

Summary
Although similar in molecular composition, synapses can exhibit strikingly distinct functional transmitter release and plasticity characteristics. To determine whether ultrastructural differences co-define this functional heterogeneity, we combine hippocampal organotypic slice cultures, high-pressure freezing, freeze substitution, and 3D-electron tomography to compare two functionally distinct synapses: hippocampal Schaffer collateral and mossy fiber synapses. We find that mossy fiber synapses, which exhibit a lower release probability and stronger short-term facilitation than Schaffer collateral synapses, harbor lower numbers of docked synaptic vesicles at active zones and a second pool of possibly tethered vesicles in their vicinity. Our data indicate that differences in the ratio of docked versus tethered vesicles at active zones contribute to distinct functional characteristics of synapses.
Keywords: synapse, active zone, synaptic vesicles, vesicle priming, vesicle docking, electron microscopy, ultrastructure, release probability, short-term plasticity, hippocampal mossy fiber synapse
Graphical Abstract
Highlights
-
•
Electron tomography enables the dissection of vesicle pools at synaptic active zones
-
•
Docked and primed vesicle availability contributes to initial release probability
-
•
The ratio of docked and tethered vesicles may co-determine short-term plasticity
-
•
Hippocampal mossy fibers contain three morphological types of docked vesicles
Distinct synapse types exhibit strikingly different morphological and functional properties. To investigate how the ultrastructural architecture of synaptic release sites contributes to such diversity, Maus et al. exploit electron tomography to correlate the nanoscale organization of synaptic vesicle pools with fundamental functional properties such as short-term synaptic plasticity characteristics.
Introduction
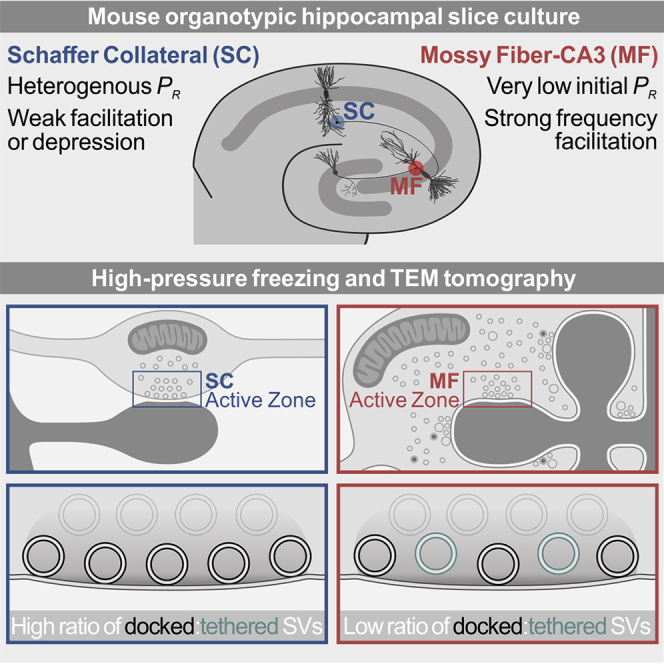
Transmitter release at presynaptic active zones (AZs) is triggered by membrane-depolarization-induced Ca2+ influx via voltage-gated Ca2+ channels. Ca2+ ions are then detected by sensor proteins, which elicit the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-mediated fusion of docked and primed, fusion-competent synaptic vesicles (SVs) (Südhof, 2013). Although distinct types of synapses in the mammalian brain employ similar sets of proteins for transmitter release, their functional characteristics can differ dramatically, particularly the initial SV fusion probability (Pr) and its short-term plasticity during and after trains of action potentials (Regehr, 2012). In extreme cases, certain synapse types, particularly those with a high initial Pr, have a phasic character and show strong synaptic depression during action potential trains, while others, often ones with a low initial Pr, are tonic and show strong frequency facilitation (Neher and Brose, 2018). In this context, the size of the readily releasable pool (RRP) of primed SVs at rest and the rate of RRP exhaustion and refilling during ongoing stimulation are parameters that synapses employ and adapt purposively to shape short-term plasticity (Neher and Brose, 2018, Regehr, 2012).
Electron tomographic analyses of high-pressure-frozen, cryo-substituted synapses showed that primed SVs are in point contact with the membrane, a state that depends on Munc13s and SNARE proteins (Hammarlund et al., 2007, Imig et al., 2014, Siksou et al., 2009). Complementary functional evidence indicates that this docked and primed state is reversible and can be generated within milliseconds (Chang et al., 2018, He et al., 2017, Miki et al., 2018). This led to the notion of a loosely docked and primed SV state that can be rapidly converted into a tightly docked and primed SV state, in which SVs fuse readily in response to an action potential. In terms of synapse function, loosely and tightly docked SVs and their rapid interconversion were proposed to explain key features of depressing and facilitating synapses, in which phasic synapses have a large tightly docked SV pool at rest, which is exhausted with ongoing stimulation to cause synaptic depression, while tonic synapses feature an initially small tightly docked SV pool that is progressively filled in an activity/Ca2+-dependent manner during ongoing stimulation to cause frequency facilitation (Neher and Brose, 2018). This model of loosely versus tightly docked SVs has similarities with functional definitions of heterogeneous SV pools, such as reluctantly/slowly versus fully/rapidly releasable (Lee et al., 2012, Neher, 2015, Neher and Brose, 2018) or primed versus superprimed SVs (Taschenberger et al., 2016), and with morphological classifications of membrane-proximal SV pools, such as tethered versus docked SVs (Imig et al., 2014, Watanabe et al., 2013).
In the present study, we tested the prediction that defined functional features of different presynapse types have an ultrastructural basis and—more concretely—that the phasic versus tonic nature of synapses might become manifest as a difference in the relative proportion of tethered and docked SVs. We used cultured hippocampal slices to compare SV pool organization at individual AZs between low-Pr (Galimberti et al., 2006, Lawrence et al., 2004) strongly facilitating (Salin et al., 1996) hippocampal mossy fiber (MF) synapses (Chicurel and Harris, 1992, Rollenhagen et al., 2007) and Schaffer collateral (SC) synapses (Harris and Stevens, 1989), which have a heterogeneous but on average substantially higher Pr (Helassa et al., 2018, Oertner et al., 2002). We show that low-Pr MF synapses at rest exhibit a lower density of docked SVs than SC synapses and a second membrane-proximal pool of tethered SVs, while the absolute number of SVs in the vicinity of individual AZs does not differ. Our data indicate that differences in the ratio of docked versus tethered SVs contribute to Pr and short-term plasticity differences between hippocampal MF and SC synapses.
Results
Intact MF Pathway in Hippocampal Organotypic Slice Cultures
We first ensured that hippocampal organotypic slice cultures recapitulate hallmark functional and morphological features of the hippocampal MF-CA3 pyramidal cell synaptic connection (Figures 1 and S1). MF synapses exhibited a higher paired-pulse ratio (PPR) and stronger short-term facilitation (Figures S1A–S1E) than SC synapses at two developmental time points, i.e., day in vitro (DIV) 14 (PPR: SC, 2.203 ± 0.116; MF, 2.981 ± 0.145; ∗∗∗p < 0.001) and DIV28 (PPR: SC, 2.195 ± 0.160; MF, 2.721 ± 0.169; ∗p = 0.04). This is in line with previous studies showing that MF synapses have a lower Pr than SC synapses in organotypic slices (Galimberti et al., 2006, Helassa et al., 2018).
Figure 1.

Ultrastructural Characterization of the MF-CA3 Synapse in Organotypic Hippocampal Slice Cultures Prepared by High-Pressure Freezing and Freeze Substitution
(A) 2D-electron micrograph of a large, presynaptic MF bouton densely packed with SVs.
(B–D) Enlarged from (A). MF boutons form three types of contacts with CA3 pyramidal cells: asymmetric spine synapses (B, white arrowhead in A), dendritic synapses (C, gray arrowhead in A; black arrowheads, postsynaptic densities), and puncta adherentia onto dendritic shafts (D, black arrowhead in A).
(E–J) Electron tomographic subvolumes of MF (E) and SC (H) synapses (green arrowheads, docked vesicles). (F and I) 3D models of synaptic profiles from MF (F) and SC (I) synapses (AZ, gray; docked vesicles, green; non-attached vesicles, gray). (G and J) Orthogonal views of MF (G) and SC (J) AZs.
(K–M) Tomographic slices illustrating the three morphologically distinct vesicle types observed docked at MF AZs: SVs (K), GVs (Ø > 60 nm; L), and DCVs (M).
a, astrocytic processes; d, dendrite; m, mitochondria; sa, spine apparatus. Scale bars: (A) 2 μm; (D) 200 nm; (E and H) 100 nm; (K–M) 50 nm.
Furthermore, Synaptoporin-positive MF boutons (Singec et al., 2002) specifically innervate the CA3 stratum lucidum at both DIV14 and DIV28 (Figures S1F–S1I), indicating that MF target specificity is preserved during development in culture. Moreover, biocytin-filled CA3 pyramidal neurons displayed thorny excrescences (Figure S1J), a hallmark postsynaptic specialization of MF synapses (Chicurel and Harris, 1992).
Electron micrographs from ultrathin sections of cryo-fixed cultured slices (Figures S1K and S1M) showed large MF boutons with excellent ultrastructural preservation and a gross morphology comparable to that of perfusion-fixed tissue (Figures S1L, S1O, and S1P) and cryo-fixed acute slices (Figure S1N). MF-CA3 contacts in cultured slices are established on complex spines (Figures 1A and 1B) and dendritic shafts (Figure 1C), whereas puncta adherentia (Figure 1D; Spacek and Harris, 1998) were observed exclusively in contact with dendritic shafts.
Electron tomographic analyses of MF (Figures 1E–1G; Video S1) and SC (Figures 1H–1J; Video S2) synapses allowed us to precisely quantify SV-AZ membrane distances and docked vesicles (green arrowheads in Figures 1E and 1H). Docked vesicles in MF boutons were composed of three morphological classes (Figures 1K–1M): (1) small clear-core SVs (diameter = 33–55 nm; Figure 1K), (2) giant clear-core vesicles (GVs; diameter = 60–120 nm; Figure 1L), and (3) dense-core vesicles (DCVs, diameter = 46–91 nm; Figure 1M). Neither GVs nor DCVs docked at SC membranes in any condition analyzed, and vesicles exhibiting these morphological features were only rarely found within 100 nm.
Comparison of SC and MF Synapses in Hippocampal Slice Cultures
We selected two developmental time points for our study, DIV14 and DIV28, to cover a period of significant morphological (Amaral and Dent, 1981) and functional maturation of MF synapses, which entails an increase in excitatory postsynaptic current (EPSC) amplitudes; an enhanced degree of low-frequency (1 Hz) facilitation, but not high-frequency (50 Hz) facilitation (Marchal and Mulle, 2004); and a shift from long-term depression to potentiation after high-frequency stimulation (Battistin and Cherubini, 1994). This time point selection was also intended to correlate our findings with existing ultrastructural (Chicurel and Harris, 1992, Rollenhagen et al., 2007, Studer et al., 2014) and functional (Hallermann et al., 2003, Jonas et al., 1993, Midorikawa and Sakaba, 2017) datasets on MF synapses.
We first compared the distribution of clear-cored vesicles within 100 nm of the AZ in SC and MF synapses at DIV14 (Figures 2A–2H; Table S1B). The spatial density of docked vesicles was 43% lower at MF than at SC synapses in the same slice (Figure 2B), as reflected by a significant reduction in the number of SVs within 5 nm of the AZ membrane (Figure 2C), while the numbers of vesicles within 40 nm (membrane proximal) were similar between synapse types (Figure 2D). Unlike SC synapses, MF synapses exhibited a second pool of vesicles accumulated ~5–15 nm from the membrane (Figures 2A and 2C), reminiscent of the tethered pool in docking- and priming-deficient mutant synapses (Imig et al., 2014, Siksou et al., 2009). Consequently, MF synapses harbored a lower proportion of docked vesicles relative to all vesicles within 40 nm of the AZ membrane compared with SC synapses (Figure 2E). MF synapses were also distinguished by an abundance of GVs in the vicinity of (<100 nm; MF, 0.545 ± 0.083; SC, 0.028 ± 0.016; ∗∗∗p < 0.001) and docked to AZs (Figures 2F and 2G). Importantly, the comparative differences in the density of docked vesicles (Figure S2B; MF, 1.128 ± 0.095; SC, 1.608 ± 0.163; ∗∗p = 0.009) and the ratio of docked and tethered vesicles were also detected in MF synapses from acutely sectioned and cryo-fixed mouse hippocampi (Figures S2E–S2G).
Figure 2.

Morphological Dissection of Distinct Vesicle Pools at MF-CA3 and SC Synapses in Organotypic Hippocampal Slice Cultures at DIV14 and DIV28
(A–P) Morphological characterization of MF and SC AZs (DIV14, N = 3 cultures: SC, n = 36 tomograms, and MF, n = 33 tomograms; DIV28, N = 3 cultures: SC, n = 29 tomograms, and MF, n = 29 tomograms).
(A and I) Distribution of vesicles within 100 nm of the AZ membrane in MF and SC synapses at DIV14 (A) and DIV28 (I).
(B and J) Mean number of docked, clear-cored vesicles (SVs and GVs) normalized to the AZ area at DIV14 (B) and DIV28 (J).
(C and K) Mean number of vesicles within bins of 5 and 10 nm from the AZ normalized to the AZ area at DIV14 (C) and DIV28 (K).
(D and L) Mean number of vesicles within 40 nm of the AZ membrane normalized to the AZ area at DIV14 (D) and DIV28 (L).
(E and M) Relative proportion of docked vesicles within 40 nm of the AZ normalized to the AZ area at DIV14 (E) and DIV28 (M).
(F and N) Diameters of all docked vesicles analyzed at DIV14 synapses (F; SC, n = 116 vesicles, and MF, n = 81 vesicles) and DIV28 synapses (N, SC, n = 167 vesicles, and MF, n = 159 vesicles).
(G and O) Respective proportions of the docked vesicle pool occupied by GVs (Ø > 60 nm) at DIV14 (G) and DIV28 (O).
(H and P) Vesicle diameters of all vesicles analyzed and their respective distances to the AZ membrane at DIV14 (H) and DIV28 (P). Values indicate mean ± SEM; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
The overall organization of ultrastructurally defined vesicle pools in the two synapse types did not differ between DIV14 and DIV28 (Figures 2I–2P; Table S1D). SC synapses exhibited a higher density of docked vesicles than MF synapses at both time points (Figures 2B and 2J), in line with our functional data demonstrating that SC synapses have a consistently higher Pr (Figures S1B and S1C). We found that the absolute density of docked vesicles was increased at both MF and SC synapses at DIV28 (Figure 2J; SC, 1.710 ± 0.139; MF, 1.061 ± 0.141; ∗∗∗p < 0.001) compared with DIV14 (Figure 2B; SC, 1.015 ± 0.112; MF, 0.576 ± 0.087; ∗∗p = 0.003). The increase in vesicle docking observed in cultured MF boutons may indicate a developmental increase in the functional RRP size at the single-AZ level, which would accompany other previously reported morphological changes during MF maturation between postnatal day (P) 14 and P21 in vivo, such as an enlargement of the presynaptic terminal, an increase in the synaptic release site number and size, and the generation of additional and more complex postsynaptic spines (Amaral and Dent, 1981). Likewise, cultured SC synapses undergo functional and structural maturation during development (Rose et al., 2013, De Simoni et al., 2003). Thus, our findings provide an important morphological aspect to the understanding of functional changes that occur upon hippocampal synapse maturation in mice. Because the general organization of vesicle pools in rat hippocampal MF boutons in vivo appears to undergo further changes between P28 and adulthood (3–4 months) (Rollenhagen et al., 2007), it would be interesting to determine the distribution of SVs at AZs in fully mature MF synapses.
Altogether, we found that low-Pr MF synapses exhibit a lower number of docked SVs than SC synapses and that this is accompanied by the presence of tethered SVs, which are adequately positioned for rapid priming to sustain the RRP of docked SVs during repetitive stimulation.
Effects of Different Fixation Protocols on Vesicle Docking
To compare effects of different fixation protocols on SV organization at individual MF release sites, we analyzed tomograms from cryo-fixed organotypic slices, from mice that had been transcardially perfused according to two protocols (Chicurel and Harris, 1992, Rollenhagen et al., 2007; Figures S2I, S2J, and S2M–S2P; Table S1E), and from organotypic slices that had been immersed in the respective perfusion fixatives (Figures S2K, S2L, and S2Q–S2S; Table S1F).
Vesicle docking densities at the surface of cryo- and immersion-fixed organotypic slices were comparable (Figure S2Q), but an analysis at greater depths from the slice surface revealed a decreased docking density for one of the aldehyde-fixation conditions (Figure S2R) that was accompanied by an increased frequency of fusion intermediates (Figure S2S). Some perfusion-fixed MF boutons also exhibited an abundance of Ω-shaped fusion intermediates (Figures S2I and S2J), which were not seen close to the surface of immersion-fixed organotypic slices (Figures S2K and S2L). The increased number of fusion intermediates in perfusion-fixed material was paralleled by a general depletion of docked and tethered vesicles (Figures S2N and S2O) and by a redistribution of AZ-proximal vesicles to more distal positions, which matches observations made in mouse cortex (Korogod et al., 2015).
Altogether, our data indicate that perfusion fixation, but not aldehyde fixation per se, perturbs pre-SV pool organization. This is likely because aldehyde crosslinking of proteins is diffusion limited and thus proceeds more slowly in the case of perfusion. In the present study, high-pressure-frozen material provided the most consistent results throughout experiments.
GVs in MF Boutons Are Not Mere By-Products of Synaptic Activity
We next focused on the nature of presynaptic GVs observed at hippocampal MF synapses. We first compared MF synaptic profiles in cryo-fixed organotypic slices with those of acutely sectioned and cryo-fixed hippocampi (Figures S2A–S2H; Table S1C) and of mice transcardially perfused with two fixatives (Chicurel and Harris, 1992, Rollenhagen et al., 2007; Figures S2I, S2J, and S2M–S2P; Table S1E). GVs were observed within 100 nm of the AZ in all samples, albeit at slightly lower numbers in perfusion-fixed preparations (fixative 1, 0.205 ± 0.073; fixative 2, 0.263 ± 0.076; p = 0.69, ns) and acute preparations (SC, 0.012 ± 0.012; MF, 0.185 ± 0.037; ∗∗∗p < 0.001) compared with organotypic slices, indicating that they are not a culture-specific phenomenon. GVs may represent precursor vesicles of somatic origin, endocytic intermediates of local SV recycling, or neurotransmitter-filled end products of a specialized SV biogenesis mode. To distinguish among these scenarios, we assessed the activity dependence of GV formation in Munc13-1/2-deficient mice, in which SV fusion (Varoqueaux et al., 2002) and exocytosis-coupled ultrafast endocytosis (Watanabe et al., 2013) are abolished (Figures 3A–3L; Table S1G). In Munc13-deficient MF boutons, we observed a complete loss of vesicle docking (Figures 3E–3G) (control, 0.861 ± 0.159; double knockout [DKO], 0 ± 0; ∗∗∗p < 0.001) and an increased number of tethered vesicles (~10–20 nm; Figure 3G). The number of vesicles within 40 nm of the AZ was unchanged (Figure 3H). SV, GV (Figure 3F), and DCV (control, 0.077 ± 0.035; DKO, 0 ± 0; p = 0.10, ns) docking was also abolished by the loss of Munc13s. The number of SVs (Figure 3J) and GVs (Figure 3K) (control, 0.229 ± 0.058; DKO, 0.315 ± 0.080; p = 39, ns) within 100 nm of the AZ was unchanged in Munc13-deficient MF boutons, but the number of DCVs increased three-fold (Figure 3L; Video S3) (control, 0.252 ± 0.061; DKO, 0.643 ± 0.095; ∗∗p = 0.002). Pharmacological blockade of slice network activity (with Tetrodotoxin/2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide/D-2-amino-5-phosphonovalerate, i.e. TTX/NBQX/APV) or presynaptic MF activity (TTX/DCG-IV) failed to eliminate GVs (Figures S3A–S3G; Table S1H).
Figure 3.

Electron Tomographic Analysis of Vesicle Pools in Munc13-Deficient and Control MF-CA3 Synapses
(A–D) Electron tomographic subvolumes of control (CTRL; A) and Munc13-1/2 (M13) DKO (C) MF synapses. (B and D) 3D models of CTRL (B) and M13 DKO (D) MF synapses.
(E) Vesicle distribution within 100 nm of the AZ membrane (N = 2 cultures; n = 15 tomograms).
(F) Mean number of docked clear-cored vesicles (SVs and GVs) in M13 CTRL and DKO MF synapses normalized to the AZ area.
(G) Mean number of vesicles within bins of 5 and 10 nm from the AZ normalized to the AZ area.
(H) Mean number of vesicles within 40 nm of the AZ membrane normalized to the AZ area.
(I) Diameters of all vesicles analyzed, and their distance to the AZ membrane.
(J) Mean number of SVs within 100 nm of the AZ membrane normalized to the AZ area.
(K) Mean number of GVs within 100 nm of the AZ membrane normalized to the AZ area.
(L) Mean number of DCVs within 100 nm of the AZ membrane normalized to the AZ area.
(M) Tomographic slice in which a docked GV is centrally localized at the AZ membrane.
(N–S) Effects of DCG-IV on mEPSCs recorded from CA3 pyramidal cells in slice cultures (N = 2 cultures; n = 28 cells).
(N) Representative traces of mEPSC events recorded from CA3 pyramidal cells in the presence of 1 μM TTX and 10 μM bicuculline (BIC) before and after the application of 2 μM DCG-IV.
(O) Histogram for mEPSC event amplitudes (≥8 pA; 2 pA bins and last bin for all events ≥ 100 pA) before (black, TTX/BIC) and after (gray, TTX/BIC/DCG-IV) application of DCG-IV. Bars represent the number of mEPSC events recorded for an indicated amplitude range in a 5 min interval (mean events per cell + SEM).
(P) Cumulative Pr plot of mEPSC amplitudes. The subtracted cumulative frequency distribution of mEPSCs removed by DCG-IV application is indicated in purple, and the dotted line marks an mEPSC amplitude of 30 pA (see STAR Methods).
(Q) Mean frequency of mEPSCs before and after application of DCG-IV.
(R) Median amplitudes of mEPSCs before and after application of DCG-IV.
(S) Relative changes in the mean amplitude and frequency after the application of DCG-IV normalized to the control condition (TTX/BIC only).
Scale bars: (A–D) and (M), 100 nm. Values indicate mean ± SEM; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Altogether, these data show that GVs are especially abundant in MF synapses, that their generation is largely independent of synaptic activity and SV recycling, and that Munc13s are required for SV, GV, and DCV docking.
GVs as Morphological Correlates of Giant mEPSCs
It was postulated that GVs in MF synapses (Figure 3M) contain glutamate and represent the morphological correlates of large-amplitude, miniature EPSCs (mEPSCs) (giant mEPSCs; ≥100 pA) recorded from CA3 pyramidal cells (Henze et al., 2002a). We measured spontaneous mEPSCs in CA3 pyramidal cells in slice cultures at DIV14 (Figures 3N–3S; Table S1I) and confirmed the existence mEPSCs of ≥100 pA (Figure 3N). Upon blockade of evoked and spontaneous MF glutamate release by the mGluR2 agonist DCG-IV (Kamiya and Ozawa, 1999, Kamiya et al., 1996, Kim and Tsien, 2008), we observed a strong reduction in mEPSC frequency (Figure 3Q) and a reduced giant mEPSC frequency (Figure 3O), as reflected by reductions in mEPSC amplitudes (Figures 3R and 3S) and by a change in the cumulative mEPSC amplitude distribution (Figure 3P). Although it is somewhat unclear to what extent DCG-IV suppresses MF mEPSCs and whether DCG-IV spares CA3-to-CA3 pyramidal cell synaptic mEPSCs in slice cultures, our data are in line with a previous study in which γ-irradiation-induced lesioning of MF-CA3 inputs in neonatal rats caused a loss of giant mEPSCs in CA3 pyramidal cells (Henze et al., 1997). This indicates that the DCG-IV effect we detected results from near-selective silencing of MF mEPSCs.
To correlate the amplitude distribution of mEPSCs with the size distribution of docked SVs, we assumed that (1) a 10 pA mEPSC (the mode of the DCG-IV-sensitive mEPSC amplitude distribution) reflects quantal glutamate release from an SV with an outer diameter of 44 nm (the mode of the docked vesicle size distribution), (2) the lumenal glutamate concentration is independent of vesicle volume, and (3) postsynaptic glutamate receptor saturation is negligible during a single mEPSC. We determined that the fusion of a vesicle with an outer diameter of 60 nm (size threshold for classification as GV) would generate an mEPSC of ~30 pA, and a GV with an outer diameter of 85 nm (mean diameter of all docked vesicles with a diameter > 60 nm) would correspond to an mEPSC of ~98 pA (see STAR Methods). The cumulative frequency distribution of DCG-IV-sensitive mEPSC amplitudes (Figure 2P, purple line) indicates that approximately 27% of all mEPSCs are ≥30 pA and could therefore originate from glutamate quanta released from GVs, which is in agreement with our finding that GVs comprise ~20% of docked clear-core vesicles (Figure 2G).
Altogether, these findings support for the notion that GVs are the morphological correlate of the large mEPSC events at MF synapses (Henze et al., 2002a).
cAMP-Dependent Changes in Transmitter Release Do Not Affect SV Docking
To test our hypothesis that the proportions of docked and tethered vesicles in the vicinity of individual AZs contribute to the Pr of a synapse, we examined whether pharmacological manipulations of vesicle fusion Pr cause changes in the ratio of docked and tethered vesicles. In MF synapses, cyclic AMP (cAMP) signaling alters presynaptic strength (Kamiya and Yamamoto, 1997) and triggers a presynaptic form of long-term plasticity via downstream effectors such as protein kinase A (PKA) (Tzounopoulos et al., 1998, Weisskopf et al., 1994). We applied the adenylyl cyclase activator forskolin or the presynaptic mGluR2 agonist DCG-IV to organotypic slices 15 min before high-pressure freezing to manipulate presynaptic cAMP levels so that neurotransmitter release from MF synapses is potentiated or depressed. To isolate the effect of these manipulations and corresponding changes in Pr on vesicle docking, all experiments were performed in the presence of TTX (Trudeau et al., 1996) to prevent spontaneous action potential firing and network activity. In contrast to a previous study, which employed tetraethylammonium (TEA) without TTX to induce a chemical form of MF long-term potentiation in hippocampal slice cultures (Zhao et al., 2012), we did not see massive changes in AZ number or size after forskolin treatment using stimulated emission depletion (STED) microscopy or ultrastructural analyses (Figures 4A–4G), and we did not see changes in MF bouton area (control, 5.053 μm2 ± 0.410; forskolin, 4.969 μm2 ± 0.352; p = 0.42, ns) and postsynaptic spine area (control, 0.435 μm2 ± 0.045; forskolin, 0.470 μm2 ± 0.0452; p = 0.52, ns). These findings indicate that our protocol does not cause massive structural changes because of increased network activity but that it would allow us to determine fine ultrastructural changes in the distribution of vesicles at AZs. However, we cannot exclude the possibility that forskolin treatment may induce structural changes of AZs under different experimental conditions or at later time points. To our surprise, neither forskolin nor DCG-IV affected docked or membrane-proximal SV and GV pools (Figures 4H–4O; Table S1L), whereas the number of docked DCVs in forskolin-treated MF synapses was increased compared with vehicle control and DCG-IV-treated slices (control, 0.045 ± 0.019; forskolin, 0.092 ± 0.022; DCG-IV, 0.014 ± 0.009; forskolin versus DCG-IV, ∗p = 0.014). We detected only a small tendency toward an increased SV docking in forskolin-treated slices (Figures 4H–4J), with no changes in the number of vesicles within 40 nm of the AZ (Figure 4K) so that the proportion of SVs within 0–40 nm from the AZ attached to the membrane was increased (Figure 4L). This trend did not reach statistical significance for the given sample size. In principle, our finding is in line with the notion that increased cAMP/PKA signaling increases Pr and the efficacy of SV fusion via improved coupling to Ca2+ channels (Midorikawa and Sakaba, 2017). Alternatively, the subtlety of the observed morphological changes supports the interpretation that cAMP-mediated PKA activation affects proteins of the SV fusion machinery at a post-docking/priming step to facilitate SV fusion, e.g., via phosphorylation of complexins (Cho et al., 2015). However, we cannot exclude the possibility that other ways of increasing presynaptic Pr, such as elevating extracellular Ca2+ concentrations or preventing SV depriming (He et al., 2017), may affect the ratio of docked versus tethered SVs in hippocampal MF synapses.
Figure 4.

Pharmacological Manipulation of Presynaptic cAMP Levels
(A–C, and E) Two-color STED images (A–C) for Synaptoporin (green; A) and Bassoon (magenta; B) obtained after immunostaining using specific primary antibodies and secondary antibodies conjugated to STAR580 and Atto647N, respectively. The amount of Bassoon staining corresponding to each Synaptoporin object was determined for both vehicle controls (VCs), or the addition of 1 μM TTX, and 25 μM forskolin (T/F) (E).
(D, F, and G) Ultrastructural 2D analysis of mitochondria and AZ release sites in vehicle control slices (VC, N = 3 cultures; n = 154 boutons) and forskolin-treated slices (T/F, N = 3; n = 144 boutons) (D). AZ number normalized to MF bouton perimeter (F). Mean AZ length per MF bouton (G).
(H) Spatial distribution of vesicles within 100 nm of the AZ membrane in MF synapses treated for 15 min with vehicle control (VC, 1 μM TTX, N = 3 cultures; n = 24 tomograms), TTX and 2 μM DCG-IV (T/D, N = 3; n = 24), or TTX and 25 μM forskolin (T/F, N = 3; n = 32).
(I) Mean number of docked clear-cored vesicles (SVs and GVs) normalized to the AZ area.
(J) Mean number of vesicles within bins of 5 and 10 nm from the AZ normalized to the AZ area.
(K) Mean number of vesicles within 40 nm of the AZ normalized to the AZ area.
(L) Proportion of docked vesicles within 40 nm of the AZ normalized to the AZ area.
(M) Diameters of all docked vesicles analyzed (VC, n = 166 vesicles; T/F, n = 355 vesicles; T/D, n = 176 vesicles).
(N) Respective proportions of the docked vesicle pool occupied by GVs (Ø > 60 nm).
(O) Diameters of all vesicles analyzed and their respective distances to the AZ membrane.
Scale bars: (C) 5 μm; (D) 2 μm. Values indicate mean ± SEM; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Tables S1H and S1J–S1M.
Discussion
Methodology
We made the important observation that chemical fixatives may deplete docked SVs at MF synapses. This is likely because of the increased osmolarity of aldehyde-supplemented buffers and the time required for fixative diffusion into the tissue (Figures S2I–S2S), but we cannot exclude the possibility that more refined fixation conditions may cause less severe effects on SV pools. Our finding, which matches previous morphological observations (Korogod et al., 2015) and functional observations (Smith and Reese, 1980), does not merely highlight a fundamental risk involved in comparing electron microscopy results obtained using different experimental parameters. Rather, it indicates (1) that ultrastructural data obtained with chemically fixed samples, while useful in numerous contexts, are not suitable for the functional interpretation of morphologically defined SV pools in tissue and (2) that a combination of high-pressure freezing and (cryo-)electron tomography should be used instead.
Synapse Ultrastructure and Short-Term Plasticity
The mechanistic basis of the strong frequency facilitation of MF boutons, a hallmark of this synapse (Salin et al., 1996) that co-determines its role as a conditional detonator synapse (Henze et al., 2002b), has long been enigmatic (Nicoll and Schmitz, 2005). Recent evidence indicates that multiple factors converge to shape Pr and short-term plasticity at MF synapses. They include a large coupling distance (~70 nm) between Ca2+ channels and SVs (Vyleta and Jonas, 2014), action potential broadening (Geiger and Jonas, 2000), Ca2+ buffer saturation (Blatow et al., 2003, Vyleta and Jonas, 2014), and specialized exocytotic Ca2+ sensors (Jackman et al., 2016). Moreover, the presence of mitochondria in presynapses has been linked to differences in Ca2+ homeostasis, transmitter release properties, and short-term plasticity (Kwon et al., 2016). In line with the notion that synapses with a higher Pr might be more prone to depression during sustained activity because of increased mitochondria-dependent Ca2+ clearance, we found that the proportional presynaptic bouton area occupied by mitochondria for synaptic profiles with mitochondria was substantially greater in SC synapses compared with MF synapses (Table S1M; SC, 15.29 ± 1.071; MF, 8.024 ± 0.341; ∗∗∗p < 0.001).
We made the important observation that ultrastructural differences in the organization of AZ-proximal SV pools likely contribute to synaptic short-term plasticity characteristics. Low-Pr MF synapses at rest exhibit a relatively low proportion of docked SVs and a second pool of possibly tethered SVs in the AZ vicinity. This is in accordance with the notion that tonic synapses at rest harbor an abundant supply of tethered, or loosely docked, SVs and that the differences in initial Pr and short-term plasticity between the synapse types we studied are caused by different ratios between docked and primed, or tightly docked, SVs and tethered, or loosely docked, SVs (Neher and Brose, 2018).
Pr, RRP, and Docked SVs
The issue as to whether the number of docked SVs per AZ can predict the Pr of a given synapse and its short-term plasticity features is the focus of substantial interest and controversy (Éltes et al., 2017, Neher and Brose, 2018, Xu-Friedman and Regehr, 2004). At its core is the question of whether docked SVs comprise the RRP of functionally primed SVs (Kaeser and Regehr, 2017, Neher and Brose, 2018, Xu-Friedman and Regehr, 2004). Priming-deficient synapses are devoid of docked SVs, which supports the notion that the number of docked SVs is a reliable proxy for the number of molecularly primed SVs, but it is also clear that in response to an action potential, only a small subset of these SVs will fuse. The biggest conundrum here is that the RRP is a relatively vague concept, usually defined functionally, in terms of SVs that can fuse in response to a given stimulus in a given synapse and is typically measured using postsynaptic responses as a proxy (Kaeser and Regehr, 2017, Neher, 2015, Neher and Brose, 2018).
We calculated mean docked vesicle numbers per AZ and per MF bouton (~320 SVs, 37 GVs, and 15 DCVs), as well as the mean membrane surface area for each vesicle type in more mature (DIV28) MF synapses (see STAR Methods). Assuming a specific membrane capacitance of 1 μF/cm2 (Hallermann et al., 2003), fusion of all docked vesicles, irrespective of their type, would change the membrane capacitance (ΔCm) by ~32 fF per MF bouton. By including the second pool of AZ-proximal, tethered vesicles in the analysis to test the notion that vesicles in an loosely docked or tethered state can rapidly be converted in a Ca2+-facilitated manner into a tightly docked and fully primed state (Chang et al., 2018, Neher and Brose, 2018), the fusion of all vesicles within 0–40 nm of the AZ would correspond to a ΔCm of ~88 fF per MF bouton. Previous studies showed that long depolarizations (30–100 ms) evoke a ΔCm of 50–100 fF in entire MF boutons, which corresponds, depending on the SV diameter used for conversion, to a functional RRP of 500–1,400 SVs per MF bouton or ~40 SVs per AZ (Hallermann et al., 2003, Midorikawa and Sakaba, 2017). This matches our calculations for the combined pools of docked and AZ-tethered vesicles (ΔCm of ~88 fF) and earlier estimates of the number of SVs with centers within 60 nm of the AZ (Rollenhagen et al., 2007). Functionally obtained RRP estimates at MF boutons are much larger than the number of docked vesicles we found, which confirms the problem that some pool-depleting stimulation protocols at a given synapse are not sufficiently refined to dissect the relative contributions of docked versus tethered SVs and cannot account for fast priming during stimulation (Neher, 2015, Neher and Brose, 2018). In essence, long depolarizations of low-Pr MF synapses may not only evoke fusion of docked vesicles but additionally induce fast and Ca2+-mediated priming of tethered vesicles and their subsequent fusion.
SVs, GVs, and DCVs at MF AZs
Our study revealed the presence of docked SVs, GVs, and DCVs as a unique feature of MF boutons and showed a loss of docking of all three vesicle types upon deletion of Munc13 priming proteins. These data indicate that Munc13s are required not only for SV docking and priming (Imig et al., 2014, Siksou et al., 2009) but also for GV and DCV docking in these synapses. This supports the notion that GVs represent neurotransmitter-containing vesicles that can fuse and release their content at MF release sites. However, their origin remains unknown, and additional or alternative functional contexts need to be considered.
We cannot exclude the possibility that cultured MF synapses accumulate more GVs than those in vivo as assessed after perfusion fixation or in acute slice preparations. However, the presence of GVs in MF terminals in vivo has been well documented in past ultrastructural studies (Chicurel and Harris, 1992, Henze et al., 2002a, Laatsch and Cowan, 1966, Rollenhagen et al., 2007). The persistence of GVs in genetically and pharmacologically silenced MF synapses in organotypic slices argues against compound fusion (He et al., 2009) or compensatory endocytic membrane retrieval (Watanabe et al., 2013) as the primary mode of GV formation, we cannot rule out the possibility that endocytic processes contribute to a subpopulation of GVs. Alternatively, GVs might originate from DCVs that have undergone non-collapse fusion, degranulation, and rapid retrieval (Laatsch and Cowan, 1966). Indeed, filamentous electron-dense material is occasionally observed in the lumen of GVs (e.g., Figures 1E and 1L). However, the accumulation of DCVs in Munc13-deficient samples implies that DCV fusion in MF boutons is dramatically impaired, yet GVs with and without filamentous luminal content remain. A final possibility is that at least some GVs in MF boutons represent vesicles of somatic origin, because granule cell axons of acute hippocampal slices contain a range of vesicle types, including large, clear-cored vesicles (Figures S3H–S3P). However, it is unclear whether such precursor vesicles would be appropriately equipped to participate in synaptic signaling. In any case, GVs are highly unlikely to be artifactual and do not appear to be a mere peculiarity without function, so further cell biological and functional studies into their role at MF synapses seem worthwhile.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken polyclonal anti-MAP2 Antibody | Novus Biologicals | Cat#NB 300-213; RRID: AB_2138178 |
| Goat anti-Chicken IgG Secondary Antibody, Alexa 488 | Thermo Fisher Scientific | Cat#A-11039; RRID: AB_2534096 |
| Goat anti-Rabbit IgG Secondary Antibody, Alexa 555 | Thermo Fisher Scientific | Cat#A21429; RRID: AB_2535851 |
| Goat anti-Mouse IgG Secondary Antibody, Alexa 555 | Thermo Fisher Scientific | Cat#A21424; RRID: AB_141780 |
| Goat anti-Rabbit IgG Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11008; RRID: AB_143165 |
| Mouse monoclonal anti-Bassoon (SAP7F407) Antibody | Enzo Life Sciences | Cat#ADI-VAM-P S003; RRID: AB_10618753 |
| Rabbit polyclonal anti-Synaptoporin Antibody | SynapticSystems | Cat#102 003; RRID: AB_2619748 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (-)-Bicuculline methochloride | Tocris Bioscience | Cat#0131 |
| (-)-Bicuculline methiodide | HelloBio | Cat#HB0893 |
| 1-Hexadecene | Sigma Aldrich | Cat#52276-5mL |
| 2,2,2,-Tribromoethanol | Alfa Aesar | Cat#A18706 |
| 2,4,6-Tris(dimethylaminomethyl)phenol (DMP-30) | Electron microscopy sciences | Cat#13600 |
| 25% Gluteraldehyde | Electron microscopy sciences | Cat#16220 |
| 2-Dodecenylsuccinic acid anhydride (DDSA) | Serva | Cat#20755.02 |
| Acetone | Electron microscopy sciences | Cat#10015 |
| Alexa Fluor 555 streptavidin conjugate | Thermo Fisher Scientific | Cat#S32355; RRID: AB_2571525 |
| Basal Medium Eagle (BME) | Thermo Fisher Scientific | Cat#41010026 |
| Biocytin hydrochloride | Sigma-Aldrich | Cat#B1758 |
| Bovine Serum Albumin (BSA) | Biomol | Cat#01400.1 |
| Bovine Serum Albumin (BSA) | Sigma Aldrich | Cat#A2153 |
| D-APV: D-(-)-2-Amino-5-phosphonopentanoic acid | Tocris Bioscience | Cat#0106 |
| DAPI: 4′,6-Diamidine-2′-phenylindole dihydrochloride | Sigma-Aldrich/Roche | Cat#10236276001 |
| DCG-IV: (2S,1’R,2′R,3′R)-2-(2,3- dicarboxycyclopropyl)glycine | Tocris Bioscience | Cat#0975 |
| Di-Sodium hydrogen phosphate dihydrate | Merck | Cat#1.06580.1000 |
| Forskolin | Sigma-Aldrich | Cat#F3917 |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat#35050038 |
| Glycidether 100 | Serva | Cat#21045.02 |
| Hank’s Balanced Salt Solution, Ca2+, Mg2+ (HBSS) | Thermo Fisher Scientific | Cat#24020091 |
| Horse Serum, heat inactivated | Thermo Fisher Scientific | Cat#26050088 |
| Kynurenic acid | Sigma-Aldrich | Cat#K3375 |
| Lead (II) Nitrate | Merck | Cat#1.07398.0100 |
| Methylnadic anhydride (MNA) | Serva | Cat#29452.02 |
| Minimum Essential Medium (MEM) | Thermo Fisher Scientific | Cat#11700077 |
| Mounting glue: Aqua-Poly/Mount | Polysciences | Cat#18606-20 |
| NBQX disodium salt | Tocris Bioscience | Cat#1044 |
| NBQX disodium salt | HelloBio | Cat#HB0443 |
| Osmium tetroxide | Electron microscopy sciences | Cat#19132 |
| Paraformaldehyde | Serva | Cat#31628.02 |
| Protein A (ProtA) couples to 10 nm gold particles | Cell Microscopy Core Products, University Medical Center Utrecht, the Netherlands | N/A |
| Sodium Cacodylate | Sigma Aldrich | Cat#C0250-100G |
| Sodium Citrate | Calbiochem | Cat#567446 |
| Sodium dihydrogen phosphate monohydrate | Merck | Cat#1.06346.0500 |
| Tannic Acid 0.1% | Sigma Aldrich | Cat#403040-100G |
| Tetrodotoxin | Tocris Bioscience | Cat#1078 |
| Uranyl Acetate | SPI Supplies | Cat#2624 |
| Experimental Models: Organisms/Strains | ||
| Mouse: M13-DKOs: Unc13atm1Bros; Unc13btm1Rmnd | Augustin et al., 1999, Varoqueaux et al., 2002 | RRID: MGI:2654056 |
| Software and Algorithms | ||
| AxoGraph X 1.3.1 | John Clements | https://axograph.com; RRID: SCR_014284 |
| GraphPad Prism 7 | GraphPad Software | http://www.graphpad.com; RRID: SCR_002798 |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij; RRID: SCR_003070 |
| IMOD software | Kremer et al., 1996 | https://bio3d.colorado.edu/imod/; RRID: SCR_003297 |
| iTEM software | Emsis GMBH | |
| Leica LAS AF | Leica Microsystems | http://www.leica-microsystems.com; RRID: SCR_013673 |
| Patchmaster v2X80 | HEKA/Harvard Bioscience | http://www.heka.com/products/products_main.html#soft_pm; RRID: SCR_000034 |
| SerialEM software | University of Colorado, Boulder, Colorado, US | http://bio3d.colorado.edu/SerialEM/ |
| Other | ||
| Millicell cell culture inserts | Merck Millipore | Cat#PICM03050 |
| Millipore membrane confetti | Merck Millipore | Cat#FHLC04700 |
| Zeiss Apotome image Z.1 | Zeiss | N/A |
| Automated freeze substitution device EM AFS2 | Leica | N/A |
| Cryostat | Leica | N/A |
| JEM 2100 | Jeol | N/A |
| High-pressure freezing device | Leica | N/A |
| Leica Reichert Jung Ultratrim device | Leica | N/A |
| Vibratome VT 1200S | Leica | N/A |
| Tissue chopper | McIlwain | N/A |
| TCS-SP5 Confocal Microscope | Leica | N/A |
| Leo912Transmission electron microscope | Zeiss | N/A |
| UC7 Ultramicrotome | Leica | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benjamin Cooper (cooper@em.mpg.de). This study did not generate new unique reagents.
Experimental Model and Subject Details
Mouse Breeding
Mouse breeding and transcardial perfusion was done with permission of the Niedersachsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES; 33.19.42502-04-15/1817 and 33.19-42502-04-18/2756). All animals were kept according to the European Union Directive 63/2010/EU and ETS 123. All wild-type animals used in this study were C57BL/6N. Mice were housed in individually ventilated cages (type II superlong, 435 cm2 floor area; TECHNIPLAST) under a 12 h light/dark cycle at 21 ± 1°C with food and water ad libitum. The health status of the animals was checked regularly by animal care technicians and a veterinarian. Mice lacking Munc13-1 (Unc13A) and Munc13-2 (Unc13B) (Augustin et al., 1999, Varoqueaux et al., 2002) and control littermates were generated from crossing animals with the genotype Unc13A+/− (Munc13-1) Unc13B+/− (Munc13-2) with Unc13A+/− Unc13B−/−. Control animals with the genotypes Unc13A+/− Unc13B+/− and Unc13A+/+ Unc13B+/− were used for tomographic analysis. Slices were prepared from wild-type animals postnatal day (P) 3-7 and from Munc13-1/2 double knockout and littermate control animals at embryonic day 18 (E18) due to the severe perinatally lethal phenotype (Varoqueaux et al., 2002). The following time points were included into the analysis: DIV14 (refers to culture experiments performed on DIV13-16), DIV28 (DIV27/28), P18 (refers to acute slice preparations performed on P17/P18), and P28 (refers to transcardial perfusions performed on P27/P28). The P18 time point for the acute preparations was chosen to match the exact age of wild-type DIV14 slice cultures that were prepared on P3-P6 (see below). Each perfusion protocol was performed on two wild-type animals. Animals of both genders were analyzed.
Tissue Culture
Hippocampal organotypic slice cultures were prepared using the interface method (Stoppini et al., 1991) according to previously published protocols (Imig et al., 2014, Studer et al., 2014). We chose the organotypic slice culture approach over other preparations because it offers several key advantages for ultrastructural analyses of distinct hippocampal synapse types: (i) Cultured slices recover from trauma induced during sectioning, (ii) slice dimensions are compatible with high-pressure freezing cryofixation, yielding a near-native preservation of synaptic ultrastructure (Imig and Cooper, 2017, Studer et al., 2014), (iii) synaptic properties, including short-term plasticity characteristics, remain largely intact (Galimberti et al., 2006, Mori et al., 2004, De Simoni et al., 2003), (iv) the functional and ultrastructural properties of synaptic connections that are difficult to reinstate in dissociated cultures, such as the hippocampal MF synapse, can be analyzed in an in situ-like environment, (v) even mouse mutants that die perinatally can be studied in detail, and (vi) pharmacological manipulations can be done in a very controlled experimental setting (Zhao et al., 2012).
Pregnant females at gestational stage E18 were anaesthetized and decapitated and embryos removed by hysterectomy. Pups were decapitated and the brain was quickly removed and placed into preparation medium (97 mL Hank’s balanced salt solution, 2.5 mL 20% glucose, and 1 mL 100 mM kynurenic acid, pH adjusted to 7.4). Both hippocampi were dissected and transferred with the entorhinal cortex attached onto a tissue chopper platform. Three hundred μm-thick hippocampal slices were cut perpendicular to the longitudinal axis of the hippocampus using a McIlwain tissue chopper and then quickly washed off the stage into preparation medium. Slices were carefully transferred onto sterile Millipore membrane confetti pieces that were placed on top of 6-well membrane inserts in culture medium (22.44 mL ddH2O, 25 mL 2xMEM, 25 mL BME, 1 mL GlutaMAX, 1.56 mL 40% Glucose, 25 mL horse serum). Residual preparation medium was removed from the inserts using a P200 pipette. A maximum of 4 hippocampal slices were cultured per membrane insert. Slice culture medium was changed 24 hours after preparation and then 2-3 times per week for the remaining culture period. Slices were cultured for either 14 or 28 days at 37°C and 5% CO2. In line with previous observations, we did not observe neurogenesis in the dentate gyrus in organotypic slices cultured in the presence of serum (Raineteau et al., 2004), as assessed by the lack of calretinin-positive immature cells (Brandt et al., 2003) in the sub-granular zone of the dentate gyrus (Figures S3Q–S3T).
Method Details
Experimental Design
Experiments from wild-type mice were performed on 3-4 independent slice cultures and from Munc13-1/2 double knockout and control cultures on two independent cultures due to the severity of the phenotype. The experimenter was blinded for the experiments that involved pharmacological treatments of organotypic slices. Electrophysiological recordings from CA3 pyramidal cells and morphological analyses from MF boutons were performed in the CA3b,c regions of the hippocampus.
High-Pressure Freezing, Automated Freeze Substitution, and Electron Microscopy Sample Preparation
Transcardial Perfusion
wild-type animals at P28 were given a lethal dose of Avertin (2,2,2,-Tribromoethanol) via intraperitoneal injection. Deeply anaesthetized animals were transcardially perfused first with 0.9% sodium chloride solution and then one of two fixatives [Fixative 1 (approximately 1900 mOsm (Hayat, 2000)): Ice-cold 4% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M phosphate buffer (PB), pH 7.4 (Rollenhagen et al., 2007); Fixative 2 (approximately 1200 mOsm (Hayat, 2000)): 37°C 2% paraformaldehyde, 2.5% glutaraldehyde, 2 mM CaCl2, in 0.1 M cacodylate buffer (Chicurel and Harris, 1992)].Fixative osmolalities were determined from literature (Hayat, 2000). Brains were removed from the animals and post-fixed in respective fixative overnight at 4°C. The brains were washed in ice-cold 0.1 M PB pH 7.4. Hundred-μm coronal hippocampal sections were cut using a Leica Vibratome (Leica VT 1200S, amplitude of 1.5 mm, cutting speed 0.1 mm/sec) in 0.1 M PB pH 7.4. Sections were stored in ice-cold 0.1 M PB and high-pressure frozen on the same day.
Acute brain slice preparation
Anaesthetized wild-type animals aged P18 were quickly decapitated and the brains were removed from the skull. One hippocampus was quickly dissected and placed onto a McIlwain tissue chopper stage. Two hundred-μm thick slices were cut perpendicular to the longitudinal axis of the hippocampus. Slices were washed into a Petri dish containing 20% bovine serum albumin (BSA) in HEPES-buffered artificial cerebrospinal fluid (ACSF). Slices were separated and the CA3 and CA1 regions were punched out of the hippocampal slice using a 1.5 mm diameter biopsy punch. The hippocampal regions were loaded into 3 mm aluminum planchettes (Leica Cat# 1677141 for type A and 1677142 for type B) and the remaining space was filled with cryoprotectant (20% BSA in ACSF) and immediately cryofixed within 5 min of decapitation. We did not attempt to recover slices after sectioning, because previous studies have demonstrated that prolonged incubation in ACSF deteriorates the freezing quality of brain tissue (Korogod et al., 2015; response to the reviewers). Only synapses that lacked morphological evidence of extensive stimulation (i.e., depletion of SVs in the terminal, endocytic pits) were analyzed.
High-pressure freezing of organotypic slices
Slices were changed to fresh slice culture medium 24 hours before high-pressure freezing. Immediately prior to freezing, untreated slices were transferred into warm, pre-equilibrated slice culture medium and excess membrane confetti were trimmed away from each slice. Slices were then transferred into non-penetrating 20% BSA cryoprotectant dissolved in culture medium. Slices were loaded into aluminum specimen carriers (type A, Leica Cat# 16770126, outer diameter 6 mm, inner cavity depth 100 μm) membrane-side up and filled with cryoprotectant. The filled carriers were loaded into middle plates of the high-pressure freezing sample holder. The flat side of type-B aluminum carriers (Leica Cat# 16770127) were coated with 1-hexadecene and placed flat-side down onto the sample-filled carrier to serve as “lids.” Excess liquid was removed with Whatman filter paper. The sample holder was then quickly assembled and loaded into the high-pressure freezing device (Leica HPM 100). Cryo-fixed samples were stored in liquid nitrogen until further processing.
Immersion fixation of Slice cultures
Hippocampal slice cultures were immersed at DIV28 in one of two fixatives [Fixative 1 (approximately 1900 mOsm (Hayat, 2000)): Ice-cold 4% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M phosphate buffer (PB), pH 7.4 (Rollenhagen et al., 2007); Fixative 2 (approximately 1200 mOsm (Hayat, 2000)): 37°C 2% paraformaldehyde, 2.5% glutaraldehyde, 2 mM CaCl2, in 0.1 M cacodylate buffer (Chicurel and Harris, 1992)].Fixative osmolalities were determined from literature (Hayat, 2000). Slices immersed in Fixative 1 were incubated overnight at 4°C with gentile agitation. Slices immersed in Fixative 2 were incubated at room temperature for 1 hour and then at 4°C overnight with gentile agitation. The slices were then washed thoroughly in 0.1 M PB (pH 7.4) and cryofixed in 20% BSA dissolved in 0.1 M PB (pH 7.4) as a cryoprotectant in tandem with untreated hippocampal slice cultures (DIV29, processed normally as described above).
Pharmacological silencing experiments
The protocol for acute pharmacological silencing of cultured slices was based on a previously published protocol for the application of drugs to organotypic slice cultures (Studer et al., 2014). Briefly, DIV14 organotypic slices were placed onto new, sterile membrane inserts in a 6-well plate containing fresh organotypic slice culture medium as well as different receptor and channel blockers: i) Vehicle control (VC, organotypic slice culture medium only); ii) T/N/A (medium containing 1 μM tetrodotoxin (TTX), 2 μM 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), and 50 μM D-(-)-2-Amino-5-phosphonopentanoic acid (D-AP5)); iii) T/D (medium containing 1 μM TTX and 2 μM (2S,2′R,3′R)-2-(2′,3′-Dicarboxycyclopropyl)glycine (DCG-IV)). Then, 50 μL drops containing T/N/A, T/D, or VC medium were pipetted onto slices and incubated at 37°C and 5% CO2 before slices were prepared for high-pressure freezing. The medium and cryoprotectant for subsequent steps (see above) always contained the respective drugs. Slices were frozen 10 min after drug exposure.
Pharmacological manipulation of presynaptic cAMP levels
DIV28 organotypic slices were transferred onto a new membrane insert in a six-well plate containing fresh organotypic slice culture medium containing: i) Vehicle control (1 μM TTX, ddH2O, and DMSO); ii) T/D (1 μM TTX, 2 μM DCG-IV, and DMSO); iii) T/F (1 μM TTX, ddH2O, and 25 μM forskolin). Slices were placed back in the incubator, exposed to the pharmacological treatment for 15 min, and were then again taken out of the incubator and prepared for high-pressure freezing (see above).
Automated freeze substitution
Automated freeze substitution was performed as previously described (Imig et al., 2014, Rostaing et al., 2006). Briefly, samples were incubated in 0.1% tannic acid in anhydrous acetone for 4 days at −90°C and then fixed with 2% osmium tetroxide in anhydrous acetone with the temperature slowly ramping up to 4°C over several days. Samples were washed in acetone and brought to room temperature for EPON infiltration and embedding. Ultimately, gel-capsules filled with 100% EPON were inverted on the sample carriers and polymerized at 60°C for 24-48 h. Polymerized blocks were removed from the glass slides and the aluminum carriers were carefully trimmed off of the blocks. With the exception of culture slices from E18 mice that fit into 3 mm aluminum carriers, blocks were further trimmed down to fit an electron microscopy grid.
Ultramicrotomy
Five hundred nm-thick sections were cut on a Leica Ultracut UCT ultramicrotome until tissue appeared and cell body lamination became apparent in semithick sections. Then, four to five 200 nm-thick semithin sections were cut and collected onto Formvar-filmed, carbon-coated and glow-discharged copper mesh grids for electron tomography. Subsequently, a few ultrathin sections (60 nm-thick) were collected and contrasted with 1% aqueous uranyl acetate and 0.3% Reynold’s lead citrate to assess the ultrastructural preservation of the sample. Protein A (ProtA, Cell Microscopy Center, Utrecht, the Netherlands) coupled to 10 nm gold particles were applied to semithin sections to serve as fiducial markers for tomographic reconstructions.
Electron Tomography and Data Analysis
MF synapses were identified by their distinct morphology (large boutons, multiple AZs) and target specificity (primary dendrites of CA3 pyramidal cells) (Chicurel and Harris, 1992). Synapses were selected for tomography when the postsynaptic density was juxtaposed to a cluster of SVs in the presynaptic compartment and the synaptic cleft was parallel to the tilt axis and clearly visible at 0° stage tilt. For the analysis of MF-CA3 pyramidal cell AZs, electron tomography is especially helpful as spines often exhibit complex shapes and highly convoluted membranes, which may confound the accurate detection of SV docking in 2D projection images acquired from ultrathin sections (Imig and Cooper, 2017). Our comparative electron tomography analysis of SC and MF AZ organization was restricted to release sites onto dendritic spines.
Glutamatergic Schaffer collateral spine synapses were identified based on their location within the CA1 region of the hippocampal slice and according to well-established ultrastructural features such as the presence of a small postsynaptic compartment lacking mitochondria or microtubules (Imig et al., 2014).
Single-axis tilt series were acquired on a JEOL JEM-2100 200kV transmission electron microscope from −60° to + 60° in 1° increments and binned by a factor of two at 30,000 times magnifications with an Orius SC1000 camera (Gatan) using SerialEM for acquiring automated tilt series (Mastronarde, 2005). Tomograms were reconstructed and binned by a factor of three (1.554 nm isotropic voxel size of the final tomogram) and segmented for analysis using the IMOD package (Kremer et al., 1996). All vesicles were segmented manually as perfect spheres with the center being placed into the tomographic slice in which the vesicle diameter appeared to be the largest (i.e., when the vesicle is cut at its midline). The sphere outline was adjusted to lie on the center of the outer leaflet of the vesicle lipid bilayer. Non-spherical organelles (e.g., endoplamisc reticulum, tubular endosomal intermediates) were occasionally observed in tomographic reconstructions and excluded from the analysis. The AZ was defined as the region of presynaptic membrane that was apposed to the postsynaptic density. In cryopreserved tissue, postsynaptic densities often appeared less electron-dense than in chemically fixed material (see for example Figure S1). Therefore we used the widening of the synaptic cleft as a second morphological feature on which to base the AZ area for segmentation. The AZ was segmented at the center of the inner leaflet of the presynaptic lipid bilayer.
In segmented models, the shortest distance between vesicle membranes and the AZ were calculated using the mtk program of the IMOD package (Kremer et al., 1996). Vesicle diameters and AZ surface areas were extracted from segmented models using the imodinfo program. A vesicle was defined as docked when there was no measurable distance between the outer leaflet of the vesicle and the inner leaflet of the lipid bilayer (i.e., when the dark pixels corresponding to the vesicular outer leaflet were contiguous with those of the inner plasma membrane leaflet). Based on the voxel size of 1.554 nm, these ‘docked’ vesicles fall into the 0-2 nm bin. The number of vesicles in discrete bins (i.e., 0-2 nm, 0-40 nm, and 0-100 nm from the AZ membrane) were normalized to the measured AZ area and reported as number of vesicles per 0.01 μm2 AZ. To allow a more direct comparison of our results to data obtained from 2D-analyses of SV docking (Chang et al., 2018), we also report the number of SVs within 5 nm of the AZ membrane for each condition analyzed. Vesicles with a diameter less than 60 nm were classified as SVs in the analysis, whereas vesicles with a diameter exceeding 60 nm and having no prominent dense core were classified as giant vesicles (giant vesicles). The 60 nm cutoff was chosen, because it exceeded three standard deviations from the mean SV diameter measured in SC synapses (mean: 43.44 nm, standard deviation: 3.92 nm). Vesicles that contained a prominent electron-dense core were considered dense-cored vesicles (DCVs). For illustrative purposes, figures depicting tomographic sub-volumes represent an overlay of seven consecutive tomographic slices (10.88 nm-thick sub-volume) unless otherwise specified and were generated using the IMOD package (Kremer et al., 1996).
2-Dimensional EM analysis
Electron microscopic images taken at 5,000 x magnification on a Leo912 transmission electron microscope (Zeiss). The CA3 region of slice cultures treated for 15 minutes with either vehicle control (VC; 1 μM TTX), TTX and forskolin (T/F; 1 μM TTX and 25 μM forskolin), or TTX and DCG-IV (T/D; 1 μM TTX and 2 μM DCG-IV) prior to cryofixation were imaged. From these images, MF boutons were identified as having large vesicle-filled presynaptic boutons with multiple AZs (marked by the presence of PSDs in the post-synaptic cell). From these images, boutons, presynaptic mitochondria, AZs, postsynaptic complex spines and bouton-spine contact areas were segmented using the IMOD package (Kremer et al., 1996). All measured parameters were then extracted from the segmented model using the imodinfo program. Closest approach of mitochondria to AZs in a given bouton was determined by the mtk program in the IMOD package (Kremer et al., 1996).
RRP Calculations
In MF-CA3 synapses, calculations of mean docked vesicle numbers per AZ and per MF bouton were based on our estimates of the mean number of docked vesicles per unit AZ area (0.9 SVs, 0.1 giant vesicles and 0.04 DCVs per 0.01 μm2), and previously published estimates of the mean MF AZ surface area (0.12 μm2) and mean AZ number (29.75 AZs) per MF bouton in P28 rat MF boutons (Rollenhagen et al., 2007). Our calculations of total docked vesicle numbers per MF bouton neglect filopodial extensions. We calculated the mean docked vesicle numbers per AZ (10.7 SVs, 1.2 giant vesicles, 0.5 DCV) and per MF bouton (~320 SVs, 37 giant vesicles, 15 DCVs) in more mature (DIV28) MF synapses as well as the mean number of total membrane-proximal (within 0-40 nm of the AZ; 24.6 SVs, 4 giant vesicles, 2.6 DCVs) vesicles per AZ. Electron tomography further enabled precise volume and membrane surface area measurements for all docked vesicles of a given morphological type (mean diameter; SV, 45.17 nm; giant vesicle, 85.77 nm; DCV 74.41 nm). We further assumed a specific membrane capacitance of 1 μF/cm2 (Hallermann et al., 2003) to determine that the fusion of all docked vesicles, irrespective of their type, would change the membrane capacitance (ΔCm) by ~32 fF per MF bouton and that the fusion of all docked and tethered vesicles together would correspond to a change in ΔCm by ~88 fF.
Electrophysiology
All recordings of mEPSCs from CA3 pyramidal cells in organotypic slice cultures were performed at DIV14. Prior to recordings, slices were incubated for 30 min in an interface chamber with carbogen-saturated ACSF (120 mM NaCl, 26 mM NaHCO3, 10 mM D-glucose, 2 mM KCl, 2 mM MgCl2, and 2 mM CaCl2, and 1 mM KH2PO4 - 304 mOsm). One or two CA3 pyramidal cells were then whole-cell voltage clamped using a glass pipette (2.5-3.0 MΩ) filled with internal solution (100 mM KCl, 50 mM K-gluconate, 10 mM HEPES, 4 mM ATP-Mg, 0.3 mM GTP-Na, 0.1 mM EGTA, and 0.4% biocytin, pH 7.4, 300 mOsm) and the holding potential was set at −70 mV using an EPC-10 amplifier [Patchmaster 2 software (HEKA/Harvard Bioscience)]. For measurements of mEPSC amplitudes and frequencies, slices were initially perfused with 1 μM TTX and 10 μM bicuculline and mEPSCs were then recorded for 10 min, after which the slices were perfused with 1 μM TTX, 10 μM bicuculline, and 2 μM DCG-IV for 15 min to record DCG-IV insensitive mEPSCs. Measurements of all mEPSCs (TTX/bicuculline) were recorded in two-5 min epochs, while measurements of non-MF mEPSCs (TTX/bicuculline/DCG-IV) were recorded in three-5 min epochs. The last epochs of each recording were used for mEPSC analysis. All electrophysiological traces were analyzed using Axograph X software (AxoGraph Scientific) using a template fit algorithm for automatic event detection (Jonas et al., 1993, Pernía-Andrade et al., 2012). After recordings, some slices were fixed and biocytin-filled CA3 pyramidal cell were stained with Alexa Fluoro-555-labeled streptavidin (see Light Microscopic Analysis section for detailed procedure).
Only cells exhibiting a reduction in the mEPSC frequency upon application of DCG-IV were included in the analysis (reduction range 16.9%–88.6%; mean 59.2%). and the threshold for mEPSC detection was set to 8 pA. The amplitude distribution of mEPSC events was plotted (1 pA bins) for epochs prior to and following the application of DCG-IV. DCG-IV-insensitive events were subtracted from the respective bins in the distribution recorded prior to drug application to isolate the DCG-IV-sensitive component of MF origin. Operating on the assumption that DCG-IV-sensitive mEPSC events result from the fusion of docked vesicles at MF AZ release sites, the statistical mode of mEPSC amplitudes (10 pA) was correlated with the statistical mode of docked vesicle diameters (44 nm). Vesicle diameters were measured between the outer leaflets of the vesicle lipid bilayer and lumenal volumes were calculated by subtracting the thickness of the lipid bilayers (each approximately 4 nm-thick as measured from center-to-center of inner and outer leaflets). Based on a previous, conceptually analogous study (Bruns et al., 2000), we assumed uniform neurotransmitter loading irrespective of vesicle size and negligible saturation of postsynaptic receptors to estimate that an mEPSC amplitude of approximately 30 pA would arise from a quantum released from a vesicle with a diameter of 60 nm.
Whole-cell voltage-clamp recordings of evoked EPSCs in CA1 and CA3 neurons of organotypic slice cultures were performed by extracellular stimulation of SCs and MFs, respectively. Briefly, short (0.1 ms) current pulses (100-1000 μA) were applied using a bipolar theta glass electrode (Science Products, Hofheim, Germany) filled with ACSF. All whole-cell recordings were made at −70mV in the presence of 100 μM picrotoxin to block inhibitory currents. Borosilicate glass pipettes (4-5 MΩ; P.Clamp Glass #0010, WPI) were filled with intracellular solution (115 mM Cs-methanesulfonate, 10 mM HEPES, 10 mM EGTA, 5 mM MgCl2, 5 mM QX-314, 4 mM Na2-ATP, 0.3 mM Na2-GTP, and 0.2% biocytin, PH 7.4). The serial resistance was compensated by 45%–80% and only cells with serial resistances below 12 MΩ were analyzed. All experiments were recorded by an EPC-10 USB double amplifier and Patch-Master (Ver. 2X73.5) software (HEKA electronics) and the obtained data were analyzed using Axograph (Ver.1.5.4) software (Axograph Scientific). Statistical analysis of data was performed with GraphPad Prism (Ver. 5 or 7) software (GraphPad Software Inc., La Jolla, CA).
Light Microscopic Analysis
To demonstrate the correct anatomical organization of the MF pathway in organotypic slices, slices were removed from culture inserts and fixed by overnight immersion in 4% paraformaldehyde in 0.1 M PB (pH 7.4). Slices were washed in 0.1 M PB (pH 7.4) and then incubated overnight at 4°C in 10% normal goat serum (NGS), 0.3% Triton X-100, and 0.1% cold water fish skin gelatin in 0.1 M PB (pH 7.4) to permeabilize membranes and block non-specific binding sites. Slices were incubated overnight at 4°C in 5% NGS, 0.3% Triton X-100 and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4) containing primary antibodies against SV clusters within the synaptic terminals of MF projections [polyclonal rabbit anti-Synaptoporin, SYnaptic SYstems (Cat# 102 003), 1:1000 dilution] and cell bodies and dendritic arborizations [polyclonal chicken anti-MAP2, Novus Biologicals (Cat# NB300-213), 1:600 dilution]. Slices were washed in 0.1 M PB (pH 7.4) and primary antibodies were visualized by a 2 hour incubation at room temperature in 5% NGS, 0.1% Triton X-100 and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4) containing goat anti–rabbit Alexa 555 [Thermo Fisher (Cat# A21429), dilution 1:1000] and goat anti-chicken Alexa 488 [Thermo Fisher(Cat# A-11039), dilution 1:1000]. Following final washing steps in 0.1 M PB (pH 7.4), slices were floated onto Superfrost glass slides with the membrane confetti in contact with the slide and Menzel-Gläser #1.5 glass coverslips were mounted using Aqua-Poly/Mount mounting medium (Polysciences, Inc., Cat# 18606-20).
To localize AZ release sites within MF boutons, slices were removed from culture inserts and fixed by overnight immersion in 4% paraformaldehyde in 0.1 M PB (pH 7.4). Slices were washed in 0.1 M PB (pH 7.4) and then cryoprotected by immersion in an increasing gradient of 10%, 20%, and 30% sucrose in 0.1 M PB (pH 7.4) until saturation. Slices were placed flat, slice-side down (confetti-side up), on the inner base of a quadratic 10 × 10 × 10 mm form made out of aluminum foil and carefully covered with liquid Tissue-Tek® OCT compound (Sakura, Cat# 4583) without introducing air bubbles. The OCT-filled form was then rapidly frozen on a liquid nitrogen-cooled aluminum block, the foil was removed and the frozen OCT block was mounted slice-side up on a specimen stub with OCT in a precooled (specimen holder, −18°C; chamber, −18°C) cryostat (Leica CM3050 S). Once the temperature of the embedded slice had equilibrated, unnecessary OCT compound was trimmed away with a razor blade and 10 μm-thick cryosections were made through the organotypic slice with the aid of a glass anti-roll plate and thaw-mounted on Superfrost slides. Slides were air-dried at room temperature for 30 min and a hydrophobic pen (DAKO, Cat# S2002) was used to delineate the border of the slide surface. Slides were washed briefly in 0.1 M PB (pH 7.4) and incubated 90 min at room temperature in 10% NGS, 0.3% Triton X-100, and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4). Slices were then incubated overnight at 4°C in 3% NGS, 0.3% Triton X-100 and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4) containing primary antibodies for the detection of SV clusters within the synaptic terminals of MF projections [polyclonal rabbit anti-Synaptoporin, SYnaptic SYstems (Cat# 102 003), 1:1000 dilution] and presynaptic AZs [monoclonal mouse anti-Bassoon, Enzo Life Sciences (Cat# SAP7F407), 1:400 dilution]. Slices were washed in 0.1 M PB (pH 7.4) and primary antibodies were visualized by 2 hr incubation at room temperature in 5% NGS, 0.1% Triton X-100 and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4) containing goat anti-rabbit Alexa 488 [Thermo Fisher (Cat# A11008), dilution 1:1000] and goat anti-mouse Alexa 555 [Thermo Fisher (Cat# A21424), dilution 1:1000]. Following a brief wash in 0.1 M PB, slides were dipped in distilled water and Menzel-Gläser #1,5 coverslips were mounted using Aqua-Poly/Mount mounting medium (Polysciences, Inc., Cat# 18606-20).
Confocal light microscopic analysis of biocytin-filled CA3 pyramidal cells was performed to validate that electrophysiological recordings were of the correct, as assessed by the anatomical location within the hippocampal subfields and morphological features (i.e, pyramidal soma, presence of large, complex spines in the proximal regions of apical dendritic arborizations) of the filled cell (see Figure S1D). Immediately following mEPSC recordings and removal of the patch pipette, biocytin-filled CA3 pyramidal cells (see Electrophysiology section above for detailed procedure) were fixed for light microscopic analysis by overnight immersion of the slice in 4% paraformaldehyde in 0.1 M PB (pH 7.4). Slices were washed in 0.1 M PB (pH 7.4) and then incubated overnight at 4°C in 10% NGS, 0.3% Triton X-100, and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4). Biocytin-filled cells were visualized by incubation of slices for 3 hr at room temperature in Steptavidin-Alexa 555 [1:500 dilution] in 5% NGS, 0.1% Triton X-100 and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4). Slices were washed in 0.1 M PB (pH 7.4) and cell nuclei were labeled by a 30 min incubation in DAPI [300 nM in 0.1 M PB]. Following final washing steps in 0.1 M PB (pH 7.4), slices were floated onto Superfrost glass slides with the membrane confetti in contact with the slide and Menzel-Gläser #1,5 glass coverslips were mounted using Aqua-Poly/Mount mounting medium (Polysciences, Inc., Cat# 18606-20).
Confocal laser scanning micrographs of were acquired with a Leica TCS-SP5 confocal microscope equipped with a tunable white light laser, a resonant scanner, hybrid GaAsP detectors, and a motorized stage. Tiled z series were acquired with (i) a HCX PL APO 40.0x (NA = 1.25) oil immersion objective to generate low magnification overviews of entire organotypic slices (pinhole = 3.0 AU, voxel size x, y, z = 0.3, 0.3, 2 μm) (Figure S1A) and reconstructions of biocytin-filled pyramidal neurons within CA3 stratum pyramidale (pinhole = 1.0 AU, voxel size x, y, z = 95, 95, 335 nm) (Figure S1D), or with (ii) a HCX PL APO CS 100x (NA = 1.4) oil immersion objective to visualize MF terminals within CA3 stratum lucidum (pinhole = 1.0 AU, voxel size x, y, z = 89, 89, 130 nm) and high magnification reconstructions of complex postsynaptic spines (thorny excrescences) emerging from the proximal dendrites of biocytin-filled CA3 pyramidal neurons (pinhole = 0.5 AU, voxel size x, y, z = 47, 47, 130 nm). For illustration purposes thorny excrescences were subjected to spatial deconvolution by use of two ImageJ (National Institutes of Health; Bethesda, MD) plugins: point spread functions (PSF) were generated using Diffraction PSF 3D plugin and iterative deconvolution was performed with the Richardson-Lucy algorithm (DeconvolutionLab plugin; Biomedical Imaging Group, EPFL; Lausanne, Switzerland).
STED microscopy
Hippocampal slice cultures were treated at DIV28 for 15 minutes with either vehicle control (1 μM TTX) or 25 μM forskolin in the presence of 1 μM TTX. Slices were transferred to a new Millipore membrane insert containing the respective drug mixtures and 50 μl of drug-containing medium was pipetted onto each slice. The plate was then placed in the incubator for 15 minutes, after which the slices adhering to the membrane confetti were removed from the inserts and were immersed in 4% paraformaldehyde in 0.1 M PB (pH 7.4) for 1 hour at 4°C. Slices were prepared for cryosectioning as described above with some exceptions. Sections (10 μm-thick) were thaw-mounted on Superfrost slides, blocked and permeabilized for 1 hour at room temperature in 5% NGS, 0.3% Triton X-100 and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4), and were then incubated overnight at 4°C in 3% NGS, 0.1% Triton X-100, and 0.1% fish skin gelatin in 0.1 M PB (pH 7.4) containing primary antibodies for Synaptoporin [polyclonal rabbit anti-Synaptoporin, SYnaptic SYstems (Cat# 102 003), 1:500 dilution] and Bassoon [monoclonal mouse anti-Bassoon, Enzo Life Sciences (Cat# SAP7F407), 1:400 dilution]. Sections were washed in 0.1 M PB (pH 7.4) to remove primary antibodies and were then incubated for 2 hours at room temperature in 3% NGS, 0.1% Triton X-100 and 0.1% FSG in 0.1 M PB (pH 7.4) containing goat anti-mouse ATTO647N [Rockland (Cat# 610-156-12), dilution 1:100] and goat anti-rabbit STAR580 [Abberior (Cat# ST580-1002-500UG), dilution 1:100]. Following a brief wash in 0.1 M PB, slides were dipped in distilled water and Menzel-Gläser #1,5 coverslips were mounted onto them, covering the sections, using Mowiol mounting medium [Merk Millipore (Cat# 475904)].
Dual STED imaging was performed on Bassoon and Synaptoporin stainings [using an Expert Line STED (Abberior) instrument based on an IX83 inverted microscope (Olympus)]. The images were analyzed with a MATLAB script (Mathworks) written by Sinem M. Sertel. The Synaptoporin and Bassoon images were thresholded to remove background, and to enable the recognition of the different objects (spots). The positions of Synaptoporin and Bassoon objects were then calculated, and for each Synaptoporin object we determined the overlap with the Basson objects found near it (within 120 nm). The total area of Bassoon objects per Synaptoporin object was then determined, and the mean object value per image was plotted using GraphPad Prism 7.
Quantification and Statistical Analysis
Data are represented as mean ± SEM unless indicated otherwise. Statistical analyses were carried out using GraphPad Prism software 7 (∗ when p < 0.05; ∗∗ when p < 0.01, and ∗∗∗ when p < 0.001). For comparisons of two conditions (i.e., SC and MF synaptic profiles from DIV14 wild-type slice cultures; Figures 2A–2G) statistical difference were determined by an unpaired t test when the dataset was normally distributed as determined by a KS normality test, and by a Mann-Whitney unpaired t test if it was not normally distributed. For pharmacological manipulation experiments, statistical significance was tested by one-way ANOVA with Bonferroni correction as a post-test if the dataset was normally distributed. If the dataset was not normally distributed, Kruskal-Wallis ANOVA test with a Dunn’s comparison of all columns was performed as a post-test to probe for statistical significance. For the electron tomography experiments, the number of AZs analyzed for each experiment (n), the number of slice cultures or animals used (N), and all electron microscopy data is summarized in Table S1. Statistics were performed based on the number of AZs for each sample with the exception of docked vesicle diameters and unattached giant vesicles and DCVs. In the latter scenarios, the number of vesicles was used for statistical analysis and is noted in parentheses. For the 2D analysis of MF synaptic profiles, the number of boutons (n), the number of slice cultures or animals used (N), and all data is summarized in Table S1. Electrophysiological of mEPSC recordings were performed on 28 cells from two independent wild-type slice cultures at DIV14. Statistical difference for electrophysiological experiments was determined by Wilcoxon matched pairs signed rank tests. For the short-term synaptic plasticity electrophysiology experiments, statistical comparisons between two groups of data were made using two-tailed unpaired Student’s t test. p values less than 0.05 were considered significant for single and multiple comparisons, respectively. Data were presented as means ± SEM.
Data and Code Availability
Any raw data supporting the current study is available from the Lead Contact upon request. This study did not generate any code.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB1286/A01 to B.H.C. and EXC 2067/1- 390729940 under Germany’s Excellence Strategy to N.B.) and the European Commission (ERC Advanced Grant SynPrime, to N.B.). We thank P. Jonas, N. Holderith, and V. O’Connor for helpful comments on the manuscript; H. Taschenberger for discussions and advice; and M. Schwark and S. Beuermann for excellent technical support.
Author Contributions
Conceptualization, N.B., C.I., and B.H.C.; Methodology, C.I. and B.H.C.; Formal Analysis, L.M., C.L., S.M.S., and K.W.; Investigation, L.M., C.L., B.A., S.M.S., C.I., and B.H.C.; Resources, J.R., S.O.R., and N.B.; Writing – Original Draft, L.M., N.B., C.I., and B.H.C.; Writing – Review & Editing, all authors; Visualization, L.M., C.L., C.I., and B.H.C.; Supervision, N.B., C.I., and B.H.C.; Funding Acquisition, N.B. and B.H.C.
Declaration of Interests
The authors declare no competing interests.
Published: March 17, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.083.
Contributor Information
Cordelia Imig, Email: imig@em.mpg.de.
Benjamin H. Cooper, Email: cooper@em.mpg.de.
Supplemental Information
(A) Comparison of the paired pulse ratio (PPR) of Schaffer collateral and MF synapses in organotypic slices at DIV14 and DIV28. Related to Figures S1B and S1C. (B) Comparison of Schaffer collateral and MF synapses in organotypic slices frozen at DIV14. Related to Figures 2A–2H. (C) Comparison of SC and MF synapses in acute hippocampal slices frozen at P18. Related to Figures S2A–S2H. (D) Comparison of Schaffer collateral and MF synapses in organotypic slices frozen at DIV28. Related to Figures 2I–2P. (E) Comparison of MF synapses in organotypic slices frozen at DIV28 (HPF) and from transcardially perfused P28 WT mice using perfusion fixative 1 (PF1; 4% PFA, 2.5% GA in 0.1 M PB) or PF2 (2% PFA, 2.5% GA, 2 mM CaCl2 in 0.1 M cacodylate buffer). Related to Figures S2M–S2P. (F) Comparison of DIV28 MF synapses in slices from the same organotypic slice culture processed by high-pressure freezing and immersion-fixed using immersion fixative 1 (IF1; 4% PFA, 2.5% GA in 0.1 M PB) or IF2 (2% PFA, 2.5% GA, 2 mM CaCl2 in 0.1 M cacodylate buffer). Related to Figures S2Q–S2S. (G) Comparison of M13 CTRL and DKO MF synapses in organotypic slices frozen at DIV28. Related to Figures 3E–3L. (H) Analysis of MF synapses in pharmacologically silenced organotypic slices frozen at DIV14. Vehicle control (VC; culture medium), 1 μM TTX, 2 μM NBQX, and 50 μM APV (T/N/A), or 1 μM TTX and 2 μM DCG-IV (T/D). Related to Figures S3A–S3G. (I) Electrophysiological recordings of mEPSC frequency and amplitude in CA3 pyramidal cells before and after application of 2 μM DCG-IV in slice cultures at DIV14. Control recordings were made in the presence of 1 μM TTX and 10 μM bicuculline. Related to Figures 3N–3S. (J) Morphometric analysis using two-color STED microscopy of Bassoon-immunoreactive AZs in Synaptoporin-reactive MF boutons. Related to Figures 4A–4C and 4E. (K) Analysis of MF synaptic bouton profiles in 2D images of ultrathin sections after pharmacological manipulation of presynaptic cAMP levels in organotypic slices frozen at DIV28. Vehicle control (VC; culture medium containing 1 μM TTX) or 1 μM TTX and 25 μM forskolin (T/F) Related to Figures 4D, 4F, and 4G. (L) Pharmacological manipulation of presynaptic cAMP levels in MF synapses of organotypic slices frozen at DIV28. Vehicle control (VC; culture medium containing 1 μM TTX), 1 μM TTX and 25 μM forskolin (T/F), or 1 μM TTX and 2 μM DCG-IV (T/D). Related to Figures 4H–4O. (M) Analysis of MF and Schaffer collateral synaptic bouton profiles in 2D images of ultrathin sections. Related to Figure 4. Abbreviations: A, APV; AZ, active zone; C, control; D, DCG-IV; DCV, dense core vesicle; DIV, days in vitro; DKO, double knock-out; F, forskolin; GA, glutaraldehyde; GV, giant, clear-cored vesicle (Ø > 60 nm); IF, immersion fixation; MF, mossy fiber; N = , number of slice cultures or mice; N, NBQX; n = , number of tomograms unless indicated otherwise; P, postnatal day; PB, phosphate buffer; PF, perfusion fixative; PFA, paraformaldehyde; PPR, paired pulse ratio; SC, Schaffer collateral; SV, small, clear-cored synaptic vesicle; T, TTX; TTX, tetrodotoxin; VC, vehicle control; versus, versus; WT, wild-type. All values are represented as mean ± SEM. The number of tomograms was used for statistical analysis unless otherwise specified in the table. Green p values: Normally distributed data sets. (Two conditions: Student’s unpaired t test; More than two conditions: One-way ANOVA plus Bonferroni’s multiple comparison post-test). Blue p values: Not normally distributed data sets. (Two conditions: Mann-Whitney U test; More than two conditions: Kruskal-Wallis test plus Dunn’s comparison post-test; Electrophysiology: Wilcoxon matched-pairs signed rank test).
References
- Amaral D.G., Dent J.A. Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J. Comp. Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- Augustin I., Rosenmund C., Südhof T.C., Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Battistin T., Cherubini E. Developmental shift from long-term depression to long-term potentiation at the mossy fibre synapses in the rat hippocampus. Eur. J. Neurosci. 1994;6:1750–1755. doi: 10.1111/j.1460-9568.1994.tb00567.x. [DOI] [PubMed] [Google Scholar]
- Blatow M., Caputi A., Burnashev N., Monyer H., Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Brandt M.D., Jessberger S., Steiner B., Kronenberg G., Reuter K., Bick-Sander A., von der Behrens W., Kempermann G. Transient calretinin expression defines early postmitotic step of neuronal differentiation in adult hippocampal neurogenesis of mice. Mol. Cell. Neurosci. 2003;24:603–613. doi: 10.1016/s1044-7431(03)00207-0. [DOI] [PubMed] [Google Scholar]
- Bruns D., Riedel D., Klingauf J., Jahn R. Quantal release of serotonin. Neuron. 2000;28:205–220. doi: 10.1016/s0896-6273(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Chang S., Trimbuch T., Rosenmund C. Synaptotagmin-1 drives synchronous Ca2+-triggered fusion by C2B-domain-mediated synaptic-vesicle-membrane attachment. Nat. Neurosci. 2018;21:33–40. doi: 10.1038/s41593-017-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicurel M.E., Harris K.M. Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J. Comp. Neurol. 1992;325:169–182. doi: 10.1002/cne.903250204. [DOI] [PubMed] [Google Scholar]
- Cho R.W., Buhl L.K., Volfson D., Tran A., Li F., Akbergenova Y., Littleton J.T. Phosphorylation of Complexin by PKA Regulates Activity-Dependent Spontaneous Neurotransmitter Release and Structural Synaptic Plasticity. Neuron. 2015;88:749–761. doi: 10.1016/j.neuron.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni A., Griesinger C.B., Edwards F.A. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J. Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Éltes T., Kirizs T., Nusser Z., Holderith N. Target Cell Type-Dependent Differences in Ca2+ Channel Function Underlie Distinct Release Probabilities at Hippocampal Glutamatergic Terminals. J. Neurosci. 2017;37:1910–1924. doi: 10.1523/JNEUROSCI.2024-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti I., Gogolla N., Alberi S., Santos A.F., Muller D., Caroni P. Long-term rearrangements of hippocampal mossy fiber terminal connectivity in the adult regulated by experience. Neuron. 2006;50:749–763. doi: 10.1016/j.neuron.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Geiger J.R.P., Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Hallermann S., Pawlu C., Jonas P., Heckmann M. A large pool of releasable vesicles in a cortical glutamatergic synapse. Proc. Natl. Acad. Sci. USA. 2003;100:8975–8980. doi: 10.1073/pnas.1432836100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M., Palfreyman M.T., Watanabe S., Olsen S., Jorgensen E.M. Open syntaxin docks synaptic vesicles. PLoS Biol. 2007;5:e198. doi: 10.1371/journal.pbio.0050198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K.M., Stevens J.K. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Xue L., Xu J., McNeil B.D., Bai L., Melicoff E., Adachi R., Wu L.-G. Compound vesicle fusion increases quantal size and potentiates synaptic transmission. Nature. 2009;459:93–97. doi: 10.1038/nature07860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat M.A. Principles and Techniques of Electron Microscopy : Biological Applications. Cambridge University Press; 2000. [Google Scholar]
- He E., Wierda K., van Westen R., Broeke J.H., Toonen R.F., Cornelisse L.N., Verhage M. Munc13-1 and Munc18-1 together prevent NSF-dependent de-priming of synaptic vesicles. Nat. Commun. 2017;8:15915. doi: 10.1038/ncomms15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helassa N., Dürst C.D., Coates C., Kerruth S., Arif U., Schulze C., Wiegert J.S., Geeves M., Oertner T.G., Török K. Ultrafast glutamate sensors resolve high-frequency release at Schaffer collateral synapses. Proc. Natl. Acad. Sci. USA. 2018;115:5594–5599. doi: 10.1073/pnas.1720648115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henze D.A., Card J.P., Barrionuevo G., Ben-Ari Y. Large amplitude miniature excitatory postsynaptic currents in hippocampal CA3 pyramidal neurons are of mossy fiber origin. J. Neurophysiol. 1997;77:1075–1086. doi: 10.1152/jn.1997.77.3.1075. [DOI] [PubMed] [Google Scholar]
- Henze D.A., McMahon D.B.T., Harris K.M., Barrionuevo G. Giant miniature EPSCs at the hippocampal mossy fiber to CA3 pyramidal cell synapse are monoquantal. J. Neurophysiol. 2002;87:15–29. doi: 10.1152/jn.00394.2001. [DOI] [PubMed] [Google Scholar]
- Henze D.A., Wittner L., Buzsáki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat. Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Imig C., Cooper B.H. 3D analysis of synaptic ultrastructure in organotypic hippocampal slice culture by high-pressure freezing and electron tomography. Methods Mol. Biol. 2017;1538:215–231. doi: 10.1007/978-1-4939-6688-2_15. [DOI] [PubMed] [Google Scholar]
- Imig C., Min S.-W., Krinner S., Arancillo M., Rosenmund C., Südhof T.C.C., Rhee J., Brose N., Cooper B.H.H. The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron. 2014;84:416–431. doi: 10.1016/j.neuron.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Jackman S.L., Turecek J., Belinsky J.E., Regehr W.G. The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature. 2016;529:88–91. doi: 10.1038/nature16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P., Major G., Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J. Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser P.S., Regehr W.G. The readily releasable pool of synaptic vesicles. Curr. Opin. Neurobiol. 2017;43:63–70. doi: 10.1016/j.conb.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H., Ozawa S. Dual mechanism for presynaptic modulation by axonal metabotropic glutamate receptor at the mouse mossy fibre-CA3 synapse. J. Physiol. 1999;518:497–506. doi: 10.1111/j.1469-7793.1999.0497p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H., Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fibre synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- Kamiya H., Shinozaki H., Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J. Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Tsien R.W. Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron. 2008;58:925–937. doi: 10.1016/j.neuron.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N., Petersen C.C., Knott G.W. Ultrastructural analysis of adult mouse neocortex comparing aldehyde perfusion with cryo fixation. eLife. 2015;4:e05793. doi: 10.7554/eLife.05793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J.R., Mastronarde D.N., McIntosh J.R.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kwon S.-K., Sando R., 3rd, Lewis T.L., Hirabayashi Y., Maximov A., Polleux F. LKB1 Regulates Mitochondria-Dependent Presynaptic Calcium Clearance and Neurotransmitter Release Properties at Excitatory Synapses along Cortical Axons. PLoS Biol. 2016;14:e1002516. doi: 10.1371/journal.pbio.1002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laatsch R.H., Cowan W.M. Electron microscopic studies of the dentate gyrus of the rat. I. Normal structure with special reference to synaptic organization. J. Comp. Neurol. 1966;128:359–395. doi: 10.1002/cne.901280305. [DOI] [PubMed] [Google Scholar]
- Lawrence J.J., Grinspan Z.M., McBain C.J. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J. Physiol. 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.S., Ho W.-K., Lee S.-H. Actin-dependent rapid recruitment of reluctant synaptic vesicles into a fast-releasing vesicle pool. Proc. Natl. Acad. Sci. USA. 2012;109:E765–E774. doi: 10.1073/pnas.1114072109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C., Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J. Physiol. 2004;561:27–37. doi: 10.1113/jphysiol.2004.069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Midorikawa M., Sakaba T. Kinetics of Releasable Synaptic Vesicles and Their Plastic Changes at Hippocampal Mossy Fiber Synapses. Neuron. 2017;96:1033–1040.e3. doi: 10.1016/j.neuron.2017.10.016. [DOI] [PubMed] [Google Scholar]
- Miki T., Nakamura Y., Malagon G., Neher E., Marty A. Two-component latency distributions indicate two-step vesicular release at simple glutamatergic synapses. Nat. Commun. 2018;9:3943. doi: 10.1038/s41467-018-06336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Abegg M.H., Gähwiler B.H., Gerber U. A frequency-dependent switch from inhibition to excitation in a hippocampal unitary circuit. Nature. 2004;431:453–456. doi: 10.1038/nature02854. [DOI] [PubMed] [Google Scholar]
- Neher E. Merits and Limitations of Vesicle Pool Models in View of Heterogeneous Populations of Synaptic Vesicles. Neuron. 2015;87:1131–1142. doi: 10.1016/j.neuron.2015.08.038. [DOI] [PubMed] [Google Scholar]
- Neher E., Brose N. Dynamically Primed Synaptic Vesicle States: Key to Understand Synaptic Short-Term Plasticity. Neuron. 2018;100:1283–1291. doi: 10.1016/j.neuron.2018.11.024. [DOI] [PubMed] [Google Scholar]
- Nicoll R.A., Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Oertner T.G., Sabatini B.L., Nimchinsky E.A., Svoboda K. Facilitation at single synapses probed with optical quantal analysis. Nat. Neurosci. 2002;5:657–664. doi: 10.1038/nn867. [DOI] [PubMed] [Google Scholar]
- Pernía-Andrade A.J., Goswami S.P., Stickler Y., Fröbe U., Schlögl A., Jonas P. A deconvolution-based method with high sensitivity and temporal resolution for detection of spontaneous synaptic currents in vitro and in vivo. Biophys. J. 2012;103:1429–1439. doi: 10.1016/j.bpj.2012.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raineteau O., Rietschin L., Gradwohl G., Guillemot F., Gähwiler B.H. Neurogenesis in hippocampal slice cultures. Mol. Cell. Neurosci. 2004;26:241–250. doi: 10.1016/j.mcn.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Regehr W.G. Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol. 2012;4:a005702. doi: 10.1101/cshperspect.a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen A., Sätzler K., Rodríguez E.P., Jonas P., Frotscher M., Lübke J.H. Structural determinants of transmission at large hippocampal mossy fiber synapses. J. Neurosci. 2007;27:10434–10444. doi: 10.1523/JNEUROSCI.1946-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose T., Schoenenberger P., Jezek K., Oertner T.G. Developmental refinement of vesicle cycling at Schaffer collateral synapses. Neuron. 2013;77:1109–1121. doi: 10.1016/j.neuron.2013.01.021. [DOI] [PubMed] [Google Scholar]
- Rostaing P., Real E., Siksou L., Lechaire J.-P.P., Boudier T., Boeckers T.M., Gertler F., Gundelfinger E.D., Triller A., Marty S. Analysis of synaptic ultrastructure without fixative using high-pressure freezing and tomography. Eur. J. Neurosci. 2006;24:3463–3474. doi: 10.1111/j.1460-9568.2006.05234.x. [DOI] [PubMed] [Google Scholar]
- Salin P.A., Scanziani M., Malenka R.C., Nicoll R.A. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc. Natl. Acad. Sci. USA. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siksou L., Varoqueaux F., Pascual O., Triller A., Brose N., Marty S. A common molecular basis for membrane docking and functional priming of synaptic vesicles. Eur. J. Neurosci. 2009;30:49–56. doi: 10.1111/j.1460-9568.2009.06811.x. [DOI] [PubMed] [Google Scholar]
- Singec I., Knoth R., Ditter M., Hagemeyer C.E., Rosenbrock H., Frotscher M., Volk B. Synaptic vesicle protein synaptoporin is differently expressed by subpopulations of mouse hippocampal neurons. J. Comp. Neurol. 2002;452:139–153. doi: 10.1002/cne.10371. [DOI] [PubMed] [Google Scholar]
- Smith J.E., Reese T.S. Use of aldehyde fixatives to determine the rate of synaptic transmitter release. J. Exp. Biol. 1980;89:19–29. doi: 10.1242/jeb.89.1.19. [DOI] [PubMed] [Google Scholar]
- Spacek J., Harris K.M. Three-dimensional organization of cell adhesion junctions at synapses and dendritic spines in area CA1 of the rat hippocampus. J. Comp. Neurol. 1998;393:58–68. doi: 10.1002/(sici)1096-9861(19980330)393:1<58::aid-cne6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Stoppini L., Buchs P.-A.A., Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Studer D., Zhao S., Chai X., Jonas P., Graber W., Nestel S., Frotscher M. Capture of activity-induced ultrastructural changes at synapses by high-pressure freezing of brain tissue. Nat. Protoc. 2014;9:1480–1495. doi: 10.1038/nprot.2014.099. [DOI] [PubMed] [Google Scholar]
- Südhof T.C.C. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H., Woehler A., Neher E. Superpriming of synaptic vesicles as a common basis for intersynapse variability and modulation of synaptic strength. Proc. Natl. Acad. Sci. USA. 2016;113:E4548–E4557. doi: 10.1073/pnas.1606383113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau L.-E., Emery D.G., Haydon P.G. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T., Janz R., Südhof T.C., Nicoll R.A., Malenka R.C. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F., Sigler A., Rhee J.-S., Brose N., Enk C., Reim K., Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. USA. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyleta N.P., Jonas P. Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science. 2014;343:665–670. doi: 10.1126/science.1244811. [DOI] [PubMed] [Google Scholar]
- Watanabe S., Rost B.R., Camacho-Pérez M., Davis M.W., Söhl-Kielczynski B., Rosenmund C., Jorgensen E.M. Ultrafast endocytosis at mouse hippocampal synapses. Nature. 2013;504:242–247. doi: 10.1038/nature12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf M.G., Castillo P.E., Zalutsky R.A., Nicoll R.A. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman M.A., Regehr W.G. Structural contributions to short-term synaptic plasticity. Physiol. Rev. 2004;84:69–85. doi: 10.1152/physrev.00016.2003. [DOI] [PubMed] [Google Scholar]
- Zhao S., Studer D., Chai X., Graber W., Brose N., Nestel S., Young C., Rodriguez E.P., Saetzler K., Frotscher M. Structural plasticity of hippocampal mossy fiber synapses as revealed by high-pressure freezing. J. Comp. Neurol. 2012;520:2340–2351. doi: 10.1002/cne.23040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Comparison of the paired pulse ratio (PPR) of Schaffer collateral and MF synapses in organotypic slices at DIV14 and DIV28. Related to Figures S1B and S1C. (B) Comparison of Schaffer collateral and MF synapses in organotypic slices frozen at DIV14. Related to Figures 2A–2H. (C) Comparison of SC and MF synapses in acute hippocampal slices frozen at P18. Related to Figures S2A–S2H. (D) Comparison of Schaffer collateral and MF synapses in organotypic slices frozen at DIV28. Related to Figures 2I–2P. (E) Comparison of MF synapses in organotypic slices frozen at DIV28 (HPF) and from transcardially perfused P28 WT mice using perfusion fixative 1 (PF1; 4% PFA, 2.5% GA in 0.1 M PB) or PF2 (2% PFA, 2.5% GA, 2 mM CaCl2 in 0.1 M cacodylate buffer). Related to Figures S2M–S2P. (F) Comparison of DIV28 MF synapses in slices from the same organotypic slice culture processed by high-pressure freezing and immersion-fixed using immersion fixative 1 (IF1; 4% PFA, 2.5% GA in 0.1 M PB) or IF2 (2% PFA, 2.5% GA, 2 mM CaCl2 in 0.1 M cacodylate buffer). Related to Figures S2Q–S2S. (G) Comparison of M13 CTRL and DKO MF synapses in organotypic slices frozen at DIV28. Related to Figures 3E–3L. (H) Analysis of MF synapses in pharmacologically silenced organotypic slices frozen at DIV14. Vehicle control (VC; culture medium), 1 μM TTX, 2 μM NBQX, and 50 μM APV (T/N/A), or 1 μM TTX and 2 μM DCG-IV (T/D). Related to Figures S3A–S3G. (I) Electrophysiological recordings of mEPSC frequency and amplitude in CA3 pyramidal cells before and after application of 2 μM DCG-IV in slice cultures at DIV14. Control recordings were made in the presence of 1 μM TTX and 10 μM bicuculline. Related to Figures 3N–3S. (J) Morphometric analysis using two-color STED microscopy of Bassoon-immunoreactive AZs in Synaptoporin-reactive MF boutons. Related to Figures 4A–4C and 4E. (K) Analysis of MF synaptic bouton profiles in 2D images of ultrathin sections after pharmacological manipulation of presynaptic cAMP levels in organotypic slices frozen at DIV28. Vehicle control (VC; culture medium containing 1 μM TTX) or 1 μM TTX and 25 μM forskolin (T/F) Related to Figures 4D, 4F, and 4G. (L) Pharmacological manipulation of presynaptic cAMP levels in MF synapses of organotypic slices frozen at DIV28. Vehicle control (VC; culture medium containing 1 μM TTX), 1 μM TTX and 25 μM forskolin (T/F), or 1 μM TTX and 2 μM DCG-IV (T/D). Related to Figures 4H–4O. (M) Analysis of MF and Schaffer collateral synaptic bouton profiles in 2D images of ultrathin sections. Related to Figure 4. Abbreviations: A, APV; AZ, active zone; C, control; D, DCG-IV; DCV, dense core vesicle; DIV, days in vitro; DKO, double knock-out; F, forskolin; GA, glutaraldehyde; GV, giant, clear-cored vesicle (Ø > 60 nm); IF, immersion fixation; MF, mossy fiber; N = , number of slice cultures or mice; N, NBQX; n = , number of tomograms unless indicated otherwise; P, postnatal day; PB, phosphate buffer; PF, perfusion fixative; PFA, paraformaldehyde; PPR, paired pulse ratio; SC, Schaffer collateral; SV, small, clear-cored synaptic vesicle; T, TTX; TTX, tetrodotoxin; VC, vehicle control; versus, versus; WT, wild-type. All values are represented as mean ± SEM. The number of tomograms was used for statistical analysis unless otherwise specified in the table. Green p values: Normally distributed data sets. (Two conditions: Student’s unpaired t test; More than two conditions: One-way ANOVA plus Bonferroni’s multiple comparison post-test). Blue p values: Not normally distributed data sets. (Two conditions: Mann-Whitney U test; More than two conditions: Kruskal-Wallis test plus Dunn’s comparison post-test; Electrophysiology: Wilcoxon matched-pairs signed rank test).
Data Availability Statement
Any raw data supporting the current study is available from the Lead Contact upon request. This study did not generate any code.