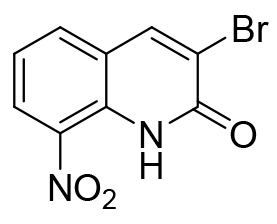 |
- 0.45 |
| 8-nitroquinolin-2(1H)-one | 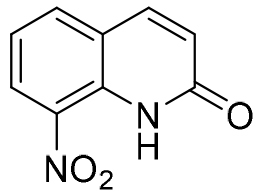 |
- 0.54 |
Cyclic voltammetry conditions: DMSO/TBAPF6, SCE/GC, 1 electron reversible reduction, values are given in V versus NHE
Thus, to understand the selective antitrypanosomal activity of 21, other parasitic targets should be explored. Among the parasitic targets that were recently reported in the literature concerning the antitrypanosomal activity of new diverse nitrohetrocycles, the S-adenosylmethionine decarboxylase (AdoMetDC) was highlighted by a high-throughput mass spectrometry-based assay conducted on 400.000 molecules [30] and is to consider.
Conclusions
An optimized Suzuki-Miyaura reaction at position 3 of 3-bromo-8-nitroquinolin-2(1H)-one led to the synthesis of 24 new derivatives. These molecules were screened in vitro against L. infantum and T. brucei brucei to evaluate their antikinetoplastid potential. Among these molecules, a new selective antitrypanosomal hit 21, bearing a para-carboxyphenyl group, was identified. Compound 21 was not cytotoxic on the HepG2 human cell line (CC50 = 120 μM), displayed a good antitrypanosomal activity (IC50 = 1.5 μM, SI = 80), better than the one of the drug eflornithine and approaching the one of fexinidazole, a 5-nitroimidazole in phase III of clinical trials against HAT. Interestingly, 21 was inactive against L. infantum showing a parasitic selectivity among kinetoplastids. Unlike fexinidazole and the previously identified hit molecule in the series, this molecule was not efficiently metabolized by the type I trypanosomal NTR, suggesting a probable additional mechanism of action in this series.
Experimental Section
Chemistry
All reagents and solvents were obtained from commercial sources (Fluorochem®, Sigma-Aldrich® or Alfa Aesar®) and used as received. The progress of the reactions was monitored by pre-coated thin layer chromatography (TLC) sheets ALUGRAM® SIL G/UV254 from Macherey-Nagel and were visualized by ultraviolet light at 254 nm. The 1H and 13C NMR spectra were recorded on a Bruker UltraShield 300 MHz, a Bruker IconNMR 400 MHz, or a Bruker Avance NEO 600 MHz instrument, at the Laboratoire de Chimie de Coordination and data are presented as follows: chemical shifts in parts per million (δ) using tetramethylsilane (TMS) as reference, coupling constant in Hertz (Hz), multiplicity by abbreviations : (s) singlet, (d) doublet, (t) triplet, (q) quartet, (dd) doublets of doublets, (m) multiplet and (br s) broad singlet. Melting points were measured on a Stuart Melting Point SMP3 instrument. High-resolution mass measurements were done on a GCT Premier Spectrometer (DCI, CH4, HRMS) or Xevo G2 QTOF (Waters, ESI+, HRMS) instrument at the Université Paul Sabatier, Toulouse. Microwave reactions were realized in a CEM Discover® microwave reactor.
3-Bromo-8-nitroquinolin-2(1H)-one was prepared according to a previsouly reported procedure.[22]
General procedure for the preparation of compounds 1-20 and 25
One equiv. of 3-bromo-8-nitroquinolin-2(1H)-one (1.1 mmol, 300 mg), 3 equiv. of cesium carbonate (3.3 mmol, 1.1 g), 0.1 equiv. of Pd(PPh3)4 (0.12 mmol, 127 mg) and 1.2 equiv. of the appropriate phenylboronic acid were added in a sealed flask of 25 mL. Under Ar atmosphere, 10 mL of dry dimethoxyethane were then added. The reaction mixture was heated at 85°C in a microwave reactor during 2 h. The reaction mixture was poured into water and extracted 3 times with dichloromethane (3x100 mL). The organic layer was washed with water, dried over anhydrous MgSO4 and evaporated in vacuo. The crude residues were purified by chromatography on silica gel using adapted eluent and recrystallized if necessary to give compounds 1-20 and 25.
8-nitro-3-phenylquinolin-2(1H)-one 1 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (97/3) as eluent, isolated and recrystallized in acetonitrile to yield a yellow solid (264 mg, 0.99 mmol, 90%); Tdec: 177°C, 1H NMR (CDCl3, 400 MHz) δ: 7.31-7.35 (m, 1H, H6), 7.41-7.50 (m, 3H, H3’, H4’ and H5’), 7.75-7.77 (m, 2H, H2’ and H6’), 7.91 (s, 1H, H4), 7.94 (dd, 1H, J=7.6 and 1.4 Hz, H5), 8.49 (dd, 1H, J=8.4 and 1.4 Hz, H7), 11.40 (br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ: 121.4 (CH), 122.7 (C), 127.3 (CH), 128.5 (2xCH), 128.7 (2xCH), 129.1 (CH), 132.7 (C), 133.1 (C), 134.5 (C), 134.6 (C), 135.6 (CH), 136.7 (CH), 161.0 (C). HRMS (DCI-CH4) calcd for C15H11N2O3 [M+H]+ = 267.0770, found 267.0762.
3-(4-methoxyphenyl)-8-nitroquinolin-2(1H)-one 2 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (95/5) as eluent, isolated and recrystallized in acetonitrile to yield an orange solid (287 mg, 0.97 mmol, 88%); mp 229°C, 1H NMR (CDCl3, 400 MHz) δ: 3.87 (s, 3H, CH3), 6.99-7.02 (m, 2H, H3’ and H5’), 7.29-7.33 (m, 1H, H6), 7.73-7.77 (m, 2H, H2’ and H6’), 7.87 (s, 1H, H4), 7.91 (dd, J=7.6 and 1.4 Hz, 1H, H5), 8.47 (dd, J=8.3 and 1.4 Hz, 1H, H7), 11.38 (br s, 1H, NH). 13C NMR (CDCl3, 75 MHz) δ: 55.4 (CH3), 114.0 (2xCH), 121.3 (CH), 122.9 (C), 125.6 (C), 126.9 (CH), 130.0 (2xCH), 132.7 (C), 132.9 (C), 134.0 (C), 135.3 (CH), 135.4 (CH), 160.3 (C), 161.2 (C). HRMS (DCI-CH4) calcd for C16H13N2O4 [M+H]+ = 297.0875, found 297.0864.
3-(4-hydroxyphenyl)-8-nitroquinolin-2(1H)-one 3 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (75/25) as eluent and isolated to yield an orange solid (115 mg, 0.41 mmol, 37%); mp 266°C, 1H NMR (DMSO-d6, 400 MHz) δ: 6.84-6.88 (m, 2H, H3’ and H5’), 7.40-7.44 (m, 1H, H6), 7.67-7.71 (m, 2H, H2’ and H6’), 8.20 (dd, J= 7.7 and 1.4 Hz, 1H, H5), 8.26 (s, 1H, H4), 8.40 (dd, J= 8.3 and 1.4 Hz, 1H, H7), 9.74 (s, 1H, OH), 11.12 (br s, 1H, NH). 13C NMR (DMSO-d6, 75 MHz) δ: 115.4 (2xCH), 122.0 (CH), 122.8 (C), 125.8 (C), 127.1 (CH), 130.4 (2xCH), 132.3 (C), 132.5 (C), 133.5 (C), 136.0 (CH), 136.1 (CH), 158.5 (C), 160.8 (C). HRMS (DCI-CH4) calcd for C15H11N2O4 [M+H]+ = 283.0719, found 283.0706.
3-(4-aminophenyl)-8-nitroquinolin-2(1H)-one 4 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (95/5) as eluent and isolated to yield an orange solid (128 mg, 0.45 mmol, 41%); mp 280°C, 1H NMR (DMSO-d6, 400 MHz) δ: 5.44 (s, 2H, NH2), 6.61-6.65 (m, 2H, H3’ and H5’), 7.37-7.41 (m, 1H, H6), 7.58-7.62 (m, 2H, H2’ and H6’), 8.15-8.18 (m, 2H, H4 and H5), 8.36 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 11.07 (br s, 1H, NH). 13C NMR (DMSO-d6, 100 MHz) δ: 113.6 (2xCH), 121.9 (CH), 122.1 (C), 123.1 (C), 126.5 (C), 129.9 (2xCH), 131.9 (C), 132.9 (C), 133.4 (C), 134.1 (CH), 135.8 (CH), 150.0 (CH), 161.0 (C). HRMS (DCI-CH4) calcd for C15H12N3O3 [M+H]+ = 282.0879, found 282.0874.
3-(4-chlorophenyl)-8-nitroquinolin-2(1H)-one 5 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (97/3) as eluent and isolated to yield a yellow solid (241 mg, 0.80 mmol, 73%); mp 222°C, 1H NMR (CDCl3, 400 MHz) δ: 7.33-7.37 (m, 1H, H6), 7.40-7.42 (m, 2H, H3’ and H5’), 7.66-7.75 (m, 2H, H2’ and H6’), 7.92 (s, 1H, H4), 7.95 (dd, 1H, J=7.6 and 1.5 Hz, H5), 8.51 (dd, 1H, J=8.4 and 1.5 Hz, H7), 11.42 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 121.6 (CH), 122.4 (C), 126.9 (CH), 127.7 (CH), 128.7 (CH), 129.1 (CH), 129.7 (CH), 132.8 (C), 133.1 (C), 133.2 (C), 134.4 (C), 135.7 (CH), 136.2 (C), 137.3 (CH), 160.6 (C). HRMS (DCI-CH4) calcd for C15H10ClN2O3 [M+H]+ = 301.0380, found 301.0373.
3-(4-fluorophenyl)-8-nitroquinolin-2(1H)-one 6 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (98/2) as eluent and isolated to yield a yellow solid (219 mg, 0.77 mmol, 70%); mp 217°C, 1H NMR (CDCl3, 400 MHz) δ: 7.12-7.20 (m, 2H, H2’ and H6’), 7.31-7.36 (m, 1H, H6), 7.73-7.80 (m, 2H, H3’ and H5’), 7.89 (s, 1H, H4), 7.94 (dd, 1H, J= 7.6 et 1.5 Hz, H5), 8.50 (dd, 1H, J= 8.4 and 1.5 Hz, H7), 11.41 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 115.5 (d, J= 21.6 Hz, 2xCH), 121.5 (CH), 122.6 (C), 124,4 (C), 127.4 (CH), 130.6 (d, J= 8.3 Hz, 2xCH), 132.7 (C), 133.1 (C), 133.5 (C), 135.5 (CH), 136.5 (CH), 160.9 (C), 163.2 (d, J= 249.2 Hz, C). HRMS (DCI-CH4) calcd for C15H10FN2O3 [M+H]+ = 285.0675, found 285.0676.
8-nitro-3-(4-trifluoromethylphenyl)quinolin-2(1H)-one 7 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (98/2) as eluent and isolated to yield a yellow solid (220 mg, 0.66 mmol, 60%); mp 168°C, 1H NMR (CDCl3, 400 MHz) δ: 7.34-7.38 (m, 1H, H6), 7.72-7.75 (m, 2H, H2’ and H6’), 7.88 (m, 2H, H3’ and H5’), 7.95-7.98 (m, 2H, H4 and H5), 8.53 (dd, 1H, J= 8.4 and 1.4 Hz, H7), 11.45 (br s, 1H, NH). 13C NMR (CDCl3, 150 MHz) δ: 121.7 (CH), 122.4 (C), 124.0 (q, J= 272.3 Hz, C), 125.4 (q, J= 3.7 Hz, 2xCH), 127.9 (CH), 129.1 (2xCH), 130.9 (q, J= 32.5 Hz, C), 132.8 (C), 133.2 (C), 133.4 (C), 135.8 (CH), 137.6 (CH), 138.0 (C), 160.5 (C). HRMS (DCI-CH4) calcd for C16H10N2O3F3 [M+H]+ = 335.0644, found 335.0629.
8-nitro-3-(thiophen-3-yl)quinolin-2(1H)-one 8 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (98/2) as eluent and isolated to yield a yellow solid (275 mg, 1.01 mmol, 92%); mp 199°C, 1H NMR (CDCl3, 400 MHz) δ: 7.31-7.35 (m, 1H, H6), 7.41-7.43 (m, 1H, H4’), 7.59 (dd, 1H, J= 5.1 Hz and 1.3 Hz, H5’), 7.94 (dd, 1H, J= 7.7 Hz and 1.4 Hz, H5), 8.05 (s, 1H, H4), 8.39 (dd, 1H, J=3.0 and 1.3 Hz, H2’), 8.48 (dd, 1H, J=8.4 and 1.4 Hz, H7), 11.42 (br s, 1H, NH). 13C NMR (CDCl3, 75 MHz) δ: 121.4 (CH), 122.6 (C), 123.6 (C), 125.6 (CH), 126.2 (CH), 127.0 (CH), 127.1 (CH), 128.6 (C), 132.4 (C), 134.1 (CH), 134.3 (C), 135.4 (CH), 160.6 (C). HRMS (DCI-CH4) calcd for C13H9N2SO3 [M+H]+ = 273.0334, found 273.0330.
3-(2-furanyl)-8-nitroquinolin-2(1H)-one 9 was purified by chromatography on silica gel using dichloromethane as eluent and isolated to yield a dark red solid (183 mg, 0.71 mmol, 65%); mp 257°C, 1H NMR (CDCl3, 300 MHz) δ: 6.58 (dd, 1H, J= 3.3 Hz and 1.9 Hz, H4’), 7.30-7.35 (m, 1H, H6), 7.55-7.56 (m, 2H, H3’ and H5’), 7.96 (dd, 1H, J= 7.6 and 1.4 Hz, H5), 8.23 (s, 1H, H4), 8.46 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 11.42 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 112.6 (CH), 114.2 (CH), 121.6 (CH), 122.5 (C), 123.6 (C), 126.9 (CH), 130.7 (CH), 132.0 (C), 132.8 (C), 135.5 (CH), 143.3 (CH), 147.7 (C), 158.7 (C). HRMS (DCI-CH4) calcd for C13H9N2O4 [M+H]+ = 257.0562, found 257.0558.
8-nitro-3-(pyridine-3-yl)quinolin-2(1H)-one 10 was purified by chromatography on silica gel using dichloromethane/ethyl acetate as eluent and isolated to yield a yellow solid (182 mg, 0.68 mmol, 62%); mp 237°C, 1H NMR (CDCl3, 400 MHz) δ: 7.35-7.39 (m, 1H, H6), 7.40-7.43 (m, 1H, H5’), 7.97-7.99 (m, 2H, H4 and H5), 8.20-8.23 (m, 1H, H6’), 8.53 (dd, 1H, J=8.4 and 1.4 Hz, H7), 8.66 (dd, 1H, J=4.9 and 1.7 Hz, H4’), 8.90 (dd, 1H, J=2.3 and 0.9 Hz, H2’), 11.45 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 121.7 (CH), 122.3 (C), 123.1 (CH), 127.9 (CH), 130.5 (C), 131.4 (C), 132.8 (C), 133.3 (C), 135.8 (CH), 136.4 (CH), 137.2 (CH), 149.0 (CH), 150.0 (CH), 160.6 (C). HRMS (DCI-CH4) calcd for C14H10N3O3 [M+H]+ = 268.0722, found 268.0712.
8-nitro-3-(pyridine-4-yl)quinolin-2(1H)-one 11 was purified by chromatography on silica gel using dichloromethane/acetone (80/20) as eluent and isolated to yield a yellow solid (209 mg, 0.78 mmol, 71%); mp 297°C, 1H NMR (CDCl3, 400 MHz) δ: 7.36-7.40 (m, 1H, H6), 7.70-7.72 (m, 2H, H2’ and H6’), 7.98 (dd, 1H, J=7.9 and 1.4 Hz, H5), 8.03 (s, 1H, H4), 8.55 (dd, 1H, J= 8.4 and 1.4 Hz, H7), 8.73-8.74 (m, 2H, H3’ and H5’), 11.46 (br s, 1H, NH). 13C NMR (TFA-d, 100 MHz) δ: 121.6 (C), 123.7 (CH), 126.1 (C), 126.7 (2xCH), 130.7 (CH), 132.4 (C), 133.4 (C), 137.5 (CH), 140.6 (2xCH), 144.1 (CH), 153.5 (C), 161.1 (C). HRMS (DCI-CH4) calcd for C14H10N3O3 [M+H]+ = 268.0722, found 268.0713.
3-(4-hydroxymethylphenyl)-8-nitroquinolin-2(1H)-one 12 was purified by chromatography on silica gel using dichloromethane/acetone (75/25) as eluent and isolated to yield a yellow solid (251 mg, 0.85 mmol, 77%); mp 219°C, 1H NMR (DMSO-d6, 400 MHz) δ: 4.56 (d, 2H, J=5.6, CH2), 5.26 (t, 1H, J= 5.6, OH), 7.41-7.46 (m, 3H, H6, H3’ and H5’), 7.76-7.79 (m, 2H, H2’ and H6’), 8.23 (dd, 1H, J=7.6 and 1.4 Hz, H5), 8.36 (s, 1H, H4), 8.43 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 11.17 (br s, 1H, NH). 13C NMR (DMSO-d6, 100 MHz) δ: 63.1 (CH2), 122.1 (CH), 122.6 (C), 126.6 (2xCH), 127.6 (CH), 128.8 (2xCH), 132.6 (C), 132.7 (C), 133.6 (C), 134.0 (C), 136.4 (CH), 137.6 (CH), 143.6 (C), 160.8 (C). HRMS (DCI-CH4) calcd for C16H13N2O4 [M+H]+ = 297.0875, found 297.0878.
3-(3-hydroxymethylphenyl)-8-nitroquinolin-2(1H)-one 13 was purified by chromatography on silica gel using dichloromethane/acetone (75/25) as eluent and isolated to yield a yellow solid (267 mg, 0.90 mmol, 82%); mp 155°C, 1H NMR (DMSO-d6, 300 MHz) δ: 4.58 (d, 2H, J=5.6 Hz, CH2), 5.27 (t, 1H, J= 5.6 Hz, OH), 7.37-7.47 (m, 3H, H6, H4’ and H5’), 7.66-7.68 (m, 1H, H6’), 7.73-7.75 (m, 1H, H2’), 8.25 (dd, 1H, J=7.8 and 1.4 Hz, H5), 8.35 (s, 1H, H4), 8.44 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 11.17 (br s, 1H, NH). 13C NMR (DMSO-d6, 75 MHz) δ: 63.3 (CH2), 122.1 (CH), 122.6 (C), 127.1 (CH), 127.2 (CH), 127.5 (CH), 127.7 (CH), 128.4 (CH), 132.7 (C), 133.0 (C), 133.7 (C), 135.1 (C), 136.5 (CH), 138.0 (CH), 142.9 (C), 160.6 (C). HRMS (DCI-CH4) calcd for C16H13N2O4 [M+H]+ = 297.0875, found 297.0861.
3-(2-hydroxymethylphenyl)-8-nitroquinolin-2(1H)-one 14 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (50/50) as eluent and isolated to yield a yellow solid (235 mg, 0.79 mmol, 72%); mp 208°C, 1H NMR (DMSO-d6, 400 MHz) δ: 4.44 (d, J=5.5 Hz, 2H, CH2), 5.09 (sl, 1H, OH), 7.28 (dd, 1H, J= 7.5 and 1.4 Hz, H3’), 7.33-7.37 (m, 1H, H6), 7.42-7.46 (m, 2H, H4’ and H5’), 7.57 (dd, 1H, J=7.5 and 1.4 Hz, H6’), 8.12 (s, 1H, H4), 8.19 (dd, 1H, J= 7.6 Hz and 1.4 Hz, H5), 8.45 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 11.19 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 63.7 (CH2), 121.9 (CH), 122.5 (C), 127.8 (CH), 128.4 (CH), 129.8 (CH), 130.1 (CH), 130.6 (CH), 132.7 (C), 133.1 (C), 133.9 (C), 135.7 (CH), 135.8 (C), 139. 2 (CH), 139.9 (C), 162.2 (C). HRMS (DCI-CH4) calcd for C16H13N2O4 [M+H]+ = 297.0875, found 297.0869.
3-(4-formylphenyl)-8-nitroquinolin-2(1H)-one 15 was purified by chromatography on silica gel using dichloromethane/cyclohexane (80/20) as eluent, isolated and recrystallized in acetonitrile to yield a yellow solid (165 mg, 0.56 mmol, 51%); mp 243°C, 1H NMR (CDCl3, 300 MHz) δ: 7.34-7.39 (m, 1H, H6), 7.93-8.00 (m, 6H, H4, H5, H2’, H3’, H5’ and H6’), 8.53 (dd, 1H, J= 8.5 Hz and 1.4 Hz, H7), 10.08 (s, 1H, CHO), 11.45 (br s, 1H, NH). 13C NMR (CDCl3, 75 MHz) δ: 121.7 (CH), 122.3 (C), 128.0 (CH), 129.4 (2xCH), 129.8 (2xCH), 132.8 (C), 133.2 (C), 133.4 (C), 135.9 (CH), 136.4 (C), 137.9 (CH), 140.5 (C), 160.5 (C), 191.7 (CH). HRMS (DCI-CH4) calcd for C16H11N2O4 [M+H]+ = 295.0719, found 295.0710.
3-(3-formylphenyl)-8-nitroquinolin-2(1H)-one 16 was purified by chromatography on silica gel using dichloromethane/acetone (75/25) as eluent and isolated to yield a yellow solid (104 mg, 0.35 mmol, 32%); mp 209°C, 1H NMR (CDCl3, 300 MHz) δ: 7.34-7.39 (m, 1H, H6), 7.63-7.69 (m, 1H, H5’), 7.94-7.99 (m, 2H, H5 and H6’), 8.01 (s, 1H, H4), 8.10-8.13 (m, 1H, H4’), 8.25-8.26 (m, 1H, H2’), 8.53 (dd, 1H, J= 8.4 and 1.4 Hz, H7), 10.10 (s, 1H, CHO), 11.5 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 121.7 (CH), 122.4 (C), 127.8 (CH), 129.2 (CH), 129.7 (CH), 130.2 (CH), 132.8 (C), 133.1 (C), 133.3 (C), 134.7 (CH), 135.6 (C), 135.8 (CH), 136.6 (C), 137.5 (CH), 160.7 (C), 192.0 (CH). HRMS (DCI-CH4) calcd for C16H11N2O4 [M+H]+ = 295.0719, found 295.0713.
3-(2-formylphenyl)-8-nitroquinolin-2(1H)-one 17 was purified by chromatography on silica gel using cyclohexane/acetone (60/40) as eluent and isolated to yield a yellow solid (210 mg, 0.71 mmol, 65%); mp 230-231°C, 1H NMR (CDCl3, 400 MHz) δ: 7.33-7.37 (m, 1H, H6), 7.44 (dd, 1H, J= 7.5 Hz and 1.3 Hz, H6’), 7.61-7.65 (m, 1H, H4’), 7.68-7.72 (m, 1H, H5’), 7.82 (s, 1H, H4), 7.93 (dd, 1H, J= 7.6 Hz and 1.4 Hz, H3’), 7.99 (dd, 1H, J= 7.8 Hz and 1.4 Hz, H5), 8.53 (dd, 1H, J= 8.4 Hz and 1.4 Hz, H7), 10.03 (s, 1H, CHO), 11.42 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 121.6 (CH), 122.3 (C), 127.8 (CH), 129.5 (CH), 130.8 (CH), 131.0 (CH), 133.0 (C), 133.7 (C), 133.9 (CH), 134.5 (C), 135.0 (C), 135.7 (CH), 136.1 (C), 138.0 (CH), 160.8 (C), 191.2 (CH). HRMS (DCI-CH4) calcd for C16H11N2O4 [M+H]+ = 295.0719, found 295.0718.
3-(4-methoxycarbonylphenyl)-8-nitroquinolin-2(1H)-one 18 was purified by chromatography on silica gel using dichloromethane as eluent, isolated and recrystallized to yield a yellow solid (257 mg, 0.79 mmol, 72%); mp 245°C, 1H NMR (CDCl3, 300 MHz) δ: 3.95 (s, 3H, CH3), 7.33-7.38 (m, 1H, H6), 7.84-7.87 (m, 2H, H2’ and H6’), 7.95-7.97 (m, 2H, H4 and H5), 8.12-8.15 (m, 2H, H3’ and H5’), 8.52 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 11.43 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 52.3 (CH3), 121.6 (CH), 122.4 (C), 127.8 (CH), 128.7 (2xCH), 129.7 (2xCH), 130.4 (C), 132.8 (C), 133.3 (C), 133.5 (C), 135.8 (CH), 137.6 (CH), 139.0 (C), 160.6 (C), 166.7 (C). HRMS (DCI-CH4) calcd for C17H13N2O5 [M+H]+ = 325.0824, found 325.0818.
3-(3-methoxycarbonylphenyl)-8-nitroquinolin-2(1H)-one 19 was purified by chromatography on silica gel using dichloromethane/cyclohexane (90/10) as eluent and isolated to yield a yellow solid (253 mg, 0.78 mmol, 71%); mp 212°C, 1H NMR (CDCl3, 400 MHz) δ: 3.95 (s, 3H, CH3) ; 7.33-7.37 (m, 1H, H6), 7.53-7.58 (m, 1H, H5’), 7.96 (dd, 1H, J= 7.7 Hz and 1.4 Hz, H5), 7.98 (s, 1H, H4), 8.05-8.11 (m, 2H, H4’ and H6’), 8.36-8.37 (m, 1H, H2’), 8.51 (dd, 1H, J= 8.4 Hz and 1.4 Hz, H7), 11.42 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 52.3 (CH3), 121.6 (CH), 122.5 (C), 127.7 (CH), 128.6 (CH), 129.6 (CH), 130.0 (CH), 130.5 (C), 132.8 (C), 133.2 (C), 133.4 (CH), 133.5 (C), 134.8 (C), 135.7 (CH), 137.3 (CH), 160.7 (C), 166.7 (C). HRMS (DCI-CH4) calcd for C17H13N2O5 [M+H]+ = 325.0824, found 325.0809.
3-(2-methoxycarbonylphenyl)-8-nitroquinolin-2(1H)-one 20 was purified by chromatography on silica gel using cyclohexane/acetone (70/30) as eluent and isolated to yield a yellow solid (257 mg, 0.79 mmol, 72%); mp 212°C, 1H NMR (CDCl3, 400 MHz) δ: 3.82 (s, 3H, CH3), 7.29-7.33 (m, 1H, H6), 7.37-7.39 (m, 1H, H6’), 7.49-7.53 (m, 1H, H4’), 7.60-7.64 (m, 1H, H5’), 7.77 (s, 1H, H4), 7.91 (d, 1H, J=7.65 Hz, H3’), 8.02 (d, 1H, J= 7.8 Hz, H5), 8.48 (d, 1H, J= 8.4 Hz, H7), 11.34 (br s, 1H, NH). 13C NMR (CDCl3, 100 MHz) δ: 52.3 (CH3), 121.4 (CH), 122.7 (C), 127.2 (CH), 129.0 (CH), 130.1 (CH), 130.6 (CH), 131.0 (C), 132.4 (CH), 132.9 (C), 133.4 (C), 135.5 (CH), 135.6 (CH), 135.8 (C), 137.0 (C), 160.9 (C), 167.5 (C). HRMS (DCI-CH4) calcd for C17H13N2O5 [M+H]+ = 325.0824, found 325.0819.
General procedure for the preparation of compounds 21-23
A mixture of 40 mL of H2O/Ethanol (2/8) was added onto 1 equiv. (200 mg) of the corresponding 3-(methoxycarbonylphenyl)-8-nitroquinolin-2(1H)-one derivative (18-20). Then, 5 equiv. of NaOH were added and the reaction mixture was stirred at 80°C for 3 h. The reaction mixture was then poured into water, acidified to pH=1 with HCl 37%, extracted twice with dichloromethane (2x50 mL) and twice with ethyl acetate (2x50 mL). The combined organic layers were washed with water, dried over anhydrous MgSO4 and evaporated in vacuo. The crude residues were purified by chromatography on silica gel using adapted eluent and recrystallized if necessary to give compounds 21-23.
3-(4-carboxyphenyl)-8-nitroquinolin-2(1H)-one 21 was washed with acid water and recrystallized in acetonitrile to yield a yellow solid (124 mg, 0.40 mmol, 65%); mp > 310°C, 1H NMR (DMSO-d6, 400 MHz) δ: 7.45-7.49 (m, 1H, H6), 7.93-7.96 (m, 2H, H2’ and H6’), 8.03-8.06 (m, 2H, H3’ and H5’), 8.26 (dd, 1H, J=7.9 and 1.4 Hz, H5), 8.46 (dd, 1H, J=8.3 and 1.4 Hz, H7), 8.49 (s, 1H, H4), 11.23 (br s, 1H, NH), 13.07 (br s, 1H, COOH). 13C NMR (DMSO-d6, 100 MHz) δ: 122.3 (CH), 122.35 (C), 128.1 (CH), 129.2 (2xCH), 129.6 (2xCH), 131.0 (C), 131.7 (C), 132.8 (C), 133.8 (C), 136.7 (CH), 139.1 (CH), 139.5 (C), 160.4 (C), 167.5 (C). HRMS (DCI-CH4) calcd for C16H11N2O5 [M+H]+ 311.0668, found 311.0669.
3-(3-carboxyphenyl)-8-nitroquinolin-2(1H)-one 22 was washed with acid water and recrystallized in acetonitrile to yield a brown solid (88 mg, 0.28 mmol, 42%); Tdec. 305-306°C, 1H NMR (CDCl3, 400 MHz) δ: 7.44-7.48 (m, 1H, H6), 7.60-7.64 (m, 1H, H5’), 8.00-8.02 (m, 1H, H6’), 8.03-8.06 (m, 1H, H4’), 8.27 (dd, 1H, J=7.8 and 1.4 Hz, H5), 8.39-8.40 (m, 1H, H2’), 8.46 (dd, 1H, J=8.3 and 1.4 Hz, H7), 8.47 (s, 1H, H4), 11.22 (br s, 1H, NH), 13.05 (br s, 1H, COOH). 13C NMR (DMSO-d6, 100 MHz) δ: 122.2 (CH), 122.5 (C), 127.9 (CH), 129.0 (CH), 129.8 (CH), 129.9 (CH), 131.3 (C), 131.8 (C), 132.8 (C), 133.4 (CH), 133.7 (C), 135.6 (C), 136.7 (CH), 138.6 (CH), 160.5 (C), 167.6 (C). HRMS (DCI-CH4) calcd for C16H11N2O4 [M+H]+ = 311.0668, found 311.0672.
3-(2-carboxyphenyl)-8-nitroquinolin-2(1H)-one 23 was purified by chromatography on silica gel using dichloromethane/ethyl acetate (50/50) as eluent and isolated to yield a yellow solid (109 mg, 0.35 mmol, 57%); T dec. 282-285°C, 1H NMR (DMSO-d6, 400 MHz) δ: 7.43-7.48 (m, 2H, H6 et H6’), 7.54-7.58 (m, 1H, H4’), 7.66-7.70 (m, 1H, H5’), 7.91 (dd, 1H, J=7.7 and 1.4 Hz, H5), 8.13 (s, 1H, H4), 8.20 (dd, 1H, J= 7.8 and 1.4 Hz, H3’), 8.42 (dd, 1H, J=8.3 and 1.4 Hz, H7) ; 11.08 (br s, 1H, NH), 12.72 (br s, 1H, COOH). 13C NMR (DMSO-d6, 100 MHz) δ: 122.1 (CH), 122.6 (C), 127.4 (CH), 129.1 (CH), 129.8 (CH), 131.3 (CH), 132.4 (CH), 132.5 (C), 132.7 (C), 133.9 (C), 136.1 (CH), 136.2 (C), 136.3 (CH), 136.4 (C), 160.7 (C), 168.4 (C). HRMS (DCI-CH4) calcd for C16H11N2O5 [M+H]+ 311.0668, found 311.0673.
Preparation of 8-nitro-3-phenylethynylquinolin-2(1H)-one 24
100 mg of 3-bromo-8-nitroquinolin-2(1H)-one previously synthesized in our team (0.37 mmol, 1 equiv.), 7 mg of CuI (0.037 mmol, 0.1 equiv.) and 43 mg of Pd(PPh3)4 (0.037 mmol, 0.1 equiv.) were added in a sealed flask of 10 mL. Under Argon atmosphere, 5 mL of dry dimethoxyethane 155 μL of Et3N (1.11 mmol, 3 equiv.), 61 μL of phenylacetylene (0.56 mmol, 1.5 equiv.) were successively added. The reaction mixture was cooled at 15°C during 1.5 hours. The reaction mixture was poured into water and extracted three times with dichloromethane. The organic layer was washed with water, dried over anhydrous MgSO4 and evaporated in vacuo. The crude residue was purified by chromatography on silica gel using dichloromethane as an eluent and isolated to yield a yellow solid (78 mg, 0.27 mmol, 72%).
Compound 24 ; mp186-187°C, 1H NMR (CDCl3, 400 MHz) δ: 7.31-7.35 (m, 1H, H6), 7.37-7.38 (m, 2H, H3’, H4’ and H5’), 7.59-7.63 (m, 2H, H2’ and H6’), 7.87 (dd, 1H, J= 7.6 and 1.5 Hz, H5), 8.06 (s, 1H, H4), 8.49 (dd, 1H, J= 8.4 and 1.4 Hz, H7), 11.40 (br s, 1H, NH). 13C NMR (CDCl3-d6, 100 MHz) δ: 83.4 (C), 97.3 (C), 119.6 (C), 121.8 (CH), 122.0 (C), 122.1 (C), 128.0 (CH), 128.4 (2xCH), 129.2 (CH), 132.1 (2xCH), 133.0 (C), 133.0 (C), 135.3 (CH), 141.6 (CH), 159.9 (C). HRMS (DCI-CH4) calcd for C17H13N2O5 [M+H]+ 291.0770, found 291.0776.
Preparation of 8-nitro-2-phenyl-furo[2,3-b]quinoline 25
100 mg of 3-bromo-8-nitroquinolin-2(1H)-one previously synthesized in our team (0.37 mmol, 1 equiv.), 7 mg of CuI (0.037 mmol, 0.1 equiv.) and 43 mg of Pd(PPh3)4 (0.037 mmol, 0.1 equiv.) were added in a sealed flask of 10 mL. Under Argon atmosphere, 5 mL of dry dimethoxyethane 155 μL of Et3N (1.11 mmol, 3 equiv.), 61 μL of phenylacetylene (0.56 mmol, 1.5 equiv.) were successively added. The reaction mixture was heated at 40°C during 30 min. The reaction mixture was poured into water and extracted three times with dichloromethane. The organic layer was washed with water, dried over anhydrous MgSO4 and evaporated in vacuo. The crude residue was purified by chromatography on silica gel using dichloromethane as an eluent and isolated to yield a grey solid (55 mg, 0.19 mmol, 51%).
Compound 25 ; mp 226°C, 1H NMR (CDCl3, 400 MHz) δ: 7.16 (s, 1H, H3), 7.45-7.55 (m, 3H, H3’, H4’ and H5’), 7.56-7.59 (m, 1H, H6), 7.99-8.02 (m, 2H, H2’ and H6’), 8.08 (dd, 1H, J= 7.5 and 1.4 Hz, H5), 8.14 (dd, 1H, J= 8.3 and 1.4 Hz, H7), 8.41 (s, 1H, H4). 13C NMR (CDCl3, 100 MHz) δ: 99.2 (CH), 123.3 (CH), 123.4 (CH), 124.0 (C), 125.9 (2xCH), 127.7 (C), 128.2 (CH), 128.6 (C), 129.1 (2xCH), 130.5 (CH), 132.0 (CH), 136.0 (C), 147.7 (C), 159.6 (C), 162.4 (C). HRMS (DCI-CH4) calcd for C17H11N2O3 [M+H]+ 291.0770, found 291.0768.
Preparation of 3-(4-carboxyphenyl)-2-methoxy-8-nitroquinoline 26
Under Ar atmosphere, 260 mg of 3-(4-methoxycarbonylphenyl)-8-nitroquinolin-2(1H)-one (0.80 mmol, 1 equiv.) were solubilized in 5 mL of dry DMF. This solution was then added onto 64 mg of 60% sodium hydride (1.6 mmol, 2 equiv.), solubilized in DMF (5 mL). After 10 min of stirring at rt, 100 μL of methyl iodide (1.6 mmol, 2 equiv.) were added dropwise. The reaction mixture was stirred at rt overnight, before being poured into ice. A precipitate was formed, filtered off and washed with water. The precipitate was solubilized into dichloromethane. The solution was dried over anhydrous MgSO4 and evaporated in vacuo. The crude residue was purified by chromatography on silica gel using dichloromethane as eluent. A white solid intermediate was isolated and solubilized into 40 mL of a mixture of H2O/Ethanol (2/8). NaOH (excess) was then added and the reaction mixture was stirred at 80°C for 2 h. The reaction mixture was then poured into water, acidified to pH=1 with HCl 37%, extracted twice with dichloromethane (2x50 mL) and twice with ethyl acetate (2x50 mL). The combined organic layers were washed with water, dried over anhydrous MgSO4 and evaporated in vacuo. Compound 26 was washed with basic water (pH = 8) to yield a white solid (87 mg, 0.27 mmol, 61%)
Compound 26 ; Tdec. 270-274°C, 1H NMR (DMSO-d6, 400 MHz) δ: 4.00 (s, 3H, CH3), 7.61-7.65 (m, 1H, H6), 7.80-7.82 (m, 2H, H2’ and H6’), 8.05-8.07 (m, 2H, H3’ and H5’), 8.23 (dd, 1H, J=7.8 and 1.4 Hz, H5), 8.27 (dd, 1H, J=8.1 and 1.4 Hz, H7), 8.56 (s, 1H, H4), 13.07 (br s, 1H, NH). 13C NMR (DMSO-d6, 100 MHz) δ: 54.5 (CH3), 124.1 (CH), 124.7 (C), 126.6 (CH), 127.0 (C), 129.7 (2xCH), 130.0 (2xCH), 130.9 (C), 132.7 (CH), 136.7 (C), 139.3 (CH), 140.0 (C), 146.2 (C), 160.8 (C), 167.5 (C). HRMS (DCI-CH4) calcd for C17H13N2O5 [M+H]+ = 325.0824, found 325.0822.
Electrochemistry
Voltammetric measurements were carried out with a potentiostat Autolab PGSTAT100 (ECO Chemie, The Netherlands) controlled by GPES 4.09 software. Experiments were performed at room temperature in a homemade airtight three–electrode cell connected to a vacuum/argon line. The reference electrode consisted of a saturated calomel electrode (SCE) separated from the solution by a bridge compartment. The counter electrode was a platinum wire of approximately 1cm² apparent surface. The working electrode was GC microdisk (1.0 mm of diameter – Bio-logic SAS). The supporting electrolyte (nBu4N)[PF6] (Fluka, 99% puriss electrochemical grade) and the solvent DMSO (Sigma-Aldrich puriss p.a. dried<0.02% water) were used as received and simply degassed under argon. The solutions used during the electrochemical studies were typically 10-3 M in compound and 0.1 M in supporting electrolyte. Before each measurement, the solutions were degassed by bubbling Ar and the working electrode was polished with a polishing machine (Presi P230). Under these experimental conditions employed in this work, the half-wave potential (E1/2) of the ferrocene Fc+/Fc couple in DMSO was E1/2 = 0.45 V vs SCE. Experimental peak potentials have been measured versus SCE and converted to NHE by adding 0.241 V.
Biology
Antileishmanial activity on L. infantum axenic amastigotes.[31]
L. infantum promastigotes (MHOM/MA/67/ITMAP-263, CNR Leishmania, Montpellier, France, expressing luciferase activity) in logarithmic phase cultivated in RPMI 1640 medium supplemented with 5% foetal calf serum (FCS), 2 mM L-glutamine and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin), were centrifuged at 900 g for 10 min. The supernatant was removed carefully and was replaced by the same volume of RPMI 1640 complete medium at pH 5.4 and incubated for 24 h at 24°C. The acidified promastigotes were incubated for 24 h at 37°C in a ventilated flask. Promastigotes were then transformed into axenic amastigotes. The effects of the tested compounds on the growth of L. infantum axenic amastigotes were assessed as follows. L. infantum amastigotes were incubated at a density of 2.106 parasites/mL in sterile 96-well plates with various concentrations of compounds dissolved in DMSO (final concentration less than 0.5% v/v), in duplicate. Appropriate controls DMSO, amphotericin B, miltefosine and fexinidazole (reference drugs purchased from Sigma Aldrich) were added to each set of experiments. After a 48 h incubation period at 37°C, each plate-well was then microscope-examined for detecting any precipitate formation. To estimate the luciferase activity of axenic amastigotes, 80 μl of each well are transferred to white 96-well plates, Steady Glow® reagent (Promega) was added according to manufacturer's instructions, and plates were incubated for 2 min. The luminescence was measured in Microbeta Luminescence Counter (PerkinElmer). Inhibitory concentration 50% (IC50) was defined as the concentration of drug required to inhibit by 50% the metabolic activity of L. infantum amastigotes compared to control. IC50 values were calculated by non-linear regression analysis processed on dose response curves, using TableCurve 2D V5 software. IC50 values represent the mean of three independent experiments.
Antitrypanosomal activity on T. brucei brucei trypomastigotes
Assays were performed on Trypanosoma brucei brucei AnTat 1.9 strain (IMTA, Antwerpen, Belgium). It was cultured in MEM with Earle’s salts, supplemented according to the protocol of Baltz et al.[32] with the following modifications, 0.5 mM mercaptoethanol (Sigma Aldrich®, France), 1.5 mM L-cysteine (Sigma Aldrich®), 0.05 mM bathocuproïne sulfate (Sigma Aldrich®) and 20% heat-inactivated horse serum (Gibco®, France), at 37°C in an atmosphere of 5% CO2. The parasites were incubated at an average density of 2000 parasites/well in sterile 96-wells plates (Mc2®, France) with various concentrations of compounds dissolved in DMSO (Sigma Aldrich®), in duplicate. Reference drugs suramin, eflornithine, and fexinidazole (purchased from Sigma Aldrich, France and Fluorochem, UK) suspended in NaCl 0.9% or DMSO, were added to each set of experiments. The effects of the tested compounds were assessed by the viability marker Alamar Blue® (Fisher, France) assay described by Räz et al.[33] After a 69 h incubation period at 37°C, 10 μL of Alamar Blue® was then added to each well, and the plates were incubated for 5h.[34] The plates were read in a PerkinElmer ENSPIRE (Germany) microplate reader using an excitation wavelength of 530 nm and an emission wavelength of 590 nm. IC50 were calculated by nonlinear regression analysis processed on dose-response curves, using GraphPad Prism software (USA). IC50 was defined as the concentration of drug necessary to inhibit by 50% the viability of T. brucei brucei compared to the control. IC50 values were calculated from three independent experiments in duplicate.
Antitrypanosomal activity on T. brucei trypomastigotes overexpressing the nitroreductase (NTR1)
Trypanosoma brucei bloodstream-form 'single marker' S427 (T7RPOL TETR NEO) and drug-resistant cell lines were cultured at 37°C in HMI9-T medium [35] supplemented with 2.5 μg/mL G418 to maintain expression of T7 RNA polymerase and the tetracycline repressor protein. Bloodstream trypanosomes overexpressing the T. brucei nitroreductase (NTR1).[36] were grown in medium supplemented with 2.5 μg/mL phleomycin and expression of NTR was induced by the addition of 1 μg/mL tetracycline. Cultures were initiated with 1 × 105 cells per mL and sub-cultured when cell densities approached 1–2 (× 106) per mL. In order to examine the effects of inhibitors on the growth of these parasites, triplicate cultures containing the inhibitor were seeded at 1 × 105 trypanosomes per mL. Cells overexpressing NTR were induced with tetracycline 48 h prior to EC50 analysis. Cell densities were determined after culture for 72 h, as previously described [37]. EC50 values were determined using the following two-parameter equation by non-linear
regression using GraFit: where the experimental data were corrected for background cell density and expressed as a percentage of the uninhibited control cell density. In this equation [I] represents inhibitor concentration and m is the slope factor.
Cytotoxic evaluation on HepG2 cell line
The evaluation of the tested molecules cytotoxicity on the HepG2 (hepatocarcinoma cell line from ECACC purchased from Sigma-Aldrich, ref 85011430-1VL certificated without mycoplasma) was done according to the method of Mosman with slight modifications.[38] Briefly, cells (1 x 105 cells/mL) in 100 μL of complete medium, [Alpha MEM Eagle from PAN BIOTECH supplemented with 10% foetal bovine serum, 2 mM L-glutamine and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin)] were seeded into each well of 96-well plates and incubated at 37°C and 5% CO2. After a 24 h incubation, 100 μL of medium with various product concentrations and appropriate controls were added and the plates were incubated for 72 h at 37°C and 5% CO2. Each plate-well was then microscope-examined for detecting possible precipitate formation before the medium was aspirated from the wells. 100 μL of MTT solution (0.5 mg/mL in Alpha MEM Eagle) were then added to each well. Cells were incubated for 2 h at 37°C and 5% CO2. After this time, the MTT solution was removed and DMSO (100 μL) was added to dissolve the resulting formazan crystals. Plates were shaken vigorously (300 rpm) for 5 min. The absorbance was measured at 570 nm with a microplate spectrophotometer (Eon BioTek). DMSO was used as blank and doxorubicin (purchased from Sigma Aldrich) as positive control. CC50 were calculated by non-linear regression analysis processed on dose response curves, using TableCurve 2D V5 software. CC50 values represent the mean value calculated from three independent experiments.
Acknowledgements
The authors thank the Université Paul Sabatier de Toulouse and the Région Occitanie / Pyrénées-Méditerranée for funding this work. A. Fairlamb and S. Wyllie are supported by funding from Wellcome Trust (WT105021). C. Bijani from the NMR facility Laboratoire de Chimie de Coordination de Toulouse, C. Claparols, N. Martins-Froment, V. Bourdon and E. Leroy from the mass spectrometry service of Institut de Chimie de Toulouse are also acknowledged for their support, respectively for NMR and HRMS experiments.
Footnotes
Lucie Paloque: https://orcid.org/0000-0003-0359-3959
Colin Bonduelle: https://orcid.org/0000-0002-7213-7861
Geneviève Pratviel: https://orcid.org/0000-0003-0688-2107
Pierre Verhaeghe: https://orcid.org/0000-0003-0238-2447
Alexis Valentin: https://orcid.org/0000-0002-2133-112X
Susan Wyllie: https://orcid.org/0000-0001-8810-5605
Alan Fairlamb: https://orcid.org/0000-0001-5134-0329
References
- [1].Capewell P, Veitch NJ, Turner CM, Turner CMR, Raper J, Berriman M, Hadjuk SL, MacLeod A. PLoS Negl Trop Dis. 2011;5:e1287. doi: 10.1371/journal.pntd.0001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Büscher P, Cecchi G, Jamonneau V, Priotto G. Lancet. 2017;390:2397–2405. doi: 10.1016/S0140-6736(17)31510-6. [DOI] [PubMed] [Google Scholar]
- [3].Pace D. J Infect. 2014;69:S10–S18. doi: 10.1016/j.jinf.2014.07.016. [DOI] [PubMed] [Google Scholar]
- [4].Ashkan MM, Rahim KM. Trop Doct. 2008;38:186–188. doi: 10.1258/td.2007.070259. [DOI] [PubMed] [Google Scholar]
- [4].http://www.who.int/neglected_diseases/diseases/en/
- [5].World Health Organization. http://www.who.int/news-room/fact-sheets/detail/leishmaniasis updated 03/2018.
- [6].World Health Organization. http://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness), updated 02/2018.
- [7].Field MC, Horn D, Fairlamb AH, Ferguson MAJ, Gray DW, Read KD, De Rycker M, Torrie LS, Wyatt PG, Wyllie S, Gilbert IH. Nat Rev Microbiol. 2017;15:217–231. doi: 10.1038/nrmicro.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eperon G, Balasegaram M, Potet J, Mowbray C, Valverde O, Chappuis F. Expert Rev Anti Infect, Ther. 2014;12:1407–1417. doi: 10.1586/14787210.2014.959496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zulfiqar B, Shelper TB, Avery VM. Drug Discov Today. 2017;22:1516–1531. doi: 10.1016/j.drudis.2017.06.004. [DOI] [PubMed] [Google Scholar]
- [10].Patterson S, Fairlamb AH. Curr Med Chem. 2018 doi: 10.2174/0929867325666180426164352. in press. [DOI] [PubMed] [Google Scholar]
- [11].Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, Rewerts C, et al. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wyllie S, Patterson S, Stojanovski L, Simeons FRC, Norval S, Kime R, Read KD, Fairlamb AH. Sci Transl Med. 2012;4:e119. doi: 10.1126/scitranslmed.3003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mesu VKBK, Kalonji WM, Bardonneau C, Mordt OV, Blesson S, Simon F, Delhomme S, Bernhard S, Kuziena W, Lubaki JF, Vuvu SL, et al. Lancet. 2018;391:144–154. doi: 10.1016/S0140-6736(17)32758-7. [DOI] [PubMed] [Google Scholar]
- [14].Patterson S, Wyllie S. Trends Parasitol. 2014;30:289–298. doi: 10.1016/j.pt.2014.04.003. Nitro drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wyllie S, Patterson S, Fairlamb AH. Antimicrob Agents Chemother. 2013;57:901–906. doi: 10.1128/AAC.01788-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wyllie S, Roberts AJ, Norval S, Patterson S, Foth B, Berriman M, Read K, Fairlamb AH. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005971. e1005971/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hall BS, Wu X, Hu L, Wilkinson SR. Antimicrob Agents Chemother. 2010;54:1193–1199. doi: 10.1128/AAC.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Verhaeghe P, Rathelot P, Rault S, Vanelle P. Lett Org Chem. 2006;3:891–897. [Google Scholar]
- [19].Paloque L, Verhaeghe P, Casanova M, Castera-Ducros C, Dumètre A, Mbatchi L, Hutter S, Kraiem-M’Rabet M, Laget M, Remusat V, Rathelot P, et al. Eur J Med Chem. 2012;54:75–86. doi: 10.1016/j.ejmech.2012.04.029. [DOI] [PubMed] [Google Scholar]
- [20].Kieffer C, Cohen A, Verhaeghe P, Hutter S, Castera-Ducros C, Laget M, Remusat V, Kraiem-M’Rabet M, Rault S, Rathelot P, Azas N, et al. Eur J Med Chem. 2015;92:282–294. doi: 10.1016/j.ejmech.2014.12.056. [DOI] [PubMed] [Google Scholar]
- [21].Kieffer C, Cohen A, Verhaeghe P, Paloque L, Hutter S, Castera-Ducros C, Laget M, Rault S, Valentin A, Rathelot P, Azas N, et al. Bioorg Med Chem. 2015;23:2377–2386. doi: 10.1016/j.bmc.2015.03.064. [DOI] [PubMed] [Google Scholar]
- [22].Pedron J, Boudot C, Hutter S, Bourgeade-Delmas S, Stigliani J-L, Sournia-Saquet A, Moreau A, Boutet-Robinet E, Paloque L, Mothes E, Laget M, et al. Eur J Med Chem. 2018;155:135–152. doi: 10.1016/j.ejmech.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Andreev VP, Nizhnnik YP. Russ J Org Chem. 2002;38:137–138. [Google Scholar]
- [24].O'Brien NJ, Brzozowski M, Wilson DJD, Deady LW, Abbott BM. Bioorg Med Chem. 2014;22:3781–3790. doi: 10.1016/j.bmc.2014.04.037. [DOI] [PubMed] [Google Scholar]
- [25].Tremblay MS, Halim M, Sames D. J Am Chem Soc. 2007;129:7570–7577. doi: 10.1021/ja070867y. [DOI] [PubMed] [Google Scholar]
- [26].Chong E, Schafer LL. Org Lett. 2013;15:6002–6005. doi: 10.1021/ol402890m. [DOI] [PubMed] [Google Scholar]
- [27].Kuduk SD, Skudlarek JW, Di Marco CN, Bruno JG, Pausch MA, O’Brien JA, Cabalu TD, Stevens J, Brunner J, Tannenbaum PL, Gotter AL, et al. Bioorg Med Chem Lett. 2014;24:1784–1789. doi: 10.1016/j.bmcl.2014.02.026. [DOI] [PubMed] [Google Scholar]
- [28].Zhou R, Wang W, Jiang Z, Wang K, Zheng X, Fu H, Chen H, Li R. Chem Commun. 2014;50:6023–6026. doi: 10.1039/c4cc00815d. [DOI] [PubMed] [Google Scholar]
- [29].Van de Poël H, Guillaumet G, Viaux-Massuard MC. Heterocycles. 2002;57:55–71. [Google Scholar]
- [30].Volkov OA, Cosner CC, Brockway AJ, Kramer M, Booker M, Zhong S, Ketcherside A, Wei S, Longgood J, McCoy MK, Richardson TE, et al. ACS Infect Dis. 2017;3:512–526. doi: 10.1021/acsinfecdis.7b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang C, Bourgeade-Delmas S, Alvarez AF, Valentin A, Hemmert C, Gornitzka H. Eur J Med Chem. 2018;143:1635–1643. doi: 10.1016/j.ejmech.2017.10.060. [DOI] [PubMed] [Google Scholar]
- [32].Baltz T, Baltz D, Giroud C, Crockett J. EMBO J. 1985;4:1273–1277. doi: 10.1002/j.1460-2075.1985.tb03772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Räz B, Iten M, Grether-Bühler Y, Kaminsky R. Acta Trop. 1997;68:139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- [34].Guillon J, Cohen A, Nath Das R, Boudot C, Gueddouda N, Moreau S, Ronga L, Savrimoutou S, Rubio S, Amaziane S, Dassonville-Klimpt A, et al. Chem Biol Drug Des. 2018;68:1–22. doi: 10.1111/cbdd.13164. [DOI] [PubMed] [Google Scholar]
- [35].Greig N, Wyllie S, Patterson S, Fairlamb AH. FEBS J. 2009;276:376–386. doi: 10.1111/j.1742-4658.2008.06788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wyllie S, Foth BJ, Kelner A, Sokolova AY, Berriman M, Fairlamb AH. Antimicrob Agents Chemother. 2016;71:625–634. doi: 10.1093/jac/dkv376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jones DC, Hallyburton I, Stojanovski L, Read KD, Frearson JA, Fairlamb AH. Biochem Pharmacol. 2010;80:1478–1486. doi: 10.1016/j.bcp.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mossman TJ. J Immunol Methods. 1983;65:55–63. [Google Scholar]