

Abstract
Heat shock protein (Hsp) 90 an emerging and attracting target in the anti-HIV drug discovery process due to the key role it plays in the pathogenicity of HIV-1 virus. In this research study, long-range all-atom molecular dynamics simulations were engaged for the bound and the unbound proteins to enhance the understanding of the molecular mechanisms of the Hsp90 dimerization and inhibition. Results evidently showed that coumermycin A1 (C–A1), a recently discovered Hsp90 inhibitor, binds at the dimer’s active site of the Hsp90 protein and leads to a substantial parting between dimeric opposed residues, which include Arg591.B, Lys594.A, Ser663.A, Thr653.B, Ala665.A, Thr649.B, Leu646.B and Asn669.A. Significant differences in magnitudes were observed in radius of gyration, root-mean-square deviation and root-mean-square fluctuation, which confirms a reasonably more flexible state in the apo conformation associated with it dimerization. In contrast, the bound conformer of Hsp90 showed less flexibility. This visibly highpoints the inhibition process resulting from the binding of the ligand. These findings were further validated by principal component analysis. We believe that the detailed dynamic analyses of Hsp90 presented in this study, would give an imperative insight and better understanding to the function and mechanisms of inhibition. Furthermore, information obtained from the binding mode of the inhibitor would be of great assistance in the design of more potent inhibitors against the HIV target Hsp90.
Electronic supplementary material
The online version of this article (doi:10.1007/s12013-016-0743-8) contains supplementary material, which is available to authorized users.
Keywords: HIV-1, Hsp90, C–A1, MD simulations, Molecular docking
Introduction
Acquired immunodeficiency syndrome (AIDS) is the most challenging disease outbreak thus far and is caused by the human immunodeficiency virus (HIV) [21]. HIV/AIDS affected over 35 million people globally, and Sub-Saharan Africa is the most affected region with approximately 68 % people affected (https://en.wikipedia.org/wiki/Epidemiology_of_HIV/AIDS#Sub-Saharan_Africa). Highly active antiretroviral therapy (HAART), approved by the Food and Drug Administration (FDA), is currently the most effective therapeutic regimen decreasing the viral load, which prolongs the survival of AID-free patients [4, 40, 58]. These drugs include inhibitors of three essential HIV enzymes protease, integrase (IN), reverse transcriptase (RT) as well as entry and progression enzyme inhibitors of in the HIV life cycle [11, 41]. In spite of all currently existing anti-HIV therapies, resistant strains still poses a problem. This creates a demanding necessity for the identification of new antivirals that do not induce drug resistance.
Hsp90 is an ATP dependent chaperone protein crucial for maintaining active forms of other proteins which are known as client proteins [28]. Hsp90 has also been reported to be responsible for the replication of nearly all kinds of viruses such as DNA, RNA viruses, and double-stranded RNA viruses [23]. Hsp90 is a large protein, forming a homodimeric complex. Each monomer shares a common domain organization which consists of a C-terminal dimerization domain (CTD), a middle domain (MD) and an N-terminal ATPase domain (NTD) (Fig. 1), which is found mostly in almost every part of eukaryotic cells [15, 35, 46]. Amongst other proteins, Hsp90 is one of the most profuse proteins in the cytoplasm, where it makes up approximately 1–2 % of the total protein levels (Anon n.d.). It is responsible for the controls of activities, such as the maturation, localization of a selected large number of substrates called “client proteins” in the cytoplasm. These clients proteins are part of several processes including transcription, translation, mitochondrial function, kinetochore assembly, inflammation, immunology, cellular antiviral defense pathways, apoptosis, centrosome function and cell cycle [18, 30, 39]. Studies have demonstrated that the replication of viruses to be sensitive to Hsp90 inhibitors at very low concentrations without affecting cellular viability, due to their dependence in Hsp90 for their survival. Hsp90 is regarded as an essential protein for malignant transformation and progression, thus target protein for anti-cancer drugs [45, 54]. A previous study in our laboratory gave a computational perspective of Hsp90 as an anti-cancer target [36]. Recently it has been reported as a potential target for anti-HIV therapy [60].
Fig. 1.
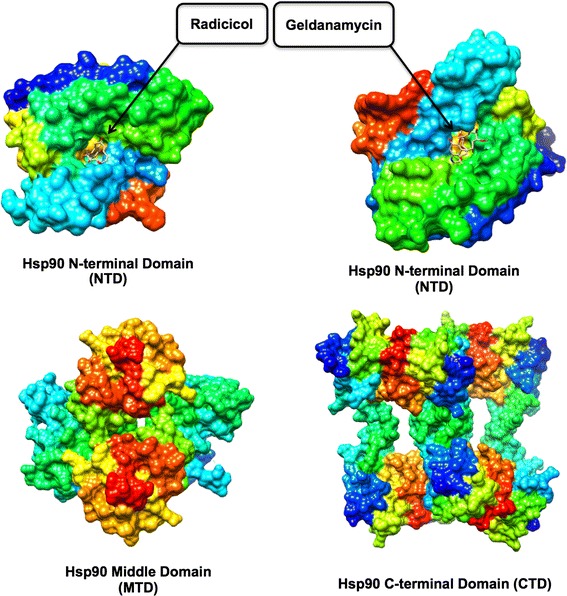
Hsp90 X-ray crystal structures accentuating different domains. The surface representations were generated using PDB co-ordinates of 1BGQ (NTD + RD), 1YET (NTD + GA), 1HK7 (MTD) and 1SF8 (CTD)
Studies have recently revealed that the CTD of Hsp90 protein has a crucial role in the dimerization of the chaperones with an additional binding site for inhibitors [52]. It has also been reported that the nucleotide binding site situated at the CTD is more favorable to guanosine-5′-triphosphate (GTP) over ATP, which would possibly enhance the selectivity of the new inhibitors against this site [9, 17, 59]. Herein we focus on the C-terminal dimerization inhibitors as this is the vital domain for the biological activity of Hsp90 [11].
To date, a broad-spectrum of antivirals have been identified, including Hsp90 inhibitors [5, 16, 23, 53]. Numerous antiviral activity of Hsp90 inhibitors have been proved against tissue culture of ill health conditions such as picornaviruses poliovirus, coxsackievirus, rhinovirus, influenza virus, paramyxoviruses, hepatitis C virus (HCV), Ebola virus, vesicular stomatitis virus, severe acquired respiratory syndrome (SARS), feline viral rhinotracheitis (FHV), HIV, vaccinia virus, and herpes viruses [14, 16, 32, 33, 44, 57, 60, 61, 64]. Gryase B inhibitors (novobiocin, coumermycin A1 (C–A1) and daunorubicin) have been reported to impair HIV-1 replication the most, by targeting Hsp90 [60]. It has been shown that they inhibit the formation Hsp90 dimer by binding in the C-terminal domain, which impairs viral gene expression [60].
The present study looked at C–A1 (Fig. 2), which has previously been screened for its antiretroviral activity by means of a chemical genetics (CG) approach [60]. This study confirmed the impairment of viral gene expression by C–A1 binding to Hsp90. C–A1 weakens both the HIV-1 integration and gene expression possesses.
Fig. 2.
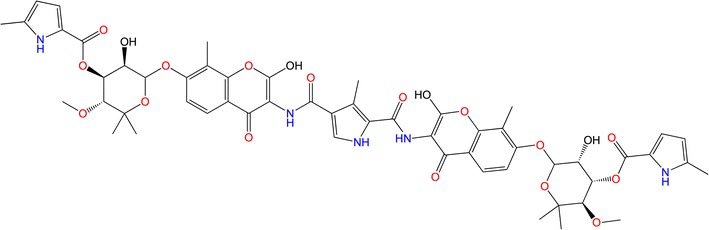
The 2D structure of coumermycin A1
Molecular dynamic simulations and post-dynamics (PD) analysis appeared as the closest methods to the actual experimental work and is of great help in the understanding of the complex biological phenomenas [12]. An application of continuous MD simulations to reveal folding and the unfolding activities of biological enzymes has made it convenient to fathom the dimerization process complexity of macromolecules [8, 20, 25, 37, 55]. The heightened utilities of the PD methods demonstrated to be effectual in comprehending the conformational background of biological macromolecules. The most extensively improved PD methodologies used to seek structural variations within distinctive biological phenomenas include principal component analysis PCA or essential dynamics (ED) [11]. Although PCA has attested its competency to be implement in the likening of motions of different macromolecules there are additional techniques, which were applied to understand such biological conformational behaviors [9, 49]. Such may include dynamic cross-correlation (DCC), which was valuable to gain insights on macromolecular motions in different biological systems [22]. To understand both the dimerization and the inhibition processes of Hsp90, virtual MD simulations were employed for the bound and the unbound conformations. To our understanding, this is the first report of such adequate computational insight on Hsp90 as a vital HIV target. Thus, we trust that this account aids as a significant computational study towards an improved understanding of the Hsp90 protein structure, dynamics and its mechanism of inhibition. Herein, we presented the structural and dynamic insight that can be executed in the discovery of drugs and the development of more effective HIV inhibitors against Hsp90.
Computational Methodologies
System Preparation
There is no available crystal structure of the human C-terminal portion of Hsp90, however structures of yeast (2CG9) [2] and E. coli (1SF8) [26] Hsp90 orthologues are available. 2CG9 was chosen for this study due to the fact there is very high sequence and structural conservation among Hsp90 proteins from prokaryotes to Homo sapien s [2]. The ligand and protein systems were prepared in the similar manner as mentioned in the previous reports [42, 43]. Auto-Dock Vina [56] was used to generate docked conformation of C–A1 inside Hsp90’s active site using the same procedure previously reported [60].
Molecular Dynamic Simulations
The unbound and C–A1 bound models of Hsp90 proteins were exposed to all-atom, continuous MD simulations in precise solvent using the GPU version PMEMD engine integrated with Amber 14 [3, 6, 7]. The ff99sb force field [19] applied with Amber 14 was employed in the description of the protein systems. To neutralize the system, missing hydrogen atoms and counter ions were added using the Leap module, integrated with Amber 14. All the systems were submerged into an orthorhombic box with TIP3P [29] water molecules in a manner that none of the protein atoms were contained in 12 Å of any box edge prior to the set up. The particle mesh Ewald (PME) [27] technique was employed to calculate the continuous electrostatic interactions and van de Waals cut-off of 12 Å. Initial system minimization for 500 steps using steepest descent was followed by 100,000 steps using conjugate gradients algorithm was performed to relax the system and remove all unfavorable clashes. CPU version of Amber14 was used to perform all the minimization but before the minimization step, a gradual heating from 0 to 300 K by means of Langevin thermostat [62] with a collision frequency of 1/ps using an official ensemble (NVT) was used. All systems were successively equilibrated at 300 K in a NPT ensemble for 100,000 ps with no restrained and to uphold the pressure at 1 bar Berendsen barostat [10] was used. To restrict the bonds of all covalently bonded atoms to hydrogen atoms for all MD simulations runs the SHAKE algorithm [34] was used. The MD was run continuously in a NPT ensemble for 100 ns with a target pressure of 1 bar and a pressure-coupling constant of 2 ps. In every 1 ps the trajectories were examined and more analyses namely RMSD, RMSF, Rg, inter atomic distances, DCC and PCA were performed using PTRAJ and CPPTRAJ modules intergrated in Amber 14. All plots and visualization were performed in Chimera molecular modeling tool [47] and Origin data analysis software (www.originlab.com) respectively.
Free Binding Energy Calculations
The docked complex of Hsp90–C–A1 was calculated to predict the binding efficiencies and confirm the HCAAR C–A1 binding to Hsp90. The binding affinity predictions were executed using the MMPBSA with Eq. (1) [38] and MMGBSA method that with Eq. (2) [24].
| 1 |
| 2 |
Where, ΔE MM is the differentiation between the minimized energies of the Hsp90–C–A1 complex and the total energies of the Hsp90 and Hsp90 inhibitor including the electrostatic and the van der Waals energies. The change in entropy of the ligand binding conformations is presented by TΔS, the variance in the P/GBSA solvation energies of the Hsp90–C–A1 complex and the summation of the solvation energies of the Hsp90 and Hsp90 inhibitor is represented by ΔG solv and ΔG SA is the difference in the surface area energies for the Hsp90 and Hsp90 inhibitor. Both MMPBSA and MMGBSA methods have been recognized to ensure the inhibitors are ranked accurately in accordance with their binding energy and thus can aid as a potent tool in the drug design research [1, 50, 63].
Principle Component Analysis (PCA)
Principle component analysis (PCA) is extensively used to effortlessly examine and identify data generated from MD simulations to highpoint principal modes accountable for changes in conformation [11]. The PTRAJ and CPPTRAJ modules were used in performing PCA C-alpha atoms [48] integrated with Amber 14 [13] The plots analysis showing the central conformational motions descriptive of each structure were created using Origin data analyses programme (www.originlab.com).
Dynamic Cross Correlation Matrix
The DCCM between fluctuations based on the residues throughout the simulation was analyzed using the module called CPPTRAJ assimilated with Amber 14. The DCCM is best defined by the equation below [31]:
Where r i(t) defines the vector of the atom’s coordinates as a function of time t, < · > t measures the time ensemble average and ∆r i(t) = r i(t) − < r i(t) >t. The backbone Cα atomic fluctuations were considered during the DCCM analysis.
Results and Discussion
Insights into Coumermycin A1 Bound with Hsp90
The lig-plot (Anon n.d.) analysis is best suited for displaying the 2D interaction between the ligand and the amino acid residue contributing to the ligand binding to the receptor. The structure of inhibitor C–A1 docked within the Hsp90 dimer’s binding site gave essential evidence on the binding of the ligand inside the active site (S1). It was observed that C–A1 formed hydrogen bonds interactions with Arg591.B, Ala595.A, Glu660.A, Lys480.B, Ser663.A and Ser666.A (S1) residues of both Hsp90 dimer subunits. Interestingly, the binding of C–A1 is at the dimer of Hsp90 possessing multiple common residues from both sub-unit A and B in its active site. The averaged values of the calculated free energy, using MM/PBSA approach; over the 100 ns MD simulations for each CAAR to the binding of C–A1 to Hsp90 are illustrated in Fig. 8 and S3. The CAAR formed hydrogen bond interactions, which is one of the key interactions favorable and vital for binding of a ligand to its receptor, possess higher binding affinities.
Fig. 8.
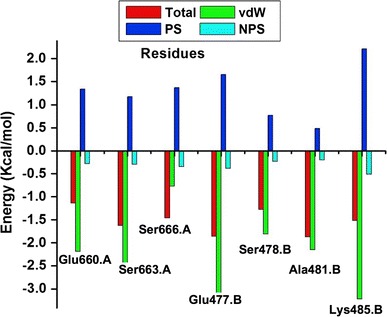
Graph showing binding energy (Total), van der Waals (vdW), polar solvation (PS) and non-polar solvation (NPS) contribution for each HCAAR
The plot of the interaction (Fig. 3) high spots the C–A1 location within the Hsp90 dimer and reciprocal residues’ active site, together with the hydrogen bond interactions shown in red dotted lines, from each subunit. The existence of hydrophobic/aromatic interactions, which promotes the appropriate binding of inhibitor into the dimer, consisting of key active site residues involved in dimer packing, including Ser595.A and Glu660.A, are visibly illustrated in the binding mode. It is assumed that binding of C–A1 in the Hsp90 dimer’s active site results to the dimer inhibition of Hsp90. The dynamic insight of the dimerization mechanism process of Hsp90 through inhibition by C–A1 was obtained from the continuous MD simulations using the structure of Hsp90–C–A1 complex (docked) (S2).
Fig. 3.
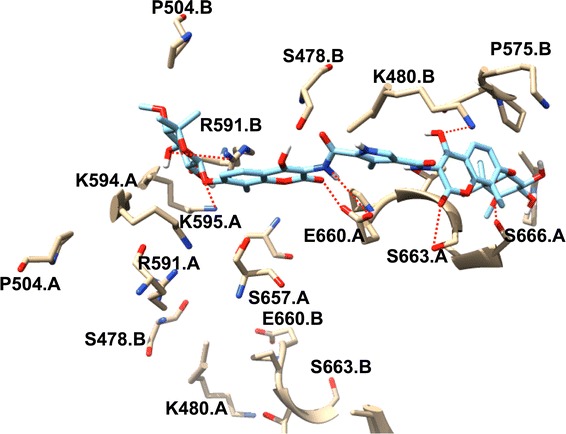
The binding mode of C–A1 to Hsp90 subunits
Molecular Dynamics Simulations and Post-Dynamics Analysis
RMSD, RMSF, and Rg
Protein dynamics, structure, and function are highly interrelated. The conformational changes of Hsp90 are particularly critical for enzyme function [51, 65]. Studying internal atomistic motions can unravel the dynamic nature of Hsp90, which is responsible for numerous biological functions as enzymes. Molecular dynamic simulations were performed for each enzyme (Apo and bound) to establish the drug-binding pattern and to better understand the interaction of C–A1 with the CTD, to ensure the stability of the MD trajectories dynamic and the stabilities dissimilarities of MD simulations. The values of the RMSD for the protein backbone atoms and side chain atoms in relation to the initial minimized structure throughout the simulations were calculated for the Cα atoms for the dimer (S4) and whole protein (Fig. 4).
Fig. 4.
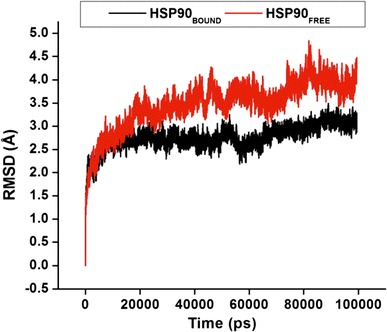
The RMSD values for the dimer atoms during continuous 100 ns MD simulation
The average structure for the complete simulation was measured by calculating values for all residues of both in Apo and bound conformations (Fig. 5) by means of the RMSF in order to obtain the system’s subset movement. Residues between 400 and 600 have the highest RMSF values, which corresponds to CTD. The most stable areas are the areas with the lowest RMSF values. Overall the bound enzyme has slightly lower RMSF values of 1.88 Å compared to the Apo with the value of 2.64 Å, which indicates that the backbone of the bound conformation is slightly more stable than that of the free enzyme.
Fig. 5.
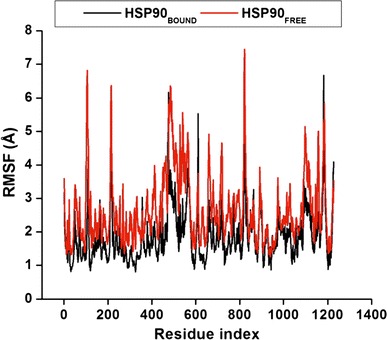
Cα atom RMSF values given for each residue number in both systems
The mass-weighted root mean square distance of a collection of atoms from their common center of mass best describes the R g. To measure for the compactness of a structure, the R g was calculated. The analysis shown in Fig. 6 and S5 provides us with an understanding into the dimensions of the dimeric cleavage and of the overall Hsp90 protein respectively. We observed the major fluctuations in both systems between 0 and 100,000 ps. The R g indicated higher structural deviations for the apo conformation in comparison to the bound conformation. The mean value of the bound is lower than that of the apo, which indicates that the bound protein is stable. This could be due to C–A1 inhibitory effect on the Hsp90 dimerization.
Fig. 6.
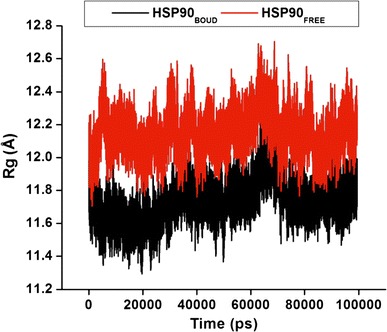
The radius of gyration (R g) of the dimeric subunit during 100 ns MD simulation
Principal Component Analysis
The trajectories’ projection obtained at 300 K onto the first two principal components (PC1, PC2) presented the motion of two proteins in phase space. Clusters were better defined in the bound conformation than the apo conformation. Furthermore, the bound conformation covered a larger region of phase space particularly along PC1 and PC2 plane than the apo conformation as depicted in Fig. 7.
Fig. 7.
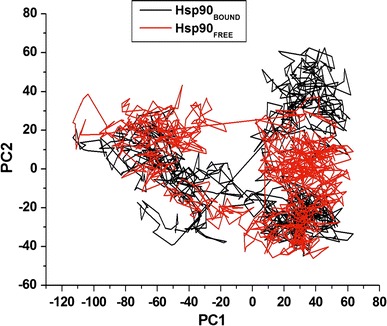
PCA projection of Cα atoms motion constructed by plotting the initial two principal components (PC1 and PC2) in conformational subspace
Per-Residue Energy Decomposition (PRED)
In an effort to offer more insight into the involvement of each amino acid residue toward the binding of C–A1, PRED was computed for the system. This helps us to observe the residues with the most contribution to the ligand binding, as seen from Fig. 8 the residues from both monomers showed interaction favorable to the ligand via different interactions. This further advocates that C–A1 inhibit dimerization. This information will ideally assist medicinal chemists in the design of inhibitors that precisely interrelate with these amino acid residues.
Dynamic Cross Correlation
The DCCM is shown in Fig. 9 and S7, which has been calculated for both bound and unbound conformations of Hsp90 dimer subunits and Hsp90 as a whole. The free state of Hsp90 displayed a more correlated motion, which further justifies the occurrence of the dimerization process resulting in a more correlated residue–residue interaction. The bound state, on the contrary, showed a much greater reduction in correlated motions during simulations. With these findings we can thus conclude that C–A1 inhibited the dimerization mechanism of Hsp90. The suitability of C–A1 to inhibit dimerization mechanism is hence confirmed as Hsp90 ground state is stabilized. The distance between the dimer residues in each dimeric subunit were calculated by measuring the interatomic distances between the Cα atoms along the simulations to gain an in-depth insight of dynamics of the dimerization process. Figure 10 and S3 highlights these residues.
Fig. 9.
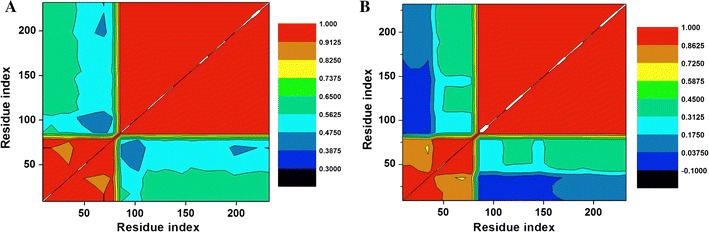
DCC matrix of the dimeric subunit during simulation taking into consideration Cα residues of Hsp90 ligand—bound (a) and free (b) conformations
Fig. 10.
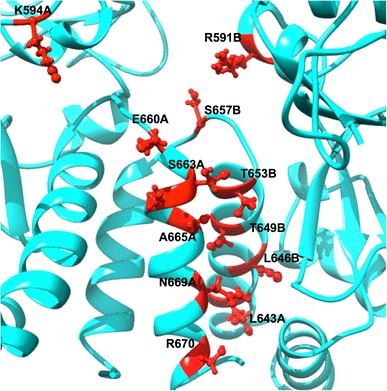
The dimeric region residues from both subunits assumed to be part of the Hsp90 dimerization process
The distances between the orthogonal residues for the apo conformation were comparatively higher than that of the bound conformation as the average Cα distances specified on Table 1 and S6. The standard deviation of distances for the apo conformation was higher than that of the bound conformation, indicating that the latter enzyme is more stable (agreeing with the RMSF, R g and PCA studies).
Table 1.
Residues involved in dimer packing and their averaged distances from each other throughout simulation
| Residues | Apo (Å) | Bound (Å) |
|---|---|---|
| S663A–T653B | 8.54 ± 0.40 | 8.46 ± 0.35 |
| A665A–T649B | 6.43 ± 0.57 | 5.55 ± 0.38 |
| N669A–L646B | 9.88 ± 0.72 | 7.32 ± 0.50 |
| K594A–R591B | 13.11 ± 0.91 | 12.44 ± 0.50 |
These substantial dissimilarities in distances between residues in the arranging of the dimer guaranteed the inhibitions of Hsp90 by C–A1 through the formation of a stable interaction with residues involved in the dimerization, preventing the dimer from opening and allowing the substrate from entering. This fact further correlates with previous biochemical analyses aimed at mapping the binding site of C–A1 in Hsp90 suggesting that the viral gene expression inhibited by C–A1 through the interference with Hsp90 dimerization [60].
Snapshots for the MD simulations for both the apo and the C–A1-bound conformations of Hsp90 were taken at specific time interval corresponding to the distances of orthogonal residues (S6) where it is evident for both conformations opening and closing. This further substantiates the lack of dimerization in the Hsp90–C–A1 complex (Fig. 11).
Fig. 11.
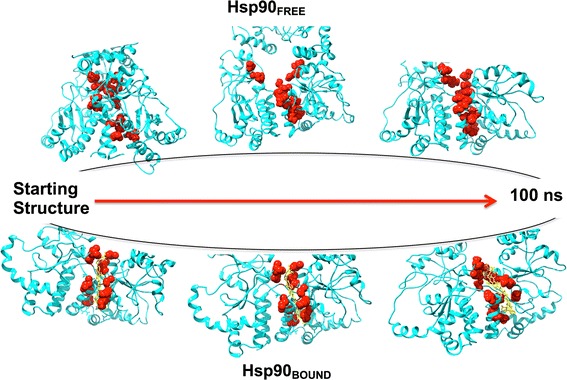
Snapshots of unbound and C–A1-bound conformations of Hsp90 at a definite frames during MD simulation
Conclusion
Molecular dynamic simulations revealed difference events of dimer mechanism of Hsp90 in its unbound and C–A1–bound conformations. Micro-molecule inhibitors such as C–A1 prevent the dimerization process of by targeting the dimer of Hsp90. We believe a more conformational rigid system was due a dimerization process being mired. This was indicated after the analysis of the RMSF and R g proving a more conformational supple nature of unbound Hsp90, whereas the C–A1-bound conformation of Hsp90 was found to be more stable with a declined dimerization throughout the MD simulation. The binding of C–A1 to the Hsp90 dimer subunits inhibits the process of dimerization. Observing the distance between the residues that are involved in the dimerization process further validates this. This is the first report emphasizing the important computational insight of the features of a crucial HIV target, which would also serve as an appropriate foundation in the process of designing new compounds against Hsp90 as future anti-HIV drugs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors would like to acknowledge the School of Health Science, University of KwaZulu-Natal for financial support. Computational resources granted by the Center for High Performance Computing (http://www.chpc.ac.za) and San Diego is highly appreciated.
Abbreviations
- RMSD
Root-mean-square deviation
- RMSF
Root-mean-square fluctuation
- Rg
Radius of gyration
- DCC
Dynamic cross correlation
- PCA
Principal component analysis
- IN
Integrase
- RNA
Ribonucleic acid
- DNA
Deoxyribonucleic acid
- ATP
Adenosine triphosphate
- CTD
C-terminal domain
- NTD
N-terminal domain
- GTP
Guanosine-5′-triphosphate
- CAAR
Contributing amino acid residues
- PRED
Per residue energy decomposition
- NP
Polar solvation
- NPS
Non-polar solvation
Compliance with Ethical Standards
Conflict of interests
The authors avow no conflict of interest to the above article.
References
- 1.Abraham DJ. Burger’s medicinal chemistry and drug discovery. Hoboken: Wiley; 2003. [Google Scholar]
- 2.Ali MMU, et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440(7087):1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anon (2015). Amber14.
- 4.Anon. (2015). Hsp90. https://en.wikipedia.org/wiki/Hsp90.
- 5.Anon (2011). Identification of broad-spectrum antiviral compounds and assessment of the druggability of their target for efficacy against respiratory syncytial virus (RSV). Proceedings of the National Academy of Sciences of the United States of America, 108(17), 6739–6744 [DOI] [PMC free article] [PubMed]
- 6.Anon (2015) LIGPLOT v.4.5.3—Operating manual. http://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/manual/. Accessed 22 Aug 2015.
- 7.Anon (2015) The Global HIV/AIDS epidemic The henry J. kaiser family foundation. http://kff.org/global-health-policy/fact-sheet/the-global-hivaids-epidemic/. Accessed 25 Sep 2015.
- 8.Beck, D.A.C., & Daggett, V., (2004). Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods (San Diego, Calif.), 34(1), 112–20. http://www.sciencedirect.com/science/article/pii/S1046202304000568. Accessed 22 July 2015. [DOI] [PubMed]
- 9.Benkovic, S.J., & Hammes-Schiffer, S. (2003). A perspective on enzyme catalysis. Science (New York), 301(5637), 1196–1202. http://www.ncbi.nlm.nih.gov/pubmed/12947189. [DOI] [PubMed]
- 10.Berendsen, H.J., et al. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684–3690. http://link.aip.org/link/JCPSA6/v81/i8/p3684/s1&Agg=doi\npapers2://publication/doi/10.1063/1.448118.
- 11.Bhakat, S., et al. (2014). An integrated molecular dynamics, principal component analysis and residue interaction network approach reveals the impact of M184V mutation on HIV reverse transcriptase resistance to lamivudine. Molecular bioSystems, 10(8), 2215–28. http://www.ncbi.nlm.nih.gov/pubmed/24931725. [DOI] [PubMed]
- 12.Carvalho, A.T., et al. (2014). Challenges in computational studies of enzyme structure, function and dynamics. Journal of molecular graphics & modelling, 54, 62–79. http://www.sciencedirect.com/science/article/pii/S1093326314001570. Accessed 30 July 2015. [DOI] [PubMed]
- 13.Case et al. (2015). Amber 14 reference manual. http://ambermd.org/doc12/Amber14.pdf.
- 14.Castorena, K.M., et al. (2007). A functional heat shock protein 90 chaperone is essential for efficient flock house virus RNA polymerase synthesis in Drosophila cells. Journal of Virology, 81(16), 8412–8420. http://www.ncbi.nlm.nih.gov/pubmed/17522196. [DOI] [PMC free article] [PubMed]
- 15.Chadli, A., et al. (2000). Dimerization and N-terminal domain proximity underlie the function of the molecular chaperone heat shock protein 90. Proceedings of the National Academy of Sciences of the United States of America, 97(23), 12524–12529. http://www.ncbi.nlm.nih.gov/pubmed/11050175. [DOI] [PMC free article] [PubMed]
- 16.Connor, J.H., et al. (2007). Antiviral activity and RNA polyinerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology, 362(1), 109–119. http://www.ncbi.nlm.nih.gov/pubmed/17258257. [DOI] [PMC free article] [PubMed]
- 17.Dixit, A., & Verkhivker, G.M. (2012). Probing molecular mechanisms of the Hsp90 chaperone: biophysical modeling identifies key regulators of functional dynamics. PloS one, 7(5), e37605. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3356286&tool=pmcentrez&rendertype=abstract. Accessed 10 Aug 2015. [DOI] [PMC free article] [PubMed]
- 18.Drysdale, M.J., et al. (2006). Targeting Hsp90 for the treatment of cancer. Current Opinion in Drug Discovery & Development, 9(4), 483–495. http://www.ncbi.nlm.nih.gov/pubmed/16889231. [PubMed]
- 19.Duan, Y., et al. (2003). A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. Journal of computational chemistry, 24, 1999–2012. http://www.ncbi.nlm.nih.gov/pubmed/14531054. [DOI] [PubMed]
- 20.Durrant, J.D., & McCammon, J.A. (2011). Molecular dynamics simulations and drug discovery. BMC biology, 9(1), 71. http://www.biomedcentral.com/1741-7007/9/71. Accessed 24 June 2015. [DOI] [PMC free article] [PubMed]
- 21.Esposito, F., et al. (2012). HIV-1 reverse transcriptase still remains a new drug target: structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Molecular biology International, p.586401. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3388302&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed]
- 22.Falconi, M., et al. (2007). Molecular dynamics simulation of human LOX-1 provides an explanation for the lack of OxLDL binding to the Trp150Ala mutant. BMC structural biology, 7, 73. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2194713&tool=pmcentrez&rendertype=abstract. Accessed 5 Aug 2015. [DOI] [PMC free article] [PubMed]
- 23.Geller, R., et al. (2012). Broad action of Hsp90 as a host chaperone required for viral replication. Biochimica Et Biophysica Acta-Molecular Cell Research, 1823(3), 698–706. http://www.ncbi.nlm.nih.gov/pubmed/22154817. [DOI] [PMC free article] [PubMed]
- 24.Greenidge PA, et al. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with mm-gbsa. Journal of chemical information and modeling. 2014 doi: 10.1021/ci5003735. [DOI] [PubMed] [Google Scholar]
- 25.Gsponer J, Caflisch A. Molecular dynamics simulations of protein folding from the transition state. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(10):6719–6724. doi: 10.1073/pnas.092686399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris, S.F., Shiau, A.K., & Agard, D.A. (2004). The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure, 12(6), 1087–1097. http://linkinghub.elsevier.com/retrieve/pii/S0969212604001261. [DOI] [PubMed]
- 27.Harvey MJ, De Fabritiis G. An implementation of the smooth particle-mesh Ewald (PME) method on GPU hardware. Journal of Chemical Theory and Computation. 2009;5:2371–2377. doi: 10.1021/ct900275y. [DOI] [PubMed] [Google Scholar]
- 28.Jhaveri, K., et al. (2012). Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochimica Et Biophysica Acta, 1823(3), 742–55. http://www.sciencedirect.com/science/article/pii/S0167488911002977. Accessed 12 July 2015. [DOI] [PMC free article] [PubMed]
- 29.Jorgensen, W.L., et al. (1983). Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79(2), 926. http://scitation.aip.org/content/aip/journal/jcp/79/2/10.1063/1.445869. Accessed 9 July 2014.
- 30.Kang, B.H., et al. (2007). Regulation of tumor cell mitochondrial Homeostasis by an organelle-specific Hsp90 chaperone network. Cell, 131(2), 257–270. http://www.ncbi.nlm.nih.gov/pubmed/17956728. [DOI] [PubMed]
- 31.Kasahara, K., et al. (2014). A novel approach of dynamic cross correlation analysis on molecular dynamics simulations and its application to Ets1 dimer-DNA complex. PloS one, 9(11), e112419. 10.1371/journal.pone.0112419. Accessed 22 June 2015. [DOI] [PMC free article] [PubMed]
- 32.Kim, M.-G., et al. (2012). Destabilization of PDK1 by Hsp90 inactivation suppresses hepatitis C virus replication through inhibition of PRK2-mediated viral RNA polymerase phosphorylation. Biochemical and Biophysical Research Communications, 421(1), 112–118. http://www.sciencedirect.com/science/article/pii/S0006291X12005967. [DOI] [PubMed]
- 33.Kowalczyk, A., et al. (2005). Heat shock protein and heat shock factor 1 expression and localization in vaccinia virus infected human monocyte derived macrophages. Journal of inflammation , 2, 12. http://MEDLINE:16246258. [DOI] [PMC free article] [PubMed]
- 34.Kräutler V, Van Gunsteren WF, Hünenberger PH. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of Computational Chemistry. 2001;22(5):501–508. doi: 10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V. [DOI] [Google Scholar]
- 35.Krukenberg KA, et al. Conformational dynamics of the molecular chaperone Hsp90. Quarterly Reviews of Biophysics. 2011;44(2):229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumalo, H.M., Bhakat, S., & Soliman, M.E. (2015). Heat-shock protein 90 (Hsp90) as anticancer target for drug discovery: an ample computational perspective. Chemical Biology & Drug Design, 86, 1131-1160. http://doi.wiley.com/10.1111/cbdd.12582. Accessed 12 July 2015. [DOI] [PubMed]
- 37.Lindorff-Larsen, K., et al. (2011). How fast-folding proteins fold. Science, 334(6055), 517–520. http://www.ncbi.nlm.nih.gov/pubmed/22034434. Accessed 25 Sep 2015. [DOI] [PubMed]
- 38.Lyne, P.D., Lamb, M.L., & Saeh, J.C. (2006). Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. Journal of Medicinal Chemistry, 49(16), 4805–4808. http://pubs.acs.org/doi/abs/10.1021/jm060522a. [DOI] [PubMed]
- 39.McClellan, A.J., et al. (2007). Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell, 131(1), 121–135. http://www.ncbi.nlm.nih.gov/pubmed/17923092. [DOI] [PubMed]
- 40.Monforte, A., et al. (2000). Insights into the reasons for discontinuation of the first highly active antiretroviral therapy (HAART) regimen in a cohort of antiretroviral naive patients. Aids, 14, 499–507. http://discovery.ucl.ac.uk/144671/. [DOI] [PubMed]
- 41.Moonsamy, S., Bhakat, S., & Soliman, M.E.S. (2014). Dynamic features of apo and bound HIV-Nef protein reveal the anti-HIV dimerization inhibition mechanism. Journal of Receptors and Signal Transduction, 9893, 1–11. http://informahealthcare.com/doi/abs/10.3109/10799893.2014.984310. [DOI] [PubMed]
- 42.Moonsamy S, Soliman MES. Dual acting HIV inhibitors: integrated rational in silico design strategy. Medicinal Chemistry Research. 2014;23(2):682–689. doi: 10.1007/s00044-013-0670-9. [DOI] [Google Scholar]
- 43.Moonsamy, S., & Soliman, M. (2014). Computer-aided perspective for the design of flexible HIV non-nucleoside reverse transcriptase inhibitors (NNRTIs): de-novo design, virtual screening and molecular dynamics simulations. Letters in Drug Design and Discovery, 513–524.
- 44.Nakagawa, S., et al. (2007). Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochemical and Biophysical Research Communications, 353(4), 882–888. http://www.ncbi.nlm.nih.gov/pubmed/17196931. [DOI] [PubMed]
- 45.Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clinical Cancer Research. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearl, L.H., & Prodromou, C. (2006). Structure and mechanism of the Hsp90 molecular chaperone machinery. Annual Review of Biochemistry. 75, 271–294. http://www.ncbi.nlm.nih.gov/pubmed/16756493. [DOI] [PubMed]
- 47.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 48.Roe, D.R., & Cheatham III, T.E. (2013). PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation, 9(7), 3084–3095. http://pubs.acs.org/doi/abs/10.1021/ct400341p. [DOI] [PubMed]
- 49.Ruiz-Pernia, J.J., Silla, E., & Tunon, I. (2007). Enzymatic effect on reactant and transition states. Chalcone isomerase. Journal American Chemistry Society, 129 (29), 9117–9124. http://pubs3.acs.org/acs/journals/doilookup?in_doi=10.1021/ja071720. [DOI] [PubMed]
- 50.Sadiq, S.K., et al. (2010). Accurate ensemble molecular dynamics binding free energy ranking of multidrug-resistant HIV-1 proteases. Journal of chemical information and modeling, 50(5), 890–905. 10.1021/ci100007w. Accessed 6 Dec 2015. [DOI] [PubMed]
- 51.Sattin, S., et al. (2015). Activation of Hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands. Chemistry (Weinheim an der Bergstrasse, Germany). http://onlinelibrary.wiley.com/doi/10.1002/chem.201502211/full. Accessed 23 Aug 2015. [DOI] [PMC free article] [PubMed]
- 52.Sgobba, M., et al. (2008). Structural models and binding site prediction of the C-terminal domain of human Hsp90: a new target for anticancer drugs. Chemical biology & drug design, 71(5), 420–33. http://www.ncbi.nlm.nih.gov/pubmed/18373550. Accessed 6 Dec 2015. [DOI] [PubMed]
- 53.Sidera, K., & Patsavoudi, E. (2014). HSP90 inhibitors: current development and potential in cancer therapy. Recent patents on anti-cancer drug discovery, 9(1), 1–20. http://www.ncbi.nlm.nih.gov/pubmed/23312026. [PubMed]
- 54.Soga, S., Akinaga, S., & Shiotsu, Y. (2013). Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Current pharmaceutical design, 19(3), 366–76. http://www.ncbi.nlm.nih.gov/pubmed/22920907. [DOI] [PubMed]
- 55.Swope, W.C., & Pitera, J.W. (2004). Describing protein folding kinetics by molecular dynamics simulations. 1. Theory †. The Journal of Physical Chemistry B, 108(21), 6571–6581. http://pubs.acs.org/doi/abs/10.1021/jp037421y.
- 56.Trott, O., & Olson, A.J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455–61. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3041641&tool=pmcentrez&rendertype=abstract. Accessed 11 July 2014. [DOI] [PMC free article] [PubMed]
- 57.Ujino, S., Shimotohno, K., & Takaku, H. (2007). 17-AAG, an Hsp90 inhibitor, suppress hepatitis C virus (HCV) replication. Antiviral Research, 74(3), A62–A63. http://linkinghub.elsevier.com/retrieve/pii/S0166354207001295.
- 58.Vento S, et al. Highly active antiretroviral therapy. Lancet. 1998;351(9108):1058–1059. doi: 10.1016/S0140-6736(05)79024-3. [DOI] [PubMed] [Google Scholar]
- 59.Verkhivker, G.M., et al. (2009). Structural and computational biology of the molecular chaperone Hsp90: from understanding molecular mechanisms to computer-based inhibitor design. Current topics in medicinal chemistry, 9(15), 1369–85. http://www.ncbi.nlm.nih.gov/pubmed/19860735. Accessed 25 Sep 2015. [DOI] [PubMed]
- 60.Vozzolo, L., et al. (2010). Gyrase B inhibitor impairs HIV-1 replication by targeting Hsp90 and the capsid protein. Journal of Biological Chemistry, 285(50), 39314–39328. http://www.ncbi.nlm.nih.gov/pubmed/20937817. [DOI] [PMC free article] [PubMed]
- 61.Weeks, S.A., et al. (2010). A targeted analysis of cellular chaperones reveals contrasting roles for heat shock protein 70 in flock house virus RNA replication. Journal of Virology, 84(1), 330–339. http://www.ncbi.nlm.nih.gov/pubmed/19828623. [DOI] [PMC free article] [PubMed]
- 62.Wu, X., & Brooks, B.R. (2003). Self-guided Langevin dynamics simulation method. Chemical Physics Letters, 381(3–4), 512–518. http://www.sciencedirect.com/science/article/pii/S0009261403017585. Accessed 16 June 2015.
- 63.Yunta MJR. Using molecular modelling to study interactions between molecules with biological activity. Rijeka: INTECH Open Access Publisher; 2012. [Google Scholar]
- 64.Zhong, M., et al. (2014). Heat-shock protein 90 promotes nuclear transport of herpes simplex virus 1 capsid protein by interacting with acetylated tubulin. Plos One, 9(6), e99425–e99425. http://MEDLINE:24901434. [DOI] [PMC free article] [PubMed]
- 65.Zuehlke, A., & Johnson, J.L. (2010). Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers, 93(3), 211–7. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2810645&tool=pmcentrez&rendertype=abstract. Accessed 23 Aug 2015. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.