

Abstract
Liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin (L-SIGN) facilitates hepatitis C virus (HCV) infection through interaction with HCV envelope protein E2. Signaling events triggered by the E2 via L-SIGN are poorly understood. Here, kinase cascades of Raf–MEK–ERK pathway were defined upon the E2 treatment in NIH3T3 cells with stable expression of L-SIGN. The E2 bound to the cells through interaction with L-SIGN and such binding subsequently resulted in phosphorylation and activation of Raf, MEK, and ERK. Blockage of L-SIGN with antibody against L-SIGN reduced the E2-induced phosphorylation of Raf, MEK, and ERK. In the cells infected with cell culture-derived HCV, phosphorylation of these kinases was enhanced by the E2. Up-regulation of Raf–MEK–ERK pathway by HCV E2 via L-SIGN provides new insights into signaling cascade of L-SIGN, and might be a potential target for control and prevention of HCV infection.
Keywords: L-SIGN, Hepatitis C virus, E2, ERK
Introduction
Dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) is a type II integral membrane protein with an extracellular C-terminal region containing a calcium-dependent carbohydrate recognition domain [1]. As a homologue of DC-SIGN, live/lymph node-specific intercellular adhesion molecule-3-grabbing integrin (L-SIGN, also termed DC-SIGN-related) exhibits 77 % amino acid sequence identity with DC-SIGN and shows similarities in the extracellular region as well as in interaction with human immunodeficiency virus (HIV) [2, 3]. L-SIGN is abundantly expressed on endothelial cells of liver and lymph nodes. DC-SIGN and L-SIGN have been demonstrated to be recognition or capture receptors for bacteria, viruses, yeast, and parasites. L-SIGN plays an important role in the pathogenesis of a variety of viruses such as HIV, Ebola virus, hepatitis C virus (HCV), hepatitis B virus, dengue virus, Sindbis virus, filoviruses, severe acute respiratory syndrome coronavirus, influenza virus, and Semliki Forest virus [2, 4–13]. In particular, L-SIGN enhances viral entry into target cells and facilitates infection through interaction with the viral envelope glycoproteins-containing carbohydrate structures [14].
Engagement of cellular receptors is required for conveying of extracellular signals to intracellular pathways comprised of kinase cascades. Modulation of some signaling pathways by DC-SIGN is involved in the induction of immune responses against numerous pathogens [15–17]. However, little is known about signal cascade of L-SIGN upon ligand binding. HCV envelope glycoprotein E2 is believed to initiate HCV attachment via cellular receptors. L-SIGN captures and delivers HCV particles to the liver through its interaction with the E2 [18, 19]. Binding of HCV E2 to target cells via relevant receptors is required not only for cell entry but also for receptors-mediated signaling. The hypothesis is that interaction of L-SIGN with HCV E2 might trigger signaling cascade responsible for HCV pathogenesis.
Aberrant regulation of mitogen-activated protein kinase (MAPK) pathways occurs during virus infection [20]. Raf–MEK–ERK, a key MAPK pathway, has been identified to be novel target-based approaches for cancer treatment [21]. Previously, we reported that the MAPK signaling was triggered by the HCV E2 protein through interaction with CD81 and low-density lipoprotein receptor on human hepatoma and lymphoma cells [22–24]. Here, we aimed to investigate role of L-SIGN in transmitting the HCV E2 to Raf–MEK–ERK pathway. In NIH3T3 cells with stable expression of L-SIGN, cascades of Raf–MEK–ERK signaling were defined upon the HCV E2 treatment. The pathway was also studied in the cells infected with cell culture-derived HCV (HCVcc). The results indicate that L-SIGN mediates early signaling events triggered by HCV E2 protein.
Materials and Methods
Materials
Soluble HCV subtype 1a E2 protein expressed in Chinese hamster ovary cells (98–99 % purity) and E2 mouse mAb were gifts of Michael Houghton (Chiron Corporation, Emeryville, CA, USA). Goat anti-HCV E2 antibody was purchased from Biodesign International (Saco, Maine, USA). Mouse anti-human DC-SIGN mAb (clone DCN46) or anti-human L-SIGN mAb (clone 120604) were purchased from BD PharMingen (San Diego, CA, USA) and R & D Systems (Minneapolis, MN, USA), respectively. Rabbit antibodies against c-Raf, MEK, ERK, phospho-Raf-1 (Ser338), phospho-MEK (Ser217/221), phospho-ERK (Thr202/Tyr204), or β-actin were purchased from Cell Signaling Technology (Beverly, MA, USA). Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG or rabbit anti-goat IgG were from Jackson ImmunoResearch (West Grove, PA, USA). Horseradish peroxidase-conjugated goat anti-rabbit IgG and alkaline phosphatase-conjugated goat anti-rabbit or anti-mouse IgG were from Vector Lab (Burlingame, CA, USA). Bovine serum albumin (BSA), 5-bromo-4-chloro-3-indolyl phosphate, and nitro blue tetrazolium were obtained from Sigma (St. Louis, MO, USA). Chemiluminescent detection reagents were from Millipore (Billerica, MA, USA).
Cells and Virus
NIH3T3 cells stably expressing L-SIGN (designated NIH3T3/L-SIGN) were obtained through the NIH AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH, USA). The NIH3T3/L-SIGN were generated by stable transduction of NIH3T3 with MLV vector MX-L-SIGN encoding human L-SIGN [25]. NIH3T3/L-SIGN and NIH3T3 were grown in Dulbecco’s-modified Eagle’s medium (DMEM) containing 10 % fetal bovine serum (HyClone, USA). In some experiments, cells were maintained for 48 h in serum-free DMEM before addition of stimuli.
J6/JFH1 HCVcc (HCV genotype 2a) was produced by transfection of FL-J6/JFH1 (kind gift from Charles M. Rice, Center for the Study of Hepatitis C, The Rockefeller University, NY, USA) into Huh7.5.1 cells in our laboratory [26]. This virus was generated by collection of culture medium from Huh7.5.1 cells infected with HCVcc stock and the virus titer was determined as described [27].
Flow Cytometry
For L-SIGN expression assessment, appropriate amount of NIH3T3/L-SIGN and NIH3T3 cells suspended in 1 % BSA were incubated for 1 h with mouse anti-L-SIGN mAb at a final concentration of 4 μg/ml. Cells were incubated with an isotype-matched mouse IgG as controls. After two washes with phosphate-buffered saline (PBS), cells were stained with FITC-conjugated goat anti-mouse IgG for 30 min at 4 °C, washed with PBS, fixed in 1 % paraformaldehyde, and subjected to flow cytometry using a FACS Calibur (BD, San Jose, CA, USA) with CellQuest software for data acquisition and analysis.
Binding of HCV E2 protein to NIH3T3/L-SIGN was evaluated. In brief, cells were incubated for 1 h at 37 °C with 6 μg/ml E2, washed twice with PBS, and incubated with 6 μg/ml mouse anti-E2 mAb for another 1 h. After two washes with PBS, cells were stained with FITC-conjugated goat anti-mouse IgG for 30 min at 4 °C, washed with PBS, and analyzed for E2 binding. Cells were incubated with the E2 mAb and the secondary antibody as controls.
For E2-binding inhibition assay, NIH3T3/L-SIGN cells were preincubated for 1 h at 37 °C with anti-L-SIGN mAb or combined with anti-DC-SIGN mAb. Cells were then washed with PBS to remove unbound mAbs followed by incubation with 6 μg/ml E2 for 1 h. After washing, cells were incubated for 1 h with 6 μg/ml goat anti-E2 Ab and the E2 binding was detected with FITC-conjugated rabbit anti-goat IgG.
Confocal Microscopy
NIH3T3/L-SIGN and NIH3T3 cells were fixed in 4 % paraformaldehyde for 30 min at 4 °C, washed with PBS, and blocked in 1 % BSA for 20 min. Cells were incubated for 1 h at 4 °C with 6 μg/ml mouse anti-L-SIGN mAb. Following three washes with PBS, cells were stained for 30 min with FITC-conjugated goat anti-mouse IgG, washed with PBS, and bound to poly-l-lysine-treated glass coverslips. Stained cells were analyzed using a Leica TCS SP2 laser scanning confocal microscope (Heidelberg, Germany) and images were captured and processed with TCS SP2 software.
HCV E2 Treatment
To verify specific effect of HCV E2, 2 μg/ml E2 was mixed with 4 μg/ml goat anti-E2 Ab or mouse anti-E2 mAb at 37 °C for 1 h and the mixtures were allowed to treat cells. Cells were serum starved for 48 h prior to treatment for 30 min with 2 μg/ml E2, the mixtures of E2-E2 Ab or E2-E2 mAb. For receptor-blocking study, cells serum-starved were preincubated for 1 h with anti-L-SIGN mAb or in combination with anti-DC-SIGN mAb prior to treatment with 2 μg/ml E2 for another 30 min. Cells were washed with PBS and cell lysates were prepared as described [22].
HCVcc Infection
Cells seeded in 6-well plates the day before were washed twice with PBS and incubated in DMEM containing 2 μg/ml HCV E2 protein for 2 h at 37 °C. After washing with PBS, HCVcc stock (1 × 105 focus-forming units/ml) was added to plates (600 μl/well) and allowed to proceed for another 2 h. Cells were washed with PBS and lysed for kinase measurement.
Western Blotting
Equal amounts of protein extracts in cell lysates were subjected to Western blot analysis. Proteins were separated by 10 % SDS-PAGE, transferred onto nitrocellulose membranes, and blocked with 5 % nonfat milk. Membranes were incubated overnight at 4 °C with rabbit antibodies against c-Raf, MEK, ERK, phospho-Raf-1, phospho-MEK, or phospho-ERK, followed by incubation with alkaline phosphatase or horseradish peroxidase-conjugated secondary antibodies. Immune complexes were visualized with 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium substrates or chemiluminescent detection reagents. In some experiments, images were captured on a GeneGnome HR image capture (Cambridge, UK) and band intensity was quantified using GeneTools software from SynGene.
Results
Expression of L-SIGN on Cells
L-SIGN is a binding receptor for HCV E2 protein. We wondered which functions are performed by L-SIGN in signaling events triggered by the HCV E2. NIH3T3/L-SIGN was chosen to be a cell line for investigation of Raf–MEK–ERK signaling in response to HCV E2 treatment. Expression of L-SIGN was first analyzed by flow cytometry. Figure 1a showed that L-SIGN was expressed on NIH3T3/L-SIGN cells at a high level, and there was no L-SIGN expression on parental NIH3T3 cells. Cellular localization of L-SIGN was also characterized by confocal microscopy. As expected, L-SIGN was indeed localized at the surface of NIH3T3/L-SIGN (Fig. 1b). In NIH3T3, L-SIGN was unobservable.
Fig. 1.
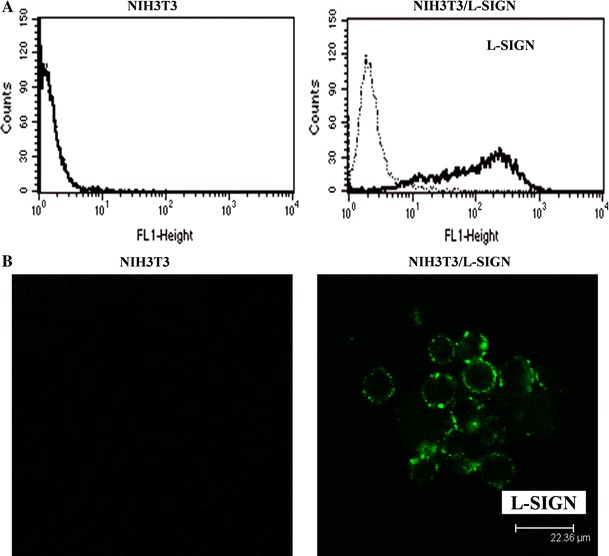
a Expression of L-SIGN on NIH3T3 and NIH3T3/L-SIGN. Cells were incubated with mouse anti-L-SIGN mAb (thick lines) or isotype control (dotted lines). Expression of L-SIGN was detected by flow cytometry using FITC-conjugated goat anti-mouse IgG. The results are representative of three experiments. b Confocal microscopy was carried out to characterize cellular localization of L-SIGN on NIH3T3 and NIH3T3/L-SIGN. Cells were stained with mouse anti-L-SIGN mAb and FITC-conjugated goat anti-mouse IgG. The results are reproducible in two experiments, and representative fields are shown
HCV E2 Binding to Cells via L-SIGN
Next, we examined whether the soluble HCV subtype 1a E2 protein bound to NIH3T3/L-SIGN cells. Figure 2a showed that NIH3T3/L-SIGN was capable of efficiently binding the E2. In contrast, NIH3T3 did not bind the E2 (data not shown). To confirm the specificity of the E2–L-SIGN interaction, antibodies against L-SIGN or DC-SIGN were assessed to inhibit the E2 binding. The cells were preincubated with the antibodies prior to the E2 incubation and the E2 binding was detected by flow cytometry. As shown in Fig. 2b, the E2 binding to NIH3T3/L-SIGN was inhibited by the anti-L-SIGN mAb in a concentration-dependent manner. The combination of anti-DC-SIGN mAb and anti-L-SIGN mAb displayed a greater inhibitory effect on the E2 binding.
Fig. 2.

a Binding of HCV E2 protein to NIH3T3/L-SIGN. Cells were treated with E2 (thick lines) or left untreated (dotted lines) and the E2 binding was detected with mouse anti-E2 mAb and FITC-conjugated goat anti-mouse IgG by flow cytometry. b Inhibition of HCV E2 binding to NIH3T3/L-SIGN by antibodies against L-SIGN or DC-SIGN. Cells were incubated with mouse anti-L-SIGN mAb at a concentration of 4 μg/ml (II), 10 μg/ml (III) or 10 μg/ml anti-DC-SIGN mAb, and 10 μg/ml anti-L-SIGN mAb (IV) before the E2 incubation. The E2 binding was detected with goat anti-E2 Ab and FITC-conjugated rabbit anti-goat IgG in the presence or absence (I) of the antibody incubation. The percentage of marker-positive cells is indicated in each case. Data are representative of three experiments
Raf, MEK, and ERK Phosphorylation Induced by HCV E2 Through Interaction with L-SIGN
To examine whether Raf–MEK–ERK pathway would be affected under the HCV E2 stimulation, kinase phosphorylation was analyzed by Western blot in NIH3T3/L-SIGN cells stimulated with the E2. Treatment of the cells with the E2 led to increased phosphorylation of Raf, MEK, and ERK, while the levels of kinase phosphorylation were lower after the E2–E2 Ab or the E2–E2 mAb treatment (Fig. 3), indicating specific phosphorylation and activation of the kinases by the E2. Total Raf, MEK, and ERK were constant in samples. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were determined as a control for protein loading.
Fig. 3.

NIH3T3/L-SIGN was treated with HCV E2, the E2-E2 mAb, or the E2-E2 Ab, and total (T-) and phosphorylated (P-) Raf, MEK, and ERK were analyzed by Western blotting. GAPDH is shown as a loading control. Similar results were obtained in four experiments, and one representative experiment is shown
Since L-SIGN-mediated the HCV E2 binding (Fig. 2), we wondered whether the activation of Raf, MEK, and ERK was induced by the E2 through interaction with L-SIGN on the cells. The phosphorylation of kinases was thus evaluated in NIH3T3/L-SIGN cells pretreated with the various concentrations of anti-L-SIGN mAb prior to the E2 treatment. Figure 4a showed that the pretreatment with anti-L-SIGN mAb reduced the phosphorylation of Raf, MEK, and ERK as compared with the E2 alone treatment. Treatment of the cells with anti-L-SIGN mAb was included as a control. Moreover, we performed the experiments to assay influence of anti-DC-SIGN mAb on the E2-induced kinase phosphorylation. As shown in Fig. 4b, pretreatment with 20 μg/ml anti-DC-SIGN mAb or the combination of 10 μg/ml anti-DC-SIGN mAb and 10 μg/ml anti-L-SIGN mAb significantly reduced the E2-induced Raf, MEK, and ERK phosphorylation in NIH3T3/L-SIGN cells, which was consistent with the E2-binding inhibition (Fig. 2b). In the case of L-SIGN-deficient NIH3T3 cells, levels of Raf, MEK, and ERK phosphorylation were not influenced by the E2. In addition, no difference in the kinase phosphorylation was observed between the E2 treatment and the pretreatment with the anti-DC-SIGN mAb or the anti-L-SIGN mAb (Fig. 4c). These results indicate that HCV E2-L-SIGN interaction leads to phosphorylation and activation of Raf, MEK, and ERK.
Fig. 4.
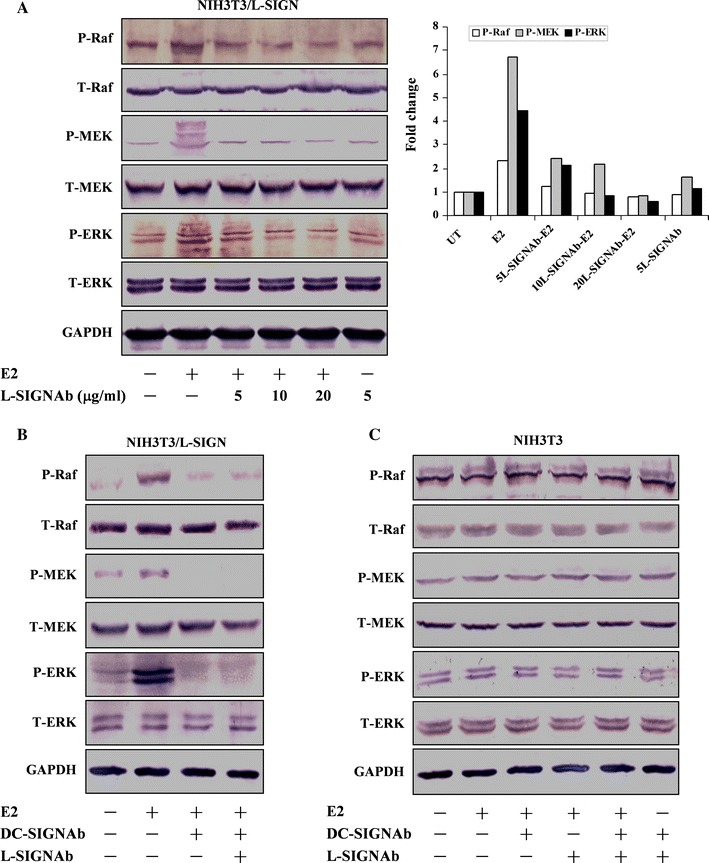
a Inhibition of HCV E2-induced Raf, MEK, and ERK phosphorylation by antibody against L-SIGN. NIH3T3/L-SIGN was treated with anti-L-SIGN mAb at the indicated concentrations before treatment with E2 protein. The signals for phosphorylated Raf, MEK, and ERK from the shown experiment were quantified. Data are expressed as fold change of the levels of phosphorylated kinase over the levels of untreated control (UT) after being normalized to the levels of GAPDH. b Inhibition of kinase phosphorylation by antibodies against DC-SIGN or L-SIGN. NIH3T3/L-SIGN was pretreated with 20 μg/ml or 10 μg/ml anti-DC-SIGN mAb and 10 μg/ml anti-L-SIGN mAb, followed by the E2 treatment. c Effect of HCV E2 on kinase phosphorylation in NIH3T3. Cells were treated with 5 μg/ml anti-DC-SIGN mAb or anti-L-SIGN mAb before treatment with the E2. Total (T-) and phosphorylated (P-) Raf, MEK, and ERK were analyzed by Western blotting. GAPDH is shown as a loading control. Representative results out of three experiments are shown
Raf–MEK–ERK Signaling Following HCVcc Infection
Based on the data obtained with uninfected cells, we further investigated Raf–MEK–ERK pathway following HCVcc infection. In HCVcc-infected NIH3T3/L-SIGN cells, levels of Raf, MEK, and ERK phosphorylation were slightly increased as compared with the uninfected cells and the E2 treatment led to strong phosphorylation of the kinases (Fig. 5). Whereas such enhancement was unobservable in HCVcc-infected NIH3T3 cells with the E2 treatment, highly implying that interaction of HCV E2 with L-SIGN accounts for the kinase phosphorylation. Actin levels were determined as a control for protein loading.
Fig. 5.
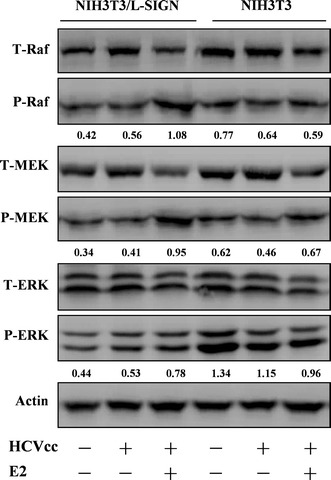
NIH3T3/L-SIGN and NIH3T3 were pretreated with HCV E2 prior to HCVcc infection. Total (T-) and phosphorylated (P-) Raf, MEK, and ERK were analyzed by Western blotting. β-actin is shown as a loading control. Changes in the levels of phosphorylated Raf, MEK, and ERK after being normalized to the levels of β-actin are shown below each blot. One representative experiment out of three is shown
Discussion
We provided evidence that L-SIGN mediates binding of the HCV E2 to target cells and such binding subsequently results in the up-regulation of Raf–MEK–ERK pathway. L-SIGN (binding and capture receptor for HCV) captures circulating HCV particles and facilitates virus infection of hepatocytes and lymphocyte subpopulations, allowing the establishment of persistent infection and the modulation of dendritic cell functions [28–30]. We previously addressed the interaction of HCV E2 protein with CD81 and low-density lipoprotein receptor (attachment and entry receptors for HCV) naturally expressed on human hepatoma and lymphoma cells. Hepatocytes are the primary target for HCV infection and that liver-derived cell lines are the standard model for HCV research. In our experiments, NIH3T3 transfected with L-SIGN (NIH3T3/L-SIGN) was used because it was generated to characterize the function of L-SIGN [25]. As a murine cell line, NIH3T3/L-SIGN does not express any other cellular receptors for HCV beyond L-SIGN, thus allowing evaluation of signaling events mediated by L-SIGN. Our data also demonstrate that NIH3T3/L-SIGN is a suitable cell line for investigation of Raf–MEK–ERK signaling following L-SIGN–HCV E2 interaction.
The soluble HCV envelope glycoprotein E2 is regarded as a surrogate to study virus-cell interaction, and putative HCV receptors including CD81, low-density lipoprotein receptor, and scavenger receptor class B type 1 have been identified using the E2 [31–33]. Indeed, we found that the HCV E2 protein expressed in Chinese hamster ovary cells was capable of efficiently binding to NIH3T3/L-SIGN cells. First, L-SIGN was shown to express at a high level on NIH3T3/L-SIGN cells (Fig. 1). The specificity of the E2–L-SIGN interaction was then confirmed by the E2-binding inhibition assay. Our data showed that the E2 binding was inhibited by the anti-L-SIGN mAb as well as by the combination of antibodies against L-SIGN or DC-SIGN (Fig. 2), indicating that these antibodies may function as competing ligands for L-SIGN and exhibit the inhibitory effects on the E2 binding. Accordingly, L-SIGN mediated the E2 binding to target cells, which is in accordance with the reports that HCV E2 protein interacts with some primary cells and cell lines expressing L-SIGN. For example, pseudotyped lentivirus particles presenting HCV glycoproteins E1 and E2 bind to BTHP1 cells expressing L-SIGN [29]. Soluble HCV E2 protein binds to HeLa cells stably expressing L-SIGN [2, 18]. HCV E2 protein binds to human monocyte-derived dendritic cells and T-REx cells expressing L-SIGN [19, 34]. As outlined above, differences including the forms of E2 and the cell types occur between the previous reports and our data.
The CD81 engagement activates Raf–MEK–ERK cascades, resulting in affecting postentry events of HCV life cycle [35]. Interaction of respiratory syncytial virus glycoprotein G with L-SIGN activates ERK [36]. However, signaling events triggered by HCV E2 through L-SIGN are poorly defined. Since L-SIGN on the cells specifically interacted with the HCV E2, we thereby investigated the consequences arising from such interaction. Our results showed that, under the stimulation of the E2, the phosphorylation of Raf, MEK, and ERK was enhanced in NIH3T3/L-SIGN cells (Fig. 3). The receptor-blocking experiments were carried out to confirm the specificity of E2–L-SIGN interaction responsible for the above effect. We found that the anti-L-SIGN mAb pretreatment reduced the phosphorylation of Raf, MEK, and ERK induced by the E2 (Fig. 4a), which was consistent with the inhibitory effects of the antibody on the E2 binding. In addition, the inhibition of E2-induced kinase phosphorylation by the anti-DC-SIGN mAb may be attributable to capacities of the antibody to cross-reactive to DC-SIGN and L-SIGN (Fig. 4b). Moreover, unspecific effects of the antibodies against L-SIGN or DC-SIGN on the E2-induced kinase phosphorylation were ruled out, as evidenced that the pretreatment with the antibody only led to the reduced kinase phosphorylation in NIH3T3/L-SIGN but not in NIH3T3 cells (Fig. 4c). Thus, in response to the E2, phosphorylation of Raf, MEK, and ERK was dependent on L-SIGN expression. It would be important to address signaling events triggered by infectious HCVcc bearing E2 protein. HCVcc infection also led to activation of Raf–MEK–ERK pathway in NIH3T3/L-SIGN cells and the E2 treatment enhanced the kinase phosphorylation (Fig. 5), suggesting the engagement of L-SIGN with the E2 protein as well as the HCVcc accounts for the pathway activation. Supporting our data, Zhang et al. [37] reported that HCV infection activated the Ras–Raf–MEK pathway. Our results demonstrate that interaction of L-SIGN with HCV E2 up-regulates Raf–MEK–ERK pathway. Further investigation into signaling events in cells endogenously expressing L-SIGN would be necessary to validate the role of L-SIGN in the promotion of infection and the pathogenesis of HCV.
Acknowledgments
This study was supported by Grants from the National Natural Science Foundation of China (30771928) and the Shanghai Leading Academic Discipline Project (B901). We thank T.D. Martin and V.N. KewalRamani (the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) for providing the NIH3T3/DC-SIGN and NIH3T3/L-SIGN cells; S. Pöhlmann, F. Baribaud, F. Kirchhoff, and R.W. Doms (the AIDS Research and Reference Reagent Program, NIH) for providing the pcDNA3-DC-SIGN plasmid; and Chiron Corporation (Emeryville, USA) for providing the HCV E2 protein and the E2 mAb.
Conflict of Interest
None.
Contributor Information
Hao Ren, Phone: +86-21-81870988, FAX: +86-21-81870988, Email: hmren@yahoo.com.
Zhong-Tian Qi, Phone: +86-21-81870988, FAX: +86-21-81870988, Email: qizt@smmu.edu.cn.
References
- 1.Geijtenbeek TB, Torensma R, van Vliet SJ, van Duijnhoven GC, Adema GJ, van Kooyk Y, et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 2000;100:575–585. doi: 10.1016/S0092-8674(00)80693-5. [DOI] [PubMed] [Google Scholar]
- 2.Gardner JP, Durso RJ, Arrigale RR, Donovan GP, Maddon PJ, Dragic T, et al. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4498–4503. doi: 10.1073/pnas.0831128100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pöhlmann S, Soilleux EJ, Baribaud F, Leslie GJ, Morris LS, Trowsdale J, et al. DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2670–2675. doi: 10.1073/pnas.051631398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bashirova AA, Geijtenbeek TB, van Duijnhoven GC, van Vliet SJ, Eilering JB, Martin MP, et al. A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. Journal of Experimental Medicine. 2001;193:671–678. doi: 10.1084/jem.193.6.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez CP, Lasala F, Carrillo J, Muñiz O, Corbí AL, Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. Journal of Virology. 2002;76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. Journal of Experimental Medicine. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klimstra WB, Nangle EM, Smith MS, Yurochko AD, Ryman KD. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. Journal of Virology. 2003;77:12022–12032. doi: 10.1128/JVI.77.22.12022-12032.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takada A, Fujioka K, Tsuiji M, Morikawa A, Higashi N, Ebihara H, et al. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. Journal of Virology. 2004;78:2943–2947. doi: 10.1128/JVI.78.6.2943-2947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Op den Brouw ML, de Jong MA, Ludwig IS, van der Molen RG, Janssen HL, Geijtenbeek TB, et al. Branched oligosaccharide structures on HBV prevent interaction with both DC-SIGN and L-SIGN. Journal of Viral Hepatitis. 2008;15:675–683. doi: 10.1111/j.1365-2893.2008.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown MG, Huang YY, Marshall JS, King CA, Hoskin DW, Anderson R. Dramatic caspase-dependent apoptosis in antibody-enhanced dengue virus infection of human mast cells. Journal of Leukocyte Biology. 2009;85:71–80. doi: 10.1189/jlb.0308167. [DOI] [PubMed] [Google Scholar]
- 12.Londrigan SL, Turville SG, Tate MD, Deng YM, Brooks AG, Reading PC. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. Journal of Virology. 2011;85:2990–3000. doi: 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Froelich S, Tai A, Kennedy K, Zubair A, Wang P. Virus-receptor mediated transduction of dendritic cells by lentiviruses enveloped with glycoproteins derived from Semliki Forest virus. PLoS ONE. 2011;6:e21491. doi: 10.1371/journal.pone.0021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lozach PY, Burleigh L, Staropoli I, Amara A. The C type lectins DC-SIGN and L-SIGN: Receptors for viral glycoproteins. Methods in Molecular Biology. 2007;379:51–68. doi: 10.1007/978-1-59745-393-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodges A, Sharrocks K, Edelmann M, Baban D, Moris A, Schwartz O, et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nature Immunology. 2007;8:569–577. doi: 10.1038/ni1470. [DOI] [PubMed] [Google Scholar]
- 16.Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity. 2007;26:605–616. doi: 10.1016/j.immuni.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 17.den Dunnen J, Gringhuis SI, Geijtenbeek TB. Innate signaling by the C-type lectin DC-SIGN dictates immune responses. Cancer Immunology Immunotherapy. 2009;58:1149–1157. doi: 10.1007/s00262-008-0615-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lozach PY, Lortat-Jacob H, de Lacroix de Lavalette A, Staropoli I, Foung S, Amara A, et al. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. The Journal of Biological Chemistry. 2003;278:20358–20366. doi: 10.1074/jbc.M301284200. [DOI] [PubMed] [Google Scholar]
- 19.Pöhlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, et al. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. Journal of Virology. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato N, Yoshida H, Ono-Nita SK, Kato J, Goto T, Otsuka M, et al. Activation of intracellular signaling by hepatitis B and C viruses: C-viral core is the most potent signal inducer. Hepatology. 2000;32:405–412. doi: 10.1053/jhep.2000.9198. [DOI] [PubMed] [Google Scholar]
- 21.Roberts PJ, Der CJ. Targeting the Raf–MEK–ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 22.Zhao LJ, Wang L, Ren H, Cao J, Li L, Ke JS, et al. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Experimental Cell Research. 2005;305:23–32. doi: 10.1016/j.yexcr.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 23.Zhao LJ, Zhang XL, Zhao P, Cao J, Cao MM, Zhu SY, et al. Up-regulation of ERK and p38 MAPK signaling pathways by hepatitis C virus E2 envelope protein in human T lymphoma cell line. Journal of Leukocyte Biology. 2006;80:424–432. doi: 10.1189/jlb.0106014. [DOI] [PubMed] [Google Scholar]
- 24.Zhao LJ, Zhao P, Chen QL, Ren H, Pan W, Qi ZT. Mitogen-activated protein kinase signaling pathways triggered by the hepatitis C virus envelope protein E2: Implications for the prevention of infection. Cell Proliferation. 2007;40:508–521. doi: 10.1111/j.1365-2184.2007.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu L, Martin TD, Vazeux R, Unutmaz D, KewalRamani VN. Functional evaluation of DC-SIGN monoclonal antibodies reveals DC-SIGN interactions with ICAM-3 do not promote human immunodeficiency virus type 1 transmission. Journal of Virology. 2002;76:5905–5914. doi: 10.1128/JVI.76.12.5905-5914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tong Y, Zhu Y, Xia X, Liu Y, Feng Y, Hua X, et al. Tupaia CD81, SR-BI, claudin-1, and occludin support hepatitis C virus infection. Journal of Virology. 2011;85:2793–2802. doi: 10.1128/JVI.01818-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludwig IS, Lekkerkerker AN, Depla E, Bosman F, Musters RJ, Depraetere S, et al. Hepatitis C virus targets DC-SIGN and L-SIGN to escape lysosomal degradation. Journal of Virology. 2004;78:8322–8332. doi: 10.1128/JVI.78.15.8322-8332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozach PY, Amara A, Bartosch B, Virelizier JL, Arenzana-Seisdedos F, Cosset FL, et al. C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. The Journal of Biological Chemistry. 2004;279:32035–32045. doi: 10.1074/jbc.M402296200. [DOI] [PubMed] [Google Scholar]
- 30.Lai WK, Sun PJ, Zhang J, Jennings A, Lalor PF, Hubscher S, et al. Expression of DC-SIGN and DC-SIGNR on human sinusoidal endothelium: A role for capturing hepatitis C virus particles. American Journal of Pathology. 2006;169:200–208. doi: 10.2353/ajpath.2006.051191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, et al. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. Journal of Virology. 1999;73:6235–6244. doi: 10.1128/jvi.73.8.6235-6244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO Journal. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wünschmann S, Medh JD, Klinzmann D, Schmidt WN, Stapleton JT. Characterization of hepatitis C virus (HCV) and HCV E2 interactions with CD81 and the low-density lipoprotein receptor. Journal of Virology. 2000;74:10055–10062. doi: 10.1128/JVI.74.21.10055-10062.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cormier EG, Durso RJ, Tsamis F, Boussemart L, Manix C, Olson WC, et al. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14067–14072. doi: 10.1073/pnas.0405695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brazzoli M, Bianchi A, Filippini S, Weiner A, Zhu Q, Pizza M, et al. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. Journal of Virology. 2008;82:8316–8329. doi: 10.1128/JVI.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson TR, McLellan JS, Graham BS. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. Journal of Virology. 2012;86:1339–1347. doi: 10.1128/JVI.06096-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Gong R, Qu J, Zhou Y, Liu W, Chen M, et al. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. Journal of Virology. 2012;86:1544–1554. doi: 10.1128/JVI.00688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]