

Abstract
Genome engineering of non-conventional microorganisms calls for the development of dedicated synthetic biology tools. Pseudomonas putida is a Gram-negative, non-pathogenic soil bacterium widely used for metabolic engineering owing to its versatile metabolism and high levels of tolerance to different types of stress. Genome editing of P. putida largely relies on homologous recombination events, assisted by helper plasmid-based expression of genes encoding DNA modifying enzymes. Plasmid curing from selected isolates is the most tedious and time-consuming step of this procedure, and implementing commonly used methods to this end in P. putida (e.g. temperature-sensitive replicons) is often impractical. To tackle this issue, we have developed a toolbox for both target- and self-curing of plasmid DNA in Pseudomonas species. Our method enables plasmid-curing in a simple cultivation step by combining in vivo digestion of vectors by the I-SceI homing nuclease with synthetic control of plasmid replication, triggered by the addition of a cheap chemical inducer (3-methylbenzoate) to the medium. The system displays an efficiency of vector curing >90% and the screening of plasmid-free clones is greatly facilitated by the use of fluorescent markers that can be selected according to the application intended. Furthermore, quick genome engineering of P. putida using self-curing plasmids is demonstrated through genome reduction of the platform strain EM42 by eliminating all genes encoding β-lactamases, the catabolic ben gene cluster, and the pyoverdine synthesis machinery. Physiological characterization of the resulting streamlined strain, P. putida SEM10, revealed advantageous features that could be exploited for metabolic engineering.
Keywords: Genome engineering, Pseudomonas putida, Metabolic engineering, Plasmid curing, Synthetic biology
Graphical abstract
Highlights
-
•
Plasmid-curing is the most time-consuming step in genome engineering approaches.
-
•
We have developed a system for easy target- and self-curing of plasmid DNA.
-
•
Synthetic control of replication and highly-specific in vivo DNA digestion were used.
-
•
Plasmid curing with this system displays an efficiency >90% in a 24-h cultivation.
-
•
Quick genome engineering facilitated genome reduction of P. putida.
1. Introduction
The scope of contemporary metabolic engineering has expanded over the years through the adoption of non-conventional microorganisms, domesticated via synthetic biology strategies (Abram and Udaondo, 2020; Calero and Nikel, 2019; Fernández-Cabezón et al., 2019; Jawed et al., 2019; Kim et al., 2016; Sánchez-Pascuala et al., 2017). Key to this development is the use of advanced genome engineering techniques (Freed et al., 2018; Kent and Dixon, 2020), often based on the temporary propagation of plasmids. Such approaches include CRISPR/Cas9 technologies (Aparicio et al., 2016; Batianis et al., 2020; Cong and Zhang, 2015; Jakočiūnas et al., 2017; Sun et al., 2018), DNA recombineering (Aparicio et al., 2020; Csörgő et al., 2016; Sharan et al., 2009), and homologous recombination-based DNA editing (Choi and Lee, 2020; Martínez-García and de Lorenzo, 2017; Wirth et al., 2020). A dedicated synthetic biology toolbox enabled the taming of Pseudomonas species (and, in particular, of P. putida strain KT2440) as robust platforms for bioproduction (Johnson et al., 2016; Loeschcke and Thies, 2015; Nikel et al., 2016; Nikel and de Lorenzo, 2014, Nikel and de Lorenzo, 2018; Poblete-Castro et al., 2020; Sánchez-Pascuala et al., 2019).
Homologous recombination is the standard principle to insert heterologous DNA fragments into (or deleting parts of) the genome of Pseudomonas species (Nikel et al., 2014). This methodology, established for P. putida by Martínez-García and de Lorenzo (2011), typically involves two rounds of recombination. A first cycle consists in the chromosomal integration of a suicide plasmid containing recognition sequence(s) for the I-SceI homing meganuclease (Gallagher and Haber, 2018; Jacquier and Dujon, 1985; Jasin, 1996). Next, a helper plasmid, encoding elements needed to introduce double-strand breaks (DSBs) in the chromosome, is transformed into co-integrants. The second recombination event, forced by DSBs (which would be otherwise lethal), uses duplicated sequences in the (co-integrated) plasmid as substrate (Pósfai et al., 1999). Such recombination-and-resolving step results in the stochastic occurrence of revertant clones (displaying the wild-type genotype) and mutant clones carrying the desired modification—e.g. deletion, insertion, or point mutation. After creating the mutation intended, the helper plasmid used for introducing DSBs should be cured prior to any subsequent experiment. Vector curing is also beneficial for successive genome engineering manipulations, as the presence of helper plasmids drastically reduces the efficiency of subsequent integration events—likely because of the basal expression of the gene encoding the DSB-inducing enzyme (Bennett et al., 1993).
The plasmid-curing step is the most time-consuming part of bacterial genome engineering protocols; generally accomplished by repetitive passaging of clones in antibiotic-free culture media (loss-by-dilution) followed by sensitivity screening against the antibiotic marker of the helper plasmid (Aparicio et al., 2019a,b; Martínez-García et al., 2017). From a broader perspective, the removal of plasmid DNA from a given bacterial host is a standard technique in microbiology, and different strategies have been implemented for this purpose (Haldimann and Wanner, 2001; Trevors, 1986). Originally designed for Escherichia coli and related species, such protocols comprise electroporation (Heery et al., 1989), use of DNA intercalating reagents (Buckner et al., 2018), adoption of conditionally-replicating plasmids and repetitive passaging under non-selective conditions (Chen et al., 2017), and CRISPR/Cas9-mediated plasmid killing (Lauritsen et al., 2017). These methodologies, however, often yield uneven results when implemented in Pseudomonas species. The most convenient technique thus far is the use of temperature-sensitive plasmids, based on the RK2 (Valla et al., 1991) and pSC101 (Hashimoto and Sekiguchi, 1976) vegetative origins of replication (oriV). Unfortunately, the narrow-host-range oriV(pSC101) does not replicate in pseudomonads, while oriV(RK2) exhibits altered temperature-sensitive characteristics depending on the species (Karunakaran et al., 1998). No temperature-sensitive oriV is known to be fully functional in P. putida, and temperature shifts could anyways be lethal to some mutants (Ito et al., 2014)—thus precluding the broad use of temperature-sensitive replicons as a strategy for plasmid curing.
On this background, here we describe a plasmid-based system designed for efficient vector curing in Pseudomonas species, based on synthetic control of plasmid replication. In particular, the replication of plasmids carrying the gene encoding the I-SceI meganuclease has been made strictly dependent on the presence of the chemical inducer 3-methylbenzoate (3-mBz). These new vectors are rapidly and irreversibly lost in the absence of 3-mBz, and screening of bacterial clones that have lost the plasmid is facilitated by using fluorescent markers. We demonstrate the ease of genome engineering with this system by deleting ten individual genomic regions in the platform P. putida strain EM42 encoding functions deemed dispensable for metabolic engineering applications (i.e. all β-lactamase–like genes, benABCD, and pvdD; together accounting for ~23 kb) towards a reduced-genome chassis of reference.
2. Materials and methods
2.1. Bacterial strains and growth conditions
All bacterial strains used in this study are listed in Table 1. Cultures of P. putida KT2440, E. coli and their derivatives were incubated at 30 °C and 37 °C, respectively. For standard applications, routine cloning procedures, and during genome engineering manipulations, cells were grown in lysogeny broth (LB) medium (10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl). All liquid cultures were agitated at 200 rpm (MaxQ™ 8000 incubator; ThermoFisher Scientific, Waltham, MA, USA). Solid culture media also contained 15 g L−1 agar. Kanamycin (Km), gentamicin (Gm), streptomycin (Str), and ampicillin (Amp) were added whenever needed at 50 μg mL−1, 10 μg mL−1, 100 μg mL−1, and 100 μg mL−1, respectively. Unless indicated otherwise, Amp was supplemented at 500 μg mL−1 in P. putida cultures. For quantification of red and green fluorescence and phenotypic characterization of reduced-genome strains, P. putida KT2440, EM42, and derivatives thereof were grown in M9 minimal medium (Nikel et al., 2008, 2015) supplemented with 0.2% (w/v) citrate in 96-well plates in a Synergy HI plate reader (BioTek Instruments; Winooski, VT, USA). The excitation and emission wavelengths (λexcitation/λemission) used for detection of mRFP/mCherry (red) and msfGFP (green) fluorescence were 488 nm/588 nm and 485 nm/528 nm, respectively.
Table 1.
Bacterial strains and plasmids used in this study.
| Name | Relevant characteristicsa | Source or reference |
|---|---|---|
| Bacterial strain | ||
| E. coli DH5α | Cloning host; F− λ–endA1 glnX44(AS) thiE1 recA1 relA1 spoT1 gyrA96(NalR) rfbC1 deoR nupG Φ80(lacZΔM15) Δ(argF-lac)U169 hsdR17(rK–mK+) | Meselson and Yuan (1968) |
| E. coli DH5α λpir | Cloning host; λpir lysogen derivative of strain DH5α | Platt et al. (2000) |
| P. putida KT2440 | Wild-type strain; derivative of P. putida mt-2 (Worsey and Williams, 1975) cured of the TOL plasmid pWW0 | Bagdasarian et al. (1981) |
| P. putida EM42 | Reduced-genome derivative of P. putida KT2440; ΔPP_4329-PP_4397 (flagellar operon) ΔPP_3849-PP_3920 (prophage I) ΔPP_3026-PP_3066 (prophage II) ΔPP_2266-PP_2297 (prophage III) ΔPP_1532-PP_1586 (prophage IV) ΔTn7 ΔendA-1 ΔendA-2 ΔhsdRMS ΔTn4652 | Martínez-García et al. (2014b) |
| P. putida SEM10 | Reduced-genome derivative of P. putida EM42; ΔPP_0052 ΔPP_0772 ΔPP_1239 ΔPP_1775 ΔPP_1952 ΔPP_2045 ΔPP_2876 ΔPP_3291 ΔbenABCD ΔpvdD | This work |
| Plasmid | ||
| pSEVA637M | Cloning vector; oriV(pBBR1), promoter-less msfGFP; GmR | Martínez-García et al. (2015) |
| pS6313·GFP | Reporter plasmid; oriV(pBBR1), PEM7→msfGFP; GmR | This work |
| pS2313·GFP | Reporter plasmid; oriV(pBBR1), PEM7→msfGFP; KmR | This work |
| pS6313·GFPs | Derivative of vector pS6313·GFP with an engineered I-SceI recognition site; GmR | This work |
| pS2313·GFPs | Derivative of vector pS2313·GFP with an engineered I-SceI recognition site; KmR | This work |
| pSEVA628S | Helper plasmid; oriV(RK2), XylS/Pm→I-SceI; GmR | Aparicio et al. (2015) |
| pSEVA228S | Helper plasmid; oriV(RK2), XylS/Pm→I-SceI; KmR | Aparicio et al. (2015) |
| pSEVA128S | Helper plasmid; oriV(RK2), XylS/Pm→I-SceI; AmpR | Aparicio et al. (2015) |
| pJBSD1 | Conditionally-replicating vector; oriV(RK2), XylS/Pm→trfA; AmpR | Karunakaran et al. (1999) |
| pS628SR | Derivative of vector pSEVA628S with P14g(BCD2)→mRFP; GmR | This work |
| pS228SR | Derivative of vector pSEVA228S with P14g(BCD2)→mRFP; KmR | This work |
| pS628SR·M | Derivative of vector pSEVA628S with PEM7→mRFP; GmR | This work |
| pS228SR·M | Derivative of vector pSEVA228S with PEM7→mRFP; KmR | This work |
| pS628SR·L | Derivative of vector pSEVA628S with PEM7→mCherry; GmR | This work |
| pS228SR·L | Derivative of vector pSEVA228S with PEM7→mCherry; KmR | This work |
| pQURE1·H | Conditionally-replicating vector; derivative of vector pJBSD1 carrying XylS/Pm→I-SceI and P14g(BCD2)→mRFP; AmpR | This work |
| pQURE2·H | Conditionally-replicating vector; derivative of vector pJBSD1 carrying XylS/Pm→I-SceI and P14g(BCD2)→mRFP; KmR | This work |
| pQURE6·L | Conditionally-replicating vector; derivative of vector pJBSD1 carrying XylS/Pm→I-SceI and P14g→mCherry; GmR | This work |
| pQURE6·M | Conditionally-replicating vector; derivative of vector pJBSD1 carrying XylS/Pm→I-SceI and P14g→mRFP; GmR | This work |
| pQURE6·H | Conditionally-replicating vector; derivative of vector pJBSD1 carrying XylS/Pm→I-SceI and P14g(BCD2)→mRFP; GmR | This work |
| pGNW2·mCherry | Suicide vector used for deletions in Gram-negative bacteria; oriT, traJ, lacZα, ori(R6K), PEM7→mCherry; KmR | Wirth et al. (2020) |
| pGNW2 | Suicide vector used for deletions in Gram-negative bacteria; oriT, traJ, lacZα, ori(R6K), PEM7→msfGFP; KmR | Wirth et al. (2020) |
| pGNW·ΔbenABCD | Derivative of vector pGNW2 carrying HRs to delete benABCD (PP_3161-PP_3164); KmR | This work |
| pGNW2·LPO | Derivative of vector pGNW2 carrying HRs to insert P14g(BCD2)→mOrange2 into a landing pad in the chromosome of P. putida KT2440; KmR | Wirth et al. (2020) |
| pGNW4 | Derivative of vector pGNW2; StrR | Wirth et al. (2020) |
| pGNW6 | Derivative of vector pGNW2; GmR | Wirth et al. (2020) |
| pSNW2 | Derivative of vector pGNW2 with P14g(BCD2)→msfGFP; KmR | This work |
| pSNW4 | Derivative of vector pGNW4 with P14g(BCD2)→msfGFP; StrR | This work |
| pSNW6 | Derivative of vector pGNW6 with P14g(BCD2)→msfGFP; GmR | This work |
| pSNW·ΔPP_1952 | Derivative of vector pSNW2 carrying HRs to delete PP_1952; KmR | This work |
| pSNW·ΔPP_3291 | Derivative of vector pSNW2 carrying HRs to delete PP_3291; KmR | This work |
| pSNW·ΔPP_2876 | Derivative of vector pSNW2 carrying HRs to delete PP_2876; KmR | This work |
| pSNW·ΔPP_2045 | Derivative of vector pSNW2 carrying HRs to delete PP_2045; KmR | This work |
| pSNW·ΔPP_0052 | Derivative of vector pSNW2 carrying HRs to delete PP_0052; KmR | This work |
| pSNW·ΔPP_1775 | Derivative of vector pSNW2 carrying HRs to delete PP_1775; KmR | This work |
| pSNW·ΔPP_1239 | Derivative of vector pSNW2 carrying HRs to delete PP_1239; KmR | This work |
| pSNW·ΔPP_0772 | Derivative of vector pSNW2 carrying HRs to delete PP_0772; KmR | This work |
| pSNW·ΔpvdD | Derivative of vector pSNW2 carrying HRs to delete pvdD (PP_4219); KmR | This work |
| pSNW·ΔbenABCD | Derivative of vector pSNW2 carrying HRs to delete benABCD (PP_3161-PP_3164); KmR | This work |
Antibiotic markers: Amp, ampicillin; Gm, gentamicin; Km, kanamycin; Nal, nalidixic acid; and Str, streptomycin. HRs, homology regions.
2.2. General cloning procedures and plasmid construction
All plasmids and oligonucleotides used in this work are listed in Table 1 and Table S1 (Supplementary Material), respectively. Unless stated otherwise, uracil-excision (USER) cloning (Cavaleiro et al., 2015) was used for the construction of all plasmids. The AMUSER tool was employed for the design of oligonucleotides (Genee et al., 2015). Phusion™ U and Phusion™ Hot Start II high-fidelity DNA polymerases (ThermoFisher Scientific) were used according to the manufacturer’s specifications for amplifications intended for USER cloning or site-directed PCR mutagenesis, respectively. For colony PCR, the commercial OneTaq™ master mix (New England BioLabs; Ipswich, MA, USA) was used according to the manufacturer’s instructions. E. coli DH5α was employed for general cloning purposes, while E. coli DH5α λpir was employed when cloning and maintaining replicons with the conditional, Π-dependent origin of replication RK6 (Table 1). Chemically-competent E. coli cells were prepared and transformed with plasmids according to well established protocols (Sambrook and Russell, 2001). P. putida was rendered electrocompetent by washing the biomass from saturated (24 h) LB medium cultures with 0.3 M sucrose, and cells were routinely transformed with plasmids by electroporation, following the protocols of Iwasaki et al. (1994) and Choi et al. (2006). Site-directed PCR mutagenesis was used for insertion of I-SceI restriction sites into selected plasmids with procedures described elsewhere (Volke et al., 2020). The identity and correctness of all plasmids and DNA constructs were confirmed by sequencing.
In the following description, the letter x is used as a place-holder in plasmid nomenclature, standing for AmpR (1), KmR (2), and GmR (6). Note that the numbers identifying these resistance determinants are coded as for the rules set in the Standard European Vector Architecture (SEVA; Silva-Rocha et al., 2013). In order to construct plasmids pSx28SR·L (low level of red fluorescence), the fragment containing mCherry under transcriptional control of the constitutive PEM7 promoter (Nikel et al., 2013) was amplified from vector pGNW2·mCherry with the primer pair RED-low-F and RED-low-R. Similarly, to construct plasmids pSx28SR·M and pSx28SR (displaying, respectively, medium and high levels of red fluorescence), the fragments containing mRFP under transcriptional control of PEM7 or P14g [a synthetic, constitutive and strong derivative of the PEM7 promoter (Zobel et al., 2015)] were amplified from plasmids pSEVA2313R and pGNW2∙LPO with the primer pairs RED-medium-F/RED-medium-R and RED-F/RED-R, respectively. In parallel, plasmids pSx28S were reverse-amplified with the oligonucleotide pair Ins-RED-F and Ins-RED-R. These fragments were used as the backbone, and stitched with the previously-generated mCherry- or mRFP-bearing DNA fragments by USER cloning to yield the corresponding plasmids (Table 1).
For the construction of plasmid pQURE1, a DNA fragment bearing both the I-SceI gene under the transcriptional control of a XylS/Pm element and the mRFP gene under control of P14g was amplified from plasmid pS228SR with oligonucleotides pQURE-F and pQURE-R. The resulting amplicon was joined with the fragment resulting from the reverse amplification of vector pJBSD1 with the primer pair Ins-pQURE-F and Ins-pQURE-R. Plasmids pQURE2 and pQURE6 were generated, respectively, by exchanging the antibiotic cassette present in plasmid pQURE1 (AmpR) by either a KmR or a GmR marker. To this end, vector pQURE1 was reverse-amplified with the primer pair Ins-pQURE-Ab-F and Ins-pQURE-Ab-R, while individual antibiotic cassettes were amplified with oligonucleotides Ab-F and Ab-R from vectors pSEVA228S and pSEVA628S. The individual fragments were then ligated by USER cloning as indicated above.
Plasmid pS6313·GFP, carrying a synthetic PEM7→msfGFP element, was constructed through site-directed PCR mutagenesis of plasmid pSEVA637M with the primer pair PEM7-Ins-F and PEM7-Ins-R. Furthermore, the I-SceI restriction site (5′-TAG GGA TAA CAG GGT AAT-3′) was engineered into this vector by a subsequent round of site-directed PCR mutagenesis with oligonucleotides I-SceI-Ins-F and I-SceI-Ins-R, yielding plasmid pS6313·GFPs. Finally, suicide vector pSNW2 and derivatives thereof, designed for quick genome engineering of Pseudomonas, were constructed by amplification of a fragment containing the P14g promoter and a bicistronic design (BCD2, adopted as a translational coupler) element from plasmid pGNW2∙LPO with the primer pair P14g-F and BCD2-R. Plasmids pGNW2, pGNW4, and pGNW6 were reverse-amplified with oligonucleotides GFP4BCD2-F and EMG4GFP-R. The resulting fragments were assembled by USER cloning, giving rise to the suicide vectors pSNW2 (KmR), pSNW4 (StrR), and pSNW6 (GmR), respectively.
2.3. Target-curing of plasmids by in vivo meganuclease digestion
The P. putida strain harboring the plasmid to be cured was co-transformed with a helper plasmid bearing the I-SceI activity (e.g. plasmid pSEVA228S) by electroporation. A 10-mL aliquot of LB medium supplemented with the appropriate antibiotics to select for both plasmids was then inoculated with the resulting strain, and the cells were grown overnight as indicated above. A 100-μL aliquot of this culture was then used to inoculate 10 mL of fresh LB medium supplemented with 2 mM 3-mBz and the antibiotic needed to select only for the I-SceI–bearing plasmid (e.g. Km, when using plasmid pSEVA228S). The culture was incubated for 24 h and aliquots were then plated on solid media and inspected for plasmid loss as specified in the text.
2.4. Quick genome engineering of P. putida using self-curing vectors
For the deletion of genes from the genome of P. putida, 500-bp DNA fragments upstream and downstream of the corresponding target to be eliminated were individually amplified from the genome of strain KT2440 with the corresponding pairs of oligonucleotides as specified in Table S1. KmR-vector pSNW2 was used in all cases, and the addition of the upstream and downstream homology regions to this backbone resulted in the corresponding suicide vectors (indicated as pSNW·Δgene in Table 1) for deleting each locus. The protocol of Wirth et al. (2020) was followed for genomic co-integration of the suicide vectors. Successful integration of the suicide pSNW plasmids into the target locus was confirmed by (i) green fluorescence of individual colonies inspected under blue light and (ii) colony PCR of selected amplicons (Martínez-García and de Lorenzo, 2011). One such colony was inoculated in 10 mL of LB medium and incubated overnight as indicated above. This culture was used to prepare competent cells by thoroughly washing the biomass with 0.3 M sucrose, and these cells were transformed by electroporation with plasmid pQURE1 or its derivatives. Cell suspensions were recovered at least for 2 h in LB medium containing 2 mM 3-mBz, and a loopfull of this culture was streaked onto LB medium agar containing 2 mM 3-mBz and the corresponding antibiotic(s), and incubated overnight or until discernible colonies appeared on the agar surface. Deletion of target gene(s) was confirmed in msfGFP– clones by colony PCR, and 5 mL of fresh LB medium (i.e. no additives) were inoculated with a single colony and incubated for 2 to 10 h under the same conditions. This culture was then diluted by streaking onto solid media (according to the nutritional needs of the mutants; solid LB medium was routinely used) to obtain isolated colonies. Non-fluorescent colonies (i.e. clones that have lost both msfGFP and mRFP/mCherry fluorescence when examined under blue light) were picked and inoculated in 5 mL of liquid LB medium. The loss of the plasmid was finally confirmed by parallel inoculation of liquid medium with the corresponding antibiotic(s), and all relevant genotypes were checked by DNA sequencing.
2.5. Ampicillin sensitivity assay
Cultures of P. putida EM42 and SEM10 were grown overnight in 10 mL of LB medium. These cultures were diluted to an optical density measured at 600 nm (OD600) of 1 before serial dilutions were prepared with fresh LB medium. A 5-μL aliquot of each dilution was then spotted on plates containing various Amp concentrations (up to 75 μg mL−1). Plates were then incubated overnight and colonies were counted in each spot to estimate antibiotic sensitivity.
3. Results and discussion
3.1. Target-curing of plasmids in P. putida through in vivo meganuclease digestion
During our current efforts to engineer P. putida for several applications, we often found it difficult to eliminate plasmids and vectors from this bacterium (especially if an engineered strain displays slow growth, rendering the plasmid loss-by-dilution approach impractical or simply impossible). Here, we chose the intron-encoded meganuclease I-SceI from yeast to selectively introduce DSBs into targeted plasmids in vivo—therefore impeding further replication. Plasmids to be removed through this protocol are easily modified by insertion of the asymmetric, 18-bp–long I-SceI recognition site into a conserved region present in all SEVA plasmids within the antibiotic resistance marker and the oriT (origin of transfer) module (Fig. 1A). A one-step, site-directed PCR mutagenesis of any SEVA vector can be used to this end.
Fig. 1.

Selective target-curing of plasmids through in vivo meganuclease digestion. (A) One-step engineering of an I-SceI restriction site into any plasmid of the Standard European Vector Collection by site-directed PCR mutagenesis with an oligonucleotide pair carrying the (split) meganuclease recognition target and a sequence homologous to the conserved region between the antibiotic resistance (green) and the oriT modules (gray). The asymmetric, 18-bp–long I-SceI restriction sequence is indicated with a pink box, and the overhangs left after digestion are highlighted. Other relevant features of SEVA vectors are identified with different colors. (B) Target curing of plasmids in P. putida. P. putida KT2440 carrying plasmids pSEVA228S (XylS/Pm→I-SceI, KmR) and either pS6313·GFP (PEM7→msfGFP, GmR) or pS6313·GFPs (PEM7→msfGFP, I-SceI site, GmR) was grown for 18 h in LB medium with (induced) or without (uninduced) 2 mM of 3-methylbenzoate (3-mBz) and Km. Aliquots of these cultures were plated onto solid LB medium, and plasmid loss was determined after 24 h by scoring the fluorescence of individual colonies under blue light. The absence of the plasmid was verified by testing colonies for Gm sensitivity. (C) Target plasmid curing over time. Cultures of P. putida KT2440 containing plasmids pSEVA228S and either pS6313·GFP (indicated as control) or pS6313·GFPs were treated with 2 mM 3-mBz for the periods indicated and aliquots of the cell suspension were plated onto non-selective solid LB medium. An uninduced control experiment was treated similarly, but 3-mBz was omitted. In all cases, average values for the percentage of plasmid-containing cells and standard deviations are presented, calculated from triplicate measurements from at least three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
In order to show the general validity of the approach, we constructed plasmid pS6313·GFP, a derivative of vector pSEVA6313 carrying a synthetic module for constitutive expression of msfGFP (i.e. PEM7→msfGFP). An I-SceI restriction site was engineered in this plasmid, giving rise to plasmid pS6313·GFPs, and this vector was electroporated into strain KT2440. P. putida KT2440 carrying either plasmid pS6313·GFP (control vector) or pS6313·GFPs (sensitive to I-SceI digestion) was co-transformed with plasmid pSEVA228S, bearing the meganuclease gene under control of the XylS/Pm expression system. Aliquots from these cultures were plated onto non-selective medium after overnight (18 h) growth in liquid LB medium added with 3-mBz (to trigger expression of I-SceI) under antibiotic selection for plasmid pSEVA228S (KmR). After an 18-h incubation, plates were examined under blue light and msfGFP+ and msfGFP– colonies were counted to estimate the percentage of plasmid loss. The resulting colonies were replicated onto LB medium plates containing Gm (the antibiotic resistance borne by plasmids pS6313·GFP and pS6313·GFPs) as a further evidence of plasmid presence or loss. Most colonies from 3-mBz–induced cultures of P. putida/pS6313·GFPs did not show any green fluorescence, while essentially all P. putida/pS6313·GFPs colonies were highly fluorescent under blue light (Fig. 1B). In comparison, roughly half of the colonies showed msfGFP fluorescence in a non-induced culture (i.e. no 3-mBz added). Considering the known leakiness of the XylS/Pm expression system in the absence of inducer (Gawin et al., 2017), the loss of fluorescence in half of these colonies (which matches plasmid loss) is assumed to result from basal expression of the I-SceI gene in plasmid pSEVA228S. Importantly, the figures for plasmid loss calculated by the amount of Gm-sensitive clones paralleled the results obtained by scoring msfGFP– colonies.
A time-course experiment was performed to determine the kinetics of target plasmid curing over 30 min using in vivo meganuclease digestion (Fig. 1C). The first time point was taken by adding the inducer to the cultures and immediately washing the treated cells with fresh LB medium before plating and colony counting. Surprisingly, exposure of P. putida to 3-mBz for less than 1 min (the minimum time needed for these manipulations) was sufficient to cure plasmid pS6313·GFPs from ~80% of all cells. More than 90% of all cells were cured of the plasmid after induction of I-SceI expression for 20 min, as indicated by the loss of msfGFP fluorescence. Compared to other plasmid curing systems described in the literature, the kinetics of plasmid loss brought about by this system are considerably fast—probably due to high levels of I-SceI activity and its simple DNA nicking mechanism, which does not require any other components (e.g. cofactors) for efficient DNA restriction (Niu et al., 2008). Taken together, these results expose the validity of the approach for quick and selective curing of plasmids by in vivo digestion, but also leave a question mark on the fate of the very plasmid bearing the I-SceI meganuclease module—an issue solved as indicated in the next section.
3.2. Insertion of red fluorescent markers in meganuclease-bearing plasmids
Target-curing of vectors can be followed by loss of a fluorescence marker (e.g. msfGFP, encoded in vector pSEVA6313·GFPs) or antibiotic sensitivity. In order to facilitate the tracking of the plasmid bearing the I-SceI meganuclease gene (pSEVA228S, KmR), easing the identification of plasmid-free cells, we designed and tested a set of synthetic modules encoding different red fluorescent proteins. As the fluorescence intensity of such reporters is known to vary immensely across microbial hosts (Piatkevich and Verkhusha, 2011), three separate constructs were created, each endowed with different expression strengths and red fluorescence levels (Fig. 2). In particular, we were interested in red markers that could support visual inspection of bacterial colonies with the naked eye to spot plasmid-free clones. Firstly, the mCherry gene, present in a number of SEVA vectors (Silva-Rocha et al., 2013), was placed under transcriptional control of the constitutive PEM7 promoter and inserted in vector pSEVA228S, which gave rise to plasmid pS228SR·L. Transformation of this low-copy-number plasmid into E. coli DH5α or P. putida KT2440 resulted in the lowest level of red fluorescence, as evaluated in colonies grown onto solid LB medium (Fig. 2A). The monomeric red fluorescent protein (mRFP; Campbell et al., 2002) was likewise placed under transcriptional control of the constitutive PEM7 promoter and added to vector pSEVA228S, resulting in plasmid pS228SR·M. Transformation of this plasmid in either bacteria enabled medium mRFP levels (Fig. 2A). Finally, in order to maximize accumulation of the red fluorescent reporter, mRFP was put under transcriptional control of the P14g promoter (a stronger and constitutive derivative of PEM7) and the gene was added with the BCD2 translational coupler (Mutalik et al., 2013; Zobel et al., 2015). Addition of this module to vector pSEVA228S resulted in plasmid pS228SR, which conferred the highest level of mRFP accumulation in both E. coli and P. putida colonies, easily spotted by naked eye on LB medium plates (Fig. 2A). To further expand this plasmid toolbox, GmR-derivatives of all three vectors were constructed (Table 1)—allowing the user to combine different vectors for target-curing of plasmids as needed while keeping red fluorescent markers for plasmid tracing. Furthermore, the use of plasmid pS228SR in P. putida KT2440 enabled the detection of plasmid-containing cells after a mere overnight incubation—whereas red fluorescent markers in other configurations typically needed a further incubation in the cold to enable chromophore maturation (Alieva et al., 2008). Direct quantification of the specific red fluorescence in liquid cultures of E. coli DH5α or P. putida KT2440 individually transformed with plasmids pS228SR·L (low), pS228SR·M (medium), and pS228SR (high) and grown in LB medium further showed the graded output conferred by the synthetic marker modules (Fig. 2B). The behavior of the chromophores tested was fairly similar in both hosts, although the low- and medium-level of red fluorescence were comparable in P. putida (exposing the importance of chromophore maturation, as these measurements were done online during growth). Importantly, plasmid pS228SR resulted in high levels of red fluorescence irrespective of the host (in the case of P. putida, for instance, the fluorescence signal was >5-fold higher than that observed when the cells were transformed either with plasmid pS228SR·L or pS228SR·M). The relevant characteristics of this set of meganuclease-bearing vectors are detailed in Table 2. With these vectors at hand, the next step was to design a protocol for self-curing of plasmids in P. putida.
Fig. 2.

Insertion of red fluorescent modules into plasmids carrying the meganuclease gene. (A) Plasmid pSEVA228S (XylS/Pm→I-SceI, KmR) was used as a template to generate a family of derivatives carrying fluorescent modules yielding low, medium, and high levels of red fluorescence. The modules contain the genes encoding either mCherry or the monomeric red fluorescent protein (mRFP) under transcriptional control of the synthetic, constitutive PEM7 or P14g promoters. Each gene is preceded by a regulatory element, indicated by a purple circle, composed of a ribosome binding site and a short spacer sequence (5′-AGG AGG AAA AAC AT-3′). For the module yielding high levels of red fluorescence, a bicistronic design (BCD2) was used as a translational coupler. E. coli DH5α and P. putida KT2440 were transformed with derivatives of plasmid pSEVA228S (Table 1) and streaked onto LB medium plates containing Km. E. coli colonies were incubated at 37 °C for 18 h and photographed afterwards, while P. putida colonies were grown at 30 °C for 18 h and plates were stored at 8 °C for a further 24 h to allow for fluorophore maturation. (B) Specific (Sp) red fluorescence in cultures of E. coli DH5α and P. putida KT2440 transformed with plasmid pS228SR·L (low), pS228SR·M (medium), or pS228SR (high). Cells were grown in LB medium added with Km for 18 h and the Sp red fluorescence, expressed as arbitrary units (AU) relative to the optical density measured at 600 nm (OD600) of the cultures, was measured after resuspending the bacterial pellets in M9 minimal medium. Each bar represents the mean value of the Sp red fluorescence in each culture ± standard deviation of quadruplicate measurements from at least three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Table 2.
New vectors designed for quick curing of plasmids and quick genome engineering of Pseudomonas.
| Plasmida | Functionality |
|||
|---|---|---|---|---|
| XylS/Pm→I-SceI | Red fluorescence | Antibiotic resistanceb | Conditional replication | |
| pSx28SR·L | Yes | mCherry; low level | Amp, Km, Gm | No |
| pSx28SR·M | Yes | mRFP; medium level | Amp, Km, Gm | No |
| pSx28SR | Yes | mRFP; high level | Km, Gm | No |
| pQUREx·L/M/H | Yes | mRFP; high level | Amp, Km, Gm | Yes (XylS/Pm→trfA) |
The letter x indicates different antibiotic resistance markers [coded as per the Standard European Vector Architecture rules (Silva-Rocha et al., 2013)], and the suffix L, M, or H specifies low, medium, or high red fluorescence (mRFP/mCherry) levels, respectively.
Antibiotic markers: Amp, ampicillin; Gm, gentamicin; and Km, kanamycin.
3.3. Self-curing of plasmids by synthetic control of plasmid replication
We next aimed at applying the plasmid removal approach through in vivo DNA digestion to cure the vector bearing I-SceI. To this end, we attempted to introduce an I-SceI recognition site into vector pSEVA228S. Even though amplification of the plasmid (Fig. 1A) and subsequent transformation of E. coli cells seemed to work well, the resulting plasmids did not contain the correct sequence or the transformants could not be sub-cultured further. We dismissed this strategy after several attempts, and we hypothesized that a plasmid carrying I-SceI and its own recognition site is genetically unstable to be maintained even under selective pressure (i.e. antibiotic resistance). This result could be due to the basal expression of I-SceI, which was evident in previous experiments (Fig. 1B), and the phenomenon is probably amplified by the physical proximity of the I-SceI meganuclease to its target, as shown for transcriptional regulators and their cognate promoters (Goñi-Moreno et al., 2017; Volke et al., 2020).
Next, we considered the use of a temperature-sensitive replicon. These replicons are routinely used in E. coli, with a well-understood mechanism of plasmid partitioning (Hashimoto-Gotoh and Ishii, 1982)—and some reports indicate their use in P. putida (Choi and Lee, 2020; Lauritsen et al., 2017; Sun et al., 2018). However, one of the difficulties in using such vectors is that P. putida KT2440 barely grows when incubated at 42 °C (Munna et al., 2016)—an incubation condition typically used as non-permissive for temperature-sensitive replicons in E. coli and other Pseudomonas species, e.g. P. aeruginosa (Prathapam and Uehara, 2018; Silo-Suh et al., 2009). Plasmids designed for recombineering in E. coli (e.g. the pSIM set of vectors and plasmid pKD46) exploit temperature-sensitive mutants of oriV(RK2) and oriV(pSC101) (Datsenko and Wanner, 2000; Thomason et al., 2014). While the pSC101 replicon has a very narrow host range and does not replicate in pseudomonads (Barth et al., 1981), the broad-host-range RK2 replicon is known to be functional in P. putida (Kolatka et al., 2008). The RK2 replication mechanism depends on the plasmid-encoded replication initiator protein (TrfA) and other elements necessary for replication encoded in the host genome (e.g. DnaA). TrfA binds to the β subunit (sliding clamp) of DNA polymerase III in E. coli (Kongsuwan et al., 2006) and, at the same time, contacts 8-17–bp repeat sequences (iterons) in the oriV(RK2), thereby opening the origin of replication. Mutants of RK2, displaying temperature-sensitivity in E. coli, seem to be stably maintained at elevated temperatures in P. putida KT2440. We verified this occurrence by culturing both E. coli DH5α and P. putida KT2440 bearing plasmid pFREE-RK2 (Lauritsen et al., 2017) in LB medium at elevated temperatures overnight and checking for plasmid loss through antibiotic sensitivity. The plasmid displayed temperature-sensitivity in E. coli but replicated in P. putida at 37 °C, yielding a virtually homogenous plasmid-containing bacterial population. This phenomenon could be due to the slow growth of P. putida at 37 °C, but also to high trfA expression levels (Pinkney et al., 1987) and/or enhanced TrfA stability, known to be affected by interactions with host components (Valla et al., 1991).
Dismissing also temperature-sensitive replicons as a strategy for quick curing of plasmids in P. putida, we focused on engineering a conditionally-replicating vector based on regulated and orthogonal control of the expression of key components of the replication machinery. Karunakaran et al. (1999) constructed plasmid pJBSD1 to generate genomic integrations in Gram-negative bacteria (Table 1), since this vector only replicates in E. coli if a chemical inducer is present in the medium. Taking inspiration on this approach, we firstly confirmed the conditional replication of this vector in P. putida KT2440 by spreading transformants onto LB medium plates with 500 μg mL−1 Amp, with or without 3-mBz as the inducer to trigger the (essential) XylS/Pm→trfA module expression. Colonies were only obtained on plates added with the chemical inducer, hinting that synthetic control of plasmid replication can be used as a general strategy to manipulate segregational stability of plasmids in P. putida. This goal was pursued as explained in the next section.
3.4. Target- and self-curing of plasmids in P. putida
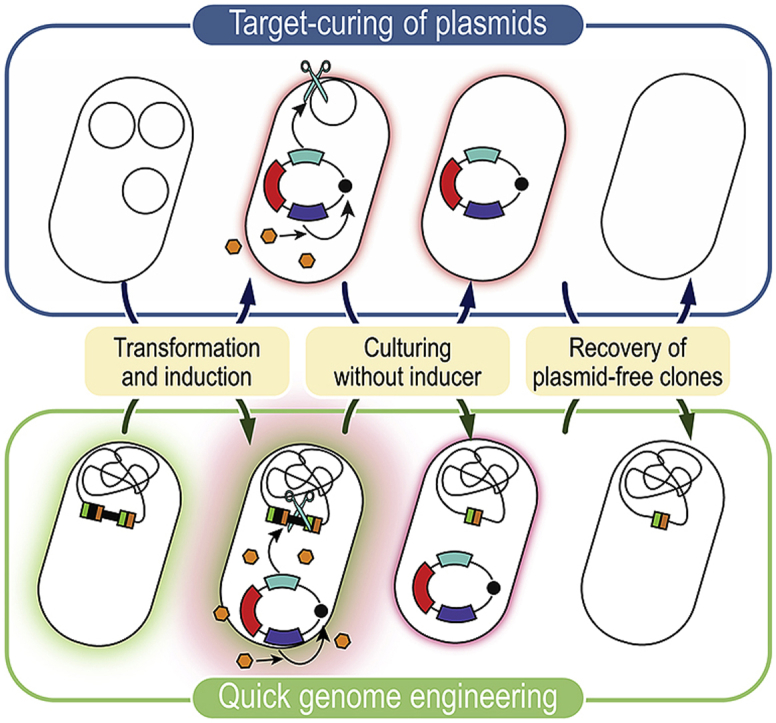
We proceeded to construct a new set of vectors, termed pQURE, in which plasmid replication is rendered fully dependent on the addition of 3-mBz to the culture medium. Furthermore, we took advantage of red fluorescent markers (Fig. 2) for plasmid tracing. Plasmid pQURE1·H (AmpR) carries the conditional replication machinery (i.e. XylS/Pm→trfA), an inducible XylS/Pm→I-SceI module, and a constitutively expressed P14g(BCD2)→mRFP fluorescence marker. pQURE vectors are designed for both target- and self-curing of plasmids by means of the I-SceI meganuclease in a 3-mBz–dependent fashion (Fig. 3A). Curing of plasmid pS6313·GFPs from P. putida was tested as a proof of concept. To this end, P. putida KT2440 was co-transformed with plasmids pS6313·GFPs and pQURE1·H. The first step (target-curing) involved growth of the cells in LB medium containing 2 mM 3-mBz for 24 h, followed by plating onto LB medium and inspection of the colonies under blue light for msfGFP fluorescence. Under these conditions, 89 ± 6% of the cells had lost plasmid pS6313·GFPs (Fig. 3B). In a consecutive step (self-curing), vector pQURE1·H was eliminated by omitting the inducer and antibiotics in the culture medium. By inoculating an msfGFP– colony into fresh LB medium and incubating the culture for another 24 h, 92 ± 7% of all cells were cured, with only ~8% of the bacterial population retaining vector pQURE1·H (Fig. 3 B). These traits, visually inspected by the presence of fluorescent markers in individual P. putida colonies, were confirmed by antibiotic sensitivity in selected clones, yielding similar figures of plasmid-free cells. Taken together, these results expose the high efficiency of the target- and self-curing process. Following the same line of reasoning, we created a whole set of pQURE vectors by combining different determinants of antibiotic resistance and red fluorescent modules (Table 1).
Fig. 3.

Target- and self-curing of plasmids by in vivo meganuclease digestion with pQURE vectors. (A) Synthetic control of plasmid replication for target- and self-curing. In pQURE vectors, the gene encoding TrfA (which binds to the vegetative origin of replication, oriV) is under transcriptional control of the XylS/Pm expression system. pQURE vectors can only replicate in the presence of 3-methylbenzoate (3-mBz), effector of the XylS transcriptional regulator. The Pm promoter also drives the expression of the gene encoding the I-SceI meganuclease. When I-SceI is expressed, the meganuclease introduces double-strand breaks in any DNA molecule containing its recognition site. In the example, a target plasmid carrying an engineered I-SceI restriction site is recognized by the meganuclease and subjected to in vivo digestion—resulting in selective plasmid loss. (B)P. putida KT2440 harboring plasmid pS6313·GFPs was co-transformed with vector pQURE1·H (XylS/Pm→trfA, AmpR). During the first curing step (indicated as 1), cell were grown in LB medium in the presence of 2 mM 3-mBz for 18 h and aliquots were plated onto solid LB medium. A single colony displaying an msfGFP– phenotype (indicative of loss of plasmid pS6313·GFPs) was inoculated into fresh LB medium without any additives, and grown for 18 h (shown as 2). After plating aliquots of this suspension, the percentage of plasmid-free cells as well as the fraction of P. putida cells carrying plasmids pS6313·GFPs and/or pQURE1·H was calculated as indicated in Fig. 1. (C) Segregational stability of vector pQURE6·H (XylS/Pm→trfA, GmR) in P. putida KT2440 over 7–8 generations in LB medium cultures added with varying concentrations (concn.) of 3-mBz in the μM range. The specific (Sp) red fluorescence of these cultures was calculated as indicated in Fig. 1. Bars represent the mean value of either the percentage of plasmid-containing cells or the Sp red fluorescence in each culture ± standard deviation of quadruplicate measurements from at least three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Segregational stability of plasmid pQURE1·H, the replication of which is subjected to the control of the XylS/Pm expression system, was studied in the presence of different 3-mBz concentrations. This regulatory system is known to be titratable, and expression levels of genes under control of the Pm promoter are also influenced by temperature (Ramos et al., 1988). We assumed that pQURE plasmids have to be duplicated at least as quickly as the cells divide in order to be stably retained. Therefore, we explored inducer concentrations in the μM range to determine the minimum amount of 3-mBz that warrants stable plasmid maintenance. Plasmid pQURE1·H was retained in P. putida KT2440 already in the presence of 8 μM 3-mBz (Fig. 3C). Higher inducer concentrations did not lead to higher fluorescence (Fig. 3C). Therefore, we assume that (i) the replication machinery is rapidly saturated (del Solar et al., 1998) and (ii) the copy number of the plasmid is not titratable, but follows an ON/OFF behaviour consistent with the replication mechanism of RK2 (Durland and Helinski, 1990). According to this model, replication of RK2 plasmids is strictly dependent on the presence of TrfA, but the machinery is already saturated at low protein concentrations and a further increase of TrfA does not lead to a higher plasmid copy number. Again, the leaky nature of the XylS/Pm expression system allowed for some replication of vector pQURE1·H even in the absence of 3-mBz, although addition of the inducer even at very low concentrations resulted in a ~2-fold increase in the red fluorescence output (Fig. 3C). This expanded plasmid toolbox (Table 2) served as the basis for practical applications in genome engineering of P. putida as indicated in the next section.
3.5. Quick genome engineering of pseudomonas with vectors pSNW and pQURE
The most widespread technique for genome engineering of Pseudomonas encompasses homologous recombination assisted by the activity of the I-SceI meganuclease (Martínez-García and de Lorenzo, 2011, 2012; Wirth et al., 2020). This method relies on plasmid-based expression of I-SceI for inflicting DSBs in the target chromosome and thus forcing DNA recombination (Fig. 4A). For most applications, the meganuclease-bearing plasmid has to be cured from mutant clones before further steps can be carried out. Failure to do so results in reduced efficiency of subsequent genome editing steps, as the unavoidable basal expression of I-SceI will force recombination while selection pressure for maintaining the resistance marker is applied. In turn, this situation usually leads to the emergence of unwanted mutants (e.g. co-integrants that cannot be resolved, when the I-SceI recognition site acquires mutations that prevent recognition by the meganuclease). The loss-by-dilution protocol routinely used for plasmid curing takes around 4–5 days with >3 passages in fresh, non-selective medium per day, followed by sensitivity screening of several tens of clones. For mutant strains displaying reduced growth (e.g. after knocking-out key metabolic genes), plasmid curing can take much longer or even being infeasible. Motivated by the ease of target- and self-curing plasmid efficacy of the conditional-replication system, we tested these vectors for fast genome engineering of P. putida. To this end, the functional elements needed for synthetic control of plasmid replication were combined together with the red fluorescent markers of pQURE vectors as indicated in Fig. 4A.
Fig. 4.

Genome engineering of Pseudomonas with self-curing vectors and genetic upgrading of suicide plasmids with highly-fluorescent markers. (A) Genome engineering in Pseudomonas. A suicide plasmid [e.g. from the pGNW or pSNW series, containing the Π-dependent ori(R6K), Table 1] is integrated into the genome of P. putida through homologous recombination. To this end, homology regions (HR) flanking the gene of interest (GOI) are assembled into the plasmid. An msfGFP+ clone, harboring a copy of the suicide plasmid co-integrated into the chromosome, is selected and transformed with the pQURE vector of choice (Table 1) and grown in the presence of 3-methylbenzoate to ensure vector replication and meganuclease activity. I-SceI introduces double-strand breaks in the chromosome and thereby enforces a second homologous recombination event. Resolution of the co-integration leads to either the wild-type or the mutant genotype; msfGP– colonies are screened for the desired alteration and cured from the pQURE vector by growing the cells in the absence of 3-methylbenzoate. (B) Genetic upgrading of suicide vector pGNW into pSNW by addition of a bicistronic design in front of msfGFP. Bacterial growth (C) and specific (Sp) green fluorescence levels (D) in P. putida KT2440 carrying either pGNW2·ΔbenABCD or pSNW2·ΔbenABCD (which only differ in the genetic architecture of the fluorescent marker added to the backbone) integrated as a single copy into the target chromosomal locus. Cells were grown in LB medium, and the Sp green fluorescence and the optical density measured at 600 nm (OD600) was calculated as indicated in Fig. 1. Data represent the mean value of each parameter ± standard deviation of triplicate measurements from at least five independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We essentially followed the genome engineering strategy by Wirth et al. (2020) for design and co-integration of suicide plasmids into the P. putida chromosome. However, we observed a high locus-dependence of the fluorescence intensity brought about by integration of the reporter msfGFP gene borne by pGNW vectors (Fig. S1 in the Supplementary Material). Depending on the genome region, in some cases we could not detect any fluorescence even when the suicide vector had landed in the correct locus. This phenomenon is not surprising, given the highly variable nature of transcriptional activity across the bacterial chromosome (Martínez-García et al., 2014a). To address this situation, we upgraded the reporter module of vector pGNW to obtain a reliable fluorescent marker even in low expression loci. The msfGFP gene was added with the strong constitutive promoter P14g, and the archetypal ribosome binding site upstream of the coding sequence was replaced by the bicistronic design BCD2 (Mutalik et al., 2013; Zobel et al., 2015) as a translational coupler (Fig. 4B). A new set of suicide plasmids for genome engineering was thus created, comprising vectors pSNW2 (KmR), pSNW4 (StrR), and pSNW6 (GmR) (Table 1).
We tested the efficiency of these new vectors for genome engineering by targeting the ben gene cluster of P. putida KT2440. Integration of a pGNW2-based suicide vector designed to delete the ben genes (i.e. plasmid pGNW·ΔbenABCD, Table 1) into this chromosomal locus (PP_3161-PP_3164) resulted in poorly fluorescent colonies, difficult to spot even when the plates were examined under blue light—given that the ben cluster displays very low expression levels in the absence of the cognate aromatic substrates (Kim et al., 2013). The pSNW2-counterpart of this suicide vector (i.e. plasmid pSNW·ΔbenABCD, Table 1) was likewise constructed and co-integrated in the chromosome. In this case, successful co-integration events were easily spotted on LB medium plates containing Km by the highly fluorescent phenotype of individual colonies. A single colony was isolated from each set of co-integrants, and bacterial growth and msfGFP fluorescence were determined in LB medium cultures over 24 h. While the growth of either P. putida co-integrant was not affected by the swapping of the fluorescence marker in the suicide plasmid (Fig. 4C), the msfGFP fluorescence intensity of the co-integrant carrying pSNW·ΔbenABCD increased by >3-fold (Fig. 4D). Considering these results, we adopted the pSNW series of plasmids for genome engineering of P. putida, and carried out deletions by means of the procedure described in the next section.
3.6. Elimination of the ben catabolic activities of P. putida KT2440
P. putida mt-2 carries the catabolic pWW0 plasmid, which endows cells with the ability of processing toluene, xylenes, and ethylbenzene via the TOL degradation pathway (de Lorenzo and Joshi, 2019). This biochemical route is encoded in two gene clusters, i.e. the upper and the lower or meta pathway for degradation of aromatic compounds (Ramos et al., 1997). These two biochemical modules convert the hydrocarbon substrate into the corresponding carboxylic acid, and the resulting benzoate(s) are further metabolized to pyruvate and acetaldehyde (Domínguez-Cuevas and Marqués, 2017). Strain mt-2 also carries a chromosomally-encoded ortho-cleavage pathway for benzoate(s) degradation (Jiménez et al., 2002). When m-xylene is processed by the upper pathway, the resulting product is 3-mBz, which can follow alternative metabolic itineraries. The products of either the plasmid-borne xyl genes of the lower TOL operon or the chromosomal ben genes mediate the transformation of 3-mBz into 3-methylcatechol (Fig. 5A). P. putida KT2440 is a derivative of mt-2 that has been cured of the catabolic pWW0 plasmid (Bagdasarian et al., 1981; Worsey and Williams, 1975), and therefore processes 3-mBz only through the ortho-cleavage pathway. From a practical point of view, spontaneous oxidation and polymerization of 3-methylcatechol gives rise to brown-coloured aggregates (Jiménez et al., 2014) that interfere with colorimetric and fluorimetry determinations (e.g. bacterial growth as assessed by optical density measurements). Additionally, 3-mBz is employed as a chemical inducer of the XylS/Pm expression system—thus hampering the selection of fluorescent colonies in 3-mBz–containing media. As such, we set to eliminate the ben gene cluster of P. putida KT2440 (Fig. 5B) using the quick genome engineering procedure assisted by self-curing vectors. To this end, the strain carrying plasmid pSNW·ΔbenABCD as a co-integration in the chromosome (Fig. 4) was subjected to the procedure outlined in Fig. 5C. Self-curing vector pQURE6·H (Table 1) was used as source of the I-SceI meganuclease. Two successive cultivation rounds (firstly, selecting for msfGFP– and mRFP+ colonies; secondly, picking clones that have lost all fluorescent markers) sufficed to isolate P. putida ΔbenABCD knock-outs in <2.5 days. The Ben– phenotype was clearly evidenced by the absence of any pigmentation in colonies grown in LB medium added with 3-mBz. Prompted by these encouraging results, we expanded the applications of fast genome engineering approaches for genome reduction of P. putida.
Fig. 5.

Elimination of the Ben catabolic activities of P. putida through quick genome engineering using self-curing pQURE vectors. (A) The Ben activities mediate the conversion of 3-methylbenzoate (3-mBz) into 3-methylcatechol, which undergoes spontaneous oxidation and polymerization into brown-coloured aggregates. (B) Genomic structure of the ben locus of P. putida KT2440. The individual genes within the cluster encode BenA, subunit α of benzoate 1,2-dioxygenase; BenB, subunit β of benzoate 1,2-dioxygenase; BenC, electron transfer component of benzoate 1,2-dioxygenase; and BenD, 1,6-dihydroxycyclohexa-2,4-diene-1-carboxylate dehydrogenase. The Δ symbol identifies the genomic region targeted for deletion. (C) Overview of the strategy to delete benABCD in P. putida KT2440 through quick genome engineering. Co-integration of the suicide plasmid pSNW·ΔbenABCD (Table 1) into the chromosome can be easily screened for by selecting clones that display a KmR and msfGFP+ phenotype. After confirming the genotype of the co-integrants by colony PCR, a clone was further transformed with vector pQURE6·H (XylS/Pm→trfA, GmR). This strain was grown for 24 h in the presence of 2 mM 3-mBz, and plated onto solid LB medium to recover msfGFP– and mRFP+ colonies. The very few colonies still displaying msfGFP fluorescence (indicated with black arrows) were discarded. Deletion of benABCD results in a Ben– phenotype, characterized by the absence of brown pigmentation of colonies in the presence of 3-mBz. To assess this phenotype, colonies were plated onto solid LB medium containing 2 mM 3-mBz and incubated at 30 °C for 24 h. The Ben+ (black arrows) or Ben– (white arrows) phenotypes were clearly spotted after storing the plates at 4 °C. The final step of the procedure is self-curing of vector pQURE6·H, accomplished by streaking a Ben– colony onto a non-selective medium plate (i.e. neither 3-mBz nor any antibiotic are added). After a 24-h incubation at 30 °C, msfGFP– and mRFP– colonies were easily spotted (white arrows) and distinguished from mRFP+ clones (still retaining vector pQURE6·H, black arrow) even with the naked eye. Relevant genotypes were confirmed by PCR amplification of the corresponding genomic regions with specific oligonucleotides and DNA sequencing. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.7. Genome reduction of P. putida strain EM42 towards a reference chassis
Capitalizing on the intrinsic physiological and metabolic strength of P. putida KT2440, Martínez-García et al. (2014b) sequentially deleted eleven chromosomal regions (comprising 300 genes) to create the reduced-genome strain EM42. This strain displays enhanced physiological properties (e.g. fast growth and increased availability of redox and energy cofactors) that are advantageous for metabolic engineering applications (Aparicio et al., 2019a; Lieder et al., 2015). By following a similar line of thought, we further streamlined strain EM42 towards a P. putida reference chassis through fast genome engineering. In particular, we wanted to delete key elements in the chromosome encoding functions that may (i) confer resistance to β-lactam antibiotics, (ii) interfere with the use of fluorescent protein markers, and (iii) hamper the use of 3-mBz as an inducer of the XylS/Pm expression system. Inspection of the genome of strain EM42 identified ten genes as obvious targets to fulfil this purpose: eight β-lactamase and β-lactamase–like genes, the siderophore gene pvdD, and the benABCD gene cluster (Table 3).
Table 3.
Targets selected for genome engineering of P. putida.
| Gene(s) number | Gene(s) name | Genomic region [bp (strand)] | Length (bp) | Annotated functiona |
|---|---|---|---|---|
| PP_1239 | 1,416,753–1,417,511 (+) | 759 | MBLFAb | |
| PP_3291 | 3,725,348–3,726,715 (+) | 1,368 | MBLFA | |
| PP_1952 | 2,208,685–2,209,632 (+) | 948 | MBLFA | |
| PP_0772 | 890,074–890,718 (+) | 645 | MBLFA | |
| PP_2876 | ampC | 3,276,978–3,278,120 (—) | 1,143 | β-lactamase |
| PP_1775 | 1,982,049–1,983,482 (+) | 1,434 | MBLFA | |
| PP_0052 | 60,831–61,715 (+) | 885 | β-lactamase domain-containing protein | |
| PP_2045 | 2,325,342–2,327,303 (—) | 1,962 | MBLFA | |
| PP_3161-PP_3164 | benABCD | 3,581,930–3,585,749 (+) | 3,819 | Benzoate catabolism gene clusterc |
| PP_4219 | pvdD | 4,768,854–4,779,266 (—) | 10,412 | Non-ribosomal peptide synthetase |
Functional annotations and genome coordinates are given according to the Pseudomonas Database (Winsor et al., 2016) and Belda et al. (2016).
MBFA, metallo-β-lactamase family protein.
The enzymes encoded in the ben gene cluster are BenA, subunit α of benzoate 1,2-dioxygenase; BenB, subunit β of benzoate 1,2-dioxygenase; BenC, electron transfer component of benzoate 1,2-dioxygenase; and BenD, 1,6-dihydroxycyclohexa-2,4-diene-1-carboxylate dehydrogenase.
P. putida exhibits a naturally high resistance to Amp, a hindrance for the use of β-lactams as a selective pressure. Both efflux pumps and β-lactamases account for the resistance of P. putida to β-lactam antibiotics. Deletion of the efflux pumps of strain KT2440 increased sensitivity towards these antibiotics, but it also impacted solvent tolerance (Martínez-García and de Lorenzo, 2011). Therefore, we decided to delete the eight genes annotated to encode β-lactamases or metallo-β-lactamase family proteins to reduce Amp resistance without interfering with solvent tolerance. Furthermore, we deleted pvdD, encoding a large non-ribosomal peptide synthetase involved in the formation of siderophores (Matilla et al., 2007). Siderophores are high-affinity iron-chelating molecules, critical for metal capture in environmental niches colonized by Pseudomonas (Cornelis, 2010). The energy- and resource-demanding biosynthesis of these secondary metabolites is not only unnecessary when sufficient iron is supplied (e.g. in laboratory setups), but their presence also interferes with fluorescence measurements. Finally, the Ben catabolic activities were targeted for the reasons explained in the preceding section. The genes encoding these features are spread over the bacterial chromosome (Fig. 6A). Again, 500-bp long homologous regions flanking the target were amplified with the corresponding set of primers for each knock-out (Table S1) and inserted into the suicide plasmid pSNW2 according to the procedure of Wirth et al. (2020) indicated in Fig. 4A. After plasmid co-integration, the resulting strains were transformed with vector pQURE6·H and cells were recovered for 2 h in LB medium with 2 mM 3-mBz. For all targets, the efficiency of co-integration resolving was >90% and, in several cases, close to 100%. Elimination of all the genes listed in Table 3 led to a ~23-kb reduction of the genome of strain EM42, giving rise to streamlined P. putida strain SEM10 (i.e. lacking 310 genes as compared to wild-type strain KT2440). To test whether off-target mutations were introduced due to the multi-step genome engineering programme, we carried out a suite of colony PCR amplifications in the parental strain (EM42) and its SEM10 derivative. All the amplicons had the expected size, and sequencing of these DNA fragments confirmed the absence of any unintended mutation (Fig. S2 in the Supplementary Material).
Fig. 6.

Streamlining reduced-genome P. putida strain EM42 by quick genome engineering using self-curing pQURE vectors. (A) Operons and genomic regions deleted in P. putida EM42. The position and the relative orientation of the eleven gene(s)/genome regions eliminated is indicated in the physical map of the chromosome (Table 3). Genes encoding β-lactamases or metallo-β-lactamase family proteins are indicated in red. (B) Ampicillin sensitivity of P. putida EM42 and its reduced-genome derivative P. putida SEM10. Ten-fold dilutions of the cultures were spotted onto LB medium plates containing ampicillin at the concentrations (concn.) indicated, and plates were photographed after incubation at 30 °C for 24 h. (C) Comparison of the growth of both strains in LB medium (left panel) and M9 minimal medium containing 0.2% (w/v) citrate (right panel). The optical density measured at 600 nm (OD600) of these cultures was recorded over 24 h, and data represent the mean value of the OD600 readings ± standard deviation of triplicate measurements from at least three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
P. putida SEM10 was then subjected to phenotypic characterization by examining growth profiles in different culture conditions and antibiotic resistance. Expectedly, the reduced-genome strain displayed increased susceptibility towards Amp (Fig. 6B). Importantly, no colonies of P. putida SEM10 were observed when dilutions of the cell suspension were spotted in LB medium plates containing 75 μg mL−1 Amp—allowing for the use of AmpR as a selection marker at antibiotic concentrations similar to those employed for E. coli. The deletion of benABCD suppressed the development of pigments derived from 3-methylcatechol in cultures of P. putida SEM10 grown in the presence of 3-mBz. Furthermore, autofluorescence of this genome-reduced strain was highly reduced due to the deletion of pvdD. Finally, no changes were observed in the growth profile of this strain in rich LB medium or minimal M9 medium containing 0.2% (w/v) citrate as compared to P. putida EM42 (Fig. 6C), indicating that the deletions introduced in this study are largely irrelevant for fitness under laboratory conditions.
4. Conclusion
In this work, we have benchmarked a toolbox for target- and self-curing of plasmids in Pseudomonas. Although the literature offers numerous examples of circuits and strategies developed to ensure plasmid maintenance in cell factories (Kroll et al., 2009; Nikel et al., 2010; Silva et al., 2012), less attention has been paid to the equally important programmable loss of plasmids. After assessing vector curing methods commonly used in other bacterial species (Hale et al., 2010; Hove-Jensen, 2008; Kamruzzaman et al., 2017), we failed to identify a straightforward procedure that can be applied to P. putida.
Recently, Lauritsen et al. (2017) developed an elegant approach to plasmid curing based on CRISPR/Cas9, reaching a vector loss efficiency of ~40–90% in E. coli within 24 h. This system relies on the Cas9 nuclease, which can cause inadvertent off-target mutations in the genome (Pattanayak et al., 2013) and is known to be a toxic protein when the cognate gene is expressed heterologously (Zhang and Voigt, 2018). On the other hand, the efficiency and editing-accuracy of CRISPR/Cas9-based systems varies even between closely related bacterial species (Vento et al., 2019). Most importantly, this system and other genome engineering tools (Choi and Lee, 2020; Sun et al., 2018), employ a temperature-sensitive oriV(RK2), which, in our hands, does not exhibit a consistent temperature-dependent replication behaviour in P. putida KT2440. The procedure developed in this study, based on in vivo digestion of vectors by the I-SceI meganuclease (i.e. target-curing) and synthetic control of plasmid replication (i.e. self-curing), is fast, plasmid-specific, and can be applied in virtually any microorganism where the use of temperature-sensitive replicons is not possible. Importantly, the plasmid-curing step during genome engineering, typically consuming several days during the routine loss-by-dilution protocol, has been brought down to a mere overnight cultivation in a simple culture medium (omitting 3-mBz in its formulation). Owing to the presence of red fluorescent markers in pQURE vectors (the output level of which can be selected according to the application), laborious sensitivity screenings are fully circumvented. Furthermore, for applications were achieving a vector loss efficiency of 100% is crucial, plasmid-free bacteria can be automatically isolated by fluorescence-activated cell sorting. Note that the procedure presented herein can be (i) scaled-up as needed, whereas temperature shifts for plasmid curing are meaningful only at the laboratory scale, and (ii) used in other Gram-negative bacteria besides Pseudomonas.
YEJPN_press_logoThe adoption of the quick genome engineering, assisted by self-curing pQURE vectors, enabled the construction of a reduced-genome, streamlined variant of P. putida KT2440, termed strain SEM10. The genome of P. putida SEM10 has been shortened by 4.76% as compared to wild-type strain KT2440, and the phenotypes of this platform bacterium (e.g. absence of siderophore-related autofluorescence and sensitivity to β-lactams) can be exploited for several applications both in the laboratory and in industrial setups. Importantly, the elimination of all β-lactamase–encoding genes [typically associated with horizontal gene transfer of antibiotic resistance and pathogenicity (San Millán et al., 2018)] enhances the biosafety of P. putida for metabolic engineering applications. Further genome reduction of strain SEM10 towards a reference chassis is currently underway in our laboratory—an overarching objective calling for quick genome engineering approaches that shorten the turnaround typically needed when manipulating non-conventional microorganisms.
CRediT authorship contribution statement
Daniel C. Volke: Conceptualization, Methodology, Investigation, Validation, Formal analysis, Data curation, Writing - original draft. Laura Friis: Methodology, Investigation, Formal analysis, Data curation. Nicolas T. Wirth: Methodology, Investigation, Formal analysis. Justine Turlin: Investigation, Data curation. Pablo I. Nikel: Project administration, Supervision, Conceptualization, Formal analysis, Writing - review & editing, Funding acquisition.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgements
We thank Rahmi Lale (NTNU, Trondheim, Norway) for supplying vector pJBSD1 and for helpful discussions. Fruitful discussions with Esteban Martínez-García and Víctor de Lorenzo (CNB–CSIC, Madrid, Spain) are likewise acknowledged. We also thank Jan Martinussen (DTU Bioengineering, Denmark) for his support and guidance of L.F. Financial support from The Novo Nordisk Foundation (NNF10CC1016517 and NNF18CC0033664), the Danish Council for Independent Research (SWEET, DFF-Research Project 8021-00039B), and the European Union’s Horizon2020 Research and Innovation Program under grant agreement No. 814418 (SinFonia) to P.I.N. is gratefully recognized. J.T. is the recipient of a fellowship from The Novo Nordisk Foundation as part of the Copenhagen Bioscience Ph.D. Programme, supported through grant NNF17CC0026768. The responsibility of this article lies with the authors; the NNF and the European Union are not responsible for any use that may be made of the information contained therein.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mec.2020.e00126.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Abram K.Z., Udaondo Z. Towards a better metabolic engineering reference: the microbial chassis. Microb. Biotechnol. 2020;13:17–18. doi: 10.1111/1751-7915.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alieva N.O., Konzen K.A., Field S.F., Meleshkevitch E.A., Hunt M.E., Beltrán-Ramírez V., Miller D.J., Wiedenmann J., Salih A., Matz M.V. Diversity and evolution of coral fluorescent proteins. PLoS One. 2008;3 doi: 10.1371/journal.pone.0002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T., de Lorenzo V., Martínez-García E. Broadening the SEVA plasmid repertoire to facilitate genomic editing of Gram-negative bacteria. In: McGenity T., Timmis K.N., Nogales B., editors. Hydrocarbon and Lipid Microbiology Protocols. Springer; Berlin, Germany: 2015. pp. 9–27. [Google Scholar]
- Aparicio T., Jensen S.I., Nielsen A.T., de Lorenzo V., Martínez-García E. The Ssr protein (T1E_1405) from Pseudomonas putida DOT-T1E enables oligonucleotide-based recombineering in platform strain P. putida EM42. Biotechnol. J. 2016;11:1309–1319. doi: 10.1002/biot.201600317. [DOI] [PubMed] [Google Scholar]
- Aparicio T., de Lorenzo V., Martínez-García E. Improved thermotolerance of genome-reduced Pseudomonas putida EM42 enables effective functioning of the PL/cI857 system. Biotechnol. J. 2019;14 doi: 10.1002/biot.201800483. e1800483. [DOI] [PubMed] [Google Scholar]
- Aparicio T., de Lorenzo V., Martínez-García E. CRISPR/Cas9-enhanced ssDNA recombineering for Pseudomonas putida. Microb. Biotechnol. 2019;12:1076–1089. doi: 10.1111/1751-7915.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T., de Lorenzo V., Martínez-García E. A broad host range plasmid-based roadmap for ssDNA-based recombineering in Gram-negative bacteria. Methods Mol. Biol. 2020;2075:383–398. doi: 10.1007/978-1-4939-9877-7_27. [DOI] [PubMed] [Google Scholar]
- Bagdasarian M., Lurz R., Rückert B., Franklin F.C.H., Bagdasarian M.M., Frey J., Timmis K.N. Specific purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene. 1981;16:237–247. doi: 10.1016/0378-1119(81)90080-9. [DOI] [PubMed] [Google Scholar]
- Barth P.T., Tobin L., Sharpe G.S. Development of broad host-range plasmid vectors. In: Levy S.B., Clowes R.C., Koenig E.L., editors. Molecular Biology, Pathogenicity, and Ecology of Bacterial Plasmids. Springer; Boston, MA, USA: 1981. pp. 439–448. [Google Scholar]
- Batianis C., Kozaeva E., Damalas S., Martín-Pascual M., Volke D.C., Nikel P.I., Martins dos Santos V.A.P. An expanded CRISPRi toolbox for tunable control of gene expression in Pseudomonas putida. Microb. Biotechnol. 2020;13:368–385. doi: 10.1111/1751-7915.13533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda E., van Heck R.G.A., López-Sánchez M.J., Cruveiller S., Barbe V., Fraser C., Klenk H.P., Petersen J., Morgat A., Nikel P.I., Vallenet D., Rouy Z., Sekowska A., Martins dos Santos V.A.P., de Lorenzo V., Danchin A., Médigue C. The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ. Microbiol. 2016;18:3403–3424. doi: 10.1111/1462-2920.13230. [DOI] [PubMed] [Google Scholar]
- Bennett C.B., Lewis A.L., Baldwin K.K., Resnick M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5613–5617. doi: 10.1073/pnas.90.12.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner M.M.C., Ciusa M.L., Piddock L.J.V. Strategies to combat antimicrobial resistance: anti-plasmid and plasmid curing. FEMS Microbiol. Rev. 2018;42:781–804. doi: 10.1093/femsre/fuy031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero P., Nikel P.I. Chasing bacterial chassis for metabolic engineering: a perspective review from classical to non-traditional microorganisms. Microb. Biotechnol. 2019;12:98–124. doi: 10.1111/1751-7915.13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell R.E., Tour O., Palmer A.E., Steinbach P.A., Baird G.S., Zacharias D.A., Tsien R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaleiro A.M., Kim S.H., Seppälä S., Nielsen M.T., Nørholm M.H. Accurate DNA assembly and genome engineering with optimized uracil excision cloning. ACS Synth. Biol. 2015;4:1042–1046. doi: 10.1021/acssynbio.5b00113. [DOI] [PubMed] [Google Scholar]
- Chen S., Larsson M., Robinson R.C., Chen S.L. Direct and convenient measurement of plasmid stability in lab and clinical isolates of E. coli. Sci. Rep. 2017;7:1–11. doi: 10.1038/s41598-017-05219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.H., Kumar A., Schweizer H.P. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods. 2006;64:391–397. doi: 10.1016/j.mimet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Choi K.R., Lee S.Y. Protocols for RecET-based markerless gene knockout and integration to express heterologous biosynthetic gene clusters in Pseudomonas putida. Microb. Biotechnol. 2020;13:199–209. doi: 10.1111/1751-7915.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Zhang F. Genome engineering using CRISPR-Cas9 system. Methods Mol. Biol. 2015;1239:197–217. doi: 10.1007/978-1-4939-1862-1_10. [DOI] [PubMed] [Google Scholar]
- Cornelis P. Iron uptake and metabolism in pseudomonads. Appl. Microbiol. Biotechnol. 2010;86:1637–1645. doi: 10.1007/s00253-010-2550-2. [DOI] [PubMed] [Google Scholar]
- Csörgő B., Nyerges A., Posfai G., Fehér T. System-level genome editing in microbes. Curr. Opin. Microbiol. 2016;33:113–122. doi: 10.1016/j.mib.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorenzo V., Joshi H. Genomic responses of Pseudomonas putida to aromatic hydrocarbons. In: Steffan R., editor. Handbook of Hydrocarbon and Lipid Microbiology (Consequences of Microbial Interactions with Hydrocarbons, Oils, and Lipids: Biodegradation and Bioremediation) Springer; Cham, Germany: 2019. pp. 1–15. [Google Scholar]
- del Solar G., Giraldo R., Ruiz-Echevarría M.J., Espinosa M., Díaz-Orejas R. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998;62:434–464. doi: 10.1128/mmbr.62.2.434-464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez-Cuevas P., Marqués S. Current view of the mechanisms controlling the transcription of the TOL plasmid aromatic degradation pathways. In: Rojo F., editor. Aerobic Utilization of Hydrocarbons, Oils and Lipids. Springer; Cham, Germany: 2017. pp. 1–22. [Google Scholar]
- Durland R.H., Helinski D.R. Replication of the broad-host-range plasmid RK2: direct measurement of intracellular concentrations of the essential TrfA replication proteins and their effect on plasmid copy number. J. Bacteriol. 1990;172:3849–3858. doi: 10.1128/jb.172.7.3849-3858.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Cabezón L., Cros A., Nikel P.I. Evolutionary approaches for engineering industrially-relevant phenotypes in bacterial cell factories. Biotechnol. J. 2019;14:1800439. doi: 10.1002/biot.201800439. [DOI] [PubMed] [Google Scholar]
- Freed E., Fenster J., Smolinski S.L., Walker J., Henard C.A., Gill R., Eckert C.A. Building a genome engineering toolbox in nonmodel prokaryotic microbes. Biotechnol. Bioeng. 2018;115:2120–2138. doi: 10.1002/bit.26727. [DOI] [PubMed] [Google Scholar]
- Gallagher D.N., Haber J.E. Repair of a site-specific DNA cleavage: old-school lessons for Cas9-mediated gene editing. ACS Chem. Biol. 2018;13:397–405. doi: 10.1021/acschembio.7b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawin A., Valla S., Brautaset T. The XylS/Pm regulator/promoter system and its use in fundamental studies of bacterial gene expression, recombinant protein production and metabolic engineering. Microb. Biotechnol. 2017;10:702–718. doi: 10.1111/1751-7915.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genee H.J., Bonde M.T., Bagger F.O., Jespersen J.B., Sommer M.O., Wernersson R., Olsen L.R. Software-supported USER cloning strategies for site-directed mutagenesis and DNA assembly. ACS Synth. Biol. 2015;4:342–349. doi: 10.1021/sb500194z. [DOI] [PubMed] [Google Scholar]
- Goñi-Moreno A., Benedetti I., Kim J., de Lorenzo V. Deconvolution of gene expression noise into spatial dynamics of transcription factor-promoter interplay. ACS Synth. Biol. 2017;6:1359–1369. doi: 10.1021/acssynbio.6b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann A., Wanner B.L. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J. Bacteriol. 2001;183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale L., Lazos O., Haines A., Thomas C. An efficient stress-free strategy to displace stable bacterial plasmids. Biotechniques. 2010;48:223–228. doi: 10.2144/000113366. [DOI] [PubMed] [Google Scholar]
- Hashimoto T., Sekiguchi M. Isolation of temperature-sensitive mutants of R plasmid by in vitro mutagenesis with hydroxylamine. J. Bacteriol. 1976;127:1561–1563. doi: 10.1128/jb.127.3.1561-1563.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto-Gotoh T., Ishii K. Temperature sensitive replication plasmids are passively distributed during cell division at non-permissive temperature: a new model for replicon duplication and partitioning. Mol. Gen. Genet. 1982;187:523–525. doi: 10.1007/BF00332639. [DOI] [PubMed] [Google Scholar]
- Heery D.M., Powell R., Gannon F., Dunican L.K. Curing of a plasmid from E. coli using high-voltage electroporation. Nucleic Acids Res. 1989;17:10131. doi: 10.1093/nar/17.23.10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Jensen B. Two-step method for curing Escherichia coli of ColE1-derived plasmids. J. Microbiol. Methods. 2008;72:208–213. doi: 10.1016/j.mimet.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Ito F., Tamiya T., Ohtsu I., Fujimura M., Fukumori F. Genetic and phenotypic characterization of the heat shock response in Pseudomonas putida. MicrobiologyOpen. 2014;3:922–936. doi: 10.1002/mbo3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki K., Uchiyama H., Yagi O., Kurabayashi T., Ishizuka K., Takamura Y. Transformation of Pseudomonas putida by electroporation. Biosci. Biotechnol. Biochem. 1994;58:851–854. doi: 10.1271/bbb.58.851. [DOI] [PubMed] [Google Scholar]
- Jacquier A., Dujon B. An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell. 1985;41:383–394. doi: 10.1016/s0092-8674(85)80011-8. [DOI] [PubMed] [Google Scholar]
- Jakočiūnas T., Jensen M.K., Keasling J.D. System-level perturbations of cell metabolism using CRISPR/Cas9. Curr. Opin. Biotechnol. 2017;46:134–140. doi: 10.1016/j.copbio.2017.03.014. [DOI] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Jawed K., Yazdani S.S., Koffas M.A.G. Advances in the development and application of microbial consortia for metabolic engineering. Metab. Eng. Commun. 2019;9 doi: 10.1016/j.mec.2019.e00095. e00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez J.I., Miñambres B., García J.L., Díaz E. Genomic analysis of the aromatic catabolic pathways from Pseudomonas putida KT2440. Environ. Microbiol. 2002;4:824–841. doi: 10.1046/j.1462-2920.2002.00370.x. [DOI] [PubMed] [Google Scholar]
- Jiménez J.I., Pérez-Pantoja D., Chavarría M., Díaz E., de Lorenzo V. A second chromosomal copy of the catA gene endows Pseudomonas putida mt-2 with an enzymatic safety valve for excess of catechol. Environ. Microbiol. 2014;16:1767–1778. doi: 10.1111/1462-2920.12361. [DOI] [PubMed] [Google Scholar]
- Johnson C.W., Salvachúa D., Khanna P., Smith H., Peterson D.J., Beckham G.T. Enhancing muconic acid production from glucose and lignin-derived aromatic compounds via increased protocatechuate decarboxylase activity. Metab. Eng. Commun. 2016;3:111–119. doi: 10.1016/j.meteno.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamruzzaman M., Shoma S., Thomas C.M., Partridge S.R., Iredell J.R. Plasmid interference for curing antibiotic resistance plasmids in vivo. PloS One. 2017;12 doi: 10.1371/journal.pone.0172913. e0172913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran P., Blatny J.M., Ertesvåg H., Valla S. Species-dependent phenotypes of replication-temperature-sensitive trfA mutants of plasmid RK2: a codon-neutral base substitution stimulates temperature sensitivity by leading to reduced levels of trfA expression. J. Bacteriol. 1998;180:3793–3798. doi: 10.1128/jb.180.15.3793-3798.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran P., Endresen D.T., Ertesvåg H., Blatny J.M., Valla S. A small derivative of the broad-host-range plasmid RK2 which can be switched from a replicating to a non-replicating state as a response to an externally added inducer. FEMS Microbiol. Lett. 1999;180:221–227. doi: 10.1111/j.1574-6968.1999.tb08799.x. [DOI] [PubMed] [Google Scholar]
- Kent R., Dixon N. Contemporary tools for regulating gene expression in bacteria. Trends Biotechnol. 2020;38:316–333. doi: 10.1016/j.tibtech.2019.09.007. [DOI] [PubMed] [Google Scholar]
- Kim J., Oliveros J.C., Nikel P.I., de Lorenzo V., Silva-Rocha R. Transcriptomic fingerprinting of Pseudomonas putida under alternative physiological regimes. Environ. Microbiol. Rep. 2013;5:883–891. doi: 10.1111/1758-2229.12090. [DOI] [PubMed] [Google Scholar]
- Kim J., Salvador M., Saunders E., González J., Avignone-Rossa C., Jiménez J.I. Properties of alternative microbial hosts used in synthetic biology: towards the design of a modular chassis. Essays Biochem. 2016;60:303–313. doi: 10.1042/EBC20160015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolatka K., Witosinska M., Pierechod M., Konieczny I. Bacterial partitioning proteins affect the subcellular location of broad-host-range plasmid RK2. Microbiology. 2008;154:2847–2856. doi: 10.1099/mic.0.2008/018762-0. [DOI] [PubMed] [Google Scholar]
- Kongsuwan K., Josh P., Picault M.J., Wijffels G., Dalrymple B. The plasmid RK2 replication initiator protein (TrfA) binds to the sliding clamp β subunit of DNA polymerase III: implication for the toxicity of a peptide derived from the amino-terminal portion of 33-kilodalton TrfA. J. Bacteriol. 2006;188:5501–5509. doi: 10.1128/JB.00231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll J., Steinle A., Reichelt R., Ewering C., Steinbüchel A. Establishment of a novel anabolism-based addiction system with an artificially introduced mevalonate pathway: complete stabilization of plasmids as universal application in white biotechnology. Metab. Eng. 2009;11:168–177. doi: 10.1016/j.ymben.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Lauritsen I., Porse A., Sommer M.O.A., Nørholm M.H.H. A versatile one-step CRISPR-Cas9 based approach to plasmid-curing. Microb. Cell Fact. 2017;16:135. doi: 10.1186/s12934-017-0748-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieder S., Nikel P.I., de Lorenzo V., Takors R. Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb. Cell Fact. 2015;14:23. doi: 10.1186/s12934-015-0207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeschcke A., Thies S. Pseudomonas putida–A versatile host for the production of natural products. Appl. Microbiol. Biotechnol. 2015;99:6197–6214. doi: 10.1007/s00253-015-6745-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E., de Lorenzo V. Engineering multiple genomic deletions in Gram-negative bacteria: analysis of the multi-resistant antibiotic profile of Pseudomonas putida KT2440. Environ. Microbiol. 2011;13:2702–2716. doi: 10.1111/j.1462-2920.2011.02538.x. [DOI] [PubMed] [Google Scholar]
- Martínez-García E., de Lorenzo V. Transposon-based and plasmid-based genetic tools for editing genomes of Gram-negative bacteria. Methods Mol. Cell Biol. 2012;813:267–283. doi: 10.1007/978-1-61779-412-4_16. [DOI] [PubMed] [Google Scholar]
- Martínez-García E., de Lorenzo V. Molecular tools and emerging strategies for deep genetic/genomic refactoring of Pseudomonas. Curr. Opin. Biotechnol. 2017;47:120–132. doi: 10.1016/j.copbio.2017.06.013. [DOI] [PubMed] [Google Scholar]
- Martínez-García E., Aparicio T., de Lorenzo V., Nikel P.I. New transposon tools tailored for metabolic engineering of Gram-negative microbial cell factories. Front. Bioeng. Biotechnol. 2014;2:46. doi: 10.3389/fbioe.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E., Nikel P.I., Aparicio T., de Lorenzo V. Pseudomonas 2.0: genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb. Cell Fact. 2014;13:159. doi: 10.1186/s12934-014-0159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E., Aparicio T., Goñi-Moreno A., Fraile S., de Lorenzo V. SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 2015;43:D1183–D1189. doi: 10.1093/nar/gku1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-García E., Aparicio T., de Lorenzo V., Nikel P.I. Engineering Gram-negative microbial cell factories using transposon vectors. Methods Mol. Biol. 2017;1498:273–293. doi: 10.1007/978-1-4939-6472-7_18. [DOI] [PubMed] [Google Scholar]
- Matilla M.A., Ramos J.L., Duque E., Alché J.D.D., Espinosa-Urgel M., Ramos-González M.I. Temperature and pyoverdine-mediated iron acquisition control surface motility of Pseudomonas putida. Environ. Microbiol. 2007;9:1842–1850. doi: 10.1111/j.1462-2920.2007.01286.x. [DOI] [PubMed] [Google Scholar]
- Meselson M., Yuan R. DNA restriction enzyme from E. coli. Nature. 1968;217:1110–1114. doi: 10.1038/2171110a0. [DOI] [PubMed] [Google Scholar]
- Munna M.S., Zeba Z., Noor R. Influence of temperature on the growth of Pseudomonas putida. J. Microbiol. 2016;5:9–12. [Google Scholar]
- Mutalik V.K., Guimaraes J.C., Cambray G., Lam C., Christoffersen M.J., Mai Q.A., Tran A.B., Paull M., Keasling J.D., Arkin A.P., Endy D. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., de Lorenzo V. Robustness of Pseudomonas putida KT2440 as a host for ethanol biosynthesis. New Biotechnol. 2014;31:562–571. doi: 10.1016/j.nbt.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., de Lorenzo V. Pseudomonas putida as a functional chassis for industrial biocatalysis: from native biochemistry to trans-metabolism. Metab. Eng. 2018;50:142–155. doi: 10.1016/j.ymben.2018.05.005. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., Pettinari M.J., Ramírez M.C., Galvagno M.A., Méndez B.S. Escherichia coli arcA mutants: metabolic profile characterization of microaerobic cultures using glycerol as a carbon source. J. Mol. Microbiol. Biotechnol. 2008;15:48–54. doi: 10.1159/000111992. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., Pettinari M.J., Galvagno M.A., Méndez B.S. Metabolic selective pressure stabilizes plasmids carrying biosynthetic genes for reduced biochemicals in Escherichia coli redox mutants. Appl. Microbiol. Biotechnol. 2010;88:563–573. doi: 10.1007/s00253-010-2774-1. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., Pérez-Pantoja D., de Lorenzo V. Why are chlorinated pollutants so difficult to degrade aerobically? Redox stress limits 1,3-dichloroprop-1-ene metabolism by Pseudomonas pavonaceae. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013;368:20120377. doi: 10.1098/rstb.2012.0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P.I., Martínez-García E., de Lorenzo V. Biotechnological domestication of pseudomonads using synthetic biology. Nat. Rev. Microbiol. 2014;12:368–379. doi: 10.1038/nrmicro3253. [DOI] [PubMed] [Google Scholar]
- Nikel P.I., Chavarría M., Fuhrer T., Sauer U., de Lorenzo V. Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the Entner-Doudoroff, Embden-Meyerhof-Parnas, and pentose phosphate pathways. J. Biol. Chem. 2015;290:25920–25932. doi: 10.1074/jbc.M115.687749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel P.I., Chavarría M., Danchin A., de Lorenzo V. From dirt to industrial applications: Pseudomonas putida as a Synthetic Biology chassis for hosting harsh biochemical reactions. Curr. Opin. Chem. Biol. 2016;34:20–29. doi: 10.1016/j.cbpa.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Niu Y., Tenney K., Li H., Gimble F.S. Engineering variants of the I-SceI homing endonuclease with strand-specific and site-specific DNA-nicking activity. J. Mol. Biol. 2008;382:188–202. doi: 10.1016/j.jmb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich K.D., Verkhusha V.V. Guide to red fluorescent proteins and biosensors for flow cytometry. Methods Cell Biol. 2011;102:431–461. doi: 10.1016/B978-0-12-374912-3.00017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkney M., Theophilus B.D.M., Warne S.R., Tacon W.C.A., Thomas C.M. Analysis of transcription from the trfA promoter of broad host range plasmid RK2 in Escherichia coli, Pseudomonas putida, and Pseudomonas aeruginosa. Plasmid. 1987;17:222–232. doi: 10.1016/0147-619x(87)90030-8. [DOI] [PubMed] [Google Scholar]
- Platt R., Drescher C., Park S.K., Phillips G.J. Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid. 2000;43:12–23. doi: 10.1006/plas.1999.1433. [DOI] [PubMed] [Google Scholar]
- Poblete-Castro I., Wittmann C., Nikel P.I. Biochemistry, genetics, and biotechnology of glycerol utilization in Pseudomonas species. Microb. Biotechnol. 2020;13:32–53. doi: 10.1111/1751-7915.13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pósfai G., Kolisnychenko V., Bereczki Z., Blattner F.R. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 1999;27:4409–4415. doi: 10.1093/nar/27.22.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prathapam R., Uehara T. A temperature-sensitive replicon enables efficient gene inactivation in Pseudomonas aeruginosa. J. Microbiol. Methods. 2018;144:47–52. doi: 10.1016/j.mimet.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., González-Carrero M., Timmis K.N. Broad-host range expression vectors containing manipulated meta-cleavage pathway regulatory elements of the TOL plasmid. FEBS Lett. 1988;226:241–246. doi: 10.1016/0014-5793(88)81431-5. [DOI] [PubMed] [Google Scholar]
- Ramos J.L., Marqués S., Timmis K.N. Transcriptional control of the Pseudomonas TOL plasmid catabolic operons is achieved through an interplay of host factors and plasmid-encoded regulators. Annu. Rev. Microbiol. 1997;51:341–373. doi: 10.1146/annurev.micro.51.1.341. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. Cold Spring Harbor Laboratory, Cold Spring Harbor; NY, USA: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- San Millán A., Toll-Riera M., Qi Q., Betts A., Hopkinson R.J., McCullagh J., MacLean R.C. Integrative analysis of fitness and metabolic effects of plasmids in Pseudomonas aeruginosa PAO1. ISME J. 2018;12:3014–3024. doi: 10.1038/s41396-018-0224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Pascuala A., de Lorenzo V., Nikel P.I. Refactoring the Embden-Meyerhof-Parnas pathway as a whole of portable GlucoBricks for implantation of glycolytic modules in Gram-negative bacteria. ACS Synth. Biol. 2017;6:793–805. doi: 10.1021/acssynbio.6b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Pascuala A., Fernández-Cabezón L., de Lorenzo V., Nikel P.I. Functional implementation of a linear glycolysis for sugar catabolism in Pseudomonas putida. Metab. Eng. 2019;54:200–211. doi: 10.1016/j.ymben.2019.04.005. [DOI] [PubMed] [Google Scholar]
- Sharan S.K., Thomason L.C., Kuznetsov S.G., Court D.L. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silo-Suh L.A., Elmore B., Ohman D.E., Suh S.J. Isolation, characterization, and utilization of a temperature-sensitive allele of a Pseudomonas replicon. J. Microbiol. Methods. 2009;78:319–324. doi: 10.1016/j.mimet.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Rocha R., Martínez-García E., Calles B., Chavarría M., Arce-Rodríguez A., de las Heras A., Páez-Espino A.D., Durante-Rodríguez G., Kim J., Nikel P.I., Platero R., de Lorenzo V. The Standard European Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013;41:D666–D675. doi: 10.1093/nar/gks1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva F., Queiroz J.A., Domingues F.C. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol. Adv. 2012;30:691–708. doi: 10.1016/j.biotechadv.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Sun J., Wang Q., Jiang Y., Wen Z., Yang L., Wu J., Yang S. Genome editing and transcriptional repression in Pseudomonas putida KT2440 via the type II CRISPR system. Microb. Cell Fact. 2018;17:41. doi: 10.1186/s12934-018-0887-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason L.C., Sawitzke J.A., Li X., Costantino N., Court D.L. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Prot. Mol. Biol. 2014;106 doi: 10.1002/0471142727.mb0116s106. 1.16.11-11.16.39. [DOI] [PubMed] [Google Scholar]
- Trevors J.T. Plasmid curing in bacteria. FEMS Microbiol. Rev. 1986;32:149–157. [Google Scholar]
- Valla S., Haugan K., Durland R., Helinski D.R. Isolation and properties of temperature-sensitive mutants of the trfA gene of the broad host range plasmid RK2. Plasmid. 1991;25:131–136. doi: 10.1016/0147-619x(91)90025-r. [DOI] [PubMed] [Google Scholar]
- Vento J.M., Crook N., Beisel C.L. Barriers to genome editing with CRISPR in bacteria. J. Ind. Microbiol. Biotechnol. 2019;46:1327–1341. doi: 10.1007/s10295-019-02195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volke D.C., Turlin J., Mol V., Nikel P.I. Physical decoupling of XylS/Pm regulatory elements and conditional proteolysis enable precise control of gene expression in Pseudomonas putida. Microb. Biotechnol. 2020;13:222–232. doi: 10.1111/1751-7915.13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor G.L., Griffiths E.J., Lo R., Dhillon B.K., Shay J.A., Brinkman F.S. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas Genome Database. Nucleic Acids Res. 2016;44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth N.T., Kozaeva E., Nikel P.I. Accelerated genome engineering of Pseudomonas putida by I-SceI–mediated recombination and CRISPR-Cas9 counterselection. Microb. Biotechnol. 2020;13:233–249. doi: 10.1111/1751-7915.13396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsey M.J., Williams P.A. Metabolism of toluene and xylenes by Pseudomonas putida (arvilla) mt-2: evidence for a new function of the TOL plasmid. J. Bacteriol. 1975;124:7–13. doi: 10.1128/jb.124.1.7-13.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Voigt C.A. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res. 2018;46:11115–11125. doi: 10.1093/nar/gky884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel S., Benedetti I., Eisenbach L., de Lorenzo V., Wierckx N., Blank L.M. Tn7-Based device for calibrated heterologous gene expression in Pseudomonas putida. ACS Synth. Biol. 2015;4:1341–1351. doi: 10.1021/acssynbio.5b00058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.