

Abstract
Respiratory syncytial virus (RSV) is amongst the most important pathogenic infections of childhood and is associated with significant morbidity and mortality. Although there have been extensive studies of epidemiology, clinical manifestations, diagnostic techniques, animal models and the immunobiology of infection, there is not yet a convincing and safe vaccine available. The major histopathologic characteristics of RSV infection are acute bronchiolitis, mucosal and submucosal edema, and luminal occlusion by cellular debris of sloughed epithelial cells mixed with macrophages, strands of fibrin, and some mucin. There is a single RSV serotype with two major antigenic subgroups, A and B. Strains of both subtypes often co-circulate, but usually one subtype predominates. In temperate climates, RSV infections reflect a distinct seasonality with onset in late fall or early winter. It is believed that most children will experience at least one RSV infection by the age of 2 years. There are several key animal models of RSV. These include a model in mice and, more importantly, a bovine model; the latter reflects distinct similarity to the human disease. Importantly, the prevalence of asthma is significantly higher amongst children who are hospitalized with RSV in infancy or early childhood. However, there have been only limited investigations of candidate genes that have the potential to explain this increase in susceptibility. An atopic predisposition appears to predispose to subsequent development of asthma and it is likely that subsequent development of asthma is secondary to the pathogenic inflammatory response involving cytokines, chemokines and their cognate receptors. Numerous approaches to the development of RSV vaccines are being evaluated, as are the use of newer antiviral agents to mitigate disease. There is also significant attention being placed on the potential impact of co-infection and defining the natural history of RSV. Clearly, more research is required to define the relationships between RSV bronchiolitis, other viral induced inflammatory responses, and asthma.
Keywords: Asthma, Respiratory syncytial virus, Animal models, Bovine respiratory syncytial virus, Cytokines, Chemokines, Infant mortality
Introduction
Human respiratory syncytial virus (hRSV, here RSV) was first isolated from chimpanzees in 1956 [1] and was subsequently recovered from infants with severe lower respiratory tract disease [2]. It is a non-segmented negative-sense single-stranded enveloped RNA virus that belongs to the family of Paramyxoviridae, genus Pneumovirus, subfamily Pneumovirinae. Its 10 genes encode 11 proteins since two overlapping open reading frames in the M2 mRNA yield two distinct matrix proteins, M2-1 and M2-2. The viral envelope contains three proteins, the G glycoprotein, the fusion (F) glycoprotein, and the small hydrophobic (SH) protein. The G protein functions in host cell attachment and the F protein is responsible for fusion and cell entry, whereas the SH protein is not required in either of these processes. The RSV virus comprises five other structural proteins, the large (L) protein, nucleocapsid (N), phosphoprotein (P), matrix (M), and M2-1, and two non-structural proteins (NS1 and NS2). Whether M2-2 also is a component of the mature assembled virus particles is currently unknown (Fig. 1).
Fig. 1.
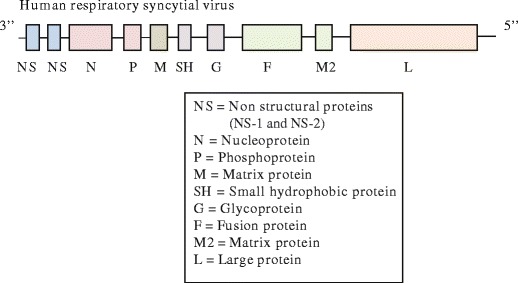
The genetic structure of RSV
Epidemiology
There is a single RSV serotype with two major antigenic subgroups, A and B. Strains of both subtypes often co-circulate, but generally one of the subtypes predominates [3, 4]. The results of molecular analyses show that several genotypes are present simultaneously in any given season and region, but even in neighboring regions, the circulating strains may differ [5, 6]. In temperate regions, RSV infections show a distinct seasonality with onset in late fall or early winter, a peak between mid-December and early February, and season offset in late spring (Fig. 2). Some areas, in particular in northern Europe, report yearly alternations between an early large outbreak and a late small outbreak. In tropical regions, the patterns are less predictable and can include two yearly peaks in spring and fall or fairly constant infection rates throughout the year. Humans are the only host for RSV, and the reasons for the near disappearance of RSV between epidemics remain unclear. Following each epidemic, mean titers of maternally derived RSV neutralizing antibodies were found to decline in consecutively hospitalized infants <6 months of age and to reach their nadir shortly before the peak of the following epidemic in Denmark [7]. This led to the hypothesis that a cyclic pattern in the level of protective RSV-specific antibodies makes a major contribution to the seasonal pattern of RSV infections in temperate climates. It is more difficult to envision how this could account for the occurrence of two yearly RSV epidemics, as has been reported from Taiwan [8].
Fig. 2.

RSV epidemic seasons (approximate for the year 2010–2011, modified from CDC website)
It is generally believed that most children will have experienced at least one RSV infection by the age of 2 years. This is largely based on results from the Houston Family Study, in which a birth cohort of 125 infants was followed prospectively during the period 1975–1980 [9]. During their first year of life, 68 % of the infants experienced a RSV infection; by the end of the second year, almost all children had been infected with RSV at least once. However, active surveillance during a RSV epidemic revealed an attack rate of 29 % in infants from Rochester, NY, though 62 % of infants within infected families [10], which is similar to the 32 % infection rate during the first year of life reported from a prospective study in the UK [11]. These rates also agree with the results of seroprevalence studies from Germany and Japan [12, 13], even when taking into account that infants may not elaborate lasting antibody responses during primary infection [14]. In contrast, seroprevalence data from China support high rates of RSV infection during the first year of life [15]. An important determinant of the infection rate appears to be the severity of the RSV epidemic, which varies from 1 year to the next, and of course the extent of individual exposure [14, 16]. In a daycare setting, essentially all children experiencing their first exposure to RSV during a major epidemic were infected, whereas the rate was only 40 % in years without a major epidemic [14].
Clinical Manifestations and Diagnosis
Primary infection with RSV is believed to be almost always symptomatic, although there are data suggesting that this may not actually be the case [17]. The clinical manifestations range from mild upper respiratory tract illness (URTI) or otitis media to severe and potentially life-threatening lower respiratory tract involvement (LRTI). The most common form of LRTI in RSV-infected infants is bronchiolitis, but pneumonia and croup are also seen. Involvement of the lower airways occurs in ∼15–50 % of infants and young children with primary RSV infection and necessitates hospitalization in 1–3 % of the annual birth cohort, with infants between 2 and 6 months of age being at the highest risk [9–11, 14, 18, 19]. However, in some regions, the highest incidence of LRTI is reported in infants aged 6–11 months or even children between 1 and 2 years of age [20, 21]. In ∼5–10 % of hospitalized infants, the disease is severe enough to require admission to the intensive care unit (ICU). Deaths are rare in previously healthy infants born in industrialized countries, but mortality rates are significantly higher in infants and children in developing countries and in patients with preexisting cardiac or lung disease or other chronic conditions [8, 21, 22].
Reinfections with RSV are observed in 30–75 % of children <2 years of age who had experienced a RSV infection during their first 12 months of life and usually occurs during the following season [9, 14]. The rate of reinfection again depends on the strength of the epidemic [14]. Even in secondary infections, the disease is usually symptomatic in young children, but its severity steadily diminishes with subsequent exposures, with fewer children experiencing bronchiolitis or other lower respiratory illness, fever, and middle-ear effusions [14]. Reinfections remain common throughout life, but in older children and adults, symptoms are generally either absent or confined to the upper respiratory tract, with LRTI (tracheobronchitis, bronchitis, wheezing) being observed in approximately one fourth of symptomatic illnesses [23]. However, immunocompromised patients, those with chronic cardiopulmonary disease, and the frail and elderly are also at risk of severe lower respiratory tract involvement, and RSV constitutes a significant cause of morbidity and mortality in these populations [24–26].
The most common manifestation of RSV LRTI is bronchiolitis, but there is no general agreement on the definition of this entity. The term is most often used to refer to an acute viral LRTI in infants or children <24 months of age characterized by rhinorrhea followed by dry, wheezy cough, tachypnea, dyspnea, and often subcostal, intercostal, and supraclavicular retractions [27]. Fever may be present, but high fever is uncommon. Apnea may be observed in very young and premature infants. These clinical signs are accompanied by wheeze and/or fine inspiratory crackles on auscultation. While North America and some parts of Europe place major emphasis on the presence of wheezing, widespread crepitations are considered to be the hallmark of bronchiolitis in the UK, Australia and other parts of Europe (while wheezing alone would be diagnosed as viral-induced wheeze). Some clinicians recommend limiting the diagnosis of bronchiolitis to the first episode of wheezing [28].
The diagnosis of acute bronchiolitis is clinical and based on presentation with the typical respiratory signs and symptoms, which may be accompanied by lethargy, irritability, and poor feeding. A chest X-ray may show hyperinflation and patchy atelectasis. Although X-ray findings may be helpful in differentiating bronchiolitis from pneumonia, X-rays are not uniformly necessary, but should be confined to cases of diagnostic uncertainty [27]. Note that many epidemiological studies, particularly from developing countries, now follow the World Health Organization (WHO) recommendation to refer to any RSV LRTI as pneumonia, since bronchiolitis and pneumonia are difficult to distinguish clinically or radiographically. In cases of diagnostic uncertainty, bacteriological testing may also be helpful, but its routine use is not indicated since the risk of serious bacterial infections is low in infants with bronchiolitis, even if they are febrile [29–33]. The likelihood of bacterial infection becomes higher in patients admitted to the intensive care unit (ICU) [34, 35]. Pulse oximetry is recommended for all patients presenting at the emergency department, although experts disagree on whether supplemental oxygen should be instituted at oxygen saturation of ≤92 or <90 %. While some recommend routine use of rapid antigen testing for RSV for guiding cohort arrangements, the guidelines proposed by the American Academy of Pediatrics (AAP) do not recommend routine virological testing since its results rarely alter management decisions, but consider it useful when cohorting is desired [27].
While antigen detection by direct or indirect immunofluorescence or ELISA and viral culture are still in common use for confirming RSV infection, similar methods are not available for all respiratory viruses. Hence, investigations of the viral etiology in epidemiological studies increasingly rely on reverse transcription-polymerase chain reaction (RT-PCR) for RNA viruses and PCR for DNA viruses. Although this does not greatly enhance the detection frequency of RSV unless it is combined with hybridization [36], it is the only method for the detection of some newly identified viruses, e.g., human metapneumovirus (hMPV) and bocaviruses, and has greatly improved the detection rate of rhinovirus, parainfluenza virus, and adenovirus (AdV) [36–39]. There is, however, continued debate over the interpretation of positive RT-PCR results since a substantial portion of samples obtained from asymptomatic subjects without evidence of recent respiratory infections are positive by RT-PCR [17, 40, 41].
Severe Disease and Hospitalizations
Total Burden of RSV Infections
The total burden and the hospitalization rates for RSV-associated respiratory infections are difficult to determine because routine testing to establish the viral etiology is not performed in an outpatient setting and, in the US guidelines, is not even recommended for hospitalized cases since the outcome of virology would not affect management [27]. Based on data from published and unpublished studies using highly varying methodologies, it has been estimated that RSV causes 33.8 million episodes of acute LRTI, 3.4 million episodes of severe acute LRTI requiring hospitalization and at least 66,000 deaths worldwide in children <5 years of age, with most of the severe cases and the fatalities occurring in children below the age of 2 years and fatalities being much more common in developing countries [21]. There is considerable variation in the reported hospitalization rates, but it is currently unclear whether this is attributable mainly to methodological differences or reflects true geographical disparities in the incidence of severe RSV infections.
In the US alone, population-based active surveillance of laboratory-confirmed RSV infections has yielded estimated hospitalization rates of 17–18.5/1,000 infants <6 months of age and of 5.1–7.4/1,000 in infants 6–11 months of age for the period 2000–2004 [42, 43]. The overall rate in children <5 years old was 3/1,000 [42] (see Table 1 for comparison with rates reported from other industrialized countries). In the same study, the rates for emergency department visits and pediatric practice visits associated with RSV infections were 28/1,000 and 80/1,000, respectively [42]. Note that there was substantial yearly and regional variation in all of these rates. Extrapolated to the entire US population, an estimated 2.1 million children <5 years of age require medical attention for RSV infection each year, with 3 % of them being hospitalized, 25 % of them being treated in emergency departments and 73 % being seen in pediatric practices. Of particular importance, more than three quarters (78 %) of patients requiring medical care for RSV infections are older than 1 year of age, and almost two thirds (61 %) of outpatient visits involve children between 2 and 5 years old.
Table 1.
Hospitalizations for RSV infections or RSV bronchiolitis (per 1,000)
| Region | Period | Method for determination of viral etiology | % bronchiolitis | Rate <6 months | 6–12 months | Rate <1 year | 1–2 years | References | |
|---|---|---|---|---|---|---|---|---|---|
| RSV-associated hospitalization | 1 county each in NY and TN | 10/2000–9/2001 | Population-based active surveillance, viral culture and RT-PCR | 79 %a | 18.5 | 7.4 | 12.9 | 3.3 | [43] |
| 1 county each in NY, TN, and OH | “During the winter months” 2000–2004 | Population-based active surveillance, viral culture and RT-PCR | 85 % | 16.9 | 5.1 | [42] | |||
| USA | 1997–2006 | RSV code | 48.9 for 0–2 months; 28.4 for 3–5 months | 13.4 | 5.0 | [46] | |||
| USA. | 2000–2001 | RSV code | 41.9 | 12.8 | 27.4 | [44] | |||
| San Sebastián, Spain | 7/2004–6/2007 | Population-based active surveillance, antigen detection and RT-PCR | n.a. | 39.4 | 10.8 | 3.0 | [493] | ||
| San Sebastián, Spain | 7/1996–6/2000 | Population-based retrospective study, RSV was laboratory confirmed in essentially all cases | 91.5 | 36.8 | 25.5 | [51] | |||
| Sweden | 1987–1998 | Population-based retrospective study, RSV was laboratory-confirmed | n.a. | 8–14b | [66] | ||||
| Germany | 1996–99 | Prospective, NPA, multiplex RT-PCR | n.a. | 12.1 | 2.35 | [73] | |||
| Taiwan | 2004–2007 | Retrospective, ICD codes | n.a. | 10.77c | [8] | ||||
| Hong Kong | 10/2003–9/2006 | Prospective, NPA, antigen detection | 43 | 23.3–31.1 | [494] | ||||
| RSV Bronchiolitis | USA | 2000–2001 | RSV code | 24.2 | [44] | ||||
| USA | 1980–1996 | RSV code | Shay | ||||||
| Norway | 1993–2000 | Population-based retrospective study, RSV was laboratory confirmed in essentially all cases | all | 21.7 | 6.8 | [70] |
n.a. not available
aThis is the overall bronchiolitis proportion for the study population, which included children up to 5 years of age, but almost 80 % were <1 year old
bThe rate was 0.8 % in years with small epidemics, and 1.4 % in years with large epidemics
cIs likely to represent an underestimate because not all hospitals have the ability to do antigen detection or viral culture
Somewhat higher RSV-associated hospitalization rates have been derived from hospital discharge data, with estimates ranging from 23.4 to 27.4/1,000 in infants <1 year [44, 45]. Significantly higher frequencies were obtained for infants <6 months old (44.5/1,000 live births) compared to infants aged 6–11 months (24.2/1,000) [44]. Another analysis yielded rates of 48.9 for infants 0–2 months of age, 28.4 for infants aged 3–5 months, and 13.4 for 6–11 month-old infants for the period 1997–2006 [46]. Of particular note, American Indian and Alaska Native infants are hospitalized due to RSV infection much more often than the general US infant population, and the highest frequencies were observed in Native infants living in Alaska and the Southwest, with rates of 70.9 and 48.2 per 1,000 for RSV hospitalizations, respectively [44]. Broken down by type of disease, the rates for RSV bronchiolitis and pneumonia hospitalizations were 24.2 and 3.0 per 1,000 births, respectively [44]. The corresponding figures for bronchiolitis in American Indian and Alaska Native infants were 54.54 and 43.4 per 1,000, respectively. At least for Alaska Native infants, this appears to represent a marked underestimate of average annual hospitalization rates, since the above data were for the period 2000–2001, during which the RSV hospitalization rates in the Yukon Delta were found to be the lowest observed between 1994 and 2004 [47]. The overall frequency for 1994–1997 was 178/1,000, which declined to 104/1,000 for 2001–2004, largely due to a reduction in RSV hospitalizations in premature high-risk infants after the introduction of palivizumab prophylaxis. Canadian Inuit infants have also been reported to have enormously high frequencies of hospitalization for bronchiolitis [48], including RSV bronchiolitis [49].
In the US, bronchiolitis of any etiology accounted for 16.4 % of hospitalizations in infants <1 year of age in 1996 (an approximately threefold increase compared to 1980) [50], while more recent data suggest a proportion of ∼19.5 % [44]. Approximately 45–85 % of RSV hospitalizations carry a discharge diagnosis of bronchiolitis [51–53], with some of the highest proportions coming from recent US surveillance and hospital discharge data [42–44]. Conversely, between ∼50 and 80 % of hospitalizations for bronchiolitis are due to RSV (see also Table 2) [51]. More specifically, a virus is detected by (RT)-PCR in ∼75–95 % of infants ≤2 years of age who are hospitalized with bronchiolitis, and RSV accounts for ∼70–75 % of the virus-positive samples [39, 54–58] (see also Table 2). Interestingly, the proportion of RSV does not vary markedly regardless of whether sampling is performed for several years [57], a single whole year [54], or one or more RSV seasons only [39, 55, 56]. This clearly illustrates that RSV is the most frequent cause of hospitalizations for bronchiolitis, but it also underscores that other viruses can cause bronchiolitis, including rhinovirus, hMPV, corona, parainfluenza and influenza viruses, bocavirus and AdV [39, 53, 54, 56, 57, 59]. The proportion with a discharge diagnosis of bronchiolitis is similar in patients with RSV, hMPV or rhinovirus infections [52, 60]. However, the overall hospitalization rates for these viruses generally are significantly lower compared to RSV-associated disease in infants <1 year of age, although some data suggest that picornaviruses, including rhinovirus, are the most common etiological agent in acute respiratory illness requiring hospitalization even in this population [61].
Table 2.
Risk factors for severe RSV infection or hospitalization
| Preterm birth |
| Chronic lung disease (CLD) of prematurity |
| Hemodynamically significant congenital heart disease (CHD) |
| Low birth weight, particularly birth weight <10th percentile |
| Infants with congenital or acquired immunodeficiencies |
| Trisomy 21 (Down syndrome) and other chromosomal abnormalities or malformations |
| Interstitial lung disease |
| Neuromuscular disease |
| Liver disease |
| Inborn errors of metabolism |
It should be noted that, while RSV generally remains the most frequent viral agent in LRTI and particularly bronchiolitis severe enough to require hospitalizations, rhinovirus is emerging as the most frequent cause of acute respiratory illness in general and LRTI in particular in prospective community-based studies from Wisconsin, Western Australia, and the UK [17, 41, 62]. Two of these cohorts consisted of children at high risk of atopy, and atopy has been shown to predispose to rhinovirus-associated wheezy respiratory tract disease [63, 64]. Consequently, these cohorts may not be fully representative of the general population. However, in unselected cohorts, picornaviruses (which include rhinovirus) also were identified as the most frequent cause of acute respiratory episodes, including LRTI [62].
Risk Factors for RSV Hospitalizations
Host Factors
Well established host risk factors for hospitalizations are preterm birth, chronic lung disease (CLD) of prematurity, and hemodynamically significant congenital heart disease (CHD) [65–75] (Table 2). A further risk factor for RSV hospitalizations is low birth weight [71, 74], particularly birth weight <10th percentile [76]. Infants with congenital or acquired immunodeficiencies are also at risk of severe disease [69, 77]. In addition, trisomy 21 (Down syndrome), other chromosomal abnormalities, malformations, interstitial lung disease, neuromuscular disease, liver disease, and inborn errors of metabolism have been associated with a ∼twofold increased risk of hospitalization overall, with individual incidence rate ratios ranging from ∼1.5 for encephalocele to 4.3 for cystic fibrosis [69, 70]. Premature birth and underlying medical conditions not only increase the risk of hospitalization, but also of more severe clinical disease manifestations, as indicated by more frequent requirement for mechanical ventilation, admission to the ICU, longer duration of hospitalization, and increased mortality [46, 58, 69, 70].
In spite of the increased hospitalization risk associated with the above risk factors, at least half of all infants hospitalized with RSV infection are previously healthy without any of these established medical risk factors [42, 65]. In this group, the most frequently and consistently identified risk factors include young age (<6 weeks to <6 months) [71, 75, 78–82]; male sex [65, 67, 70, 71, 76, 78, 81, 83, 84]; siblings or other children living in the household, particularly when they are older than the index child and already attending daycare or school [42, 65, 66, 76, 80, 81, 83–86]; the infant’s own daycare attendance [76, 83]; and exposure to environmental tobacco smoke, particularly maternal smoking during pregnancy and lactation [65, 76, 78–80, 82, 83, 87].
In the USA, ancestry or ethnicity and health insurance status also influence the hospitalization rates. As already mentioned, Alaska Native and American Indian infants are hospitalized due to RSV infection much more frequently than the general US infant population [44, 47]. However, in a Tennessee Medicaid cohort, European ancestry was an independent risk factor for hospitalization due to RSV infection [65]. Others also found that non-Hispanic patients of European extraction were more likely to be treated as inpatients than as outpatients, to have private health insurance and to be younger than 6 months of age [42]. Only younger age and prematurity were identified as independent risk factors in this investigation. Among hospitalized patients, African ancestry was protective, with black infants showing higher oxygen saturation and a shorter duration of hospitalization [79].
Most of these risk factors have been reported quite consistently in numerous studies, both in term and preterm infants; however, only a few of them are generally identified in each individual study population, and even analyses of quite similar cohorts (e.g., preterm infants born at 33–35 weeks GA and preterm infants born at 32–35 weeks of age in Spain) yield different independent predictors of RSV hospitalizations [81, 82]. There are even studies that are unable to detect a significant association of disease severity or hospitalization with any of these factors [88]. This suggests that other host factors play a primary role in such cohorts. These include the host immune response to RSV infection and the genetic susceptibility of the host, possibly including an atopic predisposition. These aspects will be discussed in greater length in later sections.
To date, only two protective factors have been identified. One is breast feeding [40, 42, 81, 83, 85]. The other is the level of maternally derived antibodies, which are present in essentially all neonates, though at vastly varying titers [89, 90]. In numerous studies, the titers of maternally derived neutralizing antibodies are inversely associated with RSV infection overall [91, 92], or with the severity of RSV disease [16, 93–96], although at least one study did not find the relationship to be linear [78].
Viral Factors
Viral Load
Whether viral load correlates with disease severity remains controversial since some analyses of hospitalized children show a significant association [97–100], whereas others do not [101, 102]. The method used for viral quantification does not account for these differences since both quantitative RT-PCR and plaque assays have yielded positive as well as negative results. In ambulatory subjects with a first episode of RSV infection, there was no significant difference in viral load between patients with bronchiolitis and those experiencing only URTI [11]. In contrast, the results from another prospectively followed birth cohort indicate a moderate correlation between viral load and disease severity in patients infected with RSV alone, but not in those co-infected with another respiratory virus [103]. However, the authors point out that the viral loads in these outpatient episodes were similar to those they had previously found in infants hospitalized with severe RSV LRTI, indicating that viral load is not the only factor determining disease severity. In a community-based surveillance study from Indonesia, viral load was associated with disease severity in children ≥1 year of age, but not in infants, even though children <1 year of age had higher viral loads and generally had more severe disease than older children [104].
Viral Subtype and Genotype
Several investigations showed RSV subtype A to be associated with more severe disease compared to subtype B, and this risk persisted after adjusting for age, prematurity, and other risk factors [3, 4, 75, 105]. Conversely, a greater severity in subtype B compared with subtype A infections has only been reported in one study [106]. To add to the confusion, other studies were unable to detect a significant association at all [98, 107, 108]. Clade or genotype may be a more important determinant of disease severity than subtype [5, 106, 109–111], although a significant association with genotype is not consistently seen either [112]. These discrepancies are likely due to the fact that several different strains generally co-circulate in any given RSV season [5, 6], and some of the studies may simply lack the statistical power to detect significant differences in disease severity between individual RSV genotypes. The results from a recent in vitro study in primary epithelial cells (ECs) and an epithelial cell lines suggest that prototypic RSV-A and RSV-B strains differ in their ability to induce nuclear factor-κB (NF-κB) activation, an important step in the cascade of events leading to the production of pro-inflammatory cytokines, and the subsequent induction of the NF-κB responsive genes IL6 and IL8, with RSV-B eliciting significantly lower responses compared to RSV-A [113]. This supports the finding of greater disease severity in subtype A compared to subtype B infections. Studies in cell culture and in vivo models also provide clear evidence that individual RSV type A isolates differ substantially in their infectivity, virulence, and immunopathogenicity [114–118]. Together, these findings strongly suggest that viral characteristics—in interaction with host susceptibility factors—determine disease phenotype, including severity.
Co-infections
The results of studies using molecular techniques for determining the viral etiology of LRTI have shown that between 9 and 44 % of infants with bronchiolitis requiring an emergency department visit or hospitalization are co-infected with 2 or more viruses [61, 119, 120] (see also Table 3). Several analyses show simultaneous infection with more than one respiratory virus to be associated with increased disease severity in infants [39, 60, 121–124], although this is not a consistent finding [53, 57]. In a Dutch community-based study, viral load was significantly associated with disease severity when RSV was the only pathogen detected, but such an association was not evident in infants co-infected with RSV and another respiratory virus [103]. In this study, it was decided to designate the viral agent that was present in higher quantities as the primary pathogen. It remains to be determined, however, whether this is a valid assumption. As a matter of fact, a detailed analysis of clinical characteristics in co-infections compared to single infections showed RSV to dictate the prevalence and severity of clinical features such as obstructive airway disease and hypoxia as well as overall duration of hospitalization, regardless of whether the co-infection involved rhinovirus or adenovirus (AdV) [125]. Only the frequency of fever was significantly higher in RSV-AdV co-infections compared to RSV alone. In contrast, dual infections involving rhinovirus in combination with AdV or influenza virus more often yielded significant differences in the prevalence of individual clinical characteristics. This suggests that simultaneous detection of two or more viruses by RT-PCR is not simply the reflection of lingering viral nucleic acids from a past infection, but truly reflects the simultaneous presence of two (or more) pathogens. Other data suggest that the localization of the virus may also play a role. A substantial portion of nonbronchoscopic bronchoalveolar lavage (BAL) samples from infants who required mechanical ventilation because of RSV bronchiolitis were found to contain hMPV RT-PCR products [126]. Although the presence of RSV had been confirmed by antigen detection in nasopharyngeal samples of all the infants, only 24/30 (80 %) of them had detectable RSV amplicons in BAL fluid, but 3 of the RSV-negative BAL samples contained hMPV amplicons. This raises the question of whether the viral agent detected in nasopharyngeal secretions (NPS) is necessarily the agent responsible for lower respiratory symptoms.
Table 3.
Viral etiologies of acute respiratory illness and bronchiolitis in hospitals and emergency departments
| Area | Period | n | Age | RSV | Parainfluenza | Influenza | AdV | Rhinovirus/(picornaviruses) | hMPV | Coronavirus | Bocavirus | None detected | Viral combinations | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acute respiratory illness | ||||||||||||||
| 1 county each in NY and TN | 10/00–9/01 | 592/812 eligible | <5 years | 19.60 % overall (32 % of those with detectable virus) | 6.8 % (11 %) | 3.4 % (5.5 %) | 4.2 % (6.9 %) | 30.1 % (49 %) | 3.2 % (5.2 %) | Not done | Not done | 39 % | Not reported | [43] |
| Hospital San Sebastián, Spain | 7/2004–6/2007 | 742 (796 episodes) | <3 years | 44.0 (57.2) | 7.54 (9.8) | 4.52 (5.9) | 3.64 (4.7) | 20.9 (27.1) | 11.3 (14.7) | 3.3 (4.2) | Not done | 23.7 | 17.2 | [493] |
| University Clinic, Istanbul | 10/2006–3/2007 | 147/165 | <5 years (but 80 % ≤2 years of age) | 20.4 overall (55.6 of those with detectable virus) | 10.2 (27.8) | 3.4 (9.3) | Not done | Not done | 4.8 (7.0) | Not done | Not done | 63.3 | 4.1 % | [495] |
| Multi-center emergency departments in 10 US states | 2–3 weeks only between 12/2005–3/2006 | 277 | <2 years | 63.5 % overall (74.3 % of those with detectable virus) | Not done | 3.9 % (4.6) | Not done | 15.9 (18.6) | 6.9 % (8.0) | Not done | Not done | 14 % | 9 % | [55] |
| Pediatric Emergency department in Dijon, France | 12/2002–4/2004 | 931 | <3 years | 28.5 | 0.5 | 6 % | Not done | 18.3 % | 6 % | Not done | Not done | 41 | n.a. | [52] |
| Pediatric Emergency Department in Caen, France | 11/2009–4/2010 | 1021 samples from 921 infants | <2 years | 34.3 (42.0) | 5.9 (7.2) | 7.7 (9.5) | 4.3 (5.3) | 23.9 (29.3) | 3.3 (4.1) | 9.3 (11.4) | Not done | 18.3 | 7.2 | [496] |
| Bronchiolitis | ||||||||||||||
| Children’s Hospital Seattle, WA, USA | 10/2003–4/2004 | 180 | <2 years | 77 (82.7) | 5.6 (6.0) | 0.56 (0.60) | 15 (16.1) | Not done | 10.6 (11.3) | 8 (8.3) | Not done | 7 | 23 | [56] |
| Madrid, Spain | 9/2005–8/2008 | 318 | <2 years | 61.3 (70.9) | 3.8 (4.4) | 1.6 (1.8) | 8.8 (10.2) | 20.1 (23.3)/Enteroviruses 1.6 (1.8) | 4.1 (4.7) | 0.3 (0.36) | 13.2 (15.3) | 34 | 24.8 | [57] |
| São Paolo, Brazil | 3/2006–6/2007 | 77 | <2 years | 63.6 [66] | 9.1 (9.7) | 2.6 (2.8) | 0 | 33.8 [34] | 15.6 (16.7) | 2.6 (2.8) | 11.7 (12.5) | 6.5 | 44 | [59] |
| Athens, Greece | 10/1999–9/2000 | 118 | <18 months | 53.4 (72.4) | 2.5 (3.5) | 2.5 (3.5) | 7.6 (10.3) | 21.2 (28.7) | Not done | 2.5 (3.5) | Not done | 26.3 | 13.6 | [54] |
| Lyon, France | 9-2003–4/2004 and 9/2004–4/2005 | 180 | <12 months | 72 [73] | 7.2 (7.5) | 3.3 (3.5) | 3.3 (3.5) | 21.7 (22.5)/Enterovirus: 3.9 (4.0) | 5.6 (5.8) | 3.9 (4.0) | Not done | 3.9 | 24.4 | [39] |
Treatment
The management of acute bronchiolitis severe enough to require hospitalization largely consists of supportive care, such as nasal suction, nasogastric or intravenous fluids, supplemental oxygen, and nasogastric feeding. It is common practice to administer bronchodilators (α and β adrenergics, anticholinergics and nebulized epinephrine), but there is no conclusive evidence that they have a positive impact on disease outcome [27]. Evidence in support of a meaningful effect of inhaled or systemic corticosteroids in the treatment of severe bronchiolitis is also lacking [127], and their routine use is not recommended. The results of a recent randomized controlled trial suggest that the combination of nebulized epinephrine with high-dose oral corticosteroids reduced the hospital admission rate in infants presenting at the emergency department [128]. There is considerable cause for concern over the high dose of dexamethasone that was used in this trial, and independent confirmation of the results should be obtained before adopting this approach. Also note that plasma cortisol levels were found to be significantly elevated in infants with mild bronchiolitis compared to healthy controls and were further elevated in patients with severe RSV bronchiolitis requiring hospitalization [129]. There was also evidence that this endogenous cortisol production was associated with the suppression of cytokines that are considered to be key mediators of antiviral responses. This strongly suggests that systemic corticosteroid treatment may not be advisable in severe RSV disease. Many infants experience recurrent wheezing episodes after hospitalization for bronchiolitis. However, inhaled corticosteroids during the acute phase of RSV bronchiolitis did not demonstrate prevention preventative effect on post-bronchiolitis wheezing [130, 131].
Ribavirin is a broad-spectrum antiviral agent approved by the FDA for use in nebulized form in the treatment of infants and children with severe bronchiolitis. The results of a meta-analysis indicate that it may be effective in reducing the duration of ventilation and length of hospitalization, but the available studies are too small and their quality is too variable to allow any firm conclusions [132]. Current AAP guidelines do not recommend its routine use because of uncertainties regarding its effectiveness, concern over potential health risk for caregivers, and its high cost [27]. However, it may be considered in high-risk infants with severe disease [27].
Despite considerable evidence that concurrent bacterial infections in infants and children hospitalized for RSV-related illness are very infrequent, antibiotics are still often prescribed [31, 34, 133]. Antibiotic therapy in randomized controlled trials has not been shown to result in improved outcome [134] and its use should be carefully evaluated because of the potential risk of adverse events and the growing threat of bacterial antibiotic resistance. In other words, a benefit–risk analysis should be performed in each case. In a mouse model, treatment with antibiotics for 15 days after RSV infection resulted in increased airway hyperresponsiveness (AHR) on the last day of treatment, but this was not seen after RSV infection alone or antibiotic treatment alone at this time point [135]. In addition, antibiotic treatment interfered with the RSV-induced upregulation of pulmonary regulatory T cells (Tregs) and immunomodulatory cytokines that have been shown to play an important role in limiting the immunopathology of RSV infection in mice [136–139].
In summary, there is great variability in the management of acute bronchiolitis not only at the international level, but also within countries and even between centers [22, 140]. This reflects the lack of conclusive evidence that any of the therapies in current use for the treatment of bronchiolitis have a positive impact on disease outcome.
Prophylaxis with Palivizumab
Palivizumab is a monoclonal antibody that is directed against the F protein of RSV and is approved by the US Food and Drug Administration (FDA) for the prevention of serious LRTI caused by RSV in high-risk infants and young children. FDA approval was based on the results of two randomized clinical trials of prophylaxis with palivizumab [141, 142]. One demonstrated an overall reduction in the rate of RSV hospitalization of 55 %, the relative decrease being 78 % in premature infants (≤35 weeks gestational age) without CLD of prematurity and 39 % in children with CLD [141]. In 1,287 infants and children ≤24 months of age with hemodynamically significant CHD, palivizumab prophylaxis was associated with a reduction in RSV hospitalization from 9.7 to 5.3 %, corresponding to a relative reduction of 45 % [142]. The results of a number of observational studies confirm that palivizumab is effective in premature infants with or without CLD and in infants with hemodynamically significant CHD [58, 143–145], although they also highlight that compliance with current recommendations for the dosing of palivizumab is suboptimal [146, 147].
The FDA approval was for administration of palivizumab in 5 monthly doses of 15 mg/kg body weight beginning with the regional start of the RSV season, which typically occurs in November or December in the US. There is considerable variation in the actual RSV epidemic season within different regions or areas of the US. In particular, the season is more difficult to predict and lasts longer in Florida, southwestern Alaska [148], and the Pacific Northwest (unpublished data).
There is international agreement that a full course of palivizumab should be given to prematurely born infants with a gestational age of <32 weeks, but considerably variation has developed in the national recommendations for the use of prophylaxis in the group born at a gestational age of between 32 and 35 weeks. Palivizumab is effective in these infants [141], but since this population comprises 3–5 % of the annual birth cohort the cost of indiscriminate treatment would be considerable. Consequently, there have been several attempts to develop models that predict which high-risk infants would benefit most from palivizumab prophylaxis [71, 149, 150]. In Canada, use of palivizumab according to the Canadian Risk-Scoring Tool [149] in infants with a GA of 32–35 weeks has been shown to be cost-effective [151] and convenient [152]. The modified recommendations of the AAP propose a maximum of 3 monthly doses, instead of the usual 5 monthly doses, in infants born at a gestational age between 32 and 35 days if they are in daycare or have siblings <5 years of age [148]. It should be noted, however, that there may be another approach to improving the effectiveness of palivizumab and simultaneously reducing cost. In almost one third of infants, trough levels after the first palivizumab dose remain below the 40 μg/ml level that is associated with a 99 % reduction in lung RSV titers in the cotton rat model [153], while trough levels after subsequent doses remain far above this value [141, 142]. A mathematical model for predicting palivizumab serum concentrations suggests that simply shortening the first interval to 23 rather than 30 days would assure adequate serum levels, and this could be followed by administrations of 10 mg/kg every 30 days and still maintain protective levels [154]. Of course, this dosage regimen would have to be tested before being adopted, but it does have the potential for enormous cost savings.
Immune Response to Primary RSV Infection
Viral Mechanisms of Immune Evasion
Reinfections with RSV are common throughout life, even though antigenic diversity among RSV strains is rather limited compared to other respiratory viruses, suggesting that protective immunity is incomplete and short-lived. This is at least partly due to the fact that RSV, like other viruses, has developed numerous mechanisms to evade or subvert the host immune response [155] (Fig. 3). The G protein, which together with the F protein is the only RSV antigen inducing neutralizing antibodies, is heavily glycosylated, and this has been shown to interfere with antibody recognition [156, 157]. Another immune evasion mechanism involves the ability to produce the G protein not only in full-length membrane-bound, but also in a truncated secreted form (Fig. 1). The secreted protein may act as a decoy for neutralizing antibodies [155]. In addition, the central conserved region of the G protein contains a CX3C motif, which endows it with the ability to signal through the CX3CR1 receptor and to exert chemotactic activity similar to that of fractalkine (CX3CL1). Whether this interferes or enhances leukocyte recruitment to the infected lung in vivo is unclear from the available data [158]. Sensitization of mice with recombinant vaccinia virus expressing the RSV G protein followed by RSV challenge activates primarily CD4+ T cells and results in the induction of eosinophilia in the context of a Th2-dominated immune response, whereas priming with the F protein activates both CD4+ and CD8+ T cells induces mainly type 1 cytokines and lung inflammation [159]. Similarly, human RSV G protein-specific T-cell lines produce interleukin (IL)-4 and IL-10, whereas the cytokine profile of F protein-specific T-cell lines is Th1-dominated and similar to that induced by live virus [160]. This implies that the RSV G protein has the potential to downregulate cellular immune responses. There are also data suggesting that the G protein can downregulate inflammatory responses by antagonizing signaling through toll-like receptors (TLRs) [155, 158]. In addition, RSV can infect dendritic cells (DCs), which results in their maturation, but nonetheless reduces their ability to present antigen and activate naïve T cells, thus altering the cytokine milieu through a variety of mechanisms. These include failure to form an immune synapse between DCs and naïve T cells, and the secretion of an inhibitory factor [161]. Furthermore, the NS1 and NS2 proteins of RSV can interfere with the production of type I IFNs, which play a vital role in viral clearance [155, 158].
Fig. 3.

Methods for evasion of host defenses
Immaturity of the Infant Immune System
Primary RSV infection most often occurs during the first few months of life. While the immune system of neonates and young infants is capable of producing adult-like responses under certain conditions, both innate and adaptive immune responses are often characterized by quantitative or functional deficiencies [162]. Compared to adults, macrophages from neonates and young infants and children frequently show markedly lower production of a variety of cytokines. This is at least in part due to the diminished expression of pathogen-associated pattern recognition receptors or their decreased upregulation upon activation.
DC numbers are reduced in neonates compared to adults and differ in their subset distribution. They show signs of impairment in antigen presentation and T cell stimulation due to decreased expression of MHC class I and II and co-stimulatory molecules and reduced production of cytokines, in particular IL-12p70. The most striking characteristic of neonatal T cell responses is their extraordinary plasticity, ranging from relative unresponsiveness to stimuli that provoke strong responses in adult T cells to the capacity to generate adult-like responses if and when the appropriate stimuli are provided. In many situations, however, CD4+ T cells exhibit a diminished capacity to produce both Th1 and Th2 cytokines. In particular, the mitogen-induced and antigen-specific production of interferon (IFN)γ does not reach adult levels for several years, possibly not until adolescence [163–166]. Despite this low IFNγ-producing capacity, Th2 skewing has not been clearly demonstrated in human viral infections [162], although it is observed in neonatal mice and in human neonatal responses to environmental allergens [167]. While neonates may exhibit deficient CD8+ T cell responses in some situations, they are clearly capable of developing mature cytotoxic T lymphocyte (CTL) responses to specific antigens, including viruses [162].
Neonatal B cell antibody production is often characterized by delayed onset, decreased peak titers, and diminished duration, although higher antibody titers to certain vaccines have been noted in some circumstances [168, 169]. In addition, infant B cells show little evidence of somatic hypermutations after encounter of their cognate antigen [170], resulting in lower affinity and decreased heterogeneity of the antibody repertoire [162]. The fact that, after the age of ∼2 years, only individuals who are immunocompromised because of immunodeficiency diseases, chemotherapy, or immunosenescence experience severe lower respiratory tract involvement with RSV infection suggests that the immaturity of the immune system in early childhood is a major factor in the frequent development of LRTI during primary RSV infection [77].
Models of Human RSV Infection
There is limited information on the normal human immune response to primary RSV infection. This is due to several factors. For one, laboratory confirmation generally is sought only in the most severe cases that require hospitalization, which may not be representative of the milder disease seen in the vast majority of patients. In addition, primary RSV infection occurs at such a young age that sampling is generally limited to the nasal lavage or nasopharyngeal aspirates (NPA) routinely taken for diagnostic or therapeutic purposes and nonbronchoscopic BAL fluid obtained from mechanically ventilated patients. Therefore, much of the information currently available on the human immune response to RSV infection comes from experimental models.
In Vitro Models
The primary targets of RSV infection are bronchial and bronchiolar ECs, particularly those that are ciliated [171, 172]. The most common in vitro models for the study of airway EC responses to RSV infection are immortalized respiratory epithelial cell lines. However, evidence is emerging that their responses to RSV infection differ substantially from those of primary airway ECs, including different kinetics of viral replication and lower viral titers, decreased cytotoxic responses and reduced production of pro-inflammatory cytokines compared to primary airway ECs [173]. In addition, primary respiratory ECs retain the donor characteristics, i.e., seem to faithfully reproduce the variability of human responses to RSV infection.
Well-differentiated human airway epithelial cultures probably represent the most faithful in vitro model for the study of the interaction of RSV with host cells. Primary human respiratory ECs, when cultured at an air–liquid interface, regrow into polarized pseudostratified airway epithelium that contains all the cell types and exhibits all the morphological and functional characteristics of normal respiratory epithelium, including mucus production, ciliary motion, and cytokine and chemokine production [174–177]. This model has been used to confirm that RSV infects the airway epithelium from the apical side only and targets almost exclusively ciliated epithelial cells [175, 177], as already seen in autopsy studies and polarized epithelial cells [171, 172]. Budding and release of the virus also occurs from the apical surface, with subsequent spread via ciliary motion [175]. This results in patchy infection, suggesting that not all ciliated cells are susceptible to RSV infection. Although there is some disagreement over the extent of cytopathology induced by RSV infection, including syncytia formation [174, 175, 177], it has been demonstrated that this model quite faithfully captures many of the features of severe RSV infection in human infants, including apoptosis and sloughing of ECs, mucus hypersecretion due to goblet cell hyperplasia or metaplasia, and the production of various chemokines and cytokines [174, 177].
The RSV receptor has not yet been identified, but nucleolin has recently been identified as a promising candidate [178]. It is known, however, that various extracellular and intracellular pattern recognition receptors, including TLRs 2, 3, 4, and 7, RNA helicases, such as retinoic acid-inducible gene I, and possibly nucleotide-binding oligomerization domain-like receptors are involved in mediating the early response to RSV via the induction of NF-κB and IFN response factors [179–181]. Upon infection in vitro, airway ECs can produce a variety of cytokines and chemokines, including macrophage inflammatory protein-1a (MIP-1a, CCL3), monocyte chemotactic protein-1 (MCP-1, CCL2), RANTES (regulated on activation, normally T cell-expressed and secreted, CCL5), eotaxin (CCL11), IL-8 (CXCL8), monokine induced by IFNγ (MIG, CXCL9), IP-10 (CXCL10), fractalkine (CX3CL1), but also the proinflammatory cytokines IL-1β, IL-6, and TNFα [117, 174, 182–186]. Conflicting results have been obtained regarding the ability of RSV to induce production of the antiviral cytokine IFNα by airway ECs [177, 187]. The discrepancies may be related to the use of epithelial cell lines compared to primary human ECs, which show differential responses to infection with the same RSV strain [173]. In addition, individual strains of RSV differ markedly in their ability to induce some of these cytokines and chemokines and [117, 118], epithelial cells from different donors show marked variability in the cytokine and chemokine responses to infection with the same RSV strain [117], and both constitutive production and upregulation following RSV infection are dependent on the localization of the epithelial cells in the airways [182, 188]. Either in response to transient infection or to activating signals received from bronchial ECs [189, 190], alveolar macrophages can further enhance the chemokine and cytokine release of bronchial ECs and can themselves contribute to their production [185, 190, 191].
Animal Models
Animal models of human RSV disease include a variety of heterologous hosts that are only semi-permissive to infection with hRSV. They comprise chimpanzees, rhesus monkeys, sheep, cotton rats, guinea pigs, and mice [192], with guinea pigs being somewhat unusual in that they develop persistent or even latent infection [193], while there is as yet no clear evidence for persistence of RSV in humans. In addition, there are several pneumoviruses with great similarities to hRSV, namely bovine RSV (bRSV), ovine RSV, and pneumonia virus of mice [192]. Over the entire genome there is approximately 80 % homology between bRSV and hRSV. Polyclonal antibodies specific for hRSV generally recognize bRSV proteins [194]. In particular, bRSV shares many of the features of hRSV, especially the occurrence of most severe disease in young (<6 months) individuals and less severe disease in older animals. The bovine model has the advantage over murine models that BRSV is a pathogen for calves and as such creates a disease that is in most ways identical to RSV in human children. The clinical and pathological features of the bRSV model are described [195].
The most widely used models of hRSV infections are inbred laboratory mouse strains, because of the ease of housing and handling them, the availability of a wide variety of transgenic and gene-deletion mice as well as immunological reagents for characterizing immunopathological pathways [192]. However, they are at best a semi-permissive hosts, their lung anatomy is much simpler compared to humans, and the more obvious clinical signs of illness in mice are non-specific and include weight loss, lethargy, and ruffled fur. Acute RSV infection in mice can result in airway obstruction and AHR that can be measured by whole body plethysmography [196–199], although there are certain strains of RSV that do not induce AHR [116, 200]. However, even the most permissive mouse strain, BALB/c, requires a very high intranasal inoculum (generally 106 or 107 PFU) in order to elicit AHR. For comparison, adult humans can develop symptomatic infections with an inoculum of as little as 1,000 PFU [201]. Peak viral titers in the airways or lungs of mice are seen 4–5 days after inoculation depending on the size of the inoculum and the virus becomes undetectable by plaque assay by day 7 or 8 postinfection [199, 202].
The Immune Response to RSV Infection in Mice
Cellular Mediators of the Immune Response
The majority of the chemokines and inflammatory cytokines produced by airway ECs in response to in vitro infection with RSV have been detected in lung tissue [136, 185, 203] or BAL fluid of RSV-infected mice, including the mouse IL-8 analog KC [198, 199]. Their induction is rapid and generally peaks early, although the individual mediators vary markedly in their kinetics and peak concentrations. Of note, depletion experiments in mice identified macrophages as important direct or indirect contributors to the production of CCL3 and CCL5 as well as TNFα, IL-6, and IFNα [204]. In addition, macrophage depletion significantly reduced the recruitment and activation of natural killer (NK) cells, but did not affect the recruitment of CD4 and CD8 T cells to the lungs. Pro-inflammatory cytokines, such as IL-1, IL-6, and TNFα induce or upregulate the expression of adhesion molecules, thereby contributing to the retention of recruited immune cells. In addition, they play a central role in the activation of effector cells. Increased levels of these cytokines have been detected BAL fluid early after RSV inoculation [198, 199]. There are indications that TNFα contributes both to viral clearance in the early stages of RSV infection and the RSV-induced immunopathology in the later phases, at which time T cells may also contribute to the production of this cytokine [205–207]. In contrast, others found a positive association between TNFα levels in BAL and viral load, while IL-6 concentrations correlated with both viral load and disease severity [199].
The concerted action of chemokines and pro-inflammatory cytokines in the initial phases of RSV infection is associated with marked changes in the cellular composition of BAL fluid and lung tissue. After inoculation of mice with hRSV, there is an early rise in the numbers of neutrophils cells in BAL and lungs [114, 196, 199, 208–215]. However, the magnitude and the kinetics of these changes vary widely between studies and also between mouse strains [216]. NK cells start appearing in the lungs and in BAL ∼2 days after infection, their numbers reaching a peak on day 4 and rapidly declining thereafter [213–215]. NK cell activity reached a maximum on day 3 and was undetectable by day 8 [213]. NK cells play an important role in viral clearance in the early phase of RSV infection [211]. Eosinophils generally constitute <1 % of total BAL cells in mice and their numbers do not significantly differ between RSV-infected and control animals [139, 196, 217]. There are, however, experimental systems in which a small but significant increase in the number of eosinophils is observed in BAL and among isolated lung cells of RSV-infected mice [218–220]. In some systems, eosinophils also constitute a minor, but significant component of the lung infiltrate [209]. Macrophages always are the major cell type in BAL of mice, and may be further upregulated by RSV infection, although some noted the largest increases by day 6 and not in the early phase [210].
The importance of some of these chemoattractants in the recruitment of effector cells to the lung has been confirmed through blockade or deletion of specific chemokines or chemokine receptors. With the exception of a study that failed to detect any effect of CCR1-deficiency on the rather unsusceptible C57BL/6 background [221], these experiments show significant attenuation of pulmonary inflammation in MIP-1α-deficient mice [203], a specific reduction in innate immune effector cells such as NK cells, neutrophils, and other leukocytes in BAL of CX3CR1-deficient mice [222], and a decrease in lung lymphocyte counts in CCR1−/− mice on the BALB/c background [186]. In addition, the RSV-induced changes in airway physiological responses were significantly reduced after RANTES blockade [223], in CCR1 deficiency [186], and in CXCR2-deficiency despite the absence of any effect of the lung infiltrate [224].
While NK cells play an important role in viral clearance during early disease [211], this role is shifted to T lymphocytes in the later stages, particularly CD8+ T cells. In BAL of RSV-infected mice, the CD4+ T cell count begins to rise early, reaches a plateau from around day 4 to day 7 or 8, then declines but remains elevated past day 20. On the other hand, CD8+ lymphocyte numbers rise steeply between day 4 and day 8 or 9, then decline, but stay increased past day 20 [204, 210, 211, 213]. Similar kinetics were observed for RSV-specific CD8+ T cells [137, 204]. Lymphocyte recruitment coincides or is preceded by a second peak in the production of certain chemokines around days 4–6, including MIG, MIP-1α, RANTES and eotaxin [185, 198, 199]. While perivascular edema and necrotic cellular debris may be evident in the mouse lung as early as 1 day after RSV inoculation [114], the neutrophils and monocytes/macrophages that dominate the early inflammation are only beginning to marginate at this time [199]. At the peak of inflammation ∼7 days postinfection, there is a dense perivascular and peribronchial infiltrate that reaches into the alveolar spaces and is composed predominantly of macrophages and lymphocytes with some neutrophils [199, 212, 225].
It is well established that both CD4 and CD8 T cells are required for efficient viral clearance in RSV-infected mice [225–227]. However, it is equally evident that both T lymphocyte subsets contribute to the pathology of RSV infection since clinical illness is markedly reduced after depletion of either CD4 or CD8 T cells and is essentially absent in mice depleted of both [225]. Conversely, the transfer of RSV-specific CTL results in rapid viral clearance in immunocompromised (irradiated) and immunocompetent animals, with the rate of viral clearance depending on the number of transferred cells [227]. At the same time, recipients of high numbers of RSV-specific CTL developed weight loss, ruffled fur, and respiratory distress in association with increased pulmonary inflammation. At the highest CTL numbers, almost all recipients died. Similar results have been obtained with less purified RSV-specific CD8+ T cells [226]. Recipients of RSV-specific CD4+ T lymphocytes also showed enhanced viral clearance, but increased lung pathology, and while passive transfer of both CD4+ and CD8+ T cells enhanced polymorphonuclear lymphocyte efflux, transfer of CD4+ lymphocytes was associated with pronounced eosinophilia. There are indications that both the perforin/granzyme pathway and Fas–FasL interactions play a role in the cytotoxic activity of RSV-specific CTL, although deletion of either of these pathways alone only delays, but does not prevent, viral clearance [206, 228].
From the currently available data, it is clear that there is an early influx of both conventional and plasmacytoid DCs (cDCs and pDCs) into the lungs and lung-draining lymph nodes of RSV-infected mice [229–232]. However, the kinetics of this influx and the persistence of individual DC subsets in the lungs remain highly controversial. Some data indicate that the increase in pDC numbers is sustained until at least day 21 postinfection [229, 231]. In marked contrast, other investigators report only a transient early increase in pDCs, whereas cDCs remained elevated until at least day 18 in the lung, though they had returned to baseline levels on day 14 in the lymph nodes [230]. Depletion of pDCs results in decreased viral clearance, enhanced inflammation in the airways and lung parenchyma, increased mucus production and prolonged RSV-induced AHR [229, 231]. This indicates that pDCs play an essential role in limiting viral replication and regulating inflammatory responses and changes in lung function. There is also controversy regarding the ability of RSV to induce production of IFNα by murine pDCs [187, 229, 230] and concerning the question of whether pDCs or bronchial ECs are the major source of IFNα production in the lungs of RSV-infected mice [187, 231]. Whatever the cellular source may be, IFNα is upregulated in BAL fluid of RSV-infected mice [187, 231, 233]. Exogenous IFNα enhanced viral clearance in normal and pDC-depleted mice [231, 234], but had no effect on AHR in either group of mice, indicating that pDCs modulate airway disease through mechanisms other than IFNα production [231].
While a major function of pDCs is to produce the antiviral type I IFNs, cDCs are vital for antigen presentation and inducing the appropriate polarization of the ensuing T cell response. It is well established that cellular immune responses and the production of IFNγ are essential for the elimination of viruses. Mice develop a strong type 1 immune response to RSV, characterized by a substantial induction of IFNγ mRNA and protein in lung and BAL fluid [198, 199, 235–238]. NK cells are the major source of this cytokine in the early phase of infection [211, 213, 214]. Since NK cells are also the major producers of granzyme B during this stage, NK cell-depleted mice show not only a pronounced reduction of IFNγ concentrations in lung tissue and BAL, but also a marked increase in viral load [211, 214]. In addition, they exhibit elevated Th2 cytokine levels (IL-4 and IL-13), tissue eosinophil infiltration, and IgE production [214], although reportedly there was no effect of NK cell depletion on the RSV-induced cytokine profile in another study [211].
Cytokines and Chemokines
In the majority of studies, induction of IL-4 and/or IL-5 is not observed in BAL or lung tissue of RSV-infected mice [198, 236–238]. If it does occur, the transcript and protein levels of these Th2 cytokines are markedly lower compared to IFNγ [199], even in experimental animals that develop eosinophilia in response to RSV infection [218, 239]. Even G protein-primed RSV-challenged mice, which show a CD4+ T cell-dominated infiltrate with marked lung eosinophilia, exhibit similar proportions of IFNγ-positive T lymphocytes in BAL fluid as F-protein or M-protein primed mice and very few IL-4 or IL-5 positive cells [240]. There is, however, at least one report of very high production of IL-4 in anti-CD3-stimulated thoracic lymph node mononuclear cells after RSV infection, but IFNγ levels were still markedly higher [241]. IFNγ deficiency was associated with more extensive airway inflammation in one study [235]. However, the increased airway resistance and the prolongation of expiratory time observed in wild-type mice were less pronounced in IFNγ-deficient mice. In another study, absence of IFNγ was associated with greater weight loss compared to wild-type controls [206]. In contrast, others found IFNγ-deficient mice not to differ significantly from wild-type mice in terms of BAL fluid composition, the extent or type of pulmonary inflammation, mucus production, or AHR [237]. In this model, a partial overlap in the functions of IFNγ and type I IFNs in viral infections could be demonstrated. Mice incapable of responding to IFN type I or type II because of STAT1 deficiency had enhanced disease with earlier peak viral load, greater inflammatory infiltrate that contained neutrophils and eosinophils instead of the predominantly lymphocytic infiltrate of wild-type mice. This was associated with decreased levels of IFNγ, increased lung tissue levels of IL-13, and a smaller and non-significant enhancement of IL-4 and IL-5 [202]. Further experiments in mice lacking the receptors for IFNα and/or IFNγ showed that STAT activation through either type I or type II IFN was required to establish a Th1 response to RSV infection and to dampen RSV-induced immunopathology [202, 216].
IL-12
IL-12, particularly in conjunction with IL-18, induces IFNγ and promotes type 1 immune responses. However, there are data indicating that IFNγ production after RSV infection of mice is independent of IL-12 and IL-18 [242]. Nonetheless, comparison of mouse strains that differ in their susceptibility to RSV-induced AHR led to the identification of IL-12 as an important mediator of protection from pulmonary inflammation, mucus production, and AHR in B6 mice, and this was confirmed through the use of neutralizing anti-IL-12 antibodies [238]. In other experiments, absence of IL-12 signaling due to IL-12 receptor deficiency on a C57BL/10 background was associated with reduced NK cell function (but similar NK cell numbers) and delayed viral clearance compared to wild-type mice [243]. In contrast, BALB/c mice with a targeted deletion of the IL-12p40 gene BAL exhibited both reduced NK cell numbers and function. However, the generation of virus-specific CTL and their functional activities were not affected by IL-12p40 deficiency. When mice were infected with a recombinant RSV expressing murine IL-18, viral clearance was enhanced in association with increased recruitment of NK cells, but weight loss was also much more pronounced [211].
IL-13
All of the above studies were conducted in adult mice. However, neonatal immune in responses in mice are also immature, and neonatally infected mice were found to elaborate significantly lower IFNγ levels in BAL fluid, whereas IL-13 concentrations in lung tissue were significantly higher compared to mice infected as weanlings [244]. Mice infected as weanlings (3 weeks old) developed AHR upon primary infection, but not when reinfected 5 weeks later despite evidence of increased airway inflammation. In contrast, mice initially infected at <1 week of age and reinfected 5 weeks later showed further enhancement of AHR compared to age-matched controls undergoing primary infection. This enhanced AHR was associated with IL-13-induced eosinophilia and mucus production, although IFNγ levels were comparable to those seen after primary or secondary infection in weanling mice.
The role of IL-13 has been investigated primarily in the context of RSV-induced AHR, because of the central role of this cytokine in allergic inflammatory airway disease [245]. There is strong evidence that individual laboratory strains and clinical isolates of RSV differ markedly in their ability to upregulate IL-13 production in the lungs of mice [114–116]. They also showed differential induction of AHR, but this did not appear to be directly correlated with the pulmonary levels of IL-13. Nonetheless, AHR after infection with IL-13 inducing RSV strains was greatly attenuated in IL-13 deficient mice [116] or after neutralization of IL-13 with polyclonal antibodies [246]. This was associated with a significant increase in IL-12 in lung supernatants and decrease in mucus production, which has been observed in other studies of RSV infection in mice [244] and is consistent with the known ability of IL-13 to induce goblet cell hyperplasia and mucus production [245]. This may explain why a RSV strain that did not enhance pulmonary IL-13 levels failed to induce AHR [116, 200, 247]. It does not explain, however, why other researchers did not see a reduction in RSV-induced AHR after inhibition of IL-13 signaling or in IL-13-deficient mice, even though the strain they used greatly upregulated IL-13 levels in the lung [209]. This is particularly noteworthy since neutralization of IL-13 decreased the levels of IL-5 in BAL, increased the concentration of IL-12 and virtually abolished the increased mucus production seen after RSV infection. In similar experimental models, IL-5-dependent eosinophilia has been implicated in RSV-induced AHR [220].
IL-10
The regulatory cytokine IL-10 is upregulated in the airways and lung parenchyma of RSV-infected mice [136, 248]. Transgenic expression of IL-10 in the nasal mucosa resulted in significantly reduced viral replication in the nasal mucosa and the lung in association with increased cytotoxic activity of nasal CD4+ T cells [249]. There is some disagreement over whether Tregs or conventional CD4+ and CD8+ T lymphocytes constitute the major source of IL-10 in the lungs of RSV-infected mice [136, 215, 248]. There is consensus, however, that the absence of IL-10 signaling either due to genetic IL-10 deficiency or IL-10 receptor (IL-10R) blockade increases disease severity and inflammation. Specifically, it enhances the recruitment of monocytes, neutrophils and effector lymphocytes [215, 248]. The numbers of RSV-specific CD8+ T cells and their IFNγ production were increased in the absence of IL-10 signaling, as were the numbers of IL-17A-producing CD4+ T cells [136]. Furthermore, IL-10R blockade was associated with a reduction in the number of Helios+ Foxp3+ CD4+ T cells, suggesting that IL-10 plays a role in maintaining natural Tregs.
Indeed, very similar effects have been observed following the depletion of Tregs. Following infection of BALB/c mice with RSV, there is a rapid and dramatic increase in the absolute and relative numbers of Foxp3+ Tregs among CD4+ T cells in the airway, lung parenchyma and lung-draining lymph nodes [138, 217]. The results of three independent depletion experiments in BALB/c [138, 217] and hybrid CB6F1 mice [137] demonstrate that these Tregs play a vital role in regulating the recruitment of innate (NK cells and neutrophils) and, particularly in the later stages of the disease, of adaptive (T and B lymphocytes) immune cells. Whether this accelerates viral clearance is somewhat controversial. In addition, Tregs dampen the production of chemokines and pro-inflammatory cytokines [137, 217], including the production of TNFα and IFNγ by virus-specific CTL [137, 138], and control the trafficking of virus-specific CD8 T cells from the lymph node into the lung [138]. The results from the hybrid CB6F1 model further suggest that Tregs also play a role in moderating disparities in epitope dominance [137]. As a result, Tregs are central in diminishing disease severity, including overall morbidity and weight loss [137, 138, 217]. Interestingly, the depletion of Tregs in CB6F1 mice was associated with increased pulmonary levels of IL-10 [137], suggesting that IL-10 alone is not sufficient for suppression of the RSV-induced immunopathology and that the effects of IL-10 deficiency or IL-10R blockade are largely mediated by the failure to maintain adequate numbers of Tregs [136]. The anti-CD25 antibody that was used in these studies for depletion of Tregs also eliminates activated CD25+ T cells. “Depletion of regulatory T cell” mice allow the specific depletion of Foxp3+ Tregs. While weight loss was even more dramatic in these mice, the overall results were similar, with the notable difference that viral load was decreased in Treg depleted mice [139]. Conversely, increasing the number of Tregs via IL-2 immune complexes decreased inflammation and accelerated recovery. Interestingly, the Tregs in the lung parenchyma and airways of RSV-infected mice were found to express granzyme B, and this was found to be essential for the regulatory function of Tregs in RSV infection.
IL-17
IL-17 designates a group of cytokines that play a central role in adaptive immune responses to bacteria and fungi, but are also able to induce pro-inflammatory responses. The major cellular sources of IL-17 are CD4+ T helper type 17 (Th17) cells, but double negative and γδ T cells can also be important contributors [250]. Mice inoculated with RSV were found to display significant upregulation of IL-17 (A), but not IL-17F in the lungs and lung-draining lymph nodes [251]. There also was an increase in the transcript levels of IL-6 and IL-23p19, which are involved in the differentiation and maintenance of Th17 cells. Intracellular expression of IL-17 was increased in CD4+ T cells, but not γδ T cells. Furthermore, IL-17 was shown to upregulate mucus production and to inhibit CD8 T cell effector functions, thereby reducing viral clearance.
Others identified macrophages, not T cells, as the major source of IL-17A in RSV infection [197]. In this study, it was shown that signaling through the receptor of the complement anaphylatoxin C3a (C3aR) induces production of the tachykinins, substance P and hemokinin-1. This signals, through their common receptor, neurokinin-1 receptor (NK-1R), resulting in the production of IL-17A and related cytokines, including, IL-6, IL-21, IL-23, and IL-1β. Mice deficient in C3aR or NK-1R did not exhibit increased airway resistance following RSV infection. This was associated with markedly reduced pulmonary inflammation and a different composition of the inflammatory infiltrate (decreased neutrophils, and increased macrophages and lymphocytes) compared to wild-type controls. Note, however, that there was still some RSV-induced production of IL-17A in C3aR-null mice, but not in TACR1-null mice, suggesting that other pathways contribute to IL-17 production.
Other Cytokines
Other studies similarly show elevated concentrations of substance P in BAL and lung tissue of RSV-infected mice and demonstrate that selective NK-1R antagonists can inhibit the development of RSV-induced AHR if given prophylactically, but not therapeutically [210]. Prophylactic blockade of NK-1R signaling was accompanied by reduced airway lymphocytic inflammation. In addition, it prevented the RSV-induced development of airway smooth muscle responsiveness to electric field stimulation. This suggests that substance P plays a dual role in the development of AHR during acute RSV infection, namely proinflammatory and neurogenic. Others found both prophylactic and therapeutic neutralization of substance P to be effective in reducing lung inflammation, and addition of anti-F protein antibodies did not significantly enhance this effect [252]. The involvement of substance P in neurogenic inflammation is supported by a variety of studies in RSV-infected rats and other animals [253]. The results from these experiments implicate the RSV-induced upregulation of nerve growth factor (NGF) and its receptors and the subsequent increase in the expression of NK-1R (at least at the transcript level) along with functional interactions between substance P-containing neurons and mast cells and their inflammatory mediators in the exaggerated neurogenic inflammation [253]. The role of cytokines and chemokines is summarized in Table 4.
Table 4.
The immunology of RSV infections
| Components | Normal functions | Evidence for effect | Relevance to RSV |
|---|---|---|---|
| Proinflammatory cytokines | |||
| IL-1, IL-6, TNF-α | IL-1, IL-6 and TNFα induce or upregulate the expression of adhesion molecules | Elevated in BAL in mouse | RSV inoculation |
| Th1 cytokines | |||
| IL-12, IL-18 and IFN-γ | Interleukin (IL)-12 and IL-18 induces IFNγ and promotes type 1 immune responses. Production of IFNγ by PBMC’s from calves with formalin-inactivated vaccine exacerbation of BRSV infection is significantly depressed when compared to sham vaccinated controls. | (See Th2 cytokines) | The Th1 and Th2 contributions to the pathogenesis of RSV bronchiolitis are not clear. It is likely a mixed response |
| Th2 cytokines | |||
| IL-4, IL-5, IL-13 | Allergic inflammation | Even G protein-primed RSV-challenged mice, which show a CD4+ T cell-dominated infiltrate with marked lung eosinophilia, exhibit similar proportions of IFNγ-positive T lymphocytes in BAL fluid as F-protein or M-protein primed mice and very few IL-4 or IL-5 positive cells. One report of high production of IL-4 in anti-CD3-stimulated thoracic lymph node mononuclear cells after RSV infection, but IFNγ levels were still markedly higher. Levels of Th2 cytokines in lymph draining the lung of BRSV-infected calves show early peak on day 4 post infection in IL-4 and IL-13 [260]. | (See Th1 cytokines) |
| Innate immunity | |||
| NK cells | Antibody independent cytotoxicity | NK cells start appearing in the lungs and in BAL ~2 days after infection, peaking on day 4 and rapidly declining thereafter. NK cell activity reached a maximum on day 3 and was undetectable by day 8. | NK cells play an important role in viral clearance in the early phase of RSV infection |
| Th17 | |||
| Th17 and IL-17 | Plays a central role in adaptive immune responses to bacteria and fungi are also able to induce pro-inflammatory responses. Cellular sources of IL-17 are Th17 cells, double negative and γδ T cells. | Mice inoculated with RSV display upregulation of IL-17 (A), but not IL-17F in the lungs and lung-draining lymph nodes. An increase in the transcript levels of IL-6 and IL-23p19are also found. Intracellular expression of IL-17 was increased in CD4+ T cells but not γδ T cells. | IL-17 upregulates mucus production and inhibits CD8 T cell effector functions, thereby reducing viral clearance |
| Dendritic cells | |||
| pDCs and cDCs | pDCs produce type I IFNs, cDCs serve as antigen presenting cells and induce polarization of the T cell response | Ability of RSV to induce production of IFNa by murine pDCs; pDCs or bronchial ECs are a possible source of IFNα production in the lungs of RSV-infected mice | Depletion of pDCs results in decreased viral clearance, enhanced inflammation in the airways and lung parenchyma, increased mucus production and prolonged RSV-induced AHR |
| T Reg function | |||
| IL-10 | IL-10R blockade was associated with a reduction in the number of Helios+ Foxp3+ CD4+ T cells, suggesting that IL-10 plays a role in maintaining natural regulatory T cells | IL-10 is upregulated in the airways and lung parenchyma of RSV-infected mice; atopy and higher IL-10 production during acute RSV infection in humans. | |
| T regulatory cells (Tregs) | Tregs dampen the production of chemokines and pro-inflammatory cytokines and control the trafficking of virus-specific CD8 T cells from the lymph node into the lung | (See IL-10) | Infection of BALB/c mice with RSVleads to a dramatic increase in the absolute and relative numbers of Foxp3+ Tregs in the airway, lung parenchyma and lung-draining lymph nodes |
| Eosinophils and leukotrienes | |||
| ECP, EDN, Leukotrienes | Eosinophilic inflammation | In some studies, eosinophil levels are more than 2SD higher than control patients. | ECP and EDN are detected at significantly higher levels and in a significantly higher proportion of patients with RSV compared to healthy controls. Levels of ECP were higher in infants and children with RSV bronchiolitis compared to those with LRTI without wheezing. |
| Pathways of apoptosis | |||
| Granzyme/perforin and Fas–FasL interactions | A role in the cytotoxic activity of RSV-specific CTL | Deletion of these pathways delays but does not prevent viral clearance | |
| Chemokines | |||
| IL-8 (CXCL8), MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4) | Neutrophil chemotaxis | Concentrations of IL-8, MIP-1α, and MCP-1 were found to directly correlate with various measures of disease severity | MCP-1 levels were inversely associated with the duration of supplemental oxygen requirement. |
| RANTES (CCL5) | Chemokines help to determine the degree of inflammation and the composition of the inflammatory infiltrate | RSV-induced changes in airway physiological responses were significantly reduced after RANTES blockade in mice. RSV infection of airway epithelial cells in vitro stimulates upregulation of RANTES | By recruiting specific leukocyte subsets, chemokines contribute to the determination of disease severity. |
| Humoral immune response | |||
| IgE | IgE-mediated response to RSV | In neonatally infected mice, virus-specific IgE plays a role in mediating enhanced AHR upon reinfection.In BRSV infected calves severity of experimental disease correlates with development of BRSV-specific IgE response [260, 497]. | Low levels of RSV-IgE are detectable in the late stages of acute infection, with peak concentrations occurring ~3 weeks after infection. |
| IgG, M, A | Host defense to infection | RSV-infected mice produce RSV-specific serum IgM, followed by IgG, predominantly of the IgG2a isotype. BAL fluid contains RSV specific IgA | |
| Neuroimmunology | |||
| Substance P | Proinflammatory and neurogenic effects | Elevated concentrations of substance P in BAL and lung tissue of RSV-infected mice. Selective NK-1R antagonists can inhibit development of RSV-induced AHR if given prophylactically. Prophylactic blockade of NK-1R signaling was accompanied by reduced airway lymphocytic inflammation, and decreased airway hyperresponsiveness. | Substance P plays a dual role in the development of AHR during acute RSV infection, namely proinflammatory and neurogenic. SP-A and SP-D can modulate TLR4 signaling |
Humoral Immune Response
The F and G proteins are the dominant RSV antigens containing epitopes that induce neutralizing antibodies. RSV-infected mice produce RSV-specific serum IgM, followed by IgG, predominantly of the IgG2a isotype, in agreement with their strong type 1 cytokine response [254, 255]. In addition, their BAL fluid contains RSV-specific IgA. In the absence of humoral responses, viral clearance is not affected, but lung inflammation is more severe and viral replication and clinical illness after rechallenge with RSV are more pronounced compared to animals with an intact B cell compartment [256]. Whether elaboration of RSV-specific antibodies or administration of passive antibodies suppresses viral replication in the lower respiratory tract only or also in the upper respiratory tract appears to depend on the specific animal model [255, 257].
Through the use of adoptive transfer experiments using B cells from wild-type, μMT mice (lacking mature B cells), and mice lacking the adaptor molecule myeloid differentiation factor 88 (MyD88) that propagates signaling downstream of TLR ligation, it could be shown that TLR stimulation in B cells is an essential step in eliciting protective, affinity-matured antibodies against RSV [258]. The role that TLRs play in this process probably hinges on their ability to induce DC maturation and T helper and B cell activation.
Some experimental mice infected with RSV develop RSV-specific IgE antibodies [259], although this was not observed in a similar experimental system [254]. However, bRSV-infected calves also exhibit RSV-specific IgE [260] and hRSV-infected guinea pigs produce RSV-specific antibodies of the IgG1 subclass, the major anaphylactic antibody in this species [261]. Guinea pigs are capable of producing IgE antibodies. Since only low levels of RSV-IgE are detectable in the late stages of acute infection, with peak concentrations occurring ∼3 weeks after infection, the production of virus-specific IgE is likely to become relevant only during reinfection. Indeed, in neonatally infected mice, virus-specific IgE was shown to play a central role in mediating enhanced AHR upon reinfection [262]. Similarly, passive sensitization of adult mice with RSV-IgE 2 days after inoculation with live virus resulted in a significant augmentation of RSV-induced AHR [259].
The Human Immune Response to RSV Infection
Chemokines
Many of the chemokines that are present early in the course of RSV infection in the lungs of mice have also been detected in non-bronchoscopic BAL fluid, tracheal aspirates, or nasal secretions obtained from severely RSV-infected infants, and are found at higher levels compared to controls. They include the CC chemokines RANTES (CCL5), MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), and eotaxin (CCL11); and the CXC chemokines IL-8 (CXCL8) and interferon inducible protein 10 (IP-10, CXCL10) [263–271]. Fewer data are available on their role in less severe RSV infection not requiring hospitalization, but some of them are also detected in nasal secretions of infants and children with URTI [184, 267, 272] and experimentally infected adults [201, 273]. Concentrations of IL-8, MIP-1α, and MCP-1 were found to directly correlate with various measures of disease severity [267–269, 274], whereas MCP-1 levels were inversely associated with the duration of supplemental oxygen requirement [275]. This suggests that chemokines, by recruiting specific leukocyte subsets, play a major role in determining the degree of inflammation and the composition of the inflammatory infiltrate and, consequently, the disease severity. Pro-inflammatory cytokines such as IL-1, IL-6, and TNFα have also been detected in nasal or nasopharyngeal secretions and tracheal aspirate of RSV-infected infants at significantly higher levels compared to controls [266, 269, 276, 277]. When nasal brush samples were investigated for the proportion of cells expressing various cytokines, IL-6-positive cells were present in both bronchiolitis and URTI, but were significantly increased compared to controls only in patients with bronchiolitis [278]. Serum or plasma concentrations of IL6 are also significantly higher during acute RSV infection [279, 280] and were found to correlate with disease severity, including a clinical severity score, duration of oxygen therapy, and duration of hospitalization [279], whereas levels in airway secretions did not [269, 279].
BAL Cellularity
In contrast to the early transient rise in the number of neutrophils in RSV-infected mice, neutrophils represent the predominant cell type found in BAL of severely ill, mechanically ventilated patients with RSV infection, making up 75–85 % of BAL cells [276, 281, 282] (see also Table 5). Their numbers are highest in the first few days after intubation and progressively decline thereafter, in parallel with a decline in the total BAL cell count [276]. Of note, the absolute and relative numbers of neutrophils in BAL from RSV-infected preterm infants were found to be significantly lower compared to RSV-infected term infants and similar to those of uninfected controls. Eosinophils are detected in a minority of RSV-infected infants and generally constitute <1 % of the total BAL cell number, which is similar to control values [276, 281, 282]. In a separate study, when bronchoscopic BAL was performed in infants and young children with RSV bronchiolitis, 6/22 patients had eosinophil proportions >2 standard deviations above normal (mean 2.3 % of BAL cells) and were classified as eosinophil positive [283]. This subgroup of patients may represent a specific subtype of RSV bronchiolitis that exhibits some of the characteristics seen in children with an acute asthma attack, including higher BAL concentrations of IL-5 and cysteinyl leukotriene levels (LTC4, LTD4, and LTE4) compared to the eosinophil-negative groups [283, 284].
Table 5.
Non-bronchoscopic bronchoalveolar fluid cellularity in RSV-infected infants
| Heidema [282] | Everard [281] | McNamara [276] | ||||
|---|---|---|---|---|---|---|
| n = 32 | 14 | 24 Term | 23 Preterm | |||
| Duration of symptoms at the time of sampling | 6.3 (2–21 days) | 5.7 | 5.4 | |||
| Method | FACS | Differential | Differential | Differential | ||
| Percentage | Absolute # (105/ml) | |||||
| Granulocytes | 76.4 | 1.5–10.9 | 85 [69–94] | 76 | 85.3 | 78 |
| Monocytes | 25 | 0.4–4.5 | not reported | 10 | 8 | 12 |
| Lymphocytes | 1.5 (range 0–5) | 9 | 6 | 8 | ||
| B cells | 0.4 | 0–0.5 | ||||
| NK cells | 0.4 | 0.1–0.6 | ||||
| CD3+ T cells | 1.9 | 0.3–5.9 | ||||
| CD4+ of CD3+ | 28.1 | 0.1–1.2 | ||||
| CD8+ of CD3+ | 62.2 | 0.2–3.0 |
As summarized in Table 5, somewhat more variable results have been obtained for monocyte and lymphocyte proportions, which may be due to differences in the time of sampling relative to the onset of symptoms. It is evident, however, that CD3+ T cells constitute a minor proportion of BAL cells. Nonetheless, a significant increase in the number of CD8+ T cells, including virus-specific CD8+ T cells, does occur in infants with severe primary RSV infection compared to uninfected controls [282]. However, due to the massive influx of neutrophils during RSV infection, the percentage of CD8+ T cells in BAL is actually lower in severe RSV disease compared to controls (1.6 vs. 8 %). NK cells represented only 0.4 % of total BAL cells [282]. In tracheal aspirates, CD3+ T lymphocytes also constituted only a small portion (1.6–2.1 %) of total cells recovered, with CD8+ T and NK cells representing 0.6–1.3 % and 0.5–1.1 % of total cells, respectively [285].
Note that non-bronchoscopic BAL or tracheal aspirate samples from patients with RSV can only be obtained from infants and young children if they require mechanical ventilation, i.e., these BAL samples may not be fully representative of infants with less severe disease. However, there appears to be a good correlation between the results obtained in non-bronchoscopic BAL samples and NPS, which also show neutrophils to constitute the major cell type, although they may represent an even larger proportion of total cells, while the proportions of lymphocytes and monocytes are somewhat lower [280, 281, 286]. Like mice, infants naturally infected with RSV showed an increase in the numbers of cDCs (also called myeloid DCs) and pDCs in nasal washings in association with a decrease in both DC subsets in peripheral blood compared to healthy controls [265, 287]. This suggests that DCs are mobilized from peripheral blood to the airway mucosa.
Histopathology
The major histopathological characteristics of RSV infection are acute bronchiolitis, mucosal and submucosal edema, and luminal occlusion by cellular debris of sloughed ECs mixed with macrophages, strands of fibrin, and some mucin [171]. Edema and the occluding material in the airway lumen, possibly in conjunction with protrusions of hyperplastic bronchiolar-associated lymphoid tissue, contribute to the obstruction of airways. Whereas older investigations highlight the importance of mucus in this process [288], mucus represents a minor component or is absent from the material occluding RSV-infected airways in recent analyses [171, 289]. The desquamation of ECs is generally deemed to reflect necrosis, but markers of apoptosis are abundant in the RSV-infected epithelium [289, 290], and recent in vitro results confirm that ECs sloughed from RSV-infected epithelium are TUNEL positive, indicative of apoptosis [177].
The infiltrate, though generally characterized as peribronchiolar, is actually centered on the bronchiolar arterioles, and it often extends into the alveoli [171, 291]. It is primarily mononuclear, with alveolar macrophages and recruited monocytes representing the major constituents [171]. In contrast to their predominance in NB-BAL fluid, neutrophils are only a minor component of the peribronchial infiltrate and are found mainly in the submucosa.
Importantly, a formalin-inactivated RSV vaccine that went into clinical trial in the 1960s provided no protection, but instead enhanced disease upon natural RSV infection and resulted in two deaths [292]. The earliest publication mentioned that postmortem examination had revealed a peribronchiolar monocytic infiltrate with “some excess in eosinophils” [292]. While this is time and again referred to as eosinophilia, the actual autopsy report describes a predominance of neutrophils and mononuclear cells in the bronchial and bronchiolar epithelium, as confirmed in a second analysis of the original postmortem tissue [293]. Eosinophils constituted 1–2 % of the inflammatory infiltrate, which agrees with the report of occasional eosinophils in the lungs of a 15-month-old child with complex congenital heart disease who died in a motor vehicle accident 1 day after being diagnosed with RSV infection as an outpatient [171].
In the same child, CD3+ T cells were a significant component of the peribronchiolar infiltrate [171]. A substantial portion of the CD3+ T cells was double negative, CD4+ T cells were rare, but CD8+ T cells were prominent particularly in the alveolar interstitial infiltrate. In contrast, a recent assessment of autopsy tissue from 9 infants with fatal RSV infection revealed the near absence of CD4+ and CD8+ T lymphocytes from the infiltrate [291], which is consistent with the observation that a majority of CD3+ T cells were double negative, but contrasts with the finding of a prominent CD8+ T cell component in the alveolar infiltrate described in the case of non-fatal RSV bronchiolitis [171]. Cells expressing CD56 (NK cells) were also detected only rarely in the nine cases with fatal RSV infection, and staining for granzyme was absent [291].
The Role of T Cells in RSV Immunopathology
Whereas animal studies clearly indicate a role for CD4+ and CD8+ T cells not only in viral clearance, but also in the lung immunopathology after RSV infection, the role of T lymphocytes in human disease is less clear. Several lines of evidence suggest that CD8 T cells do not play a major role in the immune pathology of RSV. Postmortem lung tissue obtained from infants (median age 3 months, range 1–12 months) who had died from severe RSV infection contained very few detectable CD4+ or CD8+ T cells, CD56+ NK cells, or granzyme-positive cells [291]. In contrast, lung tissue from a child with ambulatory RSV infection showed evidence of a prominent CD8+ T lymphocyte infiltrate, particularly in the alveoli [171]. Together, these findings suggest not only that these cells do not participate in the damage of the bronchiolar epithelium typical of severe RSV infection, but also that an inadequate T cell response may underlie the development of severe fatal RSV LRTI. A marked increase in the CD8+ T cell count has been observed in NB-BAL of infants with severe, but non-fatal RSV infection, and these cells exhibit an activated phenotype, including significantly higher expression of granzyme B compared to controls [282]. However, the magnitude of the T cell response did not correlate with any measure of clinical disease severity. In addition, the T cell response in peripheral blood, whether detected by RSV-specific tetramer staining or by measuring the total activated T cell population, peaked at the time of extubation, i.e., during the recovery phase approximately 9–12 days after the onset of symptoms. A further study from the same institution confirmed these findings in another group of infants with severe RSV infection and extended them to the observation that peripheral blood CD8+ T cell responses peaked well after the peak viral load, which was maximal before or on the day of admission to the ICU, and also took place after the greatest symptom severity, which occurred 2–3 days after admission to the ICU [294]. Similarly, the concentrations and activity of granzyme A and B were significantly elevated in tracheal aspirates of RSV-infected patients compared to controls without pulmonary conditions [285]. However, there was no association between granzyme levels and a marker of epithelial apoptosis or markers of disease severity. Furthermore, the most severe disease is generally observed in very young infants, but infants ≤5 months of age were found to have RSV-specific cellular cytotoxic responses much less frequently compared to children 6–24 months of age (38 vs. 67 %) during acute RSV infection [295]. Of note, detectable responses arose only 7 days or more following infection, but waned again in the youngest infants, while many of the 6–24 month-old children exhibiting sustained responses 2–3 months after the acute infection. This suggests that repeated boosting is required before cellular cytotoxic responses are sustained between RSV outbreaks.
Human Cytokine Response to RSV Infection
Whereas mice display a Th1-dominated response to RSV infection with clear upregulation of IFNγ production in the lung, investigations of the cytokine profile during human primary RSV infection have yielded variable results. In mitogen-stimulated peripheral blood mononuclear cells (PBMC), IFNγ secretion was found to be unchanged or even reduced in RSV-infected infants compared to controls [129, 296–298]. It was also considerably lower than seen after other respiratory viruses [296] regardless of whether RSV was the sole virus detected or combined with another viral agent [125]. There are conflicting data concerning the simultaneous upregulation of Th2 cytokines, with some investigations showing a Th2 dominance [297, 298], whereas others are unable to detect IL-4 or IL-5 [129]. There is also controversy regarding the precise role of IFNγ production by PBMC, with a positive correlation with disease severity seen in one study [298], whereas other data suggest a protective effect [129].
In partial contrast, in vitro restimulation of PBMC from acutely infected children with RSV was found to upregulate the intracellular production or secretion of IFNγ already in the acute phase in some infants [280, 299, 300]. This was accompanied by enhanced production of IL-2, IL-4, and IL-13 in one study, suggesting a mixed Th1 and Th2 immune response [299], but was associated with little IL-4 and IL-10 secretion in another investigation, suggesting skewing towards a Th1 immune response [280]. There were no significant differences between the cytokine responses of infants with severe and those with mild RSV disease, although the T cell response was somewhat more pronounced in the acute phase in the mild disease group. Importantly there was no evidence of Th2 skewing in the more severely affected infants. Memory responses of RSV-stimulated PBMC of adults are characterized by strong production of IFNγ as well as IL-10 and IL-6 in essentially all 60 subjects examined, but IL-13 and IL-5 in only 31 % and 8 %, respectively [301].
RSV infection is largely confined to the respiratory tract, even though RSV RNA has been detected in blood monocytes and neutrophils [302, 303]. Therefore, the responses of PBMC may not accurately reflect the local immune responses occurring in the respiratory tract, particularly since it has been shown that RSV-specific CD8+ T cells as well as CD8+ T lymphocytes specific for other respiratory viruses decrease in blood and increase in the respiratory tract even in patients who have only upper respiratory tract symptoms [304], indicating recruitment to the lung. There have been numerous investigations of the cytokine profile in airway secretions during acute RSV infections [11, 101, 267, 275, 305–308]. Note, the concentrations of mediators in samples from the upper airways generally show moderate to strong correlations with those of specimens obtained simultaneously from the lower airways of mechanically ventilated infants [101, 308]. The results are similarly conflicting as those obtained in PBMC. While some investigations show clear upregulation of IFNγ in acute RSV infection compared to healthy controls [267, 305] or non-RSV-related bronchiolitis [275], others find IFNγ to be undetectable in the majority of patients [306], or not to differ significantly compared to controls [287]. Increased concentrations of Th2 cytokines are seen in some studies [11], particularly in infants ≤3 months of age [306], whereas others found them to be undetectable or present at very low levels and not to differ significantly from controls [129, 274, 275, 305, 308, 309]. Consequently, a polarization towards a Th2 immune response has been observed in one cohort of patients with ambulatory LRTI compared to those with URTI only [11], whereas other data suggest Th2-skewing in both URTI and hypoxic bronchiolitis, but not in non-hypoxic bronchiolitis [267]. Data from an earlier study from the same institution indicate a higher IFNγ:IL-4 ratio in subjects with bronchiolitis compared to those with URTI only [308]. Accordingly, IFNγ can be (1) associated with protection from most severe disease [11, 101, 274, 275, 309], (2) have no clear role in determining disease severity [267, 307], or (3) be implicated as a major mediator of immunopathology in different patient cohorts [308]. Note that analysis of cytokine mRNA responses in NPA and BAL samples underscores that there is great inter-individual variation in the expression of Th1 and Th2 cytokines and the dominant cytokine pattern [310].
Methodological issues are likely to contribute to these discrepant results. One of the most obvious is the common practice of reporting absolute cytokine concentrations in NPA or nasal lavage specimens, even though the lavage procedure or the rinsing of the trap introduces an unknown dilution factor. Age is another factor with the potential to influence the results [306], particularly in studies that include mechanically ventilated infants since these are frequently significantly younger than non-ventilated infants [101, 309]. There is a marked age-dependence in the production of IFNγ and IL-12 and responsiveness to IL-12, an important inducer of type 1 immune responses [166, 311–313]. Even if IFNγ synthesis in RSV-infected airways does not seem to require IL-12 and IL-18 [307], which is consistent with experimental results [242], IL-12 and IL-18 may have effects that are independent of their role in the induction of IFNγ and protect from the development of bronchiolitis [278].
Data from animal studies suggest that age at first infection is a crucial determinant of the immune response pattern in later life [244, 314]. Neonatally infected mice showed decreased IFNγ responses compared to mice infected as weanlings or adults and, upon reinfection, this was associated with enhanced inflammation and further augmented AHR in the context of higher IL-4 production and lower IFNγ production. It is unclear to what extent this is relevant to RSV infection in human infants. Disease severity and particularly the involvement of the lower respiratory tract diminish after the primary infection [14] and there is little information on whether the immune response pattern is maintained or changes during secondary infections.
Concentrations of IL-10 in nasal or NPS or tracheal aspirate samples are significantly higher in some patient cohorts compared to controls [269, 315], but not significantly different in others [129, 308, 316]. Some investigators find an association between atopy and higher IL-10 production during acute RSV infection [315] while others do not [316]. Furthermore, the levels of IL-10 in nasopharyngeal or tracheal samples can be directly or inversely associated with disease severity [269, 275, 279]. These discrepancies may be attributable to small sample sizes and methodological issues, but may also relate to the fact that IL-10 has both pro-inflammatory and regulatory functions, and the timing and intensity of its production may determine whether it is protective or detrimental. In addition, animal data demonstrate that individual strains of RSV differ in their ability to induce IL-10 [116].
Other Inflammatory Mediators
In addition to chemokines and cytokines, a variety of other inflammatory mediators have been detected in the nasal wash, NPS, or tracheal aspirate samples of RSV-infected infants and young children. These include leukotrienes (LTs) and products of eosinophil degranulation, such as eosinophil cationic protein (ECP) or eosinophil-derived neurotoxin (EDN). In particular ECP and EDN are detected at significantly higher levels and in a significantly higher proportion of patients with RSV compared to healthy controls [306, 317–319]. The levels of ECP were also significantly higher in infants and children with RSV bronchiolitis compared to those with LRTI without wheezing and lowest in those with URTI only and correlated with disease severity as measured by initial oxygen saturation [318]. Other investigators detected an association between IFNγ concentrations and levels of EDN [305] or leukotriene C4 levels [308]. This may reflect the ability of IFNγ to activate eosinophils and prolong their survival. Interestingly, peripheral eosinophil counts are reduced in the majority of studies [129, 320, 321], but peripheral eosinophilia has also been reported [322, 323]. The frequent detection of high levels of products of eosinophil degranulation is particularly noteworthy given that eosinophils are rarely found in BAL fluid or nasopharyngeal samples or in postmortem lung tissue of RSV-infected infants [171, 276, 281]. This paradox is most clearly illustrated by the observation that nasopharyngeal lavage samples from infants hospitalized for RSV disease contained numerous cells that stained positive for ECP while the number of eosinophils expressing the BMK-13 marker was low [280]. It should be noted, however, that ECP may not be as specific a marker for eosinophils as is generally assumed. It has been demonstrated that neutrophils are capable of synthesizing and secreting ECP upon stimulation with specific antigens or anti-IgE antibodies [324]. Some nasopharyngeal lavage samples from RSV-infected infants contain IgE-positive cell [280], and RSV-specific IgE antibodies have been detected in NPS samples from infants with bronchiolitis [325–327], even though some failed to detect any in nasal washes [328].
While animal data suggest an important contribution of NGF- and substance P-mediated neurogenic inflammation in RSV-induced AHR [253], there are relatively few data on the contribution of these mediators to human RSV disease. The BAL fluid concentrations of NGF were found to be significantly higher in mechanically ventilated infants with severe RSV LRTI compared to controls without respiratory infections, and immunoreactivity for its receptors on bronchial ECs was strongly increased [329]. On the other hand, levels of substance P in NPA samples were found to decrease with increasing disease severity in term, but not preterm, infants hospitalized with RSV bronchiolitis, suggesting that substance P plays a protective role [101].
Human Humoral Response
Infection with RSV subtype A strains has been found to elicit antibodies that cross-react with subtype B, and vice versa [330, 331]. Although antibody responses to various RSV proteins have been detected [327, 332, 333], neutralizing antibodies are directed against the F and G protein.
In the age group that most frequently experiences primary RSV infection, the immune system is still immature and maternally derived antibodies can still be present at relatively high levels. Passively acquired maternal antibodies are detectable in essentially all neonates, though at greatly varying titers [89, 90]. Their levels decline with a half-life of ∼26 days [89] in agreement with the half-lives of endogenous and exogenous IgG antibodies. The highest risk of RSV LRTI severe enough to require hospitalization is seen at an age (2–6 months) when maternal antibodies are still present. This not only shows that RSV can cause infection in the presence of maternal antibodies, but raises the question of whether maternal antibodies may actually enhance rather than prevent disease. There are, however, numerous studies showing that the titers of maternally derived neutralizing antibodies are inversely associated with RSV infection overall [91, 92], or with the severity of RSV disease [16, 93–96]. In addition, palivizumab prophylaxis substantially reduces the risk of hospitalization for RSV LRTI in high-risk infants [141, 142]. Nonetheless, the presence of maternal antibodies can further limit the already weak ability to mount antibody responses observed in infants [331, 334], as confirmed through the use of passive antibodies in experimental animals [257]. The immaturity of the neonatal immune system also contributes to the suppression of an active serum and mucosal antibody response [331, 334]. For example, it has been shown that the postinfection IgA antibody response to the RSV F protein correlates with age, while preexisting maternally derived antibodies determine the magnitude of the IgA response to the G protein [331]. Consequently, clear (three- or fourfold) increases in serum and/or nasal wash concentrations of RSV G or F protein-specific IgG and IgA are not seen in all infants after primary RSV infection [89, 328]. The ability to elaborate neutralizing antibodies is equally limited. There are data suggesting that this relative inability to develop neutralizing antibodies contributes to the high rate of reinfections observed in infants and young children [9]. In adults, neutralizing F and G protein-specific antibody titers correlate with (incomplete) protection from reinfection, whereas the role of nasal IgA is somewhat more controversial [335, 336].
A recent analysis showed that variable gene usage in adult RSV-specific B cells was highly focused on VH3 genes, but was much broader in very young infants (<3 months of age) and included VH1 and VH4 genes [337]. In keeping with the observation that infant B cells show little evidence of somatic hypermutations after encounter of their cognate antigen [170], the antibody variable genes of RSV-specific B cells from these infants showed fewer somatic mutations compared to adults [337]. This concurs with the finding that infant RSV-specific antibodies exhibit low avidity [330], which is likely to contribute to the heightened vulnerability of infants to reinfection.
The abundant presence of B lymphocytes in the near absence of CD3+ T lymphocytes in postmortem lung tissue from cases with fatal RSV infection suggests that B cell activation in the lungs of very severe cases may be T cell-independent [338]. A variety of factors implicated in T cell-independent B cell activation (including VIP, BAFF, APRIL, and a type I interferon-induced protein) were found to be strongly expressed in tissue of patients with fatal RSV infection, but not in controls, and the concentrations of two of these mediators in NPA of infants with RSV LRTI correlated with total immunoglobulin levels and particularly with RSV-specific IgM levels.
As already mentioned, IgE antibodies recognizing whole RSV and purified F and G protein have been reported in a high proportion of NPS samples from infants with RSV bronchiolitis, but rarely in those with other RSV URTI or LRTI [325, 339]. While others have confirmed the presence of RSV-specific IgE in serum [332, 340, 341] and NPS of some RSV-infected infants, they generally detect them in a lower proportion of patients [326, 327], and some fail to detect any [328].
Genetic Susceptibility to Severe RSV Infection
An analysis of concordance for RSV hospitalization in dizygotic and monozygotic Danish twin pairs resulted in an estimated heritable contribution to susceptibility for severe RSV disease of 16 %, which rose to 22 % when more stringent documentation criteria were applied [342]. A genetic influence on disease severity is also strongly supported by the finding that the same RSV strain can cause symptoms ranging from mild upper respiratory symptoms to severe LRTI in the same season [343]. In addition, the differential responses of various mouse strains to infection with hRSV provide further evidence of a genetic contribution in susceptibility to severe disease [198, 344]. A significant association between severe RSV disease and gene polymorphisms, particularly those that result in altered expression or function of the gene product, would provide support for the involvement of the particular gene product in RSV-induced pathology.
When 347 single nucleotide polymorphisms (SNPs) were analyzed in 437 Dutch children requiring hospitalization because of RSV infection and 1,008 controls, the risk of severe RSV disease was found to be predominantly associated with innate immune genes [345]. The five genes with the highest level of significance and with associations both at the allele and the genotype level were VDR (vitamin D receptor), JUN (encoding the jun proto-oncogene, a regulator of transcription), IFNA5 (encoding IFNα5), NOS2A (the inducible nitric oxide synthase), and FCER1A (high-affinity IgE receptor α-subunit) [345]. It later became evident that preterm children were overrepresented in the case sample and that there were significant interactions between premature birth and 11 of the 347 SNPs under investigation, nine of which localized to genes coding for mediators of innate immunity [346]. Importantly, six of these SNPs showed associations with severe disease only in preterm children, with IL27, C3, NFKBIA and TGFBR1 being protective and IFNG and ADAM33 risk-enhancing. Whereas IL1RN (encoding the IL-1 receptor antagonist) was protective in the premature birth group, it augmented the risk of term children. Conversely, a variant of IFNA13 increased the risk only in term infants, while a variant of IFNAR2 was associated with protection in term infants, but not in preterm infants. The latter gene had not shown a significant association in the entire study cohort. Of note, the odds ratios (ORs) identified in these and other studies generally range between 1.2 to 1.8 for risk alleles or genotypes and rarely are smaller than 0.7 for protective alleles or genotypes, indicating that each genetic association makes only a small contribution to the overall risk. An association of severe RSV disease with the Thr1Met SNP in the VDR gene, as observed in the Dutch multigene study [345], was confirmed in black South African children, although the significance of specific genotype associations differed somewhat between the two cohorts [347]. However, an association with JUN was not detected in this African population. A summary of genetic influences in the risk and severity of RSV infection in humans is shown in Table 6.
Table 6.
Genes and polymorphisms associated with increased or protective effects on severe RSV infection
| Gene | Protein | Polymorphism or mutation | Effect or mechanisms | Population |
|---|---|---|---|---|
| VDR | Vitamin D receptor | Thr1Met SNP | Associated with severe RSV disease | Pediatric |
| JUN | Jun prot-oncogene | rs11688 | Jun is part of AP-1, a mediator of inflammation | Pediatric |
| c.750G-A | ||||
| IFNA5 | interferon alpha 5 | rs10757212 | RSV can interfere with IFN-α production | Pediatric |
| c.453C-T | ||||
| NOS2A | Inducible nitric oxide synthase | rs1060826 | iNOS has a role in airway inflammation | Pediatric |
| C2757G-A | ||||
| FCER1A | High affinity IgE receptor a-subunit | rs2251746 | Allergic diseases | Pediatric |
| c. -66T-C | ||||
| IFNG | Interferon-γ | Increased risk | In preterm children only | |
| ADAM33 | Disintegrin and metalloproteinase domain-containing protein 33 | Rs574174 | Increased risk | In preterm children only |
| c.2241-410A-G | ||||
| IL27 | Interleukin-27 | Protective | In preterm children only | |
| C3 | Complement C3 | Protective | In preterm children only | |
| TGFBR1 | Transforming growth factor, beta receptor I (activin A receptor type II-like kinase, 53 kDa) | Protective | In preterm children only | |
| IFNA13 | Interferon alpha-1/13 | Rs643070 | Increased | In term infants only |
| c. -603C-T | ||||
| IL1RN | IL-1 receptor antagonist | Variable | Protective in premature group, increased risk in term children | |
| IFNAR2 | Interferon-alpha/beta receptor beta chain | Protective | Premature group only | |
| TLR4 | TLR4 | 299Gly and 399Ile alleles | RSV ligation of TLR4 on airway epithelial cell lines enhances viral entry and replication via MyD88- and p38 MAPK-dependent mechanisms | |
| IL-18 | Interleukin-18 | Associated with severe RSV disease | In a German study | |
| CCL5 | CCL5 (RANTES) | the −403 G/A polymorphism in the promoter region of the CCL5 gene | A allele exhibited significantly higher transcriptional activity compared to the G allele, and children with the AA genotype had the highest serum RANTES concentrations, followed by those with the G/A and the G/G genotype. | Japanese patients |
| The promoter polymorphisms −403 G/A, −28 C/G and the intronic polymorphism In1.1 T/C in Japanese patients hospitalized with RSV bronchiolitis and controls showed a protective effect of the A allele and the A-containing genotypes of −403 G/A, the G allele and G-containing genotypes of −28 C/G, and the C allele and C-containing genotypes of In1.1 T/C. | ||||
| IL-4 or IL4R | Interleukin-4 or interleukin 4-receptor | rs1805011 | A risk haplotype spanning IL13, conserved non-coding sequence (CNS)-1, and IL4 carried the strongest risk, suggesting that the association signal arises from another unobserved SNP or more complex genetic interactions. | |
| Glu400Ala (in IL4R) | ||||
| IL-13 | Interleukin-13 | rs20541 polymorphism in IL13 (Arg130Gln) | Risk factor for RSV bronchiolitis. Together with IL-4, a risk haplotype as described above carries a strong risk | |
| IL-8 | CCL8 | Rs3138038 | IL-8 −251T allele was significantly associated with RSV pneumonia, as was the TT genotype and also the −251T-781C haplotype. | Chinese children |
| Lys69Gln | ||||
| IL10 | Interleukin-10 | -592C/A SNP (rs1800872) | Age-dependent difference was observed for the association between severe RSV bronchiolitis and the −592C/A SNP (rs1800872) in the promoter region of the IL10 gene, with the C allele being overrepresented in infants who were hospitalized before they reached 6 months of age, but not in older children | Pediatrics |
TLR4 Polymorphisms
The role of TLR4 in the innate immune response to RSV infection is somewhat controversial. In vitro, RSV ligation of TLR4 on airway epithelial cell lines was shown to enhance viral entry and replication via MyD88- and p38 MAPK-dependent mechanisms, with the latter playing a role in intracellular viral trafficking and virion disassembly [348]. TLR4 was also found to mediate the in vitro response of macrophages to purified RSV F protein, but not G or N protein, in a CD14-dependent manner [349]. Studies in TLR4-deficient mice have yielded conflicting results, with some showing delayed viral clearance in association with decreased NK and CD14+ cell recruitment to the lung, deficient NK cell cytotoxicity and reduced numbers of IL-12 producing cells [350], while others were unable to replicate any of these results [243]. The specific genetic background of the TLR4-deficient mice and the controls may have been a major reason for these discrepancies. In yet another TLR4-deficient mouse strain, TLR4 played a critical role in NF-κB activation in the initial response to RSV infection, though not in the later phases [351]. Activation of this transcription factor is an essential step in the induction or upregulation of numerous cytokines, chemokines and other inflammatory mediators. A subset of infants (8/26) with RSV bronchiolitis showed upregulation of TLR4 expression on peripheral blood monocytes; the procurement of samples a mean of 7.5 days after disease onset may have precluded the detection of such enhanced expression in a substantially larger proportion of patients [352]. In these eight infants, the levels of TLR4 expression correlated negatively with minimal oxygen saturation. In contrast, expression of TLR4 on BAL and peripheral neutrophils was significantly reduced in infants with RSV bronchiolitis compared to controls [353].
The TLR4 gene contains two co-segregating non-synonymous SNPs, Asp299Gly (rs4986790) and Thr399Ile (rs4986791). When bronchial ECs were transfected with all four TLR4 alleles, constructs of the minor alleles, 299Gly or 399Ile, showed markedly attenuated ability to translocate to the cell surface and to upregulate cytokine production in response to stimulation with LPS and/or RSV [354]. Similarly, PBMC from children heterozygous for Asp299Gly or Thr399Ile were characterized by lower surface expression of TLR4 and reduced cytokine responses to LPS and/or RSV stimulation. However, others failed to show any significant differences in the cytokine responses to RSV stimulation between carriers of homozygous and heterozygous TLR4 haplotypes [355].
A striking overrepresentation of heterozygosity at both of the TLR4 SNPs was reported for high-risk infants hospitalized with RSV infection compared to literature controls or control infants with non-RSV-related respiratory symptoms [356]. Most of these infants were premature, and prematurity itself has been associated with these TLR4 polymorphisms. This may be one of the reasons why heterozygosity was not identified as a risk factor in other investigations of the TLR4 SNPs in RSV disease. The 299Gly and 399Ile alleles were overrepresented in Jewish children hospitalized with RSV compared to RSV-infected outpatients [357], whereas the wild-type A allele of the Asp299Gly polymorphism tended to be more frequent, and the rare G allele less frequent (p = 0.052), in black South African patients hospitalized with RSV disease compared to healthy infants [347]. All controls carried the C allele of the Thr399Ile polymorphism, and ∼1 % of cases carried the T allele (not statistically significant). Of note, neither the Asp299Gly nor the Thr399Ile polymorphisms were present in a Japanese population [358]. In German children, no significant associations were seen at the allele or genotype level, but the G-T haplotype conferred increased risk of severe RSV disease [359].
In contrast, case–control studies in Canadian [107], Dutch [345] and Finnish children [360, 361] did not reveal significant associations between severe RSV infection and TLR4 polymorphisms. However, data from one of the Finnish studies suggest that the genetic risk associated with the TLR4 Aps299Gly polymorphism varies between RSV epidemics [360]. When subjects recruited during two epidemics that occurred several years apart were analyzed separately, the homozygous genotype was associated with protection from severe RSV disease in one epidemic, but associated with risk of severe infection in the other. This suggests that complex interactions between host genotype and viral strain may be involved in determining disease severity. It is conceivable, for example, that dampened immune responses to a mildly virulent strain may be appropriate and prevent massive tissue damage, but may be inefficient and result in more severe infection in the presence of a highly virulent strain.
Other TLRs have been implicated in mediating the immune response to RSV [181]. In a German case–control study of polymorphisms in TLR-2, -3, -5, -6, -9, and 10, only the TLR10 haplotype bearing the wild-type alleles for all 8 polymorphisms analyzed was significantly associated with severe RSV disease [362].
Since CD14 was shown to be essential for RSV F protein-induced signaling through TLR4 [349], polymorphisms in the promoter region of the CD14 gene have also been examined for potential associations with severe RSV disease. The C allele and CC genotype of the −550 C/T polymorphism was significantly associated with hospitalization for RSV bronchiolitis in a Japanese case–control study [358], whereas the −159 C/T polymorphism showed no association in Japanese, German or Jewish populations [357–359].
Surfactant Proteins
Surfactant proteins (SPs) are not only essential for proper alveolar function by maintaining appropriate surface tension, but can modulate innate immune responses [363]. In the case of RSV infection, this may involve the ability of SP-A and SP-D to modulate TLR4 signaling events as well as the capacity to interact directly with RSV, although the precise nature and effect of these interactions remains controversial [363–365]. SP-A was found to enhance uptake of RSV by PBMC and U937 macrophages, to further upregulate RSV-induced production of TNFα by PBMC, to reverse the RSV-induced suppression of TNFα production in U937 macrophages, and to augment IL-10 production in both cell types [366]. SP-A-deficient mice exhibited higher viral titers at all time-points, suggesting a role for SP-A in decreasing infectivity or increasing viral clearance or both [208]. These animals also showed significantly higher pulmonary levels of TNFα and IL-6, but these may have been produced by cell types other than alveolar macrophages. Surfactants are produced by type II pneumocytes, which can be infected by RSV [171]. This may account for the observation that concentrations of SP-A and SP-D were reduced in the lungs of infants with severe RSV LRTI; however, they were not associated with disease severity [367]. The results of three small randomized trials suggest that surfactant therapy may reduce the duration of mechanical ventilation and the length of stay in the ICU in critically ill infants [368].
Whether polymorphisms in the various SP genes influence susceptibility to severe RSV disease has been examined by transmission disequilibrium testing (TDT) [369] and in several case–control studies. Although significant associations with certain alleles and especially with particular haplotypes have been detected in all studies of SP-A [369–372], SP-B [373], SP-C [374] and SP-D [369, 371, 375], the specific haplotypes identified as risk or protection factors vary considerably between studies.
RANTES/CCL5 and CCR5
The chemokine CCL5 (RANTES) is a chemoattractant for eosinophils, basophils, and certain T lymphocyte subsets and plays a role in the activation of eosinophils, basophils, and NK cells. It is detectable not only in NPS and tracheal aspirates of patients with severe RSV LRTI [263, 264, 268, 269, 272], but also in nasal lavage samples of infants and children with URTI [184] and experimentally infected adults [201]. Tracheal aspirate CCL5 concentrations were found to correlate inversely with disease severity [269], whereas others found no correlation between concentrations in NPA and disease severity [268]. The A allele of the −403 G/A polymorphism in the promoter region of the CCL5 gene exhibited significantly higher transcriptional activity compared to the G allele, and children with the AA genotype had the highest serum RANTES concentrations, followed by those with the G/A and the G/G genotype [376].
Analyses of the promoter polymorphisms −403 G/A, −28 C/G and the intronic polymorphism In1.1 T/C in Japanese patients hospitalized with RSV bronchiolitis and controls showed a protective effect of the A allele and the A-containing genotypes of −403 G/A, the G allele and G-containing genotypes of −28 C/G, and the C allele and C-containing genotypes of In1.1 T/C [377]. Only three haplotypes were found in significant frequencies, and the A-G-C haplotype was significantly underrepresented among cases. In a Greek cohort, however, these polymorphisms were not associated with severe RSV disease at the allele or genotype level, but the combined genotype −28 C/C −403 G/A In1.1 T/T (G2) conferred increased risk [378]. In contrast, the −403 G/A polymorphism did not significantly affect the risk of hospitalization for RSV bronchiolitis in Chinese children [376]. CCR5 is one of the major receptors for CCL5 as well as MIP-1α, which has been associated with more severe RSV disease [267]. Polymorphisms in the promoter region of the CCR5 gene also showed significant associations with severe RSV bronchiolitis in case–control and TDT analyses [379].
IL-8/CXCL8
IL-8 has garnered attention primarily because it is a potent chemoattractant and activator of neutrophils, which constitute the major cell type in both the upper and the lower respiratory tract of RSV-infected infants [276, 281, 282]. Increased concentrations of IL-8 have been detected in upper and lower airway secretions of infants with severe RSV disease compared to healthy controls [269–271], but also in experimentally infected adults [201, 273]. Their concentrations in tracheal aspirates showed an inverse correlation with markers of disease severity [269].
In the UK, severe RSV disease was found to be associated with the A allele of the −251 A/T promoter polymorphism by TDT in a total of 194 nuclear families [380, 381]. Determination of the haplotype structure by identifying 8 novel SNPs in the IL8 gene and its promoter did not greatly help in refining the association of IL8 −251 A with bronchiolitis because two common haplotypes dominated in Europeans. However, the −251A +781T haplotype was associated with disease susceptibility by TDT, while a rare haplotype including −251A and +781C was not. This haplotype was shown to correlate with higher IL-8 transcript levels, and this was not attributable to the −251A polymorphism, but involved differential effects of the intronic +781 polymorphic site on transcription factor binding in the promoter region [382]. An association between disease severity and a haplotype with higher promoter activity agrees with studies showing an association between higher IL-8 protein levels in airway secretions of RSV-infected infants and increased disease severity [269–271]. In marked contrast, when comparing 101 Chinese children hospitalized with RSV-positive pneumonia to 108 children with RSV-negative pneumonia, the IL-8 −251T allele was significantly associated with RSV pneumonia, as was the TT genotype and also the −251T-781C haplotype in logistic regression analysis [383]. In a German population, no association between the −251A/T or the 781 C/T SNPs and RSV bronchiolitis was detected at the allele, genotype or haplotype level [384]. Polymorphisms in the IL8RA gene (encoding the IL-8 receptor alpha) also showed no association with RSV bronchiolitis.
Th2 Cytokine Genes
The role of Th2 cytokines such as IL-4, IL-5 and IL-13, in the immunopathology of human RSV infection remains controversial because their concentrations in nasopharyngeal secretions were higher compared to IFNγ in some cohorts [266, 267] and particularly in infants ≤3 months of age [306], whereas others found them to be undetectable or present at very low levels [274, 275, 309]. The −589C/T and −1112C/T in the IL4 and IL13 gene, respectively, are associated with altered transcriptional regulation [385, 386]. Forton et al. [387] analyzed a total of 113 SNPs in 11 genes located in the 5q31 region, which includes the type 2 cytokine gene cluster, in 780 cases with severe RSV bronchiolitis and 1045 controls, all British of European extraction, using a haplotype tagging approach. They obtained highly significant results for SNPs across the IL4 gene and its promoter, with rs2243250 (the promoter polymorphism −589C/T) yielding the highest OR at the allele and genotype level in the subset of infants without known risk factors for severe RSV disease. The rs20541 polymorphism in IL13 (Arg130Gln) also was a significant risk factor for RSV bronchiolitis. A risk haplotype spanning IL13, conserved non-coding sequence-1, and IL4 carried the strongest risk, suggesting that the association signal arises from another unobserved SNP or more complex genetic interactions. The rs2243250 polymorphism (−589C/T) in the IL4 promoter region was not identified as a risk factor in a German cohort [388]. However, all three investigated IL-13 polymorphisms were found to be in strong linkage disequilibrium with the IL-4 −589C/T SNP. And while the 1112C/T polymorphism of IL13 alone was significantly associated with severe RSV disease the inclusion of the IL4 promoter SNP in the analysis of the possible IL-13 haplotypes increased the significance of the association with RSV bronchiolitis by a factor of 10.
The −589 T variant of the promoter region of IL4 also is part of a common haplotype that was found to be associated with severe RSV disease in a Korean population [389]. However, genetic variants of IL13 or IL5 did not show any associations in this study. A Dutch study also identified the −589 T allele along with a polymorphism in the IL4RA gene (encoding the IL-4 receptor α) as risk factors for RSV hospitalization, although TDT did not yield statistically significant differences in transmission of this allele [390]. Of particular note, both in the case–control and the TDT studies, the T allele was significantly associated with severe RSV disease in infants >6 months of age, but not in younger infants.
IL-10
A similar age-dependence was observed for the association between severe RSV bronchiolitis and the −592 C/A SNP (rs1800872) in the promoter region of the IL10 gene, with the C allele being overrepresented in infants who were hospitalized before they reached 6 months of age, but not in older children [391]. The Dutch multigene study revealed a significant protective effect of the heterozygous C/A genotype at this locus [345], and a similar trend emerged in another Dutch cohort using the same controls [392]. Another polymorphisms in the IL10 promoter region, the −1082 A/G SNP (rs1800896), was not associated with severe RSV-induced bronchiolitis in Finnish infants, whereas homozygosity for the A allele was a strong risk factor for rhinovirus bronchiolitis [361]. In a British cohort, these two SNPs along with six others or the resulting haplotypes were not found to affect the risk of severe RSV bronchiolitis, although two of the SNPs were associated with the need for mechanical ventilation [393].
Interferons and IFN-inducible Proteins
In a case–control study of 156 children <2 years of age with severe RSV disease and 296 controls who had never been hospitalized for RSV infections in Germany, no association was detected between severe RSV disease and polymorphisms or haplotypes in the IFNG, IFNGR1, IFNA5, IFNAR1, ISG15 (interferon-stimulated gene 15), IFI27 (IFNα-inducible protein 27), or IFI44 (IFN-induced protein 44) [394]. Similarly, the results of a Finnish study that did not reveal any significant association between severe RSV infection and the IFNG +874 T/A genotype [361]. In contrast, the IFNG +874 T/A genotype was associated with various measures of disease severity in infants hospitalized with RSV LRTI, as were polymorphisms in the TGFB1, IL6, and IL10 genes, but not TNFA [395].
Other candidate genes that have been examined to date include IL18, which was associated with severe RSV disease in a German, but not in a Finnish case–control study [361, 396]. Also, TNFA, which showed no association in case control studies from Germany and the Netherlands [391, 397], the uteroglobulin (or Clara cell protein 10) gene, which also showed no association [398], and the immunoglobulin heavy G2 chain [399] have been studied. The homozygous IGHG2 (−n/−n) genotypes [IGHG(ga−n/ga−n), IGHG(bf−n/bf−n), and IGHG(ga−n/bf−n)] were significantly overrepresented in a cohort of 49 Norwegian children hospitalized with RSV LRTI compared to 430 healthy Swedish children, whose allele frequencies were quite similar to those found in a Finnish population. The differential susceptibility of various mouse strains to severe clinical disease and its late consequences is MHC-dependent [400, 401]. However, a genome-wide analysis of susceptibility to more severe RSV infection in F1 and F2 progeny of susceptible and resistant mouse strains did not reveal any association with MHC, but identified a single region on chromosome as a susceptibility locus, which contained several candidate genes, including the cystic fibrosis membrane conductance regulator [402]. Nonetheless, it is surprising that the association of the HLA complex, particularly class I alleles, with susceptibility to severe RSV disease in human infants has not been examined to date.
In summary, numerous associations between SNPs in immune genes and severe RSV disease have been detected, and many of them have been replicated at the gene level, but not at the level of individual risk alleles, genotypes, or haplotypes. The reported effect sizes are small, and the magnitude and even direction of the association depend on the gestational age of the study subjects [346], the chronological age [390] and the specific RSV epidemic [360]. This suggests that complex interactions between host factors, genetics being only one of them, and the genotype of the predominant viral strain during an epidemic determine the severity of RSV infections.
RSV and Asthma
There is extensive evidence that a substantial portion of infants and young children who suffer from RSV bronchiolitis severe enough to require hospitalization not only wheeze during the acute illness, but continue to experience recurrent wheezing episodes for months or even years after recovery at frequencies that are substantially higher compared to control groups who had not been hospitalized with bronchiolitis [392, 403–410]. Of note, RSV is not the only respiratory virus causing severe bronchiolitis, and wheezing may be equally common after infections with other respiratory viruses, including influenza viruses and rhinovirus [411, 412].
Many of the patients who experience recurrent wheezing after hospitalization for RSV bronchiolitis are eventually diagnosed with asthma. Key data come from a series of studies conducted by Sigurs et al. [413], who prospectively followed 47 Swedish infants who had been hospitalized because of RSV bronchiolitis at a mean age of 3.5 months and 93 controls until young adulthood [413]. At the mean ages of 1 and 3 years, physician-diagnosed asthma and parent-reported wheezing overall, but not recurrent wheezing (≥3 episodes of bronchial obstruction not verified by a physician), were significantly more frequent in the RSV compared to the control group (risk ratio 28.1). The RSV-associated risk was further increased in the presence of a family history of asthma (6/11 with such heredity compared to 5/36 without such a predisposition). The frequency of allergic sensitization, as measured by skin prick test (SPT) and/or serum allergen-specific IgE, was also significantly elevated in the RSV group, and at the age of 3 years, RSV bronchiolitis was the most important risk factor for allergic sensitization. At the 7-year follow-up, both the cumulative and current rates of asthma, any wheezing or recurrent wheezing, allergen sensitization, and allergic rhinoconjunctivitis were significantly higher among the RSV group compared to the controls [414] and these differences persisted at age 13 and into young adulthood, i.e., the age of 18 years [415, 416]. Lung function and AHR in response to isocapnic dry air hyperventilation challenge were measured in 138 of the 140 subjects originally enrolled when they were ∼13 and ∼18 years of age [415, 416]. Almost all spirometry values (FEV1, the ratio of FEV1 to FVC, and FEF25-75) were reduced, while AHR and the response to bronchodilation was increased, in the RSV compared to the control group [416].
In other cohorts, too, the prevalence of asthma is significantly higher among subjects who were hospitalized with RSV LRTI in infancy or early childhood compared to non-hospitalized controls [404, 405, 417, 418], whereas recent data from Japan do not support such an association [419]. Unlike what was observed in the Swedish cohort, the frequency of respiratory symptoms gradually declined in some of these studies, or the prevalence of asthma “caught up” in the controls, such that there were no significant differences between cases and controls by the age of 8 or 10 years [410, 420]. In other cohorts, the prevalence of wheezing and asthma was still significantly elevated compared to controls at the last follow-up at the age of ∼7 years [404, 418] or even 10 years [417]. Some of these cohorts included subjects with RSV-positive and RSV-negative bronchiolitis or LRTI, and no significant differences in the frequency of wheezing or asthma were detected between the two groups [405, 409, 418, 421]. In contrast, others observed a significantly higher prevalence of asthma after non-RSV bronchiolitis, but not after RSV bronchiolitis, compared to controls [422].
All of these studies concerned children who required hospitalization for the index episode of RSV bronchiolitis or other LRTI. However, the vast majority of infants who experience bronchiolitis are treated as outpatients. In an early retrospective study, outpatients with moderate bronchiolitis before the age of 25 months were at significantly increased risk of recurrent wheezing at the age of ∼8 years even after adjusting for atopic heredity, atopic manifestations in the children themselves, mother’s educational levels, passive smoking and other LRTI [423]. At the 13-year follow-up the frequency of wheezing and a physician’s diagnosis of asthma was still elevated, but the difference was not statistically significant compared to the control group [424]. Unfortunately, this study included children who had experienced other LRTI before the index bronchiolitis, i.e., who may have had asthma exacerbations rather than bronchiolitis. In addition, these children were compared to historical controls, and the viral etiology of the bronchiolitis episodes was not established.
The Tucson Children’s Respiratory Study is the first community-based study to prospectively examine the impact of moderately severe LRTI that is treated in an outpatient setting on subsequent wheezing [425]. A total of 888 children were prospectively followed and were examined by a pediatrician whenever they developed signs of lower respiratory tract illnesses during the first 3 years of life. Follow-up visits occurred at the ages of 6, 8, 11, and 13 years. Although 519 episodes of LRTI were identified during the first 3 years, none of the children were hospitalized. This study demonstrated that even mild RSV bronchiolitis, but not bronchiolitis of other etiologies, constituted an independent risk factor for infrequent wheeze and frequent wheeze in children up to the age of ∼11 years, but this was no longer a significant factor at the age of 13 years. Note that this differs from the results of several analyses of severe RSV (requiring hospitalization) indicating that RSV and non-RSV bronchiolitis carried the same risk of recurrent wheeze [405, 409, 418]. In the Tucson Children’s Respiratory Study, there was no significant difference in the rate of positive skin-prick reactivity or total serum IgE concentrations at 6 or 11 years of age between children who had had RSV LRTI compared to those without LRTI in early childhood, even after stratification by family history of asthma [425]. At the age of 11 years, the RSV bronchiolitis group had significantly reduced FEV1, and a significantly greater proportion responded to bronchodilators, and both were independent of the current wheezing status.
More recently, there have been two other community-based studies of the effect of early respiratory viral infections on the risk of developing childhood asthma, the Childhood Origins of ASThma (COAST) study in Wisconsin [41, 426, 427] and a study from Western Australia [428]. Both recruited neonates at high risk of atopy and determined the viral etiology of all acute RTIs (of moderate to severe nature in Wisconsin) during infancy. In both studies, rhinovirus-associated respiratory illness, particularly when accompanied by wheezing, was by far the most significant predictor of wheezing and asthma during follow-up until the age of 5 or 6 years. The associations with RSV were markedly weaker and often non-significant. Of particular note, in the Western Australian cohort, all of the associations between rhinovirus or RSV infection with later wheezing and asthma were restricted to children exhibiting sensitization to common allergens within the first 2 years of life and were not seen in those whose atopy developed at a later age [428].
Potential Mechanisms of the Association between RSV and Asthma
Allergic Sensitization
One of the most intensely debated issues in research on RSV infection and its late sequelae is the question of whether RSV bronchiolitis is causally involved in the development of asthma or whether the two entities share a common predisposition. A causal association was suggested by the early findings of Th2-skewing (of the mitogen-stimulated PBMC response) during severe RSV disease, which is characteristic of atopic asthma [296, 297]. This was further supported by the discovery that a considerable portion of infants hospitalized with RSV bronchiolitis develop virus-specific IgE and a variety of mediators implicated in asthma, including ECP, EDN, LTs, and histamine [264, 318, 325]. Furthermore, RSV-specific IgE and histamine were detected almost exclusively in patients with bronchiolitis [325], although this was not confirmed in another study [326]. In addition, virus-specific IgE was found to be associated with the development of postbronchiolitic wheezing [429]. Based on these observations, it was hypothesized that the elaboration of Th2 cytokines in response to RSV infection could promote the sensitization to inhalant allergens. Allergic sensitization usually occurs early in life and during a time that overlaps with the period of greatest risk for severe LRTI due to RSV. This would then promote atopic asthma, which is thought to be the most common form of childhood asthma in developing countries. Since the actual infection with RSV runs a self-limiting course of 1–2 weeks and RSV-specific memory responses are short-lived, this hypothesis often invokes recurrent RSV infections. This may not be necessary, however, since there is growing evidence that the immune response to RSV is not as short-lived as has long been assumed. For example, the proportions of PBMC expressing CD4, CD23 (low-affinity IgE receptor), and the activation marker CD25 were significantly increased 5 months after the bronchiolitis episode [321]. At the same time point, PBMC from subjects who wheezed produced more IL-4 in response to stimulation with house dust-mite antigen. Similarly, serum samples obtained during convalescence (10–153 days after presentation) from RSV infection were found to contain significantly elevated levels of soluble CD25 [430]. In RSV-infected mice, the levels of DCs remain elevated in the lung for at least 21 days after viral inoculation [229–232]. Both pDCs and cDCs in the lung play a central role in determining whether tolerance or allergic inflammation is induced in response to inhaled allergens [431]. Their numbers and/or activation state appear to be crucial determinants of the development of an allergic response. Therefore, the persistence of DCs in the lung for weeks after the initial RSV infection could favor the development of allergic sensitization.
In mice, RSV infection cannot only induce AHR, but when it precedes allergen sensitization, it generally increases or prolongs the AHR induced by allergen challenge [200, 219, 432, 433]. Conversely, allergen sensitization and challenge preceding RSV infection can also result in markedly enhanced or prolonged AHR compared to RSV infection alone or allergen exposure alone [218, 247]. This can be associated with the further upregulation of allergen-induced Th2 cytokines, i.e., IL-4, IL-5, and particularly IL-13[433–435], but this is not observed in all models [236] and there are even reports that RSV infection before sensitization significantly attenuated the AHR in response to allergen challenge [247] and prevented the allergen-induced upregulation of Th2 cytokines [434]. Whether RSV infection of mice also enhances allergen-specific IgE production has rarely been examined, but the available data suggest that it does not [219, 241] or actually suppresses allergen-specific IgE [434]. In contrast, bRSV-infected calves exhibited a greater increase in allergen-specific IgE after inhalation exposure to two different antigens, OVA and Alternaria, compared to mock-infected calves [436, 437].
The mechanisms implicated in the RSV-induced enhancement of allergen-induced AHR are equally variable. For example, one group of researchers showed that IL-5-dependent eosinophilia was essential for both virus-induced AHR and the enhancement of allergen-induced airway sensitization seen after RSV infection [219, 438]. Other investigators do not detect eosinophilia [115, 246] or even find decreased levels of IL-5 after RSV infection alone [209]. Instead, some data strongly implicate IL-13 and its ability to induce mucus production in both the RSV-associated AHR and the RSV-induced enhancement of allergic airway sensitization [116, 246]. While the results discussed so far suggest that that the two processes share a common mechanism, the results from another model indicate that the two events proceed via different mechanisms [209]. Specifically, through the use of IL-13-deficient mice or IL-13 neutralizing antibodies, it was demonstrated that IL-13 did not play a role in the AHR seen after RSV infection alone, even though it was strongly induced by RSV infection. It was, however an important mediator of the RSV-induced enhancement of allergic airway responses. In yet other experiments, RSV infection before allergen sensitization actually decreased the production of IL-13 and lung eosinophilia compared to OVA sensitization and challenge in mock-infected mice and thereby attenuated allergen-induced AHR [236, 247]. While some of these discrepancies can be attributed to methodological differences, it has been demonstrated that some commonly used laboratory strains of RSV do not induce pulmonary IL-13 production and upregulate IL-10 instead [114–116]. Such viral strain-specific effects in combination with host susceptibility factors may account for some of the variability seen in human studies, with allergen sensitization and its effect on lung function depending in part on the viral strains predominating in the region and during the season that cohorts are recruited.
The findings in the Swedish cohort suggest a possible causal relationship between severe RSV bronchiolitis, allergic sensitization and atopic asthma [413–416]. Remarkably, however, only two other controlled studies document a significantly increased frequency in allergic sensitization in subjects with early RSV bronchiolitis. In one case, this concerned sensitization to food allergens at the age of 1 year, while sensitization to inhaled allergens is a more important risk factor for the development of atopic asthma [407]. In a cohort that included children who had experienced RSV-positive or RSV-negative bronchiolitis in infancy, the rate of SPT positivity to common aeroallergens was significantly higher in index cases compared to controls (29 % of index cases vs. 15 % of controls) at the 5-year follow-up [409]. Note, however, that the frequency of wheezing did not differ between SPT-positive and -negative children, indicating that atopy was not a major factor in the high prevalence of wheezing observed in the bronchiolitis group. Various other cohorts do not show any evidence of significantly increased frequencies of atopy after severe RSV infection [439, 440]. Actually, in some studies, 6–10-year-old children with a history of hospitalization for RSV bronchiolitis in infancy were significantly less likely to show positive SPTs [420, 441]. Atopic manifestations (eczema and allergic rhinitis) also tended to be less common in one cohort [420], but the differences in both atopy and allergic symptoms had disappeared at the 9- to 10-year follow-up [417]. Several other investigations also found a lower rate of atopy in the RSV bronchiolitis compared to the control group, but the difference did not reach statistical significance [404] or was statistically significant in girls only [422]. Importantly, results from the prospectively followed COAST cohort yielded no evidence that wheezing during an acute viral respiratory infection was associated with an increased risk of allergen sensitization, regardless of viral etiology [427]. Even the group of researchers who first reported the detection of RSV-specific IgE and its association with postbronchiolitic wheezing did not find any association between RSV-IgE levels and the development of SPT positivity [442].
Interestingly, there are data suggesting that palivizumab prophylaxis decreases the risk of ambulatory LRTI and thereby reduces the risk of recurrent wheezing [443]. This was only observed in children without a family history of atopy or food allergies, but not in those with such a family history [444]. This suggests that RSV LRTI may have differential effects on the development of recurrent wheezing depending on the genetic predisposition. This is consistent with the existence of several asthmatic or wheezing phenotypes during childhood, as proposed on the basis of observations in the Tuscon Children’s Respiratory Study [445]. Early transient wheezing (at least 1 LRTI with wheezing during the first 3 years of life, but no wheezing at 6 years of age) was primarily associated with decreased lung function in infancy and maternal smoking during pregnancy or postnatal tobacco exposure. Persistent wheezing (at least one early LRTI with wheezing and wheezing at 6 years of age) was associated with elevated total serum IgE levels at 9 months of age, eczema, SPT positivity, maternal asthma, and male sex, whereas initial lung function was not a major risk factor. Late onset wheezing (no wheezing LRTI in early life, but wheezing at 6 years of age) was also associated with eczema, SPT positivity, maternal asthma, and male sex, but not with early elevations in total serum IgE levels or initial lung function.
Other data suggest that, rather than being caused by RSV, atopy or an atopic disposition predisposes to severe RSV infections. In a large case–control study of Danish children <18 months of age recurrent wheezing and atopic dermatitis before the hospitalization as well as parental asthma were identified as independent risk factors for hospitalization due to RSV [83]. Similar results have been obtained in several smaller studies [74, 81, 446, 447], one of which further demonstrated a significant association between a family history of atopy and disease severity, as reflected in significantly longer hospitalization [447]. In marked contrast, a family history of eczema was strongly associated with protection from hospitalization due to RSV infection in a large Canadian cohort of prematurely born infants [76]. Others failed to identify atopy or atopic sensitization as a risk factor for RSV infection [82, 427, 448, 449], although it did increase the risk of LRTI due to rhinovirus [427, 448].
Lung Function
It is also possible that RSV does not increase the risk of asthma by promoting allergic sensitization, but by inducing a strong inflammatory response that would eventually become chronic and provoke bronchial hyperreactivity. Indeed, animal data suggest that the airway obstruction and particularly the AHR induced by RSV infection persist for weeks after the inoculation and at a period when the levels of inflammatory mediators in BAL fluid have returned to baseline [198, 199, 212]. This prolonged inflammation may be attributable to the persistence of low levels of viral genomic and messenger RNA in mouse lung tissue, which has been demonstrated to last for at least 77 days after inoculation [198, 212, 450]. Some data from studies in subjects with chronic obstructive pulmonary disease suggest that RSV also can persist in humans, but this remains controversial [451]. The potential effect of the RSV-induced upregulation of NGF also needs to be considered. The BAL fluid concentrations of this neurotrophin along with immunoreactivity for its receptor on bronchial ECs were found to be strongly increased in mechanically ventilated infants with severe RSV LRTI [329]. In rats, similar increases were observed and were particularly notable in young animals [452]. NGF is critical for the normal development of the nervous system in infancy. Heightened levels of this neurotrophin during a critical time frame of neuronal plasticity may result in irreversible changes in sensorineuronal pathways that may ultimately increase AHR and asthma susceptibility.
Subjects with severe RSV bronchiolitis during infancy frequently exhibit significantly reduced lung function compared to non-hospitalized controls [410, 415, 418, 420], although there are studies not showing such impairment [409, 422, 441]. In several cohorts, lung function was decreased to a similar extent in subjects with RSV-positive and -negative bronchiolitis [405, 418]. In contrast, others found significantly reduced lung function only in the RSV-negative group, but not in the RSV-positive group [422]. Similarly, among the participants of the COAST study, RSV wheezing illness during infancy or early childhood was not associated with decreased lung function at the age of 8 years, whereas lung function was significantly reduced in children with rhinovirus wheezing illness in the first 3 years of life compared to those with non-wheezing rhinovirus illness or non-rhinovirus illnesses [453]. In the few studies with such extended follow-up, lung function impairment after severe RSV bronchiolitis in infancy was found to persist into early adulthood [416, 440]. Indications of airway obstruction are quite consistently observed, including reductions in peak expiratory flow, forced expiratory volume in 1 s (FEV1), the ratio of FEV1 to forced vital capacity (FVC), maximum expiratory flow at 75, 50, and 25 % of vital capacity. In contrast, signs of airway restriction are detected only in some studies [420, 454], while FVC is unaffected in the majority of cohorts [410, 417, 418]. Like Sigurs et al. [415, 416], other investigators report significantly increased AHR in subject with RSV bronchiolitis in early childhood [410, 420–422], an exception being the study of young adults examined 20 years after the acute RSV bronchiolitis [440].
Note, however, that data from several prospectively followed birth cohorts suggest that lung function is already decreased at birth or in the early neonatal period in those who go on to develop LRTI [455–459]. Unfortunately, the findings are difficult to compare because of the numerous different techniques used to measure premorbid lung function and the different ages at which testing took place. In addition, the aspect of early lung function that is decreased in the early neonatal period may differ between boys and girls [458] and may reach significance in one sex only [457]. Importantly, these differences in lung function between those who developed LRTI and those who did not were found to persist until the age of 10 or 11 years [456, 457], but there was no significant effect of type or frequency of early LRTI once lung function at birth was taken into account [457]. In addition, decreased neonatal function was also associated with recurrent wheezing after acute respiratory infections, particularly in those who experienced more than one episode of wheezing LRTI [460]. Finally, decrements in neonatal lung function track into adulthood [461–463], and there are numerous data suggesting that they contribute to the development of asthma [464, 465]. In another investigation of infants born to mothers with a history of physician-diagnosed asthma, neonatal AHR rather than lung function per se was significantly associated with the development of severe bronchiolitis and the development of asthma at the age of 7 years [466]. Together, these results suggest that a common host factor affects not only lung function development in utero, but also susceptibility to AHR, LRTI during early childhood, postbronchiolitic wheezing and the development of asthma. By extension, this would mean that bronchiolitis is a first manifestation of asthma.
The Risk of Recurrent Wheezing and Asthma Depends on Disease Severity
There are data suggesting that palivizumab not only prevents hospitalization for RSV LRTI, but even decreases the incidence of less severe LRTI, thereby reducing the rate of physician-documented recurrent wheezing at the age of 3–4 years [443]. Similar findings have been reported for infants who had received prophylaxis with pooled human IgG containing high titers of RSV neutralizing Abs RSV immune globulin preparation [467]. These data also support a causal association between RSV LRTI and subsequent recurrent wheezing. They also underscore, however, that even moderate bronchiolitis may constitute a risk for recurrent wheezing and asthma, as seen in the Tucson Children’s Respiratory Study [425].
Nonetheless, in a population-based retrospective birth cohort study of >90,000 infants enrolled in TennCare, the Tennessee Medicaid program, the risk of being diagnosed with asthma between 4 and 5.5 years of age rose with increasing severity of bronchiolitis during the first 12 months of life as measured by the level of health care sought, i.e., outpatient visits, emergency department visits, or hospitalization [468]. Such a “dose–response” relationship between bronchiolitis in infancy and childhood asthma suggests a causal relationship. The same group of authors claim to have provided further evidence for a causal association by showing that the highest odds ratio of developing clinically significant bronchiolitis was seen in children who were 122 days old, and the highest risk of being diagnosed with high-risk childhood asthma was observed in children who were 121 days old, at the time of the winter virus peak [469]. In contrast, age at the time of their first medical visit for bronchiolitis was not associated with asthma risk. As Kuehni and Silverman [470] so aptly comment: “If we hypothesize that … bronchiolitis confers developmental damage, this should happen when the child is ill but not when other children are ill.” Note that the viral etiology of the bronchiolitis episodes was not ascertained in these studies; therefore the contribution of RSV to the increased asthma risk cannot be determined.
Other data on the association between disease severity and the development of recurrent wheezing or asthma are conflicting. On the one hand, ICU admission as a measure of disease severity was the only significant predictor of postbronchiolitic wheezing and hospital admission for asthma in the first year after hospitalization for either RSV or influenza-associated LRTI [411]. In contrast, none of the factors reflecting the severity of the initial RSV bronchiolitis episode, which required hospital care, were significantly associated with physician-diagnosed asthma in another cohort [471]. Monozygotic twin pairs discordant for RSV hospitalization in infancy (and, it is assumed, disease severity) were not found to differ in lung function, AHR, frequency of asthma diagnosis, or allergic sensitization at a mean age of 7.6 years [472]. In addition, when asked which of the twins had the more severe disease, the parents identified the non-hospitalized twin with similar frequency as the hospitalized twin. In other cases, both twins may have been hospitalized in the expectation that their disease would eventually be of similar severity, even though one may have been less severely affected. In a study of the association between RSV hospitalization and asthma in the same 5,154 Danish twin pairs that formed the patient basis for the preceding study, modeling of the direction of causation indicated that severe RSV infection did not cause asthma, but instead was an early indicator of a shared genetic predisposition for both asthma and severe RSV disease [473].
Many of the gene polymorphisms found to be associated with severe RSV disease have also been implicated in the genetic predisposition to asthma. It should be noted, however, that the alleles or genotypes showing an association with asthma or asthmatic phenotypes in one population may be protective in another population or not show any association [474], as has also been observed for associations with severe RSV infection. Therefore, only studies comparing the genetic associations with asthma and severe RSV disease in genetically rather homogeneous populations are likely to yield meaningful results. Such studies do not suggest that there is considerable overlap in the genetic susceptibility to asthma and severe RSV infection. For example, the promoter SNP −781 C/T in the IL8 gene was associated with asthma at the genotype level, but showed no association with severe RSV disease [384]. The haplotype distribution of 3 surfactant protein (SP)-B SNPs was significantly associated with severe RSV disease, but not with asthma [373]. Statistically highly significant differences between asthma patients and RSV patients were also observed in the haplotype distribution for SP-C, with the 138Thr-186Asn haplotype being overrepresented in RSV cases, but not in asthma patients [374].
There have also been a few investigations of candidate genes with the potential to confer increased susceptibility to wheezing after RSV LRTI. While IL-10 concentrations in NPS of acutely RSV-infected infants were significantly associated with the later development of postbronchiolitic wheezing, the −592 C/A SNP in the promoter region of the IL10 gene did not significantly influence the risk of recurrent wheezing [392]. The GA genotype of the −403 G/A polymorphism in the promoter region of the RANTES gene was found to be associated with recurrent wheezing in a Chinese cohort [376]. This SNP showed no association with hospitalization for RSV bronchiolitis in this cohort, similar to what has been reported for a Greek cohort [378], whereas the A allele was protective in a Japanese population [377]. In a British cohort, TDT showed the −251A allele to be transmitted more frequently to children who wheezed after RSV bronchiolitis compared to those who did not, and this association was confirmed in a case–control study [475]. This allele was also a risk factor for hospitalization due to RSV bronchiolitis in a study from the same area, [380, 381], whereas the T allele conferred this risk in a Chinese population [383], and no association was detected in German infants [384]. Note that, in the same German cohort, the −251 T allele was associated with asthma [384, 476]. Another SNP that has been implicated in asthma susceptibility, the A38G polymorphism in the Clara cell protein 10, did not show any association with wheezing after RSV bronchiolitis [403].
RSV Vaccines
A first formalin-inactivated (FI) RSV vaccine that went into clinical trial in the 1960s not only failed to provide protection, but actually enhanced disease upon natural infection such that 80 % of the infant recipients required hospitalization during acute RSV disease and two died [292]. Histologic findings in postmortem tissue were reported to include some excess in eosinophils. It was subsequently hypothesized that the relatively better preservation of the G compared to the F protein induced a Th2 polarization and pulmonary eosinophilia, as has been observed in BALB/c mice that were challenged with RSV after immunization with FI-RSV, recombinant vaccinia virus encoding the G protein or purified G protein [477], although other data suggest upregulation of IL-10, but not of IL-4 and IL-5 [400]. However, the development of eosinophilia after G protein priming depends on the MHC haplotype in mice, and is not a feature of vaccine-enhanced disease in cotton rats [293], or bRSV-infected calves [478]. The full autopsy report apparently emphasized the predominance of neutrophils and mononuclear cells in the bronchial and bronchiolar epithelium, and these findings were confirmed in a second analysis of the original postmortem tissue [293]. Most importantly, eosinophils constituted 1–2 % of the inflammatory infiltrate. In addition, it has been shown that the G protein is not necessary in order to induce vaccine-enhanced disease [477]. Instead, the production of large amounts of non-neutralizing antibodies due to either disruption of critical epitopes or poor TLR stimulation [258] has been shown to result in the formation and tissue deposition of immune complexes [477].
It is generally felt that the disastrous consequences of the FI-RSV vaccine have hampered the development of another RSV vaccine for more than four decades. There are, however, a variety of other issues that greatly impede the generation of a safe and effective RSV vaccine [479, 480]. Since RSV infects infants at a very young age and a vaccine would have to be administered at or soon after birth, these factors include (1) the immaturity of the cellular and humoral immune system in infants, (2) the presence of maternally derived antibodies, which are known to suppress the development of serum and mucosal antibody responses, (3) the necessity to test vaccine preparations in older subjects who have already developed RSV-specific memory and are, therefore, likely to respond differently to vaccines compared to RSV-naive infants. In addition, neonates routinely receive a battery of immunizations already, and it needs to be ensured that RSV vaccines do not interfere with the efficacy of these other vaccines, and vice versa.
Formalin-inactivated RSV disease enhancement was reproduced with the bovine RSV model. Parameters significantly altered in formalin-inactivated BRSV vaccinated animals included: pulmonary function, lung pathology, arteriolar PO2 levels, and clinical sign scores [478]. Sera from vaccinated calves with and without exacerbation were examined by Western blot against BRSV and probed with both anti-IgG and anti-IgE. Serum from calves in the vaccine exacerbated group had IgE specific for BRSV proteins after the second booster vaccine and these responses became greater after infection. In contrast, serum from calves that did not show disease exacerbation had strong IgG bands and no IgE responses [481]. In addition, there was a strong correlation between BRSV-specific IgE levels and severity of clinical disease.
Numerous approaches to the development of RSV vaccines are being tested, including the classical production of live attenuated cold-passaged (cp) and temperature-sensitive (ts) mutants [479, 480, 482], the creation of recombinant RSV virus with deletions of one or more virus proteins [483], recombinant RSV expressing host cytokines in order to boost vaccine responses [211, 484], vectored vaccines, and individual virus protein subunit or peptide vaccines [485–487]. In addition, a variety of adjuvants are being examined for their ability to enhance the immunogenicity of the various preparations and to prevent vaccine-enhanced disease via Th2-skewing [181, 254, 485]. These adjuvants increasingly include CpG oligonucleotides and other TLR ligands [488] and, with the appropriate adjuvants, even formalin-inactivated preparations do not induce Th2 cytokines [181, 489]. Note that recombinant RSV vaccines are commonly derived from the RSV A2 laboratory strain. This approach may have to be reconsidered in light of evidence that recent clinical isolates differ markedly from this prototype in terms of cytopathology, replication kinetics and induction of chemokines and pro-inflammatory cytokines in primary bronchial ECs [117].
To date, many of these preparations have been shown to be highly protective in mice and occasionally in the more permissive cotton rat, but very few have progressed to clinical trials. The latter include some cp and ts mutants that were either not sufficiently attenuated, were too highly attenuated, or had an acceptable safety profile in children, but caused nasal congestion in 1–2-month-old infants [479, 480]. Currently, there still is no licensed RSV vaccine, but a live attenuated cpts RSV mutant with a deletion of the SH gene (rA2cpts 248/404/1030/ΔSH) that showed a good safety and immunogenicity profile in seronegative children is being evaluated in infants (NCT00767416) in a phase 1/2a study. In addition, a live attenuated recombinant human/bovine chimeric parainfluenza type 3 construct with the human RSV F protein engineered into the genome, was shown to be safe and immunogenic in adults and seronegative children aged 6 months to <24 months [490] and is currently in Phase 1/2a clinical trial in children 6–24 months of age and in infants 2 months of age (NCT00686075). Other ongoing clinical trials include phase 1 trials of ΔM 2-2 in adults, seropositive children and seronegative infants and children 6–24 months of age (NCT01459198) and of a RSV F particle vaccine (NCT01290419) in adults.
Summary
Many of these studies on the development of recurrent wheezing and asthma after RSV bronchiolitis have considerable methodological weaknesses. Some are retrospective and therefore based on parental recall several years after [405] or limited to review of the medical records [408]. Some of the prospective studies suffer from enormous loss to follow-up [409, 442]. Many of the publications do not provide sufficient details on the definitions of wheezing and asthma and the methods for eliciting information on their frequency from the parents. End-points are therefore highly inconsistent from study to study. Perhaps more importantly, almost all of the studies focus on a single episode of RSV infection without considering that most infants experience several episodes of acute respiratory infections, often including more than one LRTI [17, 41, 104, 426]. In addition, infants who had a RSV-associated LRTI seem prone to experience further episodes of LRTI, including episodes severe enough to require hospitalization [406, 491]. Furthermore, the number of wheezy LRTIs has been found to increase the risk of childhood asthma [428, 492], and such other wheezy LRTIs frequently are associated with rhinovirus, which has been shown to be a stronger risk factor for the development of asthma than is RSV, at least in high-risk children [426, 428].
It has further been shown that even episodes of LRTI not requiring hospitalization can significantly increase the risk of recurrent wheezing and asthma, particularly when the etiologic agent is rhinovirus [426, 428]. Hence, failure to consider LRTI following the index (hospitalization for) RSV bronchiolitis raises the question of whether the observed association with childhood asthma is truly attributable to the original RSV infection. The COAST study in Madison, WI, is a notable exception because it prospectively collected data on all moderate to severe acute respiratory illnesses during the first 3 years of life and analyzed the association between rhinovirus- and RSV-associated episodes and the subsequent development of asthma while adjusting for the occurrence of respiratory illnesses due to the other agent [426].
The potential impact of co-infections has also been neglected so far, even though up to 44 % of infants with bronchiolitis are co-infected with two or more viruses (see also Table 3), and such co-infections can be associated with increased disease severity [39, 60, 121–123]. Therefore, if disease severity has an impact on the later development of recurrent wheezing or asthma, as has been reported for bronchiolitis episodes of any etiology [468], co-infections may be one of the factors influencing the development of recurrent wheezing and asthma. Clearly, more research is needed to elucidate the relationship between RSV bronchiolitis, other viral induced LRTI and asthma.
Acknowledgments
The authors would like to thank Christopher Wilbur for assistance with the figures.
Contributor Information
M. Eric Gershwin, Phone: +1-530-7522884, FAX: +1-530-7524669.
Laurel J. Gershwin, Email: ljgershwin@ucdavis.edu
References
- 1.Blount RE, Jr, Morris JA, Savage RE. Recovery of cytopathogenic agent from chimpanzees with coryza. Proc Soc Exp Biol Med. 1956;92:544–549. doi: 10.3181/00379727-92-22538. [DOI] [PubMed] [Google Scholar]
- 2.Chanock R, Roizman B, Myers R. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). I. Isolation, properties and characterization. Am J Hyg. 1957;66:281–290. doi: 10.1093/oxfordjournals.aje.a119901. [DOI] [PubMed] [Google Scholar]
- 3.Gilca R, De Serres G, Tremblay M, et al. Distribution and clinical impact of human respiratory syncytial virus genotypes in hospitalized children over 2 winter seasons. J Infect Dis. 2006;193:54–58. doi: 10.1086/498526. [DOI] [PubMed] [Google Scholar]
- 4.Imaz MS, Sequeira MD, Videla C, et al. Clinical and epidemiologic characteristics of respiratory syncytial virus subgroups A and B infections in Santa Fe, Argentina. J Med Virol. 2000;61:76–80. doi: 10.1002/(sici)1096-9071(200005)61:1<76::aid-jmv12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 5.Fletcher JN, Smyth RL, Thomas HM, Ashby D, Hart CA. Respiratory syncytial virus genotypes and disease severity among children in hospital. Arch Dis Child. 1997;77:508–511. doi: 10.1136/adc.77.6.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peret TC, Hall CB, Hammond GW, et al. Circulation patterns of group A and B human respiratory syncytial virus genotypes in 5 communities in North America. J Infect Dis. 2000;181:1891–1896. doi: 10.1086/315508. [DOI] [PubMed] [Google Scholar]
- 7.Stensballe LG, Ravn H, Kristensen K, Meakins T, Aaby P, Simoes EA. Seasonal variation of maternally derived respiratory syncytial virus antibodies and association with infant hospitalizations for respiratory syncytial virus. J Pediatr. 2009;154:296–298. doi: 10.1016/j.jpeds.2008.07.053. [DOI] [PubMed] [Google Scholar]
- 8.Chi H, Chang IS, Tsai FY, et al. Epidemiological study of hospitalization associated with respiratory syncytial virus infection in Taiwanese children between 2004 and 2007. J Formos Med Assoc. 2011;110:388–396. doi: 10.1016/S0929-6646(11)60057-0. [DOI] [PubMed] [Google Scholar]
- 9.Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child. 1986;140:543–546. doi: 10.1001/archpedi.1986.02140200053026. [DOI] [PubMed] [Google Scholar]
- 10.Hall CB, Geiman JM, Biggar R, Kotok DI, Hogan PM, Douglas GR., Jr Respiratory syncytial virus infections within families. N Engl J Med. 1976;294:414–419. doi: 10.1056/NEJM197602192940803. [DOI] [PubMed] [Google Scholar]
- 11.Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. 2003;168:633–639. doi: 10.1164/rccm.200210-1148OC. [DOI] [PubMed] [Google Scholar]
- 12.Sastre P, Ruiz T, Schildgen O, Schildgen V, Vela C, Rueda P. Seroprevalence of human respiratory syncytial virus and human metapneumovirus in healthy population analyzed by recombinant fusion protein-based enzyme linked immunosorbent assay. Virol J. 2012;9:130. doi: 10.1186/1743-422X-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebihara T, Endo R, Kikuta H, Ishiguro N, Ishiko H, Kobayashi K. Comparison of the seroprevalence of human metapneumovirus and human respiratory syncytial virus. J Med Virol. 2004;72:304–306. doi: 10.1002/jmv.10572. [DOI] [PubMed] [Google Scholar]
- 14.Henderson FW, Collier AM, Clyde WA, Jr, Denny FW. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N Engl J Med. 1979;300:530–534. doi: 10.1056/NEJM197903083001004. [DOI] [PubMed] [Google Scholar]
- 15.Lu G, Gonzalez R, Guo L, et al. Large-scale seroprevalence analysis of human metapneumovirus and human respiratory syncytial virus infections in Beijing, China. Virol J. 2011;8:62. doi: 10.1186/1743-422X-8-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glezen WP, Paredes A, Allison JE, Taber LH, Frank AL. Risk of respiratory syncytial virus infection for infants from low-income families in relationship to age, sex, ethnic group, and maternal antibody level. J Pediatr. 1981;98:708–715. doi: 10.1016/s0022-3476(81)80829-3. [DOI] [PubMed] [Google Scholar]
- 17.Kusel MM, de Klerk NH, Holt PG, Kebadze T, Johnston SL, Sly PD. Role of respiratory viruses in acute upper and lower respiratory tract illness in the first year of life: a birth cohort study. Pediatr Infect Dis J. 2006;25:680–686. doi: 10.1097/01.inf.0000226912.88900.a3. [DOI] [PubMed] [Google Scholar]
- 18.Houben ML, Bont L, Wilbrink B, et al. Clinical prediction rule for RSV bronchiolitis in healthy newborns: prognostic birth cohort study. Pediatrics. 2011;127:35–41. doi: 10.1542/peds.2010-0581. [DOI] [PubMed] [Google Scholar]
- 19.Kim HW, Arrobio JO, Brandt CD, et al. Epidemiology of respiratory syncytial virus infection in Washington, D.C. I. Importance of the virus in different respiratory tract disease syndromes and temporal distribution of infection. Am J Epidemiol. 1973;98:216–225. doi: 10.1093/oxfordjournals.aje.a121550. [DOI] [PubMed] [Google Scholar]
- 20.Broor S, Parveen S, Bharaj P, et al. A prospective three-year cohort study of the epidemiology and virology of acute respiratory infections of children in rural India. PLoS One. 2007;2:e491. doi: 10.1371/journal.pone.0000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair H, Nokes DJ, Gessner BD, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navas L, Wang E, de Carvalho V, Robinson J. Improved outcome of respiratory syncytial virus infection in a high-risk hospitalized population of Canadian children. Pediatric Investigators Collaborative Network on Infections in Canada. J Pediatr. 1992;121:348–354. doi: 10.1016/s0022-3476(05)90000-0. [DOI] [PubMed] [Google Scholar]
- 23.Hall CB, Long CE, Schnabel KC. Respiratory syncytial virus infections in previously healthy working adults. Clin Infect Dis. 2001;33:792–796. doi: 10.1086/322657. [DOI] [PubMed] [Google Scholar]
- 24.Walsh EE, Falsey AR. Respiratory syncytial virus infection in adult populations. Infect Disord Drug Targets. 2012;12:98–102. doi: 10.2174/187152612800100116. [DOI] [PubMed] [Google Scholar]
- 25.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 26.Jansen AG, Sanders EA, Hoes AW, van Loon AM, Hak E. Influenza- and respiratory syncytial virus-associated mortality and hospitalisations. Eur Respir J. 2007;30:1158–1166. doi: 10.1183/09031936.00034407. [DOI] [PubMed] [Google Scholar]
- 27.American Academy of Pediatrics and Subcommittee on the Diagnosis and Management of Bronchiolitis Diagnosis and management of bronchiolitis. Pediatrics. 2006;118:1774–1793. doi: 10.1542/peds.2006-2223. [DOI] [PubMed] [Google Scholar]
- 28.Jartti T, Lehtinen P, Vuorinen T, Ruuskanen O. Bronchiolitis: age and previous wheezing episodes are linked to viral etiology and atopic characteristics. Pediatr Infect Dis J. 2009;28:311–317. doi: 10.1097/INF.0b013e31818ee0c1. [DOI] [PubMed] [Google Scholar]
- 29.Levine DA, Platt SL, Dayan PS, et al. Risk of serious bacterial infection in young febrile infants with respiratory syncytial virus infections. Pediatrics. 2004;113:1728–1734. doi: 10.1542/peds.113.6.1728. [DOI] [PubMed] [Google Scholar]
- 30.Randolph AG, Reder L, Englund JA. Risk of bacterial infection in previously healthy respiratory syncytial virus-infected young children admitted to the intensive care unit. Pediatr Infect Dis J. 2004;23:990–994. doi: 10.1097/01.inf.0000143647.88873.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156:322–324. doi: 10.1001/archpedi.156.4.322. [DOI] [PubMed] [Google Scholar]
- 32.Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23:267–269. doi: 10.1097/01.inf.0000116759.21252.29. [DOI] [PubMed] [Google Scholar]
- 33.Titus MO, Wright SW. Prevalence of serious bacterial infections in febrile infants with respiratory syncytial virus infection. Pediatrics. 2003;112:282–284. doi: 10.1542/peds.112.2.282. [DOI] [PubMed] [Google Scholar]
- 34.Duttweiler L, Nadal D, Frey B. Pulmonary and systemic bacterial co-infections in severe RSV bronchiolitis. Arch Dis Child. 2004;89:1155–1157. doi: 10.1136/adc.2004.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Resch B, Gusenleitner W, Mueller WD. Risk of concurrent bacterial infection in preterm infants hospitalized due to respiratory syncytial virus infection. Acta Paediatr. 2007;96:495–498. doi: 10.1111/j.1651-2227.2007.00226.x. [DOI] [PubMed] [Google Scholar]
- 36.Freymuth F, Vabret A, Galateau-Salle F, et al. Detection of respiratory syncytial virus, parainfluenzavirus 3, adenovirus and rhinovirus sequences in respiratory tract of infants by polymerase chain reaction and hybridization. Clin Diagn Virol. 1997;8:31–40. doi: 10.1016/s0928-0197(97)00060-3. [DOI] [PubMed] [Google Scholar]
- 37.Freymuth F, Vabret A, Cuvillon-Nimal D, et al. Comparison of multiplex PCR assays and conventional techniques for the diagnostic of respiratory virus infections in children admitted to hospital with an acute respiratory illness. J Med Virol. 2006;78:1498–1504. doi: 10.1002/jmv.20725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuypers J, Wright N, Ferrenberg J, et al. Comparison of real-time PCR assays with fluorescent-antibody assays for diagnosis of respiratory virus infections in children. J Clin Microbiol. 2006;44:2382–2388. doi: 10.1128/JCM.00216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richard N, Komurian-Pradel F, Javouhey E, et al. The impact of dual viral infection in infants admitted to a pediatric intensive care unit associated with severe bronchiolitis. Pediatr Infect Dis J. 2008;27:213–217. doi: 10.1097/INF.0b013e31815b4935. [DOI] [PubMed] [Google Scholar]
- 40.Bulkow LR, Singleton RJ, DeByle C, et al. Risk factors for hospitalization with lower respiratory tract infections in children in rural Alaska. Pediatrics. 2012;129:e1220–e1227. doi: 10.1542/peds.2011-1943. [DOI] [PubMed] [Google Scholar]
- 41.Lemanske RF, Jr, Jackson DJ, Gangnon RE, et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J Allergy Clin Immunol. 2005;116:571–577. doi: 10.1016/j.jaci.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 42.Hall CB, Weinberg GA, Iwane MK, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwane MK, Edwards KM, Szilagyi PG, et al. Population-based surveillance for hospitalizations associated with respiratory syncytial virus, influenza virus, and parainfluenza viruses among young children. Pediatrics. 2004;113:1758–1764. doi: 10.1542/peds.113.6.1758. [DOI] [PubMed] [Google Scholar]
- 44.Holman RC, Curns AT, Cheek JE, et al. Respiratory syncytial virus hospitalizations among American Indian and Alaska Native infants and the general United States infant population. Pediatrics. 2004;114:e437–e444. doi: 10.1542/peds.2004-0049. [DOI] [PubMed] [Google Scholar]
- 45.Zhou H, Thompson WW, Viboud CG, et al. Hospitalizations associated with influenza and respiratory syncytial virus in the United States, 1993–2008. Clin Infect Dis. 2012;54:1427–1436. doi: 10.1093/cid/cis211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stockman LJ, Curns AT, Anderson LJ, Fischer-Langley G. Respiratory syncytial virus-associated hospitalizations among infants and young children in the United States, 1997–2006. Pediatr Infect Dis J. 2012;31:5–9. doi: 10.1097/INF.0b013e31822e68e6. [DOI] [PubMed] [Google Scholar]
- 47.Singleton RJ, Bruden D, Bulkow LR, Varney G, Butler JC. Decline in respiratory syncytial virus hospitalizations in a region with high hospitalization rates and prolonged season. Pediatr Infect Dis J. 2006;25:1116–1122. doi: 10.1097/01.inf.0000245104.26996.57. [DOI] [PubMed] [Google Scholar]
- 48.Banerji A. High rates of hospitalisation for bronchiolitis in Inuit children on Baffin Island. Int J Circumpolar Health. 2001;60:375–379. [PubMed] [Google Scholar]
- 49.Orr P, McDonald S, Milley D, Brown R. Bronchiolitis in Inuit children from a Canadian central arctic community, 1995–1996. Int J Circumpolar Health. 2001;60:649–658. [PubMed] [Google Scholar]
- 50.Shay DK, Holman RC, Newman RD, Liu LL, Stout JW, Anderson LJ. Bronchiolitis-associated hospitalizations among US children, 1980–1996. JAMA. 1999;282:1440–1446. doi: 10.1001/jama.282.15.1440. [DOI] [PubMed] [Google Scholar]
- 51.Vicente D, Montes M, Cilla G, Perez-Yarza EG, Perez-Trallero E. Hospitalization for respiratory syncytial virus in the paediatric population in Spain. Epidemiol Infect. 2003;131:867–872. doi: 10.1017/s0950268803008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manoha C, Espinosa S, Aho SL, Huet F, Pothier P. Epidemiological and clinical features of hMPV, RSV and RVs infections in young children. J Clin Virol. 2007;38:221–226. doi: 10.1016/j.jcv.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 53.Wolf DG, Greenberg D, Kalkstein D, et al. Comparison of human metapneumovirus, respiratory syncytial virus and influenza A virus lower respiratory tract infections in hospitalized young children. Pediatr Infect Dis J. 2006;25:320–324. doi: 10.1097/01.inf.0000207395.80657.cf. [DOI] [PubMed] [Google Scholar]
- 54.Papadopoulos NG, Moustaki M, Tsolia M, et al. Association of rhinovirus infection with increased disease severity in acute bronchiolitis. Am J Respir Crit Care Med. 2002;165:1285–1289. doi: 10.1164/rccm.200112-118BC. [DOI] [PubMed] [Google Scholar]
- 55.Mansbach JM, McAdam AJ, Clark S, et al. Prospective multicenter study of the viral etiology of bronchiolitis in the emergency department. Acad Emerg Med. 2008;15:111–118. doi: 10.1111/j.1553-2712.2007.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stempel HE, Martin ET, Kuypers J, Englund JA, Zerr DM. Multiple viral respiratory pathogens in children with bronchiolitis. Acta Paediatr. 2009;98:123–126. doi: 10.1111/j.1651-2227.2008.01023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Calvo C, Pozo F, Garcia-Garcia ML, et al. Detection of new respiratory viruses in hospitalized infants with bronchiolitis: a three-year prospective study. Acta Paediatr. 2010;99:883–887. doi: 10.1111/j.1651-2227.2010.01714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hervás D, Reina J, Yañez A, Del Valle JM, Figuerola J, Hervás JA. Epidemiology of hospitalization for acute bronchiolitis in children: differences between RSV and non-RSV bronchiolitis. Eur J Clin Microbiol Infect Dis. 2012;31:1975–1981. doi: 10.1007/s10096-011-1529-y. [DOI] [PubMed] [Google Scholar]
- 59.Nascimento MS, Souza AV, Ferreira AV, Rodrigues JC, Abramovici S, Silva Filho LV. High rate of viral identification and coinfections in infants with acute bronchiolitis. Clinics (São Paulo) 2010;65:1133–1137. doi: 10.1590/S1807-59322010001100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foulongne V, Guyon G, Rodiere M, Segondy M. Human metapneumovirus infection in young children hospitalized with respiratory tract disease. Pediatr Infect Dis J. 2006;25:354–359. doi: 10.1097/01.inf.0000207480.55201.f6. [DOI] [PubMed] [Google Scholar]
- 61.Atmar RL, Piedra PA, Patel SM, Greenberg SB, Couch RB, Glezen WP. Picornavirus, the most common respiratory virus causing infection among patients of all ages hospitalized with acute respiratory illness. J Clin Microbiol. 2012;50:506–508. doi: 10.1128/JCM.05999-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Legg JP, Warner JA, Johnston SL, Warner JO. Frequency of detection of picornaviruses and seven other respiratory pathogens in infants. Pediatr Infect Dis J. 2005;24:611–616. doi: 10.1097/01.inf.0000168747.94999.aa. [DOI] [PubMed] [Google Scholar]
- 63.Korppi M, Kotaniemi-Syrjanen A, Waris M, Vainionpaa R, Reijonen TM. Rhinovirus-associated wheezing in infancy: comparison with respiratory syncytial virus bronchiolitis. Pediatr Infect Dis J. 2004;23:995–999. doi: 10.1097/01.inf.0000143642.72480.53. [DOI] [PubMed] [Google Scholar]
- 64.Jartti T, Kuusipalo H, Vuorinen T, et al. Allergic sensitization is associated with rhinovirus-, but not other virus-, induced wheezing in children. Pediatr Allergy Immunol. 2010;21:1008–1014. doi: 10.1111/j.1399-3038.2010.01059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boyce TG, Mellen BG, Mitchel EF, Jr, Wright PF, Griffin MR. Rates of hospitalization for respiratory syncytial virus infection among children in Medicaid. J Pediatr. 2000;137:865–870. doi: 10.1067/mpd.2000.110531. [DOI] [PubMed] [Google Scholar]
- 66.Eriksson M, Bennet R, Rotzen-Ostlund M, von Sydow M, Wirgart BZ. Population-based rates of severe respiratory syncytial virus infection in children with and without risk factors, and outcome in a tertiary care setting. Acta Paediatr. 2002;91:593–598. doi: 10.1080/080352502753711740. [DOI] [PubMed] [Google Scholar]
- 67.Deshpande SA, Northern V. The clinical and health economic burden of respiratory syncytial virus disease among children under 2 years of age in a defined geographical area. Arch Dis Child. 2003;88:1065–1069. doi: 10.1136/adc.88.12.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gouyon JB, Rozé JC, Guillermet-Fromentin C et al (2012) Hospitalizations for respiratory syncytial virus bronchiolitis in preterm infants at <33 weeks gestation without bronchopulmonary dysplasia: the CASTOR study. Epidemiol Infect 1–11 [DOI] [PMC free article] [PubMed]
- 69.Kristensen K, Hjuler T, Ravn H, Simoes EA, Stensballe LG. Chronic diseases, chromosomal abnormalities, and congenital malformations as risk factors for respiratory syncytial virus hospitalization: a population-based cohort study. Clin Infect Dis. 2012;54:810–817. doi: 10.1093/cid/cir928. [DOI] [PubMed] [Google Scholar]
- 70.Fjaerli HO, Farstad T, Bratlid D. Hospitalisations for respiratory syncytial virus bronchiolitis in Akershus, Norway, 1993–2000: a population-based retrospective study. BMC Pediatr. 2004;4:25. doi: 10.1186/1471-2431-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rietveld E, Vergouwe Y, Steyerberg EW, Huysman MW, de Groot R, Moll HA. Hospitalization for respiratory syncytial virus infection in young children: development of a clinical prediction rule. Pediatr Infect Dis J. 2006;25:201–207. doi: 10.1097/01.inf.0000202135.24485.f8. [DOI] [PubMed] [Google Scholar]
- 72.Resch B, Gusenleitner W, Muller W. The impact of respiratory syncytial virus infection: a prospective study in hospitalized infants younger than 2 years. Infection. 2002;30:193–197. doi: 10.1007/s15010-002-2122-1. [DOI] [PubMed] [Google Scholar]
- 73.Weigl JA, Puppe W, Schmitt HJ. Incidence of respiratory syncytial virus-positive hospitalizations in Germany. Eur J Clin Microbiol Infect Dis. 2001;20:452–459. doi: 10.1007/s100960100527. [DOI] [PubMed] [Google Scholar]
- 74.Park HW, Lee BS, Kim AR, et al. Epidemiology of respiratory syncytial virus infection in infants born at less than thirty-five weeks of gestational age. Pediatr Infect Dis J. 2012;31:e99–e104. doi: 10.1097/INF.0b013e318257f619. [DOI] [PubMed] [Google Scholar]
- 75.Walsh EE, McConnochie KM, Long CE, Hall CB. Severity of respiratory syncytial virus infection is related to virus strain. J Infect Dis. 1997;175:814–820. doi: 10.1086/513976. [DOI] [PubMed] [Google Scholar]
- 76.Law BJ, Langley JM, Allen U, et al. The Pediatric Investigators Collaborative Network on Infections in Canada study of predictors of hospitalization for respiratory syncytial virus infection for infants born at 33 through 35 completed weeks of gestation. Pediatr Infect Dis J. 2004;23:806–814. doi: 10.1097/01.inf.0000137568.71589.bd. [DOI] [PubMed] [Google Scholar]
- 77.Hall CB, Powell KR, MacDonald NE, et al. Respiratory syncytial viral infection in children with compromised immune function. N Engl J Med. 1986;315:77–81. doi: 10.1056/NEJM198607103150201. [DOI] [PubMed] [Google Scholar]
- 78.Nielsen HE, Siersma V, Andersen S, et al. Respiratory syncytial virus infection–risk factors for hospital admission: a case-control study. Acta Paediatr. 2003;92:1314–1321. [PubMed] [Google Scholar]
- 79.Bradley JP, Bacharier LB, Bonfiglio J, et al. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics. 2005;115:e7–e14. doi: 10.1542/peds.2004-0059. [DOI] [PubMed] [Google Scholar]
- 80.Carbonell-Estrany X, Quero J. Hospitalization rates for respiratory syncytial virus infection in premature infants born during two consecutive seasons. Pediatr Infect Dis J. 2001;20:874–879. doi: 10.1097/00006454-200109000-00010. [DOI] [PubMed] [Google Scholar]
- 81.Figueras-Aloy J, Carbonell-Estrany X, Quero J. Case-control study of the risk factors linked to respiratory syncytial virus infection requiring hospitalization in premature infants born at a gestational age of 33–35 weeks in Spain. Pediatr Infect Dis J. 2004;23:815–820. doi: 10.1097/01.inf.0000136869.21397.6b. [DOI] [PubMed] [Google Scholar]
- 82.Figueras-Aloy J, Carbonell-Estrany X, Quero-Jimenez J, et al. FLIP-2 Study: risk factors linked to respiratory syncytial virus infection requiring hospitalization in premature infants born in Spain at a gestational age of 32 to 35 weeks. Pediatr Infect Dis J. 2008;27:788–793. doi: 10.1097/INF.0b013e3181710990. [DOI] [PubMed] [Google Scholar]
- 83.Stensballe LG, Kristensen K, Simoes EA, et al. Atopic disposition, wheezing, and subsequent respiratory syncytial virus hospitalization in Danish children younger than 18 months: a nested case-control study. Pediatrics. 2006;118:e1360–e1368. doi: 10.1542/peds.2006-0907. [DOI] [PubMed] [Google Scholar]
- 84.Doering G, Gusenleitner W, Belohradsky BH, Burdach S, Resch B, Liese JG. The risk of respiratory syncytial virus-related hospitalizations in preterm infants of 29 to 35 weeks’ gestational age. Pediatr Infect Dis J. 2006;25:1188–1190. doi: 10.1097/01.inf.0000246978.58565.b5. [DOI] [PubMed] [Google Scholar]
- 85.Bulkow LR, Singleton RJ, Karron RA, Harrison LH. Risk factors for severe respiratory syncytial virus infection among Alaska native children. Pediatrics. 2002;109:210–216. doi: 10.1542/peds.109.2.210. [DOI] [PubMed] [Google Scholar]
- 86.Medrano C, Garcia-Guereta L, Grueso J, et al. Respiratory infection in congenital cardiac disease. Hospitalizations in young children in Spain during 2004 and 2005: the CIVIC Epidemiologic Study. Cardiol Young. 2007;17:360–371. doi: 10.1017/S104795110700042X. [DOI] [PubMed] [Google Scholar]
- 87.Broughton S, Roberts A, Fox G, et al. Prospective study of healthcare utilisation and respiratory morbidity due to RSV infection in prematurely born infants. Thorax. 2005;60:1039–1044. doi: 10.1136/thx.2004.037853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Somech R, Tal G, Gilad E, Mandelberg A, Tal A, Dalal I. Epidemiologic, socioeconomic, and clinical factors associated with severity of respiratory syncytial virus infection in previously healthy infants. Clin Pediatr (Phila) 2006;45:621–627. doi: 10.1177/0009922806291012. [DOI] [PubMed] [Google Scholar]
- 89.Brandenburg AH, Groen J, van Steensel-Moll HA, et al. Respiratory syncytial virus specific serum antibodies in infants under six months of age: limited serological response upon infection. J Med Virol. 1997;52:97–104. doi: 10.1002/(sici)1096-9071(199705)52:1<97::aid-jmv16>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 90.Parrott RH, Kim HW, Arrobio JO, et al. Epidemiology of respiratory syncytial virus infection in Washington, D.C. II. Infection and disease with respect to age, immunologic status, race and sex. Am J Epidemiol. 1973;98:289–300. doi: 10.1093/oxfordjournals.aje.a121558. [DOI] [PubMed] [Google Scholar]
- 91.Ochola R, Sande C, Fegan G, et al. The level and duration of RSV-specific maternal IgG in infants in Kilifi Kenya. PLoS One. 2009;4:e8088. doi: 10.1371/journal.pone.0008088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roca A, Abacassamo F, Loscertales MP, et al. Prevalence of respiratory syncytial virus IgG antibodies in infants living in a rural area of Mozambique. J Med Virol. 2002;67:616–623. doi: 10.1002/jmv.10148. [DOI] [PubMed] [Google Scholar]
- 93.Holberg CJ, Wright AL, Martinez FD, Ray CG, Taussig LM, Lebowitz MD. Risk factors for respiratory syncytial virus-associated lower respiratory illnesses in the first year of life. Am J Epidemiol. 1991;133:1135–1151. doi: 10.1093/oxfordjournals.aje.a115826. [DOI] [PubMed] [Google Scholar]
- 94.Stensballe LG, Ravn H, Kristensen K, et al. Respiratory syncytial virus neutralizing antibodies in cord blood, respiratory syncytial virus hospitalization, and recurrent wheeze. J Allergy Clin Immunol. 2009;123:398–403. doi: 10.1016/j.jaci.2008.10.043. [DOI] [PubMed] [Google Scholar]
- 95.Piedra PA, Jewell AM, Cron SG, Atmar RL, Glezen WP. Correlates of immunity to respiratory syncytial virus (RSV) associated-hospitalization: establishment of minimum protective threshold levels of serum neutralizing antibodies. Vaccine. 2003;21:3479–3482. doi: 10.1016/s0264-410x(03)00355-4. [DOI] [PubMed] [Google Scholar]
- 96.Lamprecht CL, Krause HE, Mufson MA. Role of maternal antibody in pneumonia and bronchiolitis due to respiratory syncytial virus. J Infect Dis. 1976;134:211–217. doi: 10.1093/infdis/134.3.211. [DOI] [PubMed] [Google Scholar]
- 97.El Saleeby CM, Bush AJ, Harrison LM, Aitken JA, Devincenzo JP. Respiratory syncytial virus load, viral dynamics, and disease severity in previously healthy naturally infected children. J Infect Dis. 2011;204:996–1002. doi: 10.1093/infdis/jir494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fodha I, Vabret A, Ghedira L, et al. Respiratory syncytial virus infections in hospitalized infants: association between viral load, virus subgroup, and disease severity. J Med Virol. 2007;79:1951–1958. doi: 10.1002/jmv.21026. [DOI] [PubMed] [Google Scholar]
- 99.Buckingham SC, Bush AJ, Devincenzo JP. Nasal quantity of respiratory syncytical virus correlates with disease severity in hospitalized infants. Pediatr Infect Dis J. 2000;19:113–117. doi: 10.1097/00006454-200002000-00006. [DOI] [PubMed] [Google Scholar]
- 100.DeVincenzo JP, El Saleeby CM, Bush AJ. Respiratory syncytial virus load predicts disease severity in previously healthy infants. J Infect Dis. 2005;191:1861–1868. doi: 10.1086/430008. [DOI] [PubMed] [Google Scholar]
- 101.Semple MG, Dankert HM, Ebrahimi B, et al. Severe respiratory syncytial virus bronchiolitis in infants is associated with reduced airway interferon gamma and substance P. PLoS One. 2007;2:e1038. doi: 10.1371/journal.pone.0001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wright PF, Gruber WC, Peters M, et al. Illness severity, viral shedding, and antibody responses in infants hospitalized with bronchiolitis caused by respiratory syncytial virus. J Infect Dis. 2002;185:1011–1018. doi: 10.1086/339822. [DOI] [PubMed] [Google Scholar]
- 103.Houben ML, Coenjaerts FE, Rossen JW, et al. Disease severity and viral load are correlated in infants with primary respiratory syncytial virus infection in the community. J Med Virol. 2010;82:1266–1271. doi: 10.1002/jmv.21771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Simoes EA, Mutyara K, Soh S, Agustian D, Hibberd ML, Kartasasmita CB. The epidemiology of respiratory syncytial virus lower respiratory tract infections in children less than 5 years of age in Indonesia. Pediatr Infect Dis J. 2011;30:778–784. doi: 10.1097/INF.0b013e318218ab9e. [DOI] [PubMed] [Google Scholar]
- 105.McConnochie KM, Hall CB, Walsh EE, Roghmann KJ. Variation in severity of respiratory syncytial virus infections with subtype. J Pediatr. 1990;117:52–62. doi: 10.1016/s0022-3476(05)82443-6. [DOI] [PubMed] [Google Scholar]
- 106.Hornsleth A, Klug B, Nir M, et al. Severity of respiratory syncytial virus disease related to type and genotype of virus and to cytokine values in nasopharyngeal secretions. Pediatr Infect Dis J. 1998;17:1114–1121. doi: 10.1097/00006454-199812000-00003. [DOI] [PubMed] [Google Scholar]
- 107.Paulus SC, Hirschfeld AF, Victor RE, Brunstein J, Thomas E, Turvey SE. Common human Toll-like receptor 4 polymorphisms–role in susceptibility to respiratory syncytial virus infection and functional immunological relevance. Clin Immunol. 2007;123:252–257. doi: 10.1016/j.clim.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 108.Kaplan NM, Dove W, Abd-Eldayem SA, Abu-Zeid AF, Shamoon HE, Hart CA. Molecular epidemiology and disease severity of respiratory syncytial virus in relation to other potential pathogens in children hospitalized with acute respiratory infection in Jordan. J Med Virol. 2008;80:168–174. doi: 10.1002/jmv.21067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martinello RA, Chen MD, Weibel C, Kahn JS. Correlation between respiratory syncytial virus genotype and severity of illness. J Infect Dis. 2002;186:839–842. doi: 10.1086/342414. [DOI] [PubMed] [Google Scholar]
- 110.Struck A, Forster J, Ihorst G, Werchau H, Konig W, Konig B. Respiratory syncytial virus: G gene genotype and disease severity. Pediatr Infect Dis J. 2004;23:1000–1002. doi: 10.1097/01.inf.0000145531.65979.cf. [DOI] [PubMed] [Google Scholar]
- 111.Savon C, Goyenechea A, Valdes O, et al. Respiratory syncytial virus group A and B genotypes and disease severity among Cuban children. Arch Med Res. 2006;37:543–547. doi: 10.1016/j.arcmed.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 112.Smyth RL, Mobbs KJ, O’Hea U, Ashby D, Hart CA. Respiratory syncytial virus bronchiolitis: disease severity, interleukin-8, and virus genotype. Pediatr Pulmonol. 2002;33:339–346. doi: 10.1002/ppul.10080. [DOI] [PubMed] [Google Scholar]
- 113.Wu W, Macdonald A, Hiscox JA, Barr JN. Different NF-kappaB activation characteristics of human respiratory syncytial virus subgroups A and B. Microb Pathog. 2012;52:184–191. doi: 10.1016/j.micpath.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 114.Stokes KL, Chi MH, Sakamoto K, et al. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J Virol. 2011;85:5782–5793. doi: 10.1128/JVI.01693-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moore ML, Chi MH, Luongo C, et al. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J Virol. 2009;83:4185–4194. doi: 10.1128/JVI.01853-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lukacs NW, Moore ML, Rudd BD, et al. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am J Pathol. 2006;169:977–986. doi: 10.2353/ajpath.2006.051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Villenave R, O’Donoghue D, Thavagnanam S, et al. Differential cytopathogenesis of respiratory syncytial virus prototypic and clinical isolates in primary pediatric bronchial epithelial cells. Virol J. 2011;8:43. doi: 10.1186/1743-422X-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Levitz R, Wattier R, Phillips P, et al. Induction of IL-6 and CCL5 (RANTES) in human respiratory epithelial (A549) cells by clinical isolates of respiratory syncytial virus is strain specific. Virol J. 2012;9:190. doi: 10.1186/1743-422X-9-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ursic T, Jevsnik M, Zigon N, et al. Human bocavirus and other respiratory viral infections in a 2-year cohort of hospitalized children. J Med Virol. 2012;84:99–108. doi: 10.1002/jmv.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jennings LC, Anderson TP, Werno AM, Beynon KA, Murdoch DR. Viral etiology of acute respiratory tract infections in children presenting to hospital: role of polymerase chain reaction and demonstration of multiple infections. Pediatr Infect Dis J. 2004;23:1003–1007. doi: 10.1097/01.inf.0000143648.04673.6c. [DOI] [PubMed] [Google Scholar]
- 121.Calvo C, Garcia-Garcia ML, Blanco C, et al. Multiple simultaneous viral infections in infants with acute respiratory tract infections in Spain. J Clin Virol. 2008;42:268–272. doi: 10.1016/j.jcv.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Midulla F, Scagnolari C, Bonci E, et al. Respiratory syncytial virus, human bocavirus and rhinovirus bronchiolitis in infants. Arch Dis Child. 2010;95:35–41. doi: 10.1136/adc.2008.153361. [DOI] [PubMed] [Google Scholar]
- 123.Semple MG, Cowell A, Dove W, et al. Dual infection of infants by human metapneumovirus and human respiratory syncytial virus is strongly associated with severe bronchiolitis. J Infect Dis. 2005;191:382–386. doi: 10.1086/426457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.König B, König W, Arnold R, Werchau H, Ihorst G, Forster J. Prospective study of human metapneumovirus infection in children less than 3 years of age. J Clin Microbiol. 2004;42:4632–4635. doi: 10.1128/JCM.42.10.4632-4635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aberle JH, Aberle SW, Pracher E, Hutter HP, Kundi M, Popow-Kraupp T. Single versus dual respiratory virus infections in hospitalized infants: impact on clinical course of disease and interferon-gamma response. Pediatr Infect Dis J. 2005;24:605–610. doi: 10.1097/01.inf.0000168741.59747.2d. [DOI] [PubMed] [Google Scholar]
- 126.Greensill J, McNamara PS, Dove W, Flanagan B, Smyth RL, Hart CA. Human metapneumovirus in severe respiratory syncytial virus bronchiolitis. Emerg Infect Dis. 2003;9:372–375. doi: 10.3201/eid0903.020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fernandes RM, Bialy LM, Vandermeer B et al (2010) Glucocorticoids for acute viral bronchiolitis in infants and young children. Cochrane Database Syst Rev CD004878 [DOI] [PubMed]
- 128.Plint AC, Johnson DW, Patel H, et al. Epinephrine and dexamethasone in children with bronchiolitis. N Engl J Med. 2009;360:2079–2089. doi: 10.1056/NEJMoa0900544. [DOI] [PubMed] [Google Scholar]
- 129.Pinto RA, Arredondo SM, Bono MR, Gaggero AA, Diaz PV. T helper 1/T helper 2 cytokine imbalance in respiratory syncytial virus infection is associated with increased endogenous plasma cortisol. Pediatrics. 2006;117:e878–e886. doi: 10.1542/peds.2005-2119. [DOI] [PubMed] [Google Scholar]
- 130.Blom D, Ermers M, Bont L, van Aalderen WM, van Woensel JB (2007) Inhaled corticosteroids during acute bronchiolitis in the prevention of post-bronchiolitic wheezing. Cochrane Database Syst Rev CD004881 [DOI] [PubMed]
- 131.Ermers MJ, Rovers MM, van Woensel JB, Kimpen JL, Bont LJ. The effect of high dose inhaled corticosteroids on wheeze in infants after respiratory syncytial virus infection: randomised double blind placebo controlled trial. BMJ. 2009;338:b897. doi: 10.1136/bmj.b897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ventre K, Randolph AG (2007) Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database Syst Rev CD000181 [DOI] [PubMed]
- 133.Kneyber MC, Blusse van Oud-Alblas H, van Vliet M, Uiterwaal CS, Kimpen JL, van Vught AJ. Concurrent bacterial infection and prolonged mechanical ventilation in infants with respiratory syncytial virus lower respiratory tract disease. Intensive Care Med. 2005;31:680–685. doi: 10.1007/s00134-005-2614-4. [DOI] [PubMed] [Google Scholar]
- 134.Spurling GK, Doust J, Del Mar CB, Eriksson L (2011) Antibiotics for bronchiolitis in children. Cochrane Database Syst Rev CD005189 [DOI] [PubMed]
- 135.Ni K, Li S, Xia Q, et al. Pharyngeal microflora disruption by antibiotics promotes airway hyperresponsiveness after respiratory syncytial virus infection. PLoS One. 2012;7:e41104. doi: 10.1371/journal.pone.0041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weiss KA, Christiaansen AF, Fulton RB, Meyerholz DK, Varga SM. Multiple CD4+ T cell subsets produce immunomodulatory IL-10 during respiratory syncytial virus infection. J Immunol. 2011;187:3145–3154. doi: 10.4049/jimmunol.1100764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ruckwardt TJ, Bonaparte KL, Nason MC, Graham BS. Regulatory T cells promote early influx of CD8+ T cells in the lungs of respiratory syncytial virus-infected mice and diminish immunodominance disparities. J Virol. 2009;83:3019–3028. doi: 10.1128/JVI.00036-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fulton RB, Meyerholz DK, Varga SM. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J Immunol. 2010;185:2382–2392. doi: 10.4049/jimmunol.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Loebbermann J, Thornton H, Durant L, et al. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. 2012;5:161–172. doi: 10.1038/mi.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Behrendt CE, Decker MD, Burch DJ, Watson PH. International variation in the management of infants hospitalized with respiratory syncytial virus. International RSV Study Group. Eur J Pediatr. 1998;157:215–220. doi: 10.1007/s004310050798. [DOI] [PubMed] [Google Scholar]
- 141.IMpact-RSV Study Group Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics. 1998;102:531–537. [PubMed] [Google Scholar]
- 142.Feltes TF, Cabalka AK, Meissner HC, et al. Palivizumab prophylaxis reduces hospitalization due to respiratory syncytial virus in young children with hemodynamically significant congenital heart disease. J Pediatr. 2003;143:532–540. doi: 10.1067/s0022-3476(03)00454-2. [DOI] [PubMed] [Google Scholar]
- 143.Alexander PM, Eastaugh L, Royle J, Daley AJ, Shekerdemian LS, Penny DJ. Respiratory syncytial virus immunoprophylaxis in high-risk infants with heart disease. J Paediatr Child Health. 2012;48:395–401. doi: 10.1111/j.1440-1754.2011.02219.x. [DOI] [PubMed] [Google Scholar]
- 144.Winterstein AG, Hampp C, Saidi A. Effectiveness of palivizumab prophylaxis in infants and children in Florida. Pharmacoepidemiol Drug Saf. 2012;21:53–60. doi: 10.1002/pds.2246. [DOI] [PubMed] [Google Scholar]
- 145.Checchia PA, Nalysnyk L, Fernandes AW, et al. Mortality and morbidity among infants at high risk for severe respiratory syncytial virus infection receiving prophylaxis with palivizumab: a systematic literature review and meta-analysis. Pediatr Crit Care Med. 2011;12:580–588. doi: 10.1097/PCC.0b013e3182070990. [DOI] [PubMed] [Google Scholar]
- 146.Chadha AD, Bao W, Holloway J, Mann J, Rye AK, Brown DE., 3rd Respiratory syncytial virus morbidity and outpatient palivizumab dosing in South Carolina, 2004–2009. South Med J. 2012;105:399–404. doi: 10.1097/SMJ.0b013e31825ea57d. [DOI] [PubMed] [Google Scholar]
- 147.Frogel MP, Stewart DL, Hoopes M, Fernandes AW, Mahadevia PJ. A systematic review of compliance with palivizumab administration for RSV immunoprophylaxis. J Manag Care Pharm. 2010;16:46–58. doi: 10.18553/jmcp.2010.16.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.American Academy of Pediatrics and Committee on Infectious Diseases Policy statements–Modified recommendations for use of palivizumab for prevention of respiratory syncytial virus infections. Pediatrics. 2009;124:1694–1701. doi: 10.1542/peds.2009-2345. [DOI] [PubMed] [Google Scholar]
- 149.Sampalis JS, Langley J, Carbonell-Estrany X, et al. Development and validation of a risk scoring tool to predict respiratory syncytial virus hospitalization in premature infants born at 33 through 35 completed weeks of gestation. Med Decis Making. 2008;28:471–480. doi: 10.1177/0272989X08315238. [DOI] [PubMed] [Google Scholar]
- 150.Simoes EA, Carbonell-Estrany X, Fullarton JR, et al. A predictive model for respiratory syncytial virus (RSV) hospitalisation of premature infants born at 33–35 weeks of gestational age, based on data from the Spanish FLIP Study. Respir Res. 2008;9:78. doi: 10.1186/1465-9921-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lanctôt KL, Masoud ST, Paes BA, et al. The cost-effectiveness of palivizumab for respiratory syncytial virus prophylaxis in premature infants with a gestational age of 32–35 weeks: a Canadian-based analysis. Curr Med Res Opin. 2008;24:3223–3237. doi: 10.1185/03007990802484234. [DOI] [PubMed] [Google Scholar]
- 152.Paes B, Steele S, Janes M, Pinelli J. Risk-Scoring Tool for respiratory syncytial virus prophylaxis in premature infants born at 33–35 completed weeks’ gestational age in Canada. Curr Med Res Opin. 2009;25:1585–1591. doi: 10.1185/03007990902929112. [DOI] [PubMed] [Google Scholar]
- 153.Saez-Llorens X, Castano E, Null D, et al. Safety and pharmacokinetics of an intramuscular humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. The MEDI-493 Study Group. Pediatr Infect Dis J. 1998;17:787–791. doi: 10.1097/00006454-199809000-00007. [DOI] [PubMed] [Google Scholar]
- 154.Zaaijer HL, Vandenbroucke-Grauls CM, Franssen EJ. Optimum dosage regimen of palivizumab? Ther Drug Monit. 2002;24:444–445. doi: 10.1097/00007691-200206000-00020. [DOI] [PubMed] [Google Scholar]
- 155.van Drunen Littel-van den Hurk S, Watkiss ER. Pathogenesis of respiratory syncytial virus. Curr Opin Virol. 2012;2:300–305. doi: 10.1016/j.coviro.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 156.Palomo C, Cane PA, Melero JA. Evaluation of the antibody specificities of human convalescent-phase sera against the attachment (G) protein of human respiratory syncytial virus: influence of strain variation and carbohydrate side chains. J Med Virol. 2000;60:468–474. [PubMed] [Google Scholar]
- 157.Garcia-Beato R, Melero JA. The C-terminal third of human respiratory syncytial virus attachment (G) protein is partially resistant to protease digestion and is glycosylated in a cell-type-specific manner. J Gen Virol. 2000;81:919–927. doi: 10.1099/0022-1317-81-4-919. [DOI] [PubMed] [Google Scholar]
- 158.Collins PL, Graham BS. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol. 2008;82:2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Srikiatkhachorn A, Braciale TJ. Virus-specific CD8+ T lymphocytes downregulate T helper cell type 2 cytokine secretion and pulmonary eosinophilia during experimental murine respiratory syncytial virus infection. J Exp Med. 1997;186:421–432. doi: 10.1084/jem.186.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jackson M, Scott R. Different patterns of cytokine induction in cultures of respiratory syncytial (RS) virus-specific human TH cell lines following stimulation with RS virus and RS virus proteins. J Med Virol. 1996;49:161–169. doi: 10.1002/(SICI)1096-9071(199607)49:3<161::AID-JMV2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 161.McDermott DS, Weiss KA, Knudson CJ, Varga SM. Central role of dendritic cells in shaping the adaptive immune response during respiratory syncytial virus infection. Future Virol. 2011;6:963–973. doi: 10.2217/fvl.11.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. 2004;4:553–564. doi: 10.1038/nri1394. [DOI] [PubMed] [Google Scholar]
- 163.Vekemans J, Ota MO, Wang EC, et al. T cell responses to vaccines in infants: defective IFNg production after oral polio vaccination. Clin Exp Immunol. 2002;127:495–498. doi: 10.1046/j.1365-2249.2002.01788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jaimes MC, Rojas OL, González AM, et al. Frequencies of virus-specific CD4+ and CD8+ T lymphocytes secreting gamma interferon after acute natural rotavirus infection in children and adults. J Virol. 2002;76:4741–4749. doi: 10.1128/JVI.76.10.4741-4749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Härtel C, Adam N, Strunk T, Temming P, Müller-Steinhardt M, Schultz C. Cytokine responses correlate differentially with age in infancy and early childhood. Clin Exp Immunol. 2005;142:446–453. doi: 10.1111/j.1365-2249.2005.02928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Krampera M, Vinante F, Tavecchia L, et al. Progressive polarization towards a T helper/cytotoxic type-1 cytokine pattern during age-dependent maturation of the immune response inversely correlates with CD30 cell expression and serum concentration. Clin Exp Immunol. 1999;117:291–297. doi: 10.1046/j.1365-2249.1999.00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Prescott SL, Macaubas C, Holt BJ, et al. Transplacental priming of the human immune system to environmental allergens: universal skewing of initial T cell responses toward the Th2 cytokine profile. J Immunol. 1998;160:4730–4737. [PubMed] [Google Scholar]
- 168.Siegrist CA. The challenges of vaccine responses in early life: selected examples. J Comp Pathol. 2007;137:S4–S9. doi: 10.1016/j.jcpa.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 169.Sasaki S, Jaimes MC, Holmes TH, et al. Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J Virol. 2007;81:215–228. doi: 10.1128/JVI.01957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Weitkamp JH, Lafleur BJ, Greenberg HB, Crowe JE., Jr Natural evolution of a human virus-specific antibody gene repertoire by somatic hypermutation requires both hotspot-directed and randomly-directed processes. Hum Immunol. 2005;66:666–676. doi: 10.1016/j.humimm.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 171.Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol. 2007;20:108–119. doi: 10.1038/modpathol.3800725. [DOI] [PubMed] [Google Scholar]
- 172.Wright C, Oliver KC, Fenwick FI, Smith NM, Toms GL. A monoclonal antibody pool for routine immunohistochemical detection of human respiratory syncytial virus antigens in formalin-fixed, paraffin-embedded tissue. J Pathol. 1997;182:238–244. doi: 10.1002/(SICI)1096-9896(199706)182:2<238::AID-PATH822>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 173.Fonceca AM, Flanagan BF, Trinick R, Smyth RL, McNamara PS. Primary airway epithelial cultures from children are highly permissive to respiratory syncytial virus infection. Thorax. 2012;67:42–48. doi: 10.1136/thoraxjnl-2011-200131. [DOI] [PubMed] [Google Scholar]
- 174.Tristram DA, Hicks W, Jr, Hard R. Respiratory syncytial virus and human bronchial epithelium. Arch Otolaryngol Head Neck Surg. 1998;124:777–783. doi: 10.1001/archotol.124.7.777. [DOI] [PubMed] [Google Scholar]
- 175.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol. 2002;76:5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mellow TE, Murphy PC, Carson JL, Noah TL, Zhang L, Pickles RJ. The effect of respiratory synctial virus on chemokine release by differentiated airway epithelium. Exp Lung Res. 2004;30:43–57. doi: 10.1080/01902140490252812. [DOI] [PubMed] [Google Scholar]
- 177.Villenave R, Thavagnanam S, Sarlang S, et al. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc Natl Acad Sci U S A. 2012;109:5040–5045. doi: 10.1073/pnas.1110203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med. 2011;17:1132–1135. doi: 10.1038/nm.2444. [DOI] [PubMed] [Google Scholar]
- 179.Liu P, Jamaluddin M, Li K, Garofalo RP, Casola A, Brasier AR. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J Virol. 2007;81:1401–1411. doi: 10.1128/JVI.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Okabayashi T, Kojima T, Masaki T, et al. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011;160:360–366. doi: 10.1016/j.virusres.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 181.Zeng R, Cui Y, Hai Y, Liu Y. Pattern recognition receptors for respiratory syncytial virus infection and design of vaccines. Virus Res. 2012;167:138–145. doi: 10.1016/j.virusres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 182.Olszewska-Pazdrak B, Casola A, Saito T, et al. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol. 1998;72:4756–4764. doi: 10.1128/jvi.72.6.4756-4764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mochizuki H, Todokoro M, Arakawa H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation. 2009;32:252–264. doi: 10.1007/s10753-009-9128-0. [DOI] [PubMed] [Google Scholar]
- 184.Bonville CA, Rosenberg HF, Domachowske JB. Macrophage inflammatory protein-1alpha and RANTES are present in nasal secretions during ongoing upper respiratory tract infection. Pediatr Allergy Immunol. 1999;10:39–44. doi: 10.1034/j.1399-3038.1999.101005.x. [DOI] [PubMed] [Google Scholar]
- 185.Miller AL, Bowlin TL, Lukacs NW. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J Infect Dis. 2004;189:1419–1430. doi: 10.1086/382958. [DOI] [PubMed] [Google Scholar]
- 186.Miller AL, Gerard C, Schaller M, Gruber AD, Humbles AA, Lukacs NW. Deletion of CCR1 attenuates pathophysiologic responses during respiratory syncytial virus infection. J Immunol. 2006;176:2562–2567. doi: 10.4049/jimmunol.176.4.2562. [DOI] [PubMed] [Google Scholar]
- 187.Jewell NA, Vaghefi N, Mertz SE, et al. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J Virol. 2007;81:9790–9800. doi: 10.1128/JVI.00530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Saito T, Deskin RW, Casola A, et al. Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J Infect Dis. 1997;175:497–504. doi: 10.1093/infdis/175.3.497. [DOI] [PubMed] [Google Scholar]
- 189.Morrison PT, Thomas LH, Sharland M, Friedland JS. RSV-infected airway epithelial cells cause biphasic up-regulation of CCR1 expression on human monocytes. J Leukoc Biol. 2007;81:1487–1495. doi: 10.1189/jlb.1006611. [DOI] [PubMed] [Google Scholar]
- 190.Becker S, Soukup JM. Airway epithelial cell-induced activation of monocytes and eosinophils in respiratory syncytial viral infection. Immunobiology. 1999;201:88–106. doi: 10.1016/S0171-2985(99)80049-7. [DOI] [PubMed] [Google Scholar]
- 191.Becker S, Quay J, Soukup J. Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. J Immunol. 1991;147:4307–4312. [PubMed] [Google Scholar]
- 192.Bem RA, Domachowske JB, Rosenberg HF. Animal models of human respiratory syncytial virus disease. Am J Physiol Lung Cell Mol Physiol. 2011;301:L148–L156. doi: 10.1152/ajplung.00065.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hegele RG, Hayashi S, Bramley AM, Hogg JC. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest. 1994;105:1848–1854. doi: 10.1378/chest.105.6.1848. [DOI] [PubMed] [Google Scholar]
- 194.Human respiratory syncytial virus strain hRSVs/Dibrugarh.IND/3684.12 attachment glycoprotein (G) gene, partial cds. GenBank: KC117368.2
- 195.Woolums AR, Singer RS, Boyle GA, Gershwin LJ. Interferon gamma production during bovine respiratory syncytial virus (BRSV) infection is diminished in calves vaccinated with formalin-inactivated BRSV. Vaccine. 1999;17:1293–1297. doi: 10.1016/s0264-410x(98)00379-x. [DOI] [PubMed] [Google Scholar]
- 196.van Schaik SM, Enhorning G, Vargas I, Welliver RC. Respiratory syncytial virus affects pulmonary function in BALB/c mice. J Infect Dis. 1998;177:269–276. doi: 10.1086/514208. [DOI] [PubMed] [Google Scholar]
- 197.Bera MM, Lu B, Martin TR, et al. Th17 cytokines are critical for respiratory syncytial virus-associated airway hyperreponsiveness through regulation by complement C3a and tachykinins. J Immunol. 2011;187:4245–4255. doi: 10.4049/jimmunol.1101789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chávez-Bueno S, Mejías A, Gómez AM, et al. Respiratory syncytial virus-induced acute and chronic airway disease is independent of genetic background: an experimental murine model. Virol J. 2005;2:46. doi: 10.1186/1743-422X-2-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jafri HS, Chavez-Bueno S, Mejias A, et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis. 2004;189:1856–1865. doi: 10.1086/386372. [DOI] [PubMed] [Google Scholar]
- 200.Peebles RS, Jr, Sheller JR, Johnson JE, Mitchell DB, Graham BS. Respiratory syncytial virus infection prolongs methacholine-induced airway hyperresponsiveness in ovalbumin-sensitized mice. J Med Virol. 1999;57:186–192. doi: 10.1002/(sici)1096-9071(199902)57:2<186::aid-jmv17>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 201.Noah TL, Becker S. Chemokines in nasal secretions of normal adults experimentally infected with respiratory syncytial virus. Clin Immunol. 2000;97:43–49. doi: 10.1006/clim.2000.4914. [DOI] [PubMed] [Google Scholar]
- 202.Durbin JE, Johnson TR, Durbin RK, et al. The role of IFN in respiratory syncytial virus pathogenesis. J Immunol. 2002;168:2944–2952. doi: 10.4049/jimmunol.168.6.2944. [DOI] [PubMed] [Google Scholar]
- 203.Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1alpha in lung pathology. J Virol. 2001;75:878–890. doi: 10.1128/JVI.75.2.878-890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pribul PK, Harker J, Wang B, et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. 2008;82:4441–4448. doi: 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol. 2001;31:2566–2573. doi: 10.1002/1521-4141(200109)31:9<2566::aid-immu2566>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 206.Rutigliano JA, Graham BS. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J Immunol. 2004;173:3408–3417. doi: 10.4049/jimmunol.173.5.3408. [DOI] [PubMed] [Google Scholar]
- 207.Tregoning JS, Pribul PK, Pennycook AM, et al. The chemokine MIP1alpha/CCL3 determines pathology in primary RSV infection by regulating the balance of T cell populations in the murine lung. PLoS One. 2010;5:e9381. doi: 10.1371/journal.pone.0009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.LeVine AM, Gwozdz J, Stark J, Bruno M, Whitsett J, Korfhagen T. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103:1015–1021. doi: 10.1172/JCI5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Park JW, Taube C, Yang ES, et al. Respiratory syncytial virus-induced airway hyperresponsiveness is independent of IL-13 compared with that induced by allergen. J Allergy Clin Immunol. 2003;112:1078–1087. doi: 10.1016/j.jaci.2003.08.046. [DOI] [PubMed] [Google Scholar]
- 210.Dakhama A, Park JW, Taube C, et al. Alteration of airway neuropeptide expression and development of airway hyperresponsiveness following respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol. 2005;288:L761–L770. doi: 10.1152/ajplung.00143.2004. [DOI] [PubMed] [Google Scholar]
- 211.Harker JA, Godlee A, Wahlsten JL, et al. Interleukin 18 coexpression during respiratory syncytial virus infection results in enhanced disease mediated by natural killer cells. J Virol. 2010;84:4073–4082. doi: 10.1128/JVI.02014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Estripeaut D, Torres JP, Somers CS, et al. Respiratory syncytial virus persistence in the lungs correlates with airway hyperreactivity in the mouse model. J Infect Dis. 2008;198:1435–1443. doi: 10.1086/592714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hussell T, Openshaw PJ. Intracellular IFN-gamma expression in natural killer cells precedes lung CD8+ T cell recruitment during respiratory syncytial virus infection. J Gen Virol. 1998;79(Pt 11):2593–2601. doi: 10.1099/0022-1317-79-11-2593. [DOI] [PubMed] [Google Scholar]
- 214.Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J Immunol. 2010;185:4681–4690. doi: 10.4049/jimmunol.1001758. [DOI] [PubMed] [Google Scholar]
- 215.Loebbermann J, Schnoeller C, Thornton H, et al. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS One. 2012;7:e32371. doi: 10.1371/journal.pone.0032371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Johnson TR, Mertz SE, Gitiban N, et al. Role for innate IFNs in determining respiratory syncytial virus immunopathology. J Immunol. 2005;174:7234–7241. doi: 10.4049/jimmunol.174.11.7234. [DOI] [PubMed] [Google Scholar]
- 217.Lee DC, Harker JA, Tregoning JS, et al. CD25+ natural regulatory T cells are critical in limiting innate and adaptive immunity and resolving disease following respiratory syncytial virus infection. J Virol. 2010;84:8790–8798. doi: 10.1128/JVI.00796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Mäkelä MJ, Tripp R, Dakhama A, et al. Prior airway exposure to allergen increases virus-induced airway hyperresponsiveness. J Allergy Clin Immunol. 2003;112:861–869. doi: 10.1016/s0091-6749(03)02020-7. [DOI] [PubMed] [Google Scholar]
- 219.Schwarze J, Hamelmann E, Bradley KL, Takeda K, Gelfand EW. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J Clin Invest. 1997;100:226–233. doi: 10.1172/JCI119516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Schwarze J, Cieslewicz G, Hamelmann E, et al. IL-5 and eosinophils are essential for the development of airway hyperresponsiveness following acute respiratory syncytial virus infection. J Immunol. 1999;162:2997–3004. [PubMed] [Google Scholar]
- 221.Domachowske JB, Bonville CA, Gao JL, Murphy PM, Easton AJ, Rosenberg HF. MIP-1alpha is produced but it does not control pulmonary inflammation in response to respiratory syncytial virus infection in mice. Cell Immunol. 2000;206:1–6. doi: 10.1006/cimm.2000.1730. [DOI] [PubMed] [Google Scholar]
- 222.Johnson CH, Miao C, Blanchard EG, et al. Effect of chemokine receptor CX3CR1 deficiency in a murine model of respiratory syncytial virus infection. Comp Med. 2012;62:14–20. [PMC free article] [PubMed] [Google Scholar]
- 223.Tekkanat KK, Maassab H, Miller A, Berlin AA, Kunkel SL, Lukacs NW. RANTES (CCL5) production during primary respiratory syncytial virus infection exacerbates airway disease. Eur J Immunol. 2002;32:3276–3284. doi: 10.1002/1521-4141(200211)32:11<3276::AID-IMMU3276>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 224.Miller AL, Strieter RM, Gruber AD, Ho SB, Lukacs NW. CXCR2 regulates respiratory syncytial virus-induced airway hyperreactivity and mucus overproduction. J Immunol. 2003;170:3348–3356. doi: 10.4049/jimmunol.170.6.3348. [DOI] [PubMed] [Google Scholar]
- 225.Graham BS, Bunton LA, Wright PF, Karzon DT. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin Invest. 1991;88:1026–1033. doi: 10.1172/JCI115362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Alwan WH, Record FM, Openshaw PJ. CD4+ T cells clear virus but augment disease in mice infected with respiratory syncytial virus. Comparison with the effects of CD8+ T cells. Clin Exp Immunol. 1992;88:527–536. doi: 10.1111/j.1365-2249.1992.tb06482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Cannon MJ, Openshaw PJ, Askonas BA. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med. 1988;168:1163–1168. doi: 10.1084/jem.168.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Aung S, Rutigliano JA, Graham BS. Alternative mechanisms of respiratory syncytial virus clearance in perforin knockout mice lead to enhanced disease. J Virol. 2001;75:9918–9924. doi: 10.1128/JVI.75.20.9918-9924.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol. 2006;177:6263–6270. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- 230.Guerrero-Plata A, Kolli D, Hong C, Casola A, Garofalo RP. Subversion of pulmonary dendritic cell function by paramyxovirus infections. J Immunol. 2009;182:3072–3083. doi: 10.4049/jimmunol.0802262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med. 2006;203:1153–1159. doi: 10.1084/jem.20052359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Beyer M, Bartz H, Horner K, Doths S, Koerner-Rettberg C, Schwarze J. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J Allergy Clin Immunol. 2004;113:127–133. doi: 10.1016/j.jaci.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 233.Guerrero-Plata A, Casola A, Garofalo RP. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J Virol. 2005;79:14992–14997. doi: 10.1128/JVI.79.23.14992-14997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Guerrero-Plata A, Baron S, Poast JS, Adegboyega PA, Casola A, Garofalo RP. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J Virol. 2005;79:10190–10199. doi: 10.1128/JVI.79.16.10190-10199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.van Schaik SM, Obot N, Enhorning G, et al. Role of interferon gamma in the pathogenesis of primary respiratory syncytial virus infection in BALB/c mice. J Med Virol. 2000;62:257–266. doi: 10.1002/1096-9071(200010)62:2<257::aid-jmv19>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 236.Peebles RS, Jr, Sheller JR, Collins RD, et al. Respiratory syncytial virus infection does not increase allergen-induced type 2 cytokine production, yet increases airway hyperresponsiveness in mice. J Med Virol. 2001;63:178–188. [PubMed] [Google Scholar]
- 237.Lee YM, Miyahara N, Takeda K, et al. IFN-gamma production during initial infection determines the outcome of reinfection with respiratory syncytial virus. Am J Respir Crit Care Med. 2008;177:208–218. doi: 10.1164/rccm.200612-1890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Tekkanat KK, Maassab H, Berlin AA, et al. Role of interleukin-12 and stat-4 in the regulation of airway inflammation and hyperreactivity in respiratory syncytial virus infection. Am J Pathol. 2001;159:631–638. doi: 10.1016/S0002-9440(10)61734-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Schwarze J, Cieslewicz G, Joetham A, Ikemura T, Hamelmann E, Gelfand EW. CD8 T cells are essential in the development of respiratory syncytial virus-induced lung eosinophilia and airway hyperresponsiveness. J Immunol. 1999;162:4207–4211. [PubMed] [Google Scholar]
- 240.Spender LC, Hussell T, Openshaw PJ. Abundant IFN-gamma production by local T cells in respiratory syncytial virus-induced eosinophilic lung disease. J Gen Virol. 1998;79(Pt 7):1751–1758. doi: 10.1099/0022-1317-79-7-1751. [DOI] [PubMed] [Google Scholar]
- 241.Matsuse H, Behera AK, Kumar M, Rabb H, Lockey RF, Mohapatra SS. Recurrent respiratory syncytial virus infections in allergen-sensitized mice lead to persistent airway inflammation and hyperresponsiveness. J Immunol. 2000;164:6583–6592. doi: 10.4049/jimmunol.164.12.6583. [DOI] [PubMed] [Google Scholar]
- 242.Wang SZ, Bao YX, Rosenberger CL, Tesfaigzi Y, Stark JM, Harrod KS. IL-12p40 and IL-18 modulate inflammatory and immune responses to respiratory syncytial virus infection. J Immunol. 2004;173:4040–4049. doi: 10.4049/jimmunol.173.6.4040. [DOI] [PubMed] [Google Scholar]
- 243.Ehl S, Bischoff R, Ostler T, et al. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur J Immunol. 2004;34:1146–1153. doi: 10.1002/eji.200324449. [DOI] [PubMed] [Google Scholar]
- 244.Dakhama A, Park JW, Taube C, et al. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J Immunol. 2005;175:1876–1883. doi: 10.4049/jimmunol.175.3.1876. [DOI] [PubMed] [Google Scholar]
- 245.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev. 2004;202:175–190. doi: 10.1111/j.0105-2896.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 246.Tekkanat KK, Maassab HF, Cho DS, et al. IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J Immunol. 2001;166:3542–3548. doi: 10.4049/jimmunol.166.5.3542. [DOI] [PubMed] [Google Scholar]
- 247.Peebles RS, Jr, Hashimoto K, Collins RD, et al. Immune interaction between respiratory syncytial virus infection and allergen sensitization critically depends on timing of challenges. J Infect Dis. 2001;184:1374–1379. doi: 10.1086/324429. [DOI] [PubMed] [Google Scholar]
- 248.Sun J, Cardani A, Sharma AK, et al. Autocrine regulation of pulmonary inflammation by effector T-cell derived IL-10 during infection with respiratory syncytial virus. PLoS Pathog. 2011;7:e1002173. doi: 10.1371/journal.ppat.1002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ruan Y, Okamoto Y, Matsuzaki Z, et al. Suppressive effect of locally produced interleukin-10 on respiratory syncytial virus infection. Immunology. 2001;104:355–360. doi: 10.1046/j.1365-2567.2001.01318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology. 2011;134:8–16. doi: 10.1111/j.1365-2567.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Mukherjee S, Lindell DM, Berlin AA, et al. IL-17-induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. Am J Pathol. 2011;179:248–258. doi: 10.1016/j.ajpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Haynes LM, Tonkin J, Anderson LJ, Tripp RA. Neutralizing anti-F glycoprotein and anti-substance P antibody treatment effectively reduces infection and inflammation associated with respiratory syncytial virus infection. J Virol. 2002;76:6873–6881. doi: 10.1128/JVI.76.14.6873-6881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Piedimonte G. Contribution of neuroimmune mechanisms to airway inflammation and remodeling during and after respiratory syncytial virus infection. Pediatr Infect Dis J. 2003;22:S66–S74. doi: 10.1097/01.inf.0000053888.67311.1d. [DOI] [PubMed] [Google Scholar]
- 254.Kamphuis T, Meijerhof T, Stegmann T, Lederhofer J, Wilschut J, de Haan A. Immunogenicity and protective capacity of a virosomal respiratory syncytial virus vaccine adjuvanted with monophosphoryl lipid A in mice. PLoS One. 2012;7:e36812. doi: 10.1371/journal.pone.0036812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Singleton R, Etchart N, Hou S, Hyland L. Inability to evoke a long-lasting protective immune response to respiratory syncytial virus infection in mice correlates with ineffective nasal antibody responses. J Virol. 2003;77:11303–11311. doi: 10.1128/JVI.77.21.11303-11311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Graham BS, Bunton LA, Rowland J, Wright PF, Karzon DT. Respiratory syncytial virus infection in anti-mu-treated mice. J Virol. 1991;65:4936–4942. doi: 10.1128/jvi.65.9.4936-4942.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Crowe JE, Jr, Firestone CY, Murphy BR. Passively acquired antibodies suppress humoral but not cell-mediated immunity in mice immunized with live attenuated respiratory syncytial virus vaccines. J Immunol. 2001;167:3910–3918. doi: 10.4049/jimmunol.167.7.3910. [DOI] [PubMed] [Google Scholar]
- 258.Delgado MF, Coviello S, Monsalvo AC, et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. 2009;15:34–41. doi: 10.1038/nm.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Dakhama A, Park JW, Taube C, et al. The role of virus-specific immunoglobulin E in airway hyperresponsiveness. Am J Respir Crit Care Med. 2004;170:952–959. doi: 10.1164/rccm.200311-1610OC. [DOI] [PubMed] [Google Scholar]
- 260.Gershwin LJ, Gunther RA, Anderson ML, et al. Bovine respiratory syncytial virus-specific IgE is associated with interleukin-2 and -4, and interferon-gamma expression in pulmonary lymph of experimentally infected calves. Am J Vet Res. 2000;61:291–298. doi: 10.2460/ajvr.2000.61.291. [DOI] [PubMed] [Google Scholar]
- 261.Dakhama A, Vitalis TZ, Hegele RG. Persistence of respiratory syncytial virus (RSV) infection and development of RSV-specific IgG1 response in a guinea-pig model of acute bronchiolitis. Eur Respir J. 1997;10:20–26. doi: 10.1183/09031936.97.10010020. [DOI] [PubMed] [Google Scholar]
- 262.Dakhama A, Lee YM, Ohnishi H, et al. Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J Allergy Clin Immunol. 2009;123(138–145):e135. doi: 10.1016/j.jaci.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 263.McNamara PS, Flanagan BF, Hart CA, Smyth RL. Production of chemokines in the lungs of infants with severe respiratory syncytial virus bronchiolitis. J Infect Dis. 2005;191:1225–1232. doi: 10.1086/428855. [DOI] [PubMed] [Google Scholar]
- 264.Harrison AM, Bonville CA, Rosenberg HF, Domachowske JB. Respiratory syncytical virus-induced chemokine expression in the lower airways: eosinophil recruitment and degranulation. Am J Respir Crit Care Med. 1999;159:1918–1924. doi: 10.1164/ajrccm.159.6.9805083. [DOI] [PubMed] [Google Scholar]
- 265.Gill MA, Long K, Kwon T, et al. Differential recruitment of dendritic cells and monocytes to respiratory mucosal sites in children with influenza virus or respiratory syncytial virus infection. J Infect Dis. 2008;198:1667–1676. doi: 10.1086/593018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bermejo-Martin JF, Garcia-Arevalo MC, De Lejarazu RO, et al. Predominance of Th2 cytokines, CXC chemokines and innate immunity mediators at the mucosal level during severe respiratory syncytial virus infection in children. Eur Cytokine Netw. 2007;18:162–167. doi: 10.1684/ecn.2007.0096. [DOI] [PubMed] [Google Scholar]
- 267.Garofalo RP, Patti J, Hintz KA, Hill V, Ogra PL, Welliver RC. Macrophage inflammatory protein-1alpha (not T helper type 2 cytokines) is associated with severe forms of respiratory syncytial virus bronchiolitis. J Infect Dis. 2001;184:393–399. doi: 10.1086/322788. [DOI] [PubMed] [Google Scholar]
- 268.Garofalo RP, Hintz KH, Hill V, Patti J, Ogra PL, Welliver RC., Sr A comparison of epidemiologic and immunologic features of bronchiolitis caused by influenza virus and respiratory syncytial virus. J Med Virol. 2005;75:282–289. doi: 10.1002/jmv.20268. [DOI] [PubMed] [Google Scholar]
- 269.Sheeran P, Jafri H, Carubelli C, et al. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr Infect Dis J. 1999;18:115–122. doi: 10.1097/00006454-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 270.Abu-Harb M, Bell F, Finn A, et al. IL-8 and neutrophil elastase levels in the respiratory tract of infants with RSV bronchiolitis. Eur Respir J. 1999;14:139–143. doi: 10.1034/j.1399-3003.1999.14a23.x. [DOI] [PubMed] [Google Scholar]
- 271.Gern JE, Martin MS, Anklam KA, et al. Relationships among specific viral pathogens, virus-induced interleukin-8, and respiratory symptoms in infancy. Pediatr Allergy Immunol. 2002;13:386–393. doi: 10.1034/j.1399-3038.2002.01093.x. [DOI] [PubMed] [Google Scholar]
- 272.Murai H, Terada A, Mizuno M, et al. IL-10 and RANTES are elevated in nasopharyngeal secretions of children with respiratory syncytial virus infection. Allergol Int. 2007;56:157–163. doi: 10.2332/allergolint.O-06-454. [DOI] [PubMed] [Google Scholar]
- 273.DeVincenzo JP, Wilkinson T, Vaishnaw A, et al. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182:1305–1314. doi: 10.1164/rccm.201002-0221OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Garcia C, Soriano-Fallas A, Lozano J, et al. Decreased innate immune cytokine responses correlate with disease severity in children with respiratory syncytial virus and human rhinovirus bronchiolitis. Pediatr Infect Dis J. 2012;31:86–89. doi: 10.1097/INF.0b013e31822dc8c1. [DOI] [PubMed] [Google Scholar]
- 275.Bennett BL, Garofalo RP, Cron SG, et al. Immunopathogenesis of respiratory syncytial virus bronchiolitis. J Infect Dis. 2007;195:1532–1540. doi: 10.1086/515575. [DOI] [PubMed] [Google Scholar]
- 276.McNamara PS, Ritson P, Selby A, Hart CA, Smyth RL. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch Dis Child. 2003;88:922–926. doi: 10.1136/adc.88.10.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Matsuda K, Tsutsumi H, Okamoto Y, Chiba C. Development of interleukin 6 and tumor necrosis factor alpha activity in nasopharyngeal secretions of infants and children during infection with respiratory syncytial virus. Clin Diagn Lab Immunol. 1995;2:322–324. doi: 10.1128/cdli.2.3.322-324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.van Benten IJ, van Drunen CM, Koopman LP, et al. RSV-induced bronchiolitis but not upper respiratory tract infection is accompanied by an increased nasal IL-18 response. J Med Virol. 2003;71:290–297. doi: 10.1002/jmv.10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Vieira RA, Diniz EM, Ceccon ME. Correlation between inflammatory mediators in the nasopharyngeal secretion and in the serum of children with lower respiratory tract infection caused by respiratory syncytial virus and disease severity. J Bras Pneumol. 2010;36:59–66. doi: 10.1590/s1806-37132010000100011. [DOI] [PubMed] [Google Scholar]
- 280.Brandenburg AH, Kleinjan A, van Het Land B, et al. Type 1-like immune response is found in children with respiratory syncytial virus infection regardless of clinical severity. J Med Virol. 2000;62:267–277. [PubMed] [Google Scholar]
- 281.Everard ML, Swarbrick A, Wrightham M, et al. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child. 1994;71:428–432. doi: 10.1136/adc.71.5.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Heidema J, Lukens MV, van Maren WW, et al. CD8+ T cell responses in bronchoalveolar lavage fluid and peripheral blood mononuclear cells of infants with severe primary respiratory syncytial virus infections. J Immunol. 2007;179:8410–8417. doi: 10.4049/jimmunol.179.12.8410. [DOI] [PubMed] [Google Scholar]
- 283.Kim CK, Kim SW, Park CS, Kim BI, Kang H, Koh YY. Bronchoalveolar lavage cytokine profiles in acute asthma and acute bronchiolitis. J Allergy Clin Immunol. 2003;112:64–71. doi: 10.1067/mai.2003.1618. [DOI] [PubMed] [Google Scholar]
- 284.Kim CK, Koh JY, Han TH, Kim do K, Kim BI, Koh YY. Increased levels of BAL cysteinyl leukotrienes in acute [corrected] RSV bronchiolitis. Acta Paediatr. 2006;95:479–485. doi: 10.1080/08035250600554268. [DOI] [PubMed] [Google Scholar]
- 285.Bem RA, Bos AP, Bots M, et al. Activation of the granzyme pathway in children with severe respiratory syncytial virus infection. Pediatr Res. 2008;63:650–655. doi: 10.1203/PDR.0b013e31816fdc32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Smith PK, Wang SZ, Dowling KD, Forsyth KD. Leucocyte populations in respiratory syncytial virus-induced bronchiolitis. J Paediatr Child Health. 2001;37:146–151. doi: 10.1046/j.1440-1754.2001.00618.x. [DOI] [PubMed] [Google Scholar]
- 287.Gill MA, Palucka AK, Barton T, et al. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J Infect Dis. 2005;191:1105–1115. doi: 10.1086/428589. [DOI] [PubMed] [Google Scholar]
- 288.Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. Pathological changes in virus infections of the lower respiratory tract in children. J Clin Pathol. 1970;23:7–18. doi: 10.1136/jcp.23.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Reed JL, Brewah YA, Delaney T, et al. Macrophage impairment underlies airway occlusion in primary respiratory syncytial virus bronchiolitis. J Infect Dis. 2008;198:1783–1793. doi: 10.1086/593173. [DOI] [PubMed] [Google Scholar]
- 290.Welliver TP, Reed JL, Welliver RC., Sr Respiratory syncytial virus and influenza virus infections: observations from tissues of fatal infant cases. Pediatr Infect Dis J. 2008;27:S92–S96. doi: 10.1097/INF.0b013e318168b706. [DOI] [PubMed] [Google Scholar]
- 291.Welliver TP, Garofalo RP, Hosakote Y, et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195:1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kim HW, Canchola JG, Brandt CD, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89:422–434. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 293.Prince GA, Curtis SJ, Yim KC, Porter DD. Vaccine-enhanced respiratory syncytial virus disease in cotton rats following immunization with Lot 100 or a newly prepared reference vaccine. J Gen Virol. 2001;82:2881–2888. doi: 10.1099/0022-1317-82-12-2881. [DOI] [PubMed] [Google Scholar]
- 294.Lukens MV, van de Pol AC, Coenjaerts FE, et al. A systemic neutrophil response precedes robust CD8+ T-cell activation during natural respiratory syncytial virus infection in infants. J Virol. 2010;84:2374–2383. doi: 10.1128/JVI.01807-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Chiba Y, Higashidate Y, Suga K, Honjo K, Tsutsumi H, Ogra PL. Development of cell-mediated cytotoxic immunity to respiratory syncytial virus in human infants following naturally acquired infection. J Med Virol. 1989;28:133–139. doi: 10.1002/jmv.1890280304. [DOI] [PubMed] [Google Scholar]
- 296.Aberle JH, Aberle SW, Rebhandl W, Pracher E, Kundi M, Popow-Kraupp T. Decreased interferon-gamma response in respiratory syncytial virus compared to other respiratory viral infections in infants. Clin Exp Immunol. 2004;137:146–150. doi: 10.1111/j.1365-2249.2004.02504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Román M, Calhoun WJ, Hinton KL, et al. Respiratory syncytial virus infection in infants is associated with predominant Th-2-like response. Am J Respir Crit Care Med. 1997;156:190–195. doi: 10.1164/ajrccm.156.1.9611050. [DOI] [PubMed] [Google Scholar]
- 298.Bendelja K, Gagro A, Bace A, et al. Predominant type-2 response in infants with respiratory syncytial virus (RSV) infection demonstrated by cytokine flow cytometry. Clin Exp Immunol. 2000;121:332–338. doi: 10.1046/j.1365-2249.2000.01297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Tripp RA, Moore D, Barskey A, IV, et al. Peripheral blood mononuclear cells from infants hospitalized because of respiratory syncytial virus infection express T helper-1 and T helper-2 cytokines and CC chemokine messenger RNA. J Infect Dis. 2002;185:1388–1394. doi: 10.1086/340505. [DOI] [PubMed] [Google Scholar]
- 300.Lee FE, Walsh EE, Falsey AR, et al. Human infant respiratory syncytial virus (RSV)-specific type 1 and 2 cytokine responses ex vivo during primary RSV infection. J Infect Dis. 2007;195:1779–1788. doi: 10.1086/518249. [DOI] [PubMed] [Google Scholar]
- 301.Douville RN, Bastien N, Li Y, Pochard P, Simons FE, HayGlass KT. Human metapneumovirus elicits weak IFN-gamma memory responses compared with respiratory syncytial virus. J Immunol. 2006;176:5848–5855. doi: 10.4049/jimmunol.176.10.5848. [DOI] [PubMed] [Google Scholar]
- 302.O’Donnell DR, McGarvey MJ, Tully JM, Balfour-Lynn IM, Openshaw PJ. Respiratory syncytial virus RNA in cells from the peripheral blood during acute infection. J Pediatr. 1998;133:272–274. doi: 10.1016/s0022-3476(98)70234-3. [DOI] [PubMed] [Google Scholar]
- 303.Halfhide CP, Flanagan BF, Brearey SP, et al. Respiratory syncytial virus binds and undergoes transcription in neutrophils from the blood and airways of infants with severe bronchiolitis. J Infect Dis. 2011;204:451–458. doi: 10.1093/infdis/jir280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Heidema J, Rossen JW, Lukens MV, et al. Dynamics of human respiratory virus-specific CD8+ T cell responses in blood and airways during episodes of common cold. J Immunol. 2008;181:5551–5559. doi: 10.4049/jimmunol.181.8.5551. [DOI] [PubMed] [Google Scholar]
- 305.Kim CK, Callaway Z, Koh YY, Kim SH, Fujisawa T. Airway IFN-gamma production during RSV bronchiolitis is associated with eosinophilic inflammation. Lung. 2012;190:183–188. doi: 10.1007/s00408-011-9349-5. [DOI] [PubMed] [Google Scholar]
- 306.Kristjansson S, Bjarnarson SP, Wennergren G, et al. Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local TH2-like response. J Allergy Clin Immunol. 2005;116:805–811. doi: 10.1016/j.jaci.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 307.Garofalo RP, Hintz KH, Hill V, Ogra PL, Welliver RC., Sr Production of interferon gamma in respiratory syncytial virus infection of humans is not associated with interleukins 12 and 18. J Med Virol. 2004;73:289–294. doi: 10.1002/jmv.20089. [DOI] [PubMed] [Google Scholar]
- 308.van Schaik SM, Tristram DA, Nagpal IS, Hintz KM, Welliver RC, 2nd, Welliver RC. Increased production of IFN-gamma and cysteinyl leukotrienes in virus-induced wheezing. J Allergy Clin Immunol. 1999;103:630–636. doi: 10.1016/s0091-6749(99)70235-6. [DOI] [PubMed] [Google Scholar]
- 309.Bont L, Heijnen CJ, Kavelaars A, et al. Local interferon-gamma levels during respiratory syncytial virus lower respiratory tract infection are associated with disease severity. J Infect Dis. 2001;184:355–358. doi: 10.1086/322035. [DOI] [PubMed] [Google Scholar]
- 310.Mobbs KJ, Smyth RL, O’Hea U, Ashby D, Ritson P, Hart CA. Cytokines in severe respiratory syncytial virus bronchiolitis. Pediatr Pulmonol. 2002;33:449–452. doi: 10.1002/ppul.10101. [DOI] [PubMed] [Google Scholar]
- 311.Buck RH, Cordle CT, Thomas DJ, Winship TR, Schaller JP, Dugle JE. Longitudinal study of intracellular T cell cytokine production in infants compared to adults. Clin Exp Immunol. 2002;128:490–497. doi: 10.1046/j.1365-2249.2002.01851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Motegi A, Kinoshita M, Sato K, et al. An in vitro Shwartzman reaction-like response is augmented age-dependently in human peripheral blood mononuclear cells. J Leukoc Biol. 2006;79:463–472. doi: 10.1189/jlb.0705396. [DOI] [PubMed] [Google Scholar]
- 313.Yerkovich ST, Wikström ME, Suriyaarachchi D, Prescott SL, Upham JW, Holt PG. Postnatal development of monocyte cytokine responses to bacterial lipopolysaccharide. Pediatr Res. 2007;62:547–552. doi: 10.1203/PDR.0b013e3181568105. [DOI] [PubMed] [Google Scholar]
- 314.Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med. 2002;196:1381–1386. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Chung HL, Kim WT, Kim JK, et al. Relationship between atopic status and nasal interleukin 10 and 11 levels in infants with respiratory syncytial virus bronchiolitis. Ann Allergy Asthma Immunol. 2005;94:267–272. doi: 10.1016/S1081-1206(10)61307-5. [DOI] [PubMed] [Google Scholar]
- 316.Joshi P, Shaw A, Kakakios A, Isaacs D. Interferon-gamma levels in nasopharyngeal secretions of infants with respiratory syncytial virus and other respiratory viral infections. Clin Exp Immunol. 2003;131:143–147. doi: 10.1046/j.1365-2249.2003.02039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Dimova-Yaneva D, Russell D, Main M, Brooker RJ, Helms PJ. Eosinophil activation and cysteinyl leukotriene production in infants with respiratory syncytial virus bronchiolitis. Clin Exp Allergy. 2004;34:555–558. doi: 10.1111/j.1365-2222.2004.1918.x. [DOI] [PubMed] [Google Scholar]
- 318.Garofalo R, Kimpen JL, Welliver RC, Ogra PL. Eosinophil degranulation in the respiratory tract during naturally acquired respiratory syncytial virus infection. J Pediatr. 1992;120:28–32. doi: 10.1016/s0022-3476(05)80592-x. [DOI] [PubMed] [Google Scholar]
- 319.Rakes GP, Arruda E, Ingram JM, et al. Rhinovirus and respiratory syncytial virus in wheezing children requiring emergency care. IgE and eosinophil analyses. Am J Respir Crit Care Med. 1999;159:785–790. doi: 10.1164/ajrccm.159.3.9801052. [DOI] [PubMed] [Google Scholar]
- 320.Øymar K, Elsayed S, Bjerknes R. Serum eosinophil cationic protein and interleukin-5 in children with bronchial asthma and acute bronchiolitis. Pediatr Allergy Immunol. 1996;7:180–186. doi: 10.1111/j.1399-3038.1996.tb00130.x. [DOI] [PubMed] [Google Scholar]
- 321.Renzi PM, Turgeon JP, Yang JP, et al. Cellular immunity is activated and a TH-2 response is associated with early wheezing in infants after bronchiolitis. J Pediatr. 1997;130:584–593. doi: 10.1016/s0022-3476(97)70243-9. [DOI] [PubMed] [Google Scholar]
- 322.Ehlenfield DR, Cameron K, Welliver RC. Eosinophilia at the time of respiratory syncytial virus bronchiolitis predicts childhood reactive airway disease. Pediatrics. 2000;105:79–83. doi: 10.1542/peds.105.1.79. [DOI] [PubMed] [Google Scholar]
- 323.Kim CK, Choi J, Callaway Z, et al. Clinical and epidemiological comparison of human metapneumovirus and respiratory syncytial virus in Seoul, Korea, 2003–2008. J Korean Med Sci. 2010;25:342–347. doi: 10.3346/jkms.2010.25.3.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Monteseirin J, Vega A, Chacon P, et al. Neutrophils as a novel source of eosinophil cationic protein in IgE-mediated processes. J Immunol. 2007;179:2634–2641. doi: 10.4049/jimmunol.179.4.2634. [DOI] [PubMed] [Google Scholar]
- 325.Welliver RC, Wong DT, Sun M, Middleton E, Jr, Vaughan RS, Ogra PL. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305:841–846. doi: 10.1056/NEJM198110083051501. [DOI] [PubMed] [Google Scholar]
- 326.Russi JC, Delfraro A, Borthagaray MD, Velazquez B, Garcia-Barreno B, Hortal M. Evaluation of immunoglobulin E-specific antibodies and viral antigens in nasopharyngeal secretions of children with respiratory syncytial virus infections. J Clin Microbiol. 1993;31:819–823. doi: 10.1128/jcm.31.4.819-823.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Rabatic S, Gagro A, Lokar-Kolbas R, et al. Increase in CD23+ B cells in infants with bronchiolitis is accompanied by appearance of IgE and IgG4 antibodies specific for respiratory syncytial virus. J Infect Dis. 1997;175:32–37. doi: 10.1093/infdis/175.1.32. [DOI] [PubMed] [Google Scholar]
- 328.De Alarcon A, Walsh EE, Carper HT, et al. Detection of IgA and IgG but not IgE antibody to respiratory syncytial virus in nasal washes and sera from infants with wheezing. J Pediatr. 2001;138:311–317. doi: 10.1067/mpd.2001.111277. [DOI] [PubMed] [Google Scholar]
- 329.Tortorolo L, Langer A, Polidori G, et al. Neurotrophin overexpression in lower airways of infants with respiratory syncytial virus infection. Am J Respir Crit Care Med. 2005;172:233–237. doi: 10.1164/rccm.200412-1693OC. [DOI] [PubMed] [Google Scholar]
- 330.Meurman O, Waris M, Hedman K. Immunoglobulin G antibody avidity in patients with respiratory syncytial virus infection. J Clin Microbiol. 1992;30:1479–1484. doi: 10.1128/jcm.30.6.1479-1484.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Murphy BR, Alling DW, Snyder MH, et al. Effect of age and preexisting antibody on serum antibody response of infants and children to the F and G glycoproteins during respiratory syncytial virus infection. J Clin Microbiol. 1986;24:894–898. doi: 10.1128/jcm.24.5.894-898.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Wilczynski J, Lukasik B, Torbicka E, Tranda I, Brzozowska-Binda A. The immune response of small children by antibodies of different classes to respiratory syncytial virus (RSV) proteins. Acta Microbiol Pol. 1994;43:369–379. [PubMed] [Google Scholar]
- 333.Popow-Kraupp T, Lakits E, Kellner G, Kunz C. Immunoglobulin-class-specific immune response to respiratory syncytial virus structural proteins in infants, children, and adults. J Med Virol. 1989;27:215–223. doi: 10.1002/jmv.1890270307. [DOI] [PubMed] [Google Scholar]
- 334.Murphy BR, Graham BS, Prince GA, et al. Serum and nasal-wash immunoglobulin G and A antibody response of infants and children to respiratory syncytial virus F and G glycoproteins following primary infection. J Clin Microbiol. 1986;23:1009–1014. doi: 10.1128/jcm.23.6.1009-1014.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. 1991;163:693–698. doi: 10.1093/infdis/163.4.693. [DOI] [PubMed] [Google Scholar]
- 336.Walsh EE, Falsey AR. Humoral and mucosal immunity in protection from natural respiratory syncytial virus infection in adults. J Infect Dis. 2004;190:373–378. doi: 10.1086/421524. [DOI] [PubMed] [Google Scholar]
- 337.Williams JV, Weitkamp JH, Blum DL, LaFleur BJ, Crowe JE., Jr The human neonatal B cell response to respiratory syncytial virus uses a biased antibody variable gene repertoire that lacks somatic mutations. Mol Immunol. 2009;47:407–414. doi: 10.1016/j.molimm.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Reed JL, Welliver TP, Sims GP, et al. Innate immune signals modulate antiviral and polyreactive antibody responses during severe respiratory syncytial virus infection. J Infect Dis. 2009;199:1128–1138. doi: 10.1086/597386. [DOI] [PubMed] [Google Scholar]
- 339.Welliver RC, Sun M, Hildreth SW, Arumugham R, Ogra PL. Respiratory syncytial virus-specific antibody responses in immunoglobulin A and E isotypes to the F and G proteins and to intact virus after natural infection. J Clin Microbiol. 1989;27:295–299. doi: 10.1128/jcm.27.2.295-299.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Aberle JH, Aberle SW, Dworzak MN, et al. Reduced interferon-gamma expression in peripheral blood mononuclear cells of infants with severe respiratory syncytial virus disease. Am J Respir Crit Care Med. 1999;160:1263–1268. doi: 10.1164/ajrccm.160.4.9812025. [DOI] [PubMed] [Google Scholar]
- 341.Bui RH, Molinaro GA, Kettering JD, Heiner DC, Imagawa DT, St Geme JW., Jr Virus-specific IgE and IgG4 antibodies in serum of children infected with respiratory syncytial virus. J Pediatr. 1987;110:87–90. doi: 10.1016/s0022-3476(87)80295-0. [DOI] [PubMed] [Google Scholar]
- 342.Thomsen SF, Stensballe LG, Skytthe A, Kyvik KO, Backer V, Bisgaard H. Increased concordance of severe respiratory syncytial virus infection in identical twins. Pediatrics. 2008;121:493–496. doi: 10.1542/peds.2007-1889. [DOI] [PubMed] [Google Scholar]
- 343.Venter M, Collinson M, Schoub BD. Molecular epidemiological analysis of community circulating respiratory syncytial virus in rural South Africa: Comparison of viruses and genotypes responsible for different disease manifestations. J Med Virol. 2002;68:452–461. doi: 10.1002/jmv.10225. [DOI] [PubMed] [Google Scholar]
- 344.Tregoning JS, Yamaguchi Y, Wang B, et al. Genetic susceptibility to the delayed sequelae of neonatal respiratory syncytial virus infection is MHC dependent. J Immunol. 2010;185:5384–5391. doi: 10.4049/jimmunol.1001594. [DOI] [PubMed] [Google Scholar]
- 345.Janssen R, Bont L, Siezen CL, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J Infect Dis. 2007;196:826–834. doi: 10.1086/520886. [DOI] [PubMed] [Google Scholar]
- 346.Siezen CL, Bont L, Hodemaekers HM, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis in preterm children is associated with airway remodeling genes and innate immune genes. Pediatr Infect Dis J. 2009;28:333–335. doi: 10.1097/INF.0b013e31818e2aa9. [DOI] [PubMed] [Google Scholar]
- 347.Kresfelder TL, Janssen R, Bont L, Pretorius M, Venter M. Confirmation of an association between single nucleotide polymorphisms in the VDR gene with respiratory syncytial virus related disease in South African children. J Med Virol. 2011;83:1834–1840. doi: 10.1002/jmv.22179. [DOI] [PubMed] [Google Scholar]
- 348.Marchant D, Singhera GK, Utokaparch S, et al. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J Virol. 2010;84:11359–11373. doi: 10.1128/JVI.00804-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 350.Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. 2001;75:10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Haeberle HA, Takizawa R, Casola A, et al. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis. 2002;186:1199–1206. doi: 10.1086/344644. [DOI] [PubMed] [Google Scholar]
- 352.Gagro A, Tominac M, Krsulovic-Hresic V, et al. Increased Toll-like receptor 4 expression in infants with respiratory syncytial virus bronchiolitis. Clin Exp Immunol. 2004;135:267–272. doi: 10.1111/j.1365-2249.2004.02364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Halfhide CP, Brearey SP, Flanagan BF, et al. Neutrophil TLR4 expression is reduced in the airways of infants with severe bronchiolitis. Thorax. 2009;64:798–805. doi: 10.1136/thx.2008.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Tulic MK, Hurrelbrink RJ, Prele CM, et al. TLR4 polymorphisms mediate impaired responses to respiratory syncytial virus and lipopolysaccharide. J Immunol. 2007;179:132–140. doi: 10.4049/jimmunol.179.1.132. [DOI] [PubMed] [Google Scholar]
- 355.Douville RN, Lissitsyn Y, Hirschfeld AF, et al. TLR4 Asp299Gly and Thr399Ile polymorphisms: no impact on human immune responsiveness to LPS or respiratory syncytial virus. PLoS One. 2010;5:e12087. doi: 10.1371/journal.pone.0012087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Awomoyi AA, Rallabhandi P, Pollin TI, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. 2007;179:3171–3177. doi: 10.4049/jimmunol.179.5.3171. [DOI] [PubMed] [Google Scholar]
- 357.Tal G, Mandelberg A, Dalal I, et al. Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis. 2004;189:2057–2063. doi: 10.1086/420830. [DOI] [PubMed] [Google Scholar]
- 358.Inoue Y, Shimojo N, Suzuki Y, et al. CD14–550 C/T, which is related to the serum level of soluble CD14, is associated with the development of respiratory syncytial virus bronchiolitis in the Japanese population. J Infect Dis. 2007;195:1618–1624. doi: 10.1086/516790. [DOI] [PubMed] [Google Scholar]
- 359.Puthothu B, Forster J, Heinzmann A, Krueger M. TLR-4 and CD14 polymorphisms in respiratory syncytial virus associated disease. Dis Markers. 2006;22:303–308. doi: 10.1155/2006/865890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Löfgren J, Marttila R, Renko M, Rämet M, Hallman M. Toll-like receptor 4 Asp299Gly polymorphism in respiratory syncytial virus epidemics. Pediatr Pulmonol. 2010;45:687–692. doi: 10.1002/ppul.21248. [DOI] [PubMed] [Google Scholar]
- 361.Helminen M, Nuolivirta K, Virta M, et al. IL-10 gene polymorphism at -1082 A/G is associated with severe rhinovirus bronchiolitis in infants. Pediatr Pulmonol. 2008;43:391–395. doi: 10.1002/ppul.20793. [DOI] [PubMed] [Google Scholar]
- 362.Mailaparambil B, Krueger M, Heinze J, Forster J, Heinzmann A. Polymorphisms of toll like receptors in the genetics of severe RSV associated diseases. Dis Markers. 2008;25:59–65. doi: 10.1155/2008/619595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Rämet M, Korppi M, Hallman M. Pattern recognition receptors and genetic risk for RSV infection: value for clinical decision-making? Pediatr Pulmonol. 2011;46:101–110. doi: 10.1002/ppul.21348. [DOI] [PubMed] [Google Scholar]
- 364.Hickling TP, Malhotra R, Bright H, McDowell W, Blair ED, Sim RB. Lung surfactant protein A provides a route of entry for respiratory syncytial virus into host cells. Viral Immunol. 2000;13:125–135. doi: 10.1089/vim.2000.13.125. [DOI] [PubMed] [Google Scholar]
- 365.Ghildyal R, Hartley C, Varrasso A, et al. Surfactant protein A binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J Infect Dis. 1999;180:2009–2013. doi: 10.1086/315134. [DOI] [PubMed] [Google Scholar]
- 366.Barr FE, Pedigo H, Johnson TR, Shepherd VL. Surfactant protein-A enhances uptake of respiratory syncytial virus by monocytes and U937 macrophages. Am J Respir Cell Mol Biol. 2000;23:586–592. doi: 10.1165/ajrcmb.23.5.3771. [DOI] [PubMed] [Google Scholar]
- 367.Kerr MH, Paton JY. Surfactant protein levels in severe respiratory syncytial virus infection. Am J Respir Crit Care Med. 1999;159:1115–1118. doi: 10.1164/ajrccm.159.4.9709065. [DOI] [PubMed] [Google Scholar]
- 368.Jat KR, Chawla D. Surfactant therapy for bronchiolitis in critically ill infants. Cochrane Database Syst Rev. 2012;9:CD009194. doi: 10.1002/14651858.CD009194.pub2. [DOI] [PubMed] [Google Scholar]
- 369.Thomas NJ, DiAngelo S, Hess JC, et al. Transmission of surfactant protein variants and haplotypes in children hospitalized with respiratory syncytial virus. Pediatr Res. 2009;66:70–73. doi: 10.1203/PDR.0b013e3181a1d768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Löfgren J, Rämet M, Renko M, Marttila R, Hallman M. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J Infect Dis. 2002;185:283–289. doi: 10.1086/338473. [DOI] [PubMed] [Google Scholar]
- 371.Ampuero S, Luchsinger V, Tapia L, Palomino MA, Larranaga CE. SP-A1, SP-A2 and SP-D gene polymorphisms in severe acute respiratory syncytial infection in Chilean infants. Infect Genet Evol. 2011;11:1368–1377. doi: 10.1016/j.meegid.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 372.El Saleeby CM, Li R, Somes GW, Dahmer MK, Quasney MW, DeVincenzo JP. Surfactant protein A2 polymorphisms and disease severity in a respiratory syncytial virus-infected population. J Pediatr. 2010;156:409–414. doi: 10.1016/j.jpeds.2009.09.043. [DOI] [PubMed] [Google Scholar]
- 373.Puthothu B, Forster J, Heinze J, Heinzmann A, Krueger M. Surfactant protein B polymorphisms are associated with severe respiratory syncytial virus infection, but not with asthma. BMC Pulm Med. 2007;7:6. doi: 10.1186/1471-2466-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Puthothu B, Krueger M, Heinze J, Forster J, Heinzmann A. Haplotypes of surfactant protein C are associated with common paediatric lung diseases. Pediatr Allergy Immunol. 2006;17:572–577. doi: 10.1111/j.1399-3038.2006.00467.x. [DOI] [PubMed] [Google Scholar]
- 375.Lahti M, Lofgren J, Marttila R, et al. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res. 2002;51:696–699. doi: 10.1203/00006450-200206000-00006. [DOI] [PubMed] [Google Scholar]
- 376.Tian M, Liu F, Wen GY, Shi SY, Chen RH, Zhao DY. Effect of variation in RANTES promoter on serum RANTES levels and risk of recurrent wheezing after RSV bronchiolitis in children from Han, Southern China. Eur J Pediatr. 2009;168:963–967. doi: 10.1007/s00431-008-0870-3. [DOI] [PubMed] [Google Scholar]
- 377.Hattori S, Shimojo N, Mashimo T, et al. Relationship between RANTES polymorphisms and respiratory syncytial virus bronchiolitis in a Japanese infant population. Jpn J Infect Dis. 2011;64:242–245. [PubMed] [Google Scholar]
- 378.Amanatidou V, Sourvinos G, Apostolakis S, et al. RANTES promoter gene polymorphisms and susceptibility to severe respiratory syncytial virus-induced bronchiolitis. Pediatr Infect Dis J. 2008;27:38–42. doi: 10.1097/INF.0b013e31814d4e42. [DOI] [PubMed] [Google Scholar]
- 379.Hull J, Rowlands K, Lockhart E, Moore C, Sharland M, Kwiatkowski D. Variants of the chemokine receptor CCR5 are associated with severe bronchiolitis caused by respiratory syncytial virus. J Infect Dis. 2003;188:904–907. doi: 10.1086/377587. [DOI] [PubMed] [Google Scholar]
- 380.Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. 2000;55:1023–1027. doi: 10.1136/thorax.55.12.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Hull J, Ackerman H, Isles K, et al. Unusual haplotypic structure of IL8, a susceptibility locus for a common respiratory virus. Am J Hum Genet. 2001;69:413–419. doi: 10.1086/321291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Hacking D, Knight JC, Rockett K, et al. Increased in vivo transcription of an IL-8 haplotype associated with respiratory syncytial virus disease-susceptibility. Genes Immun. 2004;5:274–282. doi: 10.1038/sj.gene.6364067. [DOI] [PubMed] [Google Scholar]
- 383.Lu A, Wang L, Zhang X. Haplotype of IL-8–251T and 781C is associated with the susceptibility to respiratory syncytial virus. J Trop Pediatr. 2010;56:242–246. doi: 10.1093/tropej/fmp101. [DOI] [PubMed] [Google Scholar]
- 384.Puthothu B, Krueger M, Heinze J, Forster J, Heinzmann A. Impact of IL8 and IL8-receptor alpha polymorphisms on the genetics of bronchial asthma and severe RSV infections. Clin Mol Allergy. 2006;4:2. doi: 10.1186/1476-7961-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Rosenwasser LJ, Klemm DJ, Dresback JK, et al. Promoter polymorphisms in the chromosome 5 gene cluster in asthma and atopy. Clin Exp Allergy. 1995;25(Suppl 2):74–78. doi: 10.1111/j.1365-2222.1995.tb00428.x. [DOI] [PubMed] [Google Scholar]
- 386.van der Pouw Kraan TC, van Veen A, Boeije LC, et al. An IL-13 promoter polymorphism associated with increased risk of allergic asthma. Genes Immun. 1999;1:61–65. doi: 10.1038/sj.gene.6363630. [DOI] [PubMed] [Google Scholar]
- 387.Forton JT, Rowlands K, Rockett K, et al. Genetic association study for RSV bronchiolitis in infancy at the 5q31 cytokine cluster. Thorax. 2009;64:345–352. doi: 10.1136/thx.2008.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Puthothu B, Krueger M, Forster J, Heinzmann A. Association between severe respiratory syncytial virus infection and IL13/IL4 haplotypes. J Infect Dis. 2006;193:438–441. doi: 10.1086/499316. [DOI] [PubMed] [Google Scholar]
- 389.Choi EH, Lee HJ, Yoo T, Chanock SJ. A common haplotype of interleukin-4 gene IL4 is associated with severe respiratory syncytial virus disease in Korean children. J Infect Dis. 2002;186:1207–1211. doi: 10.1086/344310. [DOI] [PubMed] [Google Scholar]
- 390.Hoebee B, Rietveld E, Bont L, et al. Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor alpha polymorphisms. J Infect Dis. 2003;187:2–11. doi: 10.1086/345859. [DOI] [PubMed] [Google Scholar]
- 391.Hoebee B, Bont L, Rietveld E, et al. Influence of promoter variants of interleukin-10, interleukin-9, and tumor necrosis factor-alpha genes on respiratory syncytial virus bronchiolitis. J Infect Dis. 2004;189:239–247. doi: 10.1086/380908. [DOI] [PubMed] [Google Scholar]
- 392.Schuurhof A, Janssen R, de Groot H, et al. Local interleukin-10 production during respiratory syncytial virus bronchiolitis is associated with post-bronchiolitis wheeze. Respir Res. 2011;12:121. doi: 10.1186/1465-9921-12-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Wilson J, Rowlands K, Rockett K, et al. Genetic variation at the IL10 gene locus is associated with severity of respiratory syncytial virus bronchiolitis. J Infect Dis. 2005;191:1705–1709. doi: 10.1086/429636. [DOI] [PubMed] [Google Scholar]
- 394.Mailaparambil B, Jochum J, Forster J, Heinze J, Krueger M, Heinzmann A. Polymorphisms of interferons and their receptors in the genetics of severe RSV-associated diseases. Arch Virol. 2008;153:2133–2137. doi: 10.1007/s00705-008-0232-5. [DOI] [PubMed] [Google Scholar]
- 395.Gentile DA, Doyle WJ, Zeevi A, et al. Cytokine gene polymorphisms moderate illness severity in infants with respiratory syncytial virus infection. Hum Immunol. 2003;64:338–344. doi: 10.1016/s0198-8859(02)00827-3. [DOI] [PubMed] [Google Scholar]
- 396.Puthothu B, Krueger M, Forster J, Heinze J, Weckmann M, Heinzmann A. Interleukin (IL)-18 polymorphism 133C/G is associated with severe respiratory syncytial virus infection. Pediatr Infect Dis J. 2007;26:1094–1098. doi: 10.1097/INF.0b013e3181453579. [DOI] [PubMed] [Google Scholar]
- 397.Puthothu B, Bierbaum S, Kopp MV, et al. Association of TNF-alpha with severe respiratory syncytial virus infection and bronchial asthma. Pediatr Allergy Immunol. 2009;20:157–163. doi: 10.1111/j.1399-3038.2008.00751.x. [DOI] [PubMed] [Google Scholar]
- 398.Hashimoto K, Katayose M, Sakuma H, et al. Uteroglobulin-related protein 1 and severity of respiratory syncytial virus infection in children admitted to hospital. J Med Virol. 2011;83:1086–1092. doi: 10.1002/jmv.22073. [DOI] [PubMed] [Google Scholar]
- 399.Aurivillius M, Oymar K, Oxelius VA. Immunoglobulin heavy G2 chain (IGHG2) gene restriction in the development of severe respiratory syncytial virus infection. Acta Paediatr. 2005;94:414–418. doi: 10.1111/j.1651-2227.2005.tb01910.x. [DOI] [PubMed] [Google Scholar]
- 400.Hussell T, Georgiou A, Sparer TE, Matthews S, Pala P, Openshaw PJ. Host genetic determinants of vaccine-induced eosinophilia during respiratory syncytial virus infection. J Immunol. 1998;161:6215–6222. [PubMed] [Google Scholar]
- 401.Jessen B, Faller S, Krempl CD, Ehl S. Major histocompatibility complex-dependent cytotoxic T lymphocyte repertoire and functional avidity contribute to strain-specific disease susceptibility after murine respiratory syncytial virus infection. J Virol. 2011;85:10135–10143. doi: 10.1128/JVI.00816-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Stark JM, Barmada MM, Winterberg AV, et al. Genomewide association analysis of respiratory syncytial virus infection in mice. J Virol. 2010;84:2257–2269. doi: 10.1128/JVI.00584-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Overödder H, Navér L. Clara cell protein 10 polymorphism is not associated with severe respiratory syncytial virus infection. Acta Paediatr. 2012;101:34–37. doi: 10.1111/j.1651-2227.2011.02408.x. [DOI] [PubMed] [Google Scholar]
- 404.Henderson J, Hilliard TN, Sherriff A, Stalker D, Al Shammari N, Thomas HM. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr Allergy Immunol. 2005;16:386–392. doi: 10.1111/j.1399-3038.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 405.Fjærli HO, Farstad T, Rød G, Ufert GK, Gulbrandsen P, Nakstad B. Acute bronchiolitis in infancy as risk factor for wheezing and reduced pulmonary function by seven years in Akershus County, Norway. BMC Pediatr. 2005;5:31. doi: 10.1186/1471-2431-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Singleton RJ, Redding GJ, Lewis TC, et al. Sequelae of severe respiratory syncytial virus infection in infancy and early childhood among Alaska Native children. Pediatrics. 2003;112:285–290. doi: 10.1542/peds.112.2.285. [DOI] [PubMed] [Google Scholar]
- 407.Schauer U, Hoffjan S, Bittscheidt J, et al. RSV bronchiolitis and risk of wheeze and allergic sensitisation in the first year of life. Eur Respir J. 2002;20:1277–1283. doi: 10.1183/09031936.02.00019902. [DOI] [PubMed] [Google Scholar]
- 408.Osundwa VM, Dawod ST, Ehlayel M. Recurrent wheezing in children with respiratory syncytial virus (RSV) bronchiolitis in Qatar. Eur J Pediatr. 1993;152:1001–1003. doi: 10.1007/BF01957225. [DOI] [PubMed] [Google Scholar]
- 409.Murray M, Webb MS, O’Callaghan C, Swarbrick AS, Milner AD. Respiratory status and allergy after bronchiolitis. Arch Dis Child. 1992;67:482–487. doi: 10.1136/adc.67.4.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Sims DG, Downham MA, Gardner PS, Webb JK, Weightman D. Study of 8-year-old children with a history of respiratory syncytial virus bronchiolitis in infancy. Br Med J. 1978;1:11–14. doi: 10.1136/bmj.1.6104.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Eriksson M, Bennet R, Nilsson A. Wheezing following lower respiratory tract infections with respiratory syncytial virus and influenza A in infancy. Pediatr Allergy Immunol. 2000;11:193–197. doi: 10.1034/j.1399-3038.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- 412.Kotaniemi-Syrjänen A, Vainionpää R, Reijonen TM, Waris M, Korhonen K, Korppi M. Rhinovirus-induced wheezing in infancy—the first sign of childhood asthma? J Allergy Clin Immunol. 2003;111:66–71. doi: 10.1067/mai.2003.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B, Bjorksten B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics. 1995;95:500–505. [PubMed] [Google Scholar]
- 414.Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am J Respir Crit Care Med. 2000;161:1501–1507. doi: 10.1164/ajrccm.161.5.9906076. [DOI] [PubMed] [Google Scholar]
- 415.Sigurs N, Gustafsson PM, Bjarnason R, et al. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am J Respir Crit Care Med. 2005;171:137–141. doi: 10.1164/rccm.200406-730OC. [DOI] [PubMed] [Google Scholar]
- 416.Sigurs N, Aljassim F, Kjellman B, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65:1045–1052. doi: 10.1136/thx.2009.121582. [DOI] [PubMed] [Google Scholar]
- 417.Noble V, Murray M, Webb MS, Alexander J, Swarbrick AS, Milner AD. Respiratory status and allergy nine to 10 years after acute bronchiolitis. Arch Dis Child. 1997;76:315–319. doi: 10.1136/adc.76.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Mok JY, Simpson H. Outcome of acute lower respiratory tract infection in infants: preliminary report of seven-year follow-up study. Br Med J (Clin Res Ed) 1982;285:333–337. doi: 10.1136/bmj.285.6338.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Narita A, Nishimura N, Arakawa Y, et al. Relationship between lower respiratory tract infections caused by respiratory syncytial virus and subsequent development of asthma in Japanese children. Jpn J Infect Dis. 2011;64:433–435. [PubMed] [Google Scholar]
- 420.Pullan CR, Hey EN. Wheezing, asthma, and pulmonary dysfunction 10 years after infection with respiratory syncytial virus in infancy. Br Med J (Clin Res Ed) 1982;284:1665–1669. doi: 10.1136/bmj.284.6330.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Mok JY, Simpson H. Symptoms, atopy, and bronchial reactivity after lower respiratory infection in infancy. Arch Dis Child. 1984;59:299–305. doi: 10.1136/adc.59.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Mikalsen IB, Halvorsen T, Oymar K. The outcome after severe bronchiolitis is related to gender and virus. Pediatr Allergy Immunol. 2012;23:391–398. doi: 10.1111/j.1399-3038.2012.01283.x. [DOI] [PubMed] [Google Scholar]
- 423.McConnochie KM, Roghmann KJ. Bronchiolitis as a possible cause of wheezing in childhood: new evidence. Pediatrics. 1984;74:1–10. [PubMed] [Google Scholar]
- 424.McConnochie KM, Roghmann KJ. Wheezing at 8 and 13 years: changing importance of bronchiolitis and passive smoking. Pediatr Pulmonol. 1989;6:138–146. doi: 10.1002/ppul.1950060303. [DOI] [PubMed] [Google Scholar]
- 425.Stein RT, Sherrill D, Morgan WJ, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet. 1999;354:541–545. doi: 10.1016/S0140-6736(98)10321-5. [DOI] [PubMed] [Google Scholar]
- 426.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178:667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Jackson DJ, Evans MD, Gangnon RE, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am J Respir Crit Care Med. 2012;185:281–285. doi: 10.1164/rccm.201104-0660OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Kusel MM, de Klerk NH, Kebadze T, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119:1105–1110. doi: 10.1016/j.jaci.2006.12.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Welliver RC, Sun M, Rinaldo D, Ogra PL. Predictive value of respiratory syncytial virus-specific IgE responses for recurrent wheezing following bronchiolitis. J Pediatr. 1986;109:776–780. doi: 10.1016/s0022-3476(86)80692-8. [DOI] [PubMed] [Google Scholar]
- 430.Smyth RL, Fletcher JN, Thomas HM, Hart CA. Immunological responses to respiratory syncytial virus infection in infancy. Arch Dis Child. 1997;76:210–214. doi: 10.1136/adc.76.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Gill MA. The role of dendritic cells in asthma. J Allergy Clin Immunol. 2012;129:889–901. doi: 10.1016/j.jaci.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 432.John AE, Gerard CJ, Schaller M, et al. Respiratory syncytial virus-induced exaggeration of allergic airway disease is dependent upon CCR1-associated immune responses. Eur J Immunol. 2005;35:108–116. doi: 10.1002/eji.200425439. [DOI] [PubMed] [Google Scholar]
- 433.Lukacs NW, Tekkanat KK, Berlin A, et al. Respiratory syncytial virus predisposes mice to augmented allergic airway responses via IL-13-mediated mechanisms. J Immunol. 2001;167:1060–1065. doi: 10.4049/jimmunol.167.2.1060. [DOI] [PubMed] [Google Scholar]
- 434.Liu B, Kimura Y. Respiratory syncytial virus protects against the subsequent development of Japanese cedar pollen-induced allergic responses. J Med Virol. 2007;79:1600–1605. doi: 10.1002/jmv.20944. [DOI] [PubMed] [Google Scholar]
- 435.Barends M, Boelen A, de Rond L, et al. Respiratory syncytial virus enhances respiratory allergy in mice despite the inhibitory effect of virus-induced interferon-gamma. J Med Virol. 2003;69:156–162. doi: 10.1002/jmv.10252. [DOI] [PubMed] [Google Scholar]
- 436.Gershwin LJ, Anderson ML, Wang C, Berghaus LJ, Kenny TP, Gunther RA. Assessment of IgE response and cytokine gene expression in pulmonary efferent lymph collected after ovalbumin inhalation during experimental infection of calves with bovine respiratory syncytial virus. Am J Vet Res. 2011;72:134–145. doi: 10.2460/ajvr.72.1.134. [DOI] [PubMed] [Google Scholar]
- 437.Kalina WV, Anderson ML, Gershwin LJ. Alternaria aerosol during a bovine respiratory syncytial virus infection alters the severity of subsequent re-infection and enhances IgE production. Comp Immunol Microbiol Infect Dis. 2006;29:138–156. doi: 10.1016/j.cimid.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 438.Schwarze J, Cieslewicz G, Joetham A, et al. Critical roles for interleukin-4 and interleukin-5 during respiratory syncytial virus infection in the development of airway hyperresponsiveness after airway sensitization. Am J Respir Crit Care Med. 2000;162:380–386. doi: 10.1164/ajrccm.162.2.9903057. [DOI] [PubMed] [Google Scholar]
- 439.Sims DG, Gardner PS, Weightman D, Turner MW, Soothill JF. Atopy does not predispose to RSV bronchiolitis or postbronchiolitic wheezing. Br Med J (Clin Res Ed) 1981;282:2086–2088. doi: 10.1136/bmj.282.6282.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Korppi M, Piippo-Savolainen E, Korhonen K, Remes S. Respiratory morbidity 20 years after RSV infection in infancy. Pediatr Pulmonol. 2004;38:155–160. doi: 10.1002/ppul.20058. [DOI] [PubMed] [Google Scholar]
- 441.Juntti H, Kokkonen J, Dunder T, Renko M, Niinimaki A, Uhari M. Association of an early respiratory syncytial virus infection and atopic allergy. Allergy. 2003;58:878–884. doi: 10.1034/j.1398-9995.2003.00233.x. [DOI] [PubMed] [Google Scholar]
- 442.Welliver RC, Duffy L. The relationship of RSV-specific immunoglobulin E antibody responses in infancy, recurrent wheezing, and pulmonary function at age 7–8 years. Pediatr Pulmonol. 1993;15:19–27. doi: 10.1002/ppul.1950150104. [DOI] [PubMed] [Google Scholar]
- 443.Simoes EA, Groothuis JR, Carbonell-Estrany X, et al. Palivizumab prophylaxis, respiratory syncytial virus, and subsequent recurrent wheezing. J Pediatr. 2007;151:34–42. doi: 10.1016/j.jpeds.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 444.Simoes EA, Carbonell-Estrany X, Rieger CH, Mitchell I, Fredrick L, Groothuis JR. The effect of respiratory syncytial virus on subsequent recurrent wheezing in atopic and nonatopic children. J Allergy Clin Immunol. 2010;126:256–262. doi: 10.1016/j.jaci.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, Morgan WJ. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. N Engl J Med. 1995;332:133–138. doi: 10.1056/NEJM199501193320301. [DOI] [PubMed] [Google Scholar]
- 446.Laing I, Reidel F, Yap PL, Simpson H. Atopy predisposing to acute bronchiolitis during an epidemic of respiratory syncytial virus. Br Med J (Clin Res Ed) 1982;284:1070–1072. doi: 10.1136/bmj.284.6322.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Trefny P, Stricker T, Baerlocher C, Sennhauser FH. Family history of atopy and clinical course of RSV infection in ambulatory and hospitalized infants. Pediatr Pulmonol. 2000;30:302–306. doi: 10.1002/1099-0496(200010)30:4<302::aid-ppul5>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 448.Carroll KN, Gebretsadik T, Minton P, et al. Influence of maternal asthma on the cause and severity of infant acute respiratory tract infections. J Allergy Clin Immunol. 2012;129:1236–1242. doi: 10.1016/j.jaci.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Simoes EA, Carbonell-Estrany X, Fullarton JR, Rossi GA, Barberi I, Lanari M. European risk factors’ model to predict hospitalization of premature infants born 33–35 weeks’ gestational age with respiratory syncytial virus: validation with Italian data. J Matern Fetal Neonatal Med. 2011;24:152–157. doi: 10.3109/14767058.2010.482610. [DOI] [PubMed] [Google Scholar]
- 450.Schwarze J, O’Donnell DR, Rohwedder A, Openshaw PJ. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am J Respir Crit Care Med. 2004;169:801–805. doi: 10.1164/rccm.200308-1203OC. [DOI] [PubMed] [Google Scholar]
- 451.Sikkel MB, Quint JK, Mallia P, Wedzicha JA, Johnston SL. Respiratory syncytial virus persistence in chronic obstructive pulmonary disease. Pediatr Infect Dis J. 2008;27:S63–S70. doi: 10.1097/INF.0b013e3181684d67. [DOI] [PubMed] [Google Scholar]
- 452.Hu C, Wedde-Beer K, Auais A, Rodriguez MM, Piedimonte G. Nerve growth factor and nerve growth factor receptors in respiratory syncytial virus-infected lungs. Am J Physiol Lung Cell Mol Physiol. 2002;283:L494–L502. doi: 10.1152/ajplung.00414.2001. [DOI] [PubMed] [Google Scholar]
- 453.Guilbert TW, Singh AM, Danov Z, et al. Decreased lung function after preschool wheezing rhinovirus illnesses in children at risk to develop asthma. J Allergy Clin Immunol. 2011;128:532–538. doi: 10.1016/j.jaci.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Hyvärinen MK, Kotaniemi-Syrjänen A, Reijonen TM, Korhonen K, Korppi MO. Lung function and bronchial hyper-responsiveness 11 years after hospitalization for bronchiolitis. Acta Paediatr. 2007;96:1464–1469. doi: 10.1111/j.1651-2227.2007.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Young S, O’Keeffe PT, Arnott J, Landau LI. Lung function, airway responsiveness, and respiratory symptoms before and after bronchiolitis. Arch Dis Child. 1995;72:16–24. doi: 10.1136/adc.72.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Turner SW, Young S, Landau LI, Le Souef PN. Reduced lung function both before bronchiolitis and at 11 years. Arch Dis Child. 2002;87:417–420. doi: 10.1136/adc.87.5.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Haland G, Lodrup Carlsen KC, Mowinckel P, et al. Lung function at 10 yr is not impaired by early childhood lower respiratory tract infections. Pediatr Allergy Immunol. 2009;20:254–260. doi: 10.1111/j.1399-3038.2008.00781.x. [DOI] [PubMed] [Google Scholar]
- 458.Martinez FD, Morgan WJ, Wright AL, Holberg CJ, Taussig LM. Diminished lung function as a predisposing factor for wheezing respiratory illness in infants. N Engl J Med. 1988;319:1112–1117. doi: 10.1056/NEJM198810273191702. [DOI] [PubMed] [Google Scholar]
- 459.Drysdale SB, Wilson T, Alcazar M, et al. Lung function prior to viral lower respiratory tract infections in prematurely born infants. Thorax. 2011;66:468–473. doi: 10.1136/thx.2010.148023. [DOI] [PubMed] [Google Scholar]
- 460.Martinez FD, Morgan WJ, Wright AL, Holberg C, Taussig LM. Initial airway function is a risk factor for recurrent wheezing respiratory illnesses during the first three years of life. Group Health Medical Associates. Am Rev Respir Dis. 1991;143:312–316. doi: 10.1164/ajrccm/143.2.312. [DOI] [PubMed] [Google Scholar]
- 461.Haland G, Carlsen KH, Devulapalli CS, Pettersen M, Mowinckel P, Lodrup Carlsen KC. Lung function development in the first 2 yr of life is independent of allergic diseases by 2 yr. Pediatr Allergy Immunol. 2007;18:528–534. doi: 10.1111/j.1399-3038.2007.00555.x. [DOI] [PubMed] [Google Scholar]
- 462.Stern DA, Morgan WJ, Wright AL, Guerra S, Martinez FD. Poor airway function in early infancy and lung function by age 22 years: a non-selective longitudinal cohort study. Lancet. 2007;370:758–764. doi: 10.1016/S0140-6736(07)61379-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Sears MR, Greene JM, Willan AR, et al. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N Engl J Med. 2003;349:1414–1422. doi: 10.1056/NEJMoa022363. [DOI] [PubMed] [Google Scholar]
- 464.Bisgaard H, Jensen SM, Bonnelykke K. Interaction between asthma and lung function growth in early life. Am J Respir Crit Care Med. 2012;185:1183–1189. doi: 10.1164/rccm.201110-1922OC. [DOI] [PubMed] [Google Scholar]
- 465.van der Gugten AC, Uiterwaal CS, van Putte-Katier N, Koopman M, Verheij TJ, van der Ent CK (2012) Reduced neonatal lung function and wheezing illnesses during the first five years of life. Eur Respir J [DOI] [PubMed]
- 466.Chawes BL, Poorisrisak P, Johnston SL, Bisgaard H. Neonatal bronchial hyperresponsiveness precedes acute severe viral bronchiolitis in infants. J Allergy Clin Immunol. 2012;130:354–361. doi: 10.1016/j.jaci.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Wenzel SE, Gibbs RL, Lehr MV, Simoes EA. Respiratory outcomes in high-risk children 7 to 10 years after prophylaxis with respiratory syncytial virus immune globulin. Am J Med. 2002;112:627–633. doi: 10.1016/s0002-9343(02)01095-1. [DOI] [PubMed] [Google Scholar]
- 468.Carroll KN, Wu P, Gebretsadik T, et al. The severity-dependent relationship of infant bronchiolitis on the risk and morbidity of early childhood asthma. J Allergy Clin Immunol. 2009;123:1055–1061. doi: 10.1016/j.jaci.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Wu P, Dupont WD, Griffin MR, et al. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am J Respir Crit Care Med. 2008;178:1123–1129. doi: 10.1164/rccm.200804-579OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Kuehni CE, Spycher BD, Silverman M. Causal links between RSV infection and asthma: no clear answers to an old question. Am J Respir Crit Care Med. 2009;179:1079–1080. doi: 10.1164/rccm.200904-0567ED. [DOI] [PubMed] [Google Scholar]
- 471.Bacharier LB, Cohen R, Schweiger T, et al. Determinants of asthma after severe respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol. 2012;130(91–100):e103. doi: 10.1016/j.jaci.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Poorisrisak P, Halkjaer LB, Thomsen SF, et al. Causal direction between respiratory syncytial virus bronchiolitis and asthma studied in monozygotic twins. Chest. 2010;138:338–344. doi: 10.1378/chest.10-0365. [DOI] [PubMed] [Google Scholar]
- 473.Thomsen SF, van der Sluis S, Stensballe LG, et al. Exploring the association between severe respiratory syncytial virus infection and asthma: a registry-based twin study. Am J Respir Crit Care Med. 2009;179:1091–1097. doi: 10.1164/rccm.200809-1471OC. [DOI] [PubMed] [Google Scholar]
- 474.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Goetghebuer T, Isles K, Moore C, Thomson A, Kwiatkowski D, Hull J. Genetic predisposition to wheeze following respiratory syncytial virus bronchiolitis. Clin Exp Allergy. 2004;34:801–803. doi: 10.1111/j.1365-2222.2004.1947.x. [DOI] [PubMed] [Google Scholar]
- 476.Heinzmann A, Ahlert I, Kurz T, Berner R, Deichmann KA. Association study suggests opposite effects of polymorphisms within IL8 on bronchial asthma and respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol. 2004;114:671–676. doi: 10.1016/j.jaci.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 477.Polack FP, Teng MN, Collins PL, et al. A role for immune complexes in enhanced respiratory syncytial virus disease. J Exp Med. 2002;196:859–865. doi: 10.1084/jem.20020781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Gershwin LJ, Schelegle ES, Gunther RA, et al. A bovine model of vaccine enhanced respiratory syncytial virus pathophysiology. Vaccine. 1998;16:1225–1236. doi: 10.1016/s0264-410x(98)80123-0. [DOI] [PubMed] [Google Scholar]
- 479.Murata Y. Respiratory syncytial virus vaccine development. Clin Lab Med. 2009;29:725–739. doi: 10.1016/j.cll.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Schickli JH, Dubovsky F, Tang RS. Challenges in developing a pediatric RSV vaccine. Hum Vaccin. 2009;5:582–591. doi: 10.4161/hv.9131. [DOI] [PubMed] [Google Scholar]
- 481.Kalina WV, Woolums AR, Berghaus RD, Gershwin LJ. Formalin-inactivated bovine RSV vaccine enhances a Th2 mediated immune response in infected cattle. Vaccine. 2004;22:1465–1474. doi: 10.1016/j.vaccine.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 482.Wright PF, Karron RA, Belshe RB, et al. Evaluation of a live, cold-passaged, temperature-sensitive, respiratory syncytial virus vaccine candidate in infancy. J Infect Dis. 2000;182:1331–1342. doi: 10.1086/315859. [DOI] [PubMed] [Google Scholar]
- 483.Widjojoatmodjo MN, Boes J, van Bers M, van Remmerden Y, Roholl PJ, Luytjes W. A highly attenuated recombinant human respiratory syncytial virus lacking the G protein induces long-lasting protection in cotton rats. Virol J. 2010;7:114. doi: 10.1186/1743-422X-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Bukreyev A, Whitehead SS, Bukreyeva N, Murphy BR, Collins PL. Interferon gamma expressed by a recombinant respiratory syncytial virus attenuates virus replication in mice without compromising immunogenicity. Proc Natl Acad Sci U S A. 1999;96:2367–2372. doi: 10.1073/pnas.96.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Garlapati S, Garg R, Brownlie R, et al. Enhanced immune responses and protection by vaccination with respiratory syncytial virus fusion protein formulated with CpG oligodeoxynucleotide and innate defense regulator peptide in polyphosphazene microparticles. Vaccine. 2012;30:5206–5214. doi: 10.1016/j.vaccine.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 486.Nguyen TN, Power UF, Robert A, et al. The respiratory syncytial virus G protein conserved domain induces a persistent and protective antibody response in rodents. PLoS One. 2012;7:e34331. doi: 10.1371/journal.pone.0034331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Shao HY, Lin YW, Yu SL, et al. Immunoprotectivity of HLA-A2 CTL peptides derived from respiratory syncytial virus fusion protein in HLA-A2 transgenic mouse. PLoS One. 2011;6:e25500. doi: 10.1371/journal.pone.0025500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Shafique M, Wilschut J, de Haan A. Induction of mucosal and systemic immunity against respiratory syncytial virus by inactivated virus supplemented with TLR9 and NOD2 ligands. Vaccine. 2012;30:597–606. doi: 10.1016/j.vaccine.2011.11.054. [DOI] [PubMed] [Google Scholar]
- 489.Lindell DM, Morris SB, White MP, et al. A novel inactivated intranasal respiratory syncytial virus vaccine promotes viral clearance without Th2 associated vaccine-enhanced disease. PLoS One. 2011;6:e21823. doi: 10.1371/journal.pone.0021823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Bernstein DI, Malkin E, Abughali N, Falloon J, Yi T, Dubovsky F. Phase 1 study of the safety and immunogenicity of a live, attenuated respiratory syncytial virus and parainfluenza virus type 3 vaccine in seronegative children. Pediatr Infect Dis J. 2012;31:109–114. doi: 10.1097/INF.0b013e31823386f1. [DOI] [PubMed] [Google Scholar]
- 491.Weber MW, Milligan P, Giadom B, et al. Respiratory illness after severe respiratory syncytial virus disease in infancy in The Gambia. J Pediatr. 1999;135:683–688. doi: 10.1016/s0022-3476(99)70085-5. [DOI] [PubMed] [Google Scholar]
- 492.Oddy WH, de Klerk NH, Sly PD, Holt PG. The effects of respiratory infections, atopy, and breastfeeding on childhood asthma. Eur Respir J. 2002;19:899–905. doi: 10.1183/09031936.02.00103602. [DOI] [PubMed] [Google Scholar]
- 493.Cilla G, Onate E, Perez-Yarza EG, Montes M, Vicente D, Perez-Trallero E. Hospitalization rates for human metapneumovirus infection among 0- to 3-year-olds in Gipuzkoa (Basque Country), Spain. Epidemiol Infect. 2009;137:66–72. doi: 10.1017/S0950268808000666. [DOI] [PubMed] [Google Scholar]
- 494.Chiu SS, Chan KH, Chen H, et al. Virologically confirmed population-based burden of hospitalization caused by respiratory syncytial virus, adenovirus, and parainfluenza viruses in children in Hong Kong. Pediatr Infect Dis J. 2010;29:1088–1092. doi: 10.1097/INF.0b013e3181e9de24. [DOI] [PubMed] [Google Scholar]
- 495.Hatipoglu N, Somer A, Badur S, et al. Viral etiology in hospitalized children with acute lower respiratory tract infection. Turk J Pediatr. 2011;53:508–516. [PubMed] [Google Scholar]
- 496.Laurent C, Dugue AE, Brouard J, et al. Viral epidemiology and severity of respiratory infections in infants in 2009: a prospective study. Pediatr Infect Dis J. 2012;31:827–831. doi: 10.1097/INF.0b013e3182566005. [DOI] [PubMed] [Google Scholar]
- 497.Stewart RS, Gershwin LJ. Detection of IgE antibodies to bovine respiratory syncytial virus. Vet Immunol Immunopathol. 1989;20:313–323. doi: 10.1016/0165-2427(89)90077-9. [DOI] [PubMed] [Google Scholar]