

Abstract
Activation of the Toll-like receptor 4 (TLR4) complex, a receptor of the innate immune system, may underpin the pathophysiology of many human diseases, including asthma, cardiovascular disorder, diabetes, obesity, metabolic syndrome, autoimmune disorders, neuroinflammatory disorders, schizophrenia, bipolar disorder, autism, clinical depression, chronic fatigue syndrome, alcohol abuse, and toluene inhalation. TLRs are pattern recognition receptors that recognize damage-associated molecular patterns and pathogen-associated molecular patterns, including lipopolysaccharide (LPS) from gram-negative bacteria. Here we focus on the environmental factors, which are known to trigger TLR4, e.g., ozone, atmosphere particulate matter, long-lived reactive oxygen intermediate, pentachlorophenol, ionizing radiation, and toluene. Activation of the TLR4 pathways may cause chronic inflammation and increased production of reactive oxygen and nitrogen species (ROS/RNS) and oxidative and nitrosative stress and therefore TLR-related diseases. This implies that drugs or substances that modify these pathways may prevent or improve the abovementioned diseases. Here we review some of the most promising drugs and agents that have the potential to attenuate TLR-mediated inflammation, e.g., anti-LPS strategies that aim to neutralize LPS (synthetic anti-LPS peptides and recombinant factor C) and TLR4/MyD88 antagonists, including eritoran, CyP, EM-163, epigallocatechin-3-gallate, 6-shogaol, cinnamon extract, N-acetylcysteine, melatonin, and molecular hydrogen. The authors posit that activation of the TLR radical (ROS/RNS) cycle is a common pathway underpinning many “civilization” disorders and that targeting the TLR radical cycle may be an effective method to treat many inflammatory disorders.
Keywords: Toll-like receptor, LPS, Inflammation, Oxidative and nitrosative stress, Cytokines, Depression, Chronic fatigue
Introduction
A substantial review of receptors and regulation of the innate immune system emphasized that pattern recognition receptors (PRRs), including the Toll-like receptors (TLRs), play a key role in the host defense system [1]. Innate immune responses are initiated when damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) are recognized by PRRs [1]. A classical PAMP is lipopolysaccharide (LPS) from gram-negative bacteria. In addition, TLR4 recognizes a broad variety of substances from viruses, fungus, and mycoplasma [1, 2]. The same is true for TLR2 [3]. Typical DAMPs acting as TLR4 agonists are endogenous substances, which appear following injury and inflammation, including OxPL, OxLDL, β-defensin, high-mobility group protein 1 (HMGB1), HSPs, ECM, LL37, hyaluronic acid, heparin sulfate, substance P, and others [4–6].
Dimerization of two receptor molecules precedes TLR activation. Best understood is the mechanism by which LPS triggers TLR4. In a complex process, the LPS-binding protein transfers LPS to the TLR4 accessory protein cluster of differentiation 14 (CD14). In a second step, LPS is transferred to the next TLR4 accessory molecule named myeloid differential protein-2 (MD-2). At least two TLR4/MD-2/LPS complexes are necessary. They can form a dimer, which initiates the cell internal signal pathway. The TLR4 pathway consists of two different signaling pathways, the myeloid differentiating primary response gene 88 (MyD88)-dependent and the MyD88-independent pathway. The TLR2 pathway, on the contrary, shows only the MyD88-dependent pathway, but is more complex in activation, since it forms heterodimers with TLR1 and TLR6. Figure 1 shows the interactions between DAMPs, PAMPs, LPS, and the TLR4 and TLR2 pathways.
Fig. 1.

Toll-like receptor (TLR) activation. Toll-like receptors TLR2 and TLR4 are part of the innate immune system. Both recognize primary bacterial components, i.e., TLR2 recognizes lipoproteins and TLR4 lipopolysaccharides (LPS). TLR activation occurs through receptor dimerization. TLR4 builds homodimers, while TLR2 pairs either with TLR1 or TLR6. TLR4 activation ensues when LPS binds to lipopolysaccharide-binding protein (LPB). Cluster of differentiation 14 (CD14) and myeloid differentiation factor-2 (MD-2) are required for TLR4 dimerization. TLR4 signaling can follow two different intracellular pathways. The MyD88-dependent pathway via TIRAP induces the transcription factor nuclear factor-κB (NF-κB) resulting in the release of inflammatory cytokines, e.g., interleukin 6 and tumor necrosis factor-α. Enhanced amounts of reactive oxygen species (ROS) will be produced. Alternatively, the MyD88-independent pathway via TRAM and TRAF leads to the release of type 1 interferons
Antibody and T-cell-mediated immunity have both the biological disadvantage that several days are needed until a specific immune response is established. In contrast, the phylogenetic much older TLR system reacts immediately on invading microbes like viruses, bacteria, and fungal infections. The Nobel Prize laureate Nüsslein-Vollhardt was the first to discover the Toll protein in Drosophila melanogaster, the fruit fly. TLRs are strongly conserved in all vertebrates and even more in mammals. In humans, nine different TLRs recognize different microbiological substances, including LPS, lipoproteins, DNA, RNA, etc. TLRs are not only restricted to immune cells, but are also present, e.g., on epithelial cells in the lung or skin. After activation through dimerization, a cell internal cascade is activated leading to the release of several interleukins, interferons, and other signaling substances. These signals attract macrophages, natural killer cells, mast cells, etc., which in turn may release reactive oxygen species (ROS) and reactive nitrogen species (RNS). In the case of microbiological pathogen invasions, the increased production of ROS/RNS helps kill the microbes by destroying pathogen substances, such as proteins, lipids, carbohydrates, and nucleic acids. In contrast, the release of ROS/RNS following “sterile infections,” e.g., those where DAMPs activate TLRs, may be more harmful because the radicals may damage the hosts tissues. Obviously, the very archaic TLR system has only this simple program to react to triggers. Moreover, as described below, many environmental substances do have the ability to activate TLRs. Probably, the resulting release of radicals is the major problem in many civilization diseases.
A systematic review of the TLR literature shows that TLR4 is involved in many civilization disorders. For example, asthma—the most common chronic disease among children (WHO 2012)—is associated with TLRs [7]. Even if the etiology of asthma is still poorly understood, the inhalation of particles and substances that modify TLR2 and TLR4 may be major causes. For example, Kerkhof et al. [7] showed a significantly enhanced susceptibility for childhood asthma depending on single nucleotide polymorphisms of TLR2 and TLR4. Also, the susceptibility to develop chronic obstructive pulmonary disease (COPD) [8] correlates with single nucleotide polymorphisms of TLR2 and TLR4. Arteriosclerosis is an inflammatory and oxidative stress disorder of the blood vessels with involvement of TLRs either due to chronic infectious or sterile processes [9, 10]. Sterile inflammation seems to occur in atherosclerosis through an unusual dimerization of TLR4 with TLR6 [11]. TLRs may play a role in the tissue damage following cardiac infarction [9]. Likewise, TLRs also play an important role in tissue damage that occurs after an initial stroke [12]. TLR2 and TLR4 may be involved in some cancers. Thus, TLR2 and TLR4 single nucleotide polymorphisms are associated with a 3–5-fold increased risk for colorectal cancer [13].
TLR4 seems to be directly involved in the pathophysiology of type 2 diabetes [14]. Obese mice developing type 2 diabetes following hyperglycemia showed a 5.6-fold increased expression of TLR4 and a 37-fold increase in interleukin-6 (IL-6) as compared with normal lean mice. TLR4 plays a direct role in β-cell dysfunctions because LPS may inhibit the secretion of insulin [14, 15]. The prevalence of metabolic syndrome in the USA is estimated to be 24.0 % for men and 23.4 % for women [16]. Metabolic syndrome is not only accompanied by inflammation, as indicated by increased levels of circulating C-reactive protein, IL-1, IL-6, IL-8, and tumor necrosis factor-α (TNF-α), but also by an enhanced expression of TLR2 and TLR4 in monocytes [17].
Also psychiatric diseases are associated with dysfunctions in TLRs. A systematic analysis of TLRs showed an enhanced response in TLR2 and TLR4 activation in both schizophrenic and bipolar patients [18]. Also, in autism, an enhanced TLR2 and TLR4 responsivity can be measured, while TLR9 showed a decreased response in the blood [19]. The TLR4 complex is also involved in several diseases associated with drug abuse, including alcohol abuse and toluene sniffing. In an experimental model, the damage of the liver through alcohol consumption can be inhibited by the suppression of the TLR signal molecule MyD88 [20].
There is now also evidence that the TLR4 complex may play a role in a large number of diseases, which are related to bacterial translocation of the gram-negative enterobacteria, which contain LPS in their bacterial wall. Thus, through increased gut hyperpermeability, these gut commensals may translocate and activate the immunocyte TLR4 complex either in the blood stream or in the mesenteric lymph nodes. This process, in turn, causes activation of intracellular signaling pathways, such as nuclear factor-κB (NF-κB), which increases the production of ROS/RNS and pro-inflammatory cytokines [21]. Therefore, this gut-derived inflammatory pathway may be associated with the onset of a number of inflammatory and oxidative and nitrosative stress (IO&NS) diseases or intensify and perpetuate the IO&NS pathways in these diseases. This process is thought to play a role in clinical depression, chronic fatigue syndrome (CFS), IBD, rheumatoid arthritis, cardiovascular disorders, psoriasis, HIV infection, Parkinson’s and Alzheimer’s disease, multiple sclerosis, and chronic alcoholism [22–36].
The TLR4 and TLR2 pathways are not only activated by PAMPs and DAMPs, but are also activated or modified by environmental factors, including ozone, nanoparticles, volatilic organic compounds, metals, organophosphate pesticides, dioxins, and ionizing radiation. Moreover, these factors have a common feature, i.e., they all generate free chemical radicals in tissues. The aim of this paper is to review these factors that trigger or modify TLR4 and TLR2 pathways and the most promising drugs and agents that have the potential to attenuate these pathways and thus may have a clinical efficacy preventing or treating these disorders.
Environmentally Relevant Substances and Radiation as TLR Pathway Activators
Ozone
Ozone is an oxygen molecule consisting of three oxygen atoms (O3) instead of the usual oxygen, which is build up by two atoms (O2). This makes ozone a very instable and highly reactive molecule. Usually occurring in the higher layers of the atmosphere, ozone is also present close to the ground during summer days with high UV radiation. The inhalation of ozone causes inflammation of the lung and is characterized by the accumulation of macrophages [37, 38]. Since no pathogens are involved, this process is termed “sterile inflammation.” Ozone may increase the immune responses to TLR2 in the lungs [39]. Following ozone exposure, macrophages produce pro-inflammatory mediators, e.g., NF-κB, and release cytotoxic substances. These, in turn, may cause injury to the surrounding tissues [40]. For example, TLR4-deficient mice show no increased NF-κB activation following ozone administration, suggesting that a functional TLR4 receptor is essential for ozone-induced sterile inflammation [40]. There are two mechanisms that may explain the effects of ozone activating the TLR4 complex. Firstly, ozone degrades hyaluronan to fragments, which are potent TLR4 agonists [41]. In a murine model of ozone-induced airway hyperresponsiveness (AHR), wild-type, TLR4-deficient, MyD88-deficient, and Toll–interleukin 1 receptor (TIR) domain-containing adapter protein (TIRAP)-deficient mice were exposed to ozone or stimulated with hyaluronan fragments [42]. Ozone-exposed mice lacking functional TLR4, MyD88, or TIRAP showed reduced AHR and lower levels of pro-inflammatory cytokines. Challenging mice directly with hyaluronan resulted in AHR in wild-type, but not in TLR4-deficient, MyD88-deficient, and TIRAP-deficient mice [42]. The conclusion is that AHR induced by ozone depends on the fragmentation of hyaluronan and the TLR4–TIRAP–MyD88 pathway [42]. Ozone can induce AHR in wild-type, but not in TLR2-deficient, TLR4-deficient, and MyD88-deficient mice [38]. Therefore, also TLR2 plays an important role in ozone-induced inflammation. The authors found that gene expression of TLR2, TLR4, MyD88, and IL-6 increased in a time-dependent manner with the duration of ozone exposition. Second, ozone has the ability to oxidize organic compounds, e.g., pathways of cholesterol oxidation [43]. This oxidation without enzymatic catalysis is termed “autoxidation” [43]. Also, free radicals like ROS have the ability to autoxidize cholesterol. Autoxidation of the body’s own substances may be the key for the generation of several DAMPs.
Atmospheric Particulate Matter or Particulates
There are many types of particular matter (PM) that may be found in the atmosphere, including allergens, viruses, bacteria, dust, oil smoke, smog, and gaseous contaminants. PM 2.5 is the PM fraction of airborne nanoparticles with a diameter <2.5 μm. In many cases, PM 2.5 comprises volatile organic compounds (VOC). These are condensation products of organic matter of different origin. Outside VOC result from plant or combustion processes (industry, traffic), whereas inside VOC mainly result from heaters, gas stoves (at home), and laser printer as well as photocopying machines (in the office). The numbers of VOC during printing process can be extraordinary high. Lee and Hsu [44] reported peak particle number concentrations at 1 × 108 particles/cm3 during photocopying. The physiological reaction to chronic fine particulate matter exposure has been described in detail by Kampfrath et al. [45]. Chronic exposure to PM 2.5 causes inflammation and activated O&NS pathways, such as increased release of ROS via NADPH oxidase. These processes lead to markedly increased oxidized phospholipid derivatives and thus probably TLR4 activation. Increased PM in urban areas contributes to premature mortality, asthma, pulmonary infections, cardiovascular disease, etc. [45, 46]. Moreover, PM may downregulate TLR4 and TLR2 on dendritic cells in the lungs, a process that may interfere with an adequate response to bacterial and viral infections thus worsening lung infections [47].
Aerosol Particles in Combination with Ozone
Surprisingly, it was found that ozone in combination with aerosol particles can result in long-lived reactive oxygen intermediates (ROIs) [48]. These chemical intermediates are shown to nitrate proteins via radical reaction mechanisms [48]. Even after 10 min, those reactions were not attenuated [48]. These authors conclude that long-lived ROIs are a key to adverse health effects and may explain the increased incidence of allergic reactions, for example, by nitrating pollen which in turn causes inflammatory reactions [48, 49]. Activation of TLRs was not investigated in this publication [48].
Nanoparticles
Titanium dioxide (TiO2) nanoparticles are used in cosmetic and pigment manufacturing [50] and as additives in many toners for laser printer and copying machines (e.g., titanium oxide; CAS no. 13463-67-7; weight 1–5 %, see Material Safety Data Sheet Kyocera No. TK-17-KME-05). TiO2 nanoparticles can trigger TLR4 and cause inflammation as well as brain and lung problems [51]. The administration of TiO2 nanoparticles leads to a significant dose-dependent increased expression of TLR4 and TLR2 mRNA and protein [52]. A characteristic of many nanoparticles (e.g., carbon black, TiO2, ZnO, gold) is in their ability to generate free radicals in their immediate surroundings [53–57]. Administration of TiO2 nanoparticles for 60 days leads to behavioral changes in animals in association with significant changes in ion balance, altered enzyme activities, and changes in various neurotransmitters in the brain [58]. For example, inhalation of TiO2 nanoparticles for 30 days causes morphological changes of hippocampal neurons in the rodent in association with the presence of TiO2 nanoparticles [59]. In the brain, increased oxidative stress, lipid peroxidation, increased catalase activity, and the release of glutamic acid and NO2 were detectable [59]. Moreover, an accumulation of solid inorganic nanoparticles, e.g., TiO2 and SiO2, may occur in the body. Long-term effects from prolonged exposure are to be anticipated. For example, one nanoparticle can generate many radicals until excreted. Smaller nanoparticles (5 nm) induce a stronger innate immune response than larger (28 nm) nanoparticles of the same material [60]. One explanation for the size dependence could be that the smaller nanoparticles produce more hydrogen peroxide than the larger ones [60]. Silver nanoparticles may induce dose-dependent effects on different cell types in terms of both cytotoxicity and TLR2 expression [61]. Silver nanoparticle-induced apoptosis is reduced after treatment with TLR2 siRNA [61]. Functional blocking of TLR2 with anti-TLR2 antibodies inhibits silver nanoparticle-mediated cytotoxicity [61]. Nanoparticles are of great interest for drug delivery especially in asthma. In this context, several publications emphasize the opportunities of nanoparticles for this purpose. But the reports are in part contradictory, e.g., it is reported that silver nanoparticles can reduce mucus hypersecretion in an allergic airway inflammation [62] and that TiO2 nanoparticle conglomerates resolve lung inflammation in asthmatic rats [63]. On the other hand, Chen et al. [64] found that many respiratory diseases like COPD and asthma may be associated with exposure to nanoparticles. These authors reported that TiO2 nanoparticles stimulate mucin secretion in human bronchial epithelial cells.
Metals
A recent publication by Schmidt et al. [65] caused a paradigm shift presenting data that contact allergy to nickel depends on TLR4 functions. Normally, nickel cannot evoke a nickel allergy in mice. But when in mice—lacking the functional mice TLR4—the human TLR4 is expressed, they become susceptible to contact allergy to nickel [65]. In a personal communication, Marc Schmidt emphasized that TLR4 is necessary, but not sufficient for the development of nickel allergy. Also, cells of the adaptive immune system are involved in specific nickel allergy. Interestingly, nickel and cobalt facilitate TLR4 homodimerization independent of MD-2 [66]. These metal-induced TLR responses may be inhibited by soluble expressed TLRs, opening new therapeutic perspectives [66]. To induce an artificial allergy to nickel in mice, several components from different microorganisms may serve as adjuvant. Interestingly, not only TLR4 stimulation by bacterial LPS but also several TLR2 stimulating microbial components may promote an allergic response to nickel in mice [67]. Lerner [68] reports an association between aluminum and Crohn’s disease (CD). He suggests that aluminum adjuvant activity may modulate the aberrations of innate and adaptive immune responses occurring in CD. Due to the occurrence of aluminum in food, air, water, waste, the earth’s surface, and pharmaceuticals, the whole human population may be exposed, and therefore, the abovementioned effects of aluminum could have an enormous impact on public health. Many metals, such as arsenic, cadmium, chromium, cobalt, lead, mercury, and nickel, are known for their adverse health effects, e.g., carcinogenic effects. Exposition occurs mostly occupational or environmental [69]. Both in in vivo and in vitro systems, metals induce the production of ROS and RNS (e.g., nitric oxide, peroxynitrite and S-nitrosothiols), causing O&NS damage to DNA, proteins, and lipids [69]. Iron, another metal, is the central atom of hemoglobin. During hemorrhage, the antioxidant vitamin C may become in combination with iron a strong prooxidant leading to serious brain damage [70].
Adjuvants
Adjuvants are substances which are co-administered with immunizing agents to induce a stronger response. Vaccination usually is done using a mild immunizing agent in combination with an adjuvant. While the provoked generation of specific antibodies must be regarded as a reaction of the adaptive immune system, efficient adjuvants trigger TLR4 and other receptors of the innate immune system. For human vaccines, very common adjuvants are aluminum phosphate and aluminum hydroxide. Apparently, aluminum hydroxide itself does not trigger TLR4 [71], but it becomes very potent in combination with TLR4 agonists [72].
Organophosphate Pesticides, Dioxin, and Wood Preservation
Currently, there are only a few data linking pesticides or dioxin with TLR activation. On the other hand, many publications show associations between these pesticides and dioxin and the generation of ROS. Poovala et al. [73] showed that Bidrint®, a pesticide, leads to enhanced lipid peroxidation in cell culture experiments. The authors provided evidence that lipid peroxidation is due to ROS, produced by the exposed cells. Alluwaimi and Hussein [74] studied the cellular and humoral immune responses to the organophosphate pesticide diazinon and found gradual changes in the levels of various cytokines in diazinon-intoxicated mice. Singh and Jiang [75] found that chronic low level exposure to acephate (ACE), an organophosphate insecticide, impairs the response to LPS in rats. ACE-exposed rats exhibit an abnormal cytokine production compared with the control animals. Pestka and Zhou [76] reported that a dioxin (2,3,7,8-tetrachlorodibenzo-p-dioxin) can trigger TLR4. Probably, the very toxic effects of dioxins may result from the ability to activate the aryl hydrocarbon receptor (AhR). For example, the dioxin 2,3,7,8-tetrachlorodibenzo-p-dioxin is a very potent AhR ligand [77]. AhR can be regarded as a “higher authority,” since activation of AhR leads to a modification of pro-inflammatory cytokines in general [77]. Direct modulation of TLR response through AhR is described by Masuda et al. [78].
A common substance employed for wood preservation is pentachlorophenol (PCP). Zhu and Shan [79] showed that metabolites of PCP generate hydroxyl radicals. The authors conclude that this generation of free chemical radicals explains the toxicity of this important pollutant. Ohnishi et al. [80] showed directed interference of PCP and other organic substances (e.g., atrazine, bisphenol A) with the TLR4 signaling pathway.
Ionizing Radiation
Ionizing radiation X-ray, radioactivity, and UV light have in common the ability to generate free radicals in tissues. Exposure to ionizing radiation occurs mostly by sunlight, radiotherapy, and radioactive spill through nuclear power accidents or atomic bombs. There are papers reporting that UV light activates the TLR4 complex and pro-inflammatory cytokines and O&NS pathways as well [81]. Radiotherapy and chemoradiation cause neuroinflammation and peripheral ROS generation and the release of pro-inflammatory cytokines, such as IL-6, TNF-α, IL-1, and IL-12 [82, 83]. Shan et al. [83] reported that ionizing radiation caused an increased TLR4–MD-2, CD14, and MyD88 expression in association with a sustained induction of NF-κB and increased production of pro-inflammatory cytokines, including IL-12. The secretion of different cytokines additionally increased in a dose-dependent manner between 0.05 and 4 Gy [83]. The authors concluded that the radiation-induced cytokine production depends on TLR-like activation. Interestingly, Hayashi et al. [84] examined the long-lasting effects of radiation in 442 atomic bomb survivors. They found that radiation had long-term effects increasing inflammatory biomarkers even 60 years after exposure. Heyman et al. [85] examined the effects of iodinated contrast media in kidneys in vitro as well as in vivo. They found that administration of contrast media enhanced the levels of ROS and hypoxia. Importantly, radiation exposure even when applied locally on the body of rats caused neuroinflammatory reactions in the brain [87]. These authors examined the effects of radiation exposure (cobalt gamma up to 15 Gy) while the head was shielded against radiation and detected that several cytokines were increased in the brain. The same authors established that resection of the vagus nerve before irradiation prevented the onset of neuroinflammation [86]. This suggests that irradiation-induced peripheral inflammation had induced neuroinflammation via bottom-up signaling through the vagus nerve [87]. Since ROS may cause TLR4 activation, it may be hypothesized that radioactive exposure activates the TLR4 complex through ROS effects. For example, CFS is a very common symptom in cancer patients receiving radiotherapy [88]. CFS is characterized by increased ROS/RNS levels and a chronic inflammatory response [89].
On the other hand, there are many data that show that TLR4 activation has radioprotective effects. Shakhov et al. [90] showed that administration of a synthetic lipopeptide (sLP), which activates TLR2, can help mice to survive otherwise lethal doses of radiation. The sLP must be given in a time window between 48 h before and 24 h after exposure to radiation. In this case, activation of TLR2 is the essential step. No radioprotective effect of sLP was found in irradiated TLR2-deficient mice. Riehl et al. [91] determined the radioprotective effect of hyaluronic acid (HA). HA fragments are known to activate TLR4. Wild-type mice administered HA before radiation exposure were able to survive an 1.8-fold dose of radiation. TLR4-deficient and cyclooxygenase-2 (COX-2)-deficient mice showed no radioprotective advancement by the administration of HA.
Toluene
Toluene sniffing is a medical problem in many countries around the world because it may induce parkinsonism and encephalopathy with brain abnormalities, including atrophy and demyelination [92]. In rodent models, it has been shown that toluene significantly upregulates mRNA production of TLR4 and NF-κB and that this may play a role in its neurotoxic effects [93]. The exposure to different concentrations of toluene of wild-type mice and TLR4-deficient mice revealed that TLR4 and NF-κB expression in the hippocampus is significantly upregulated in the wild-type, but not in the TLR-deficient mice. Heat shock protein 70, an agonist of TLR4, was increased in toluene-exposed wild-type mice. The authors conclude that the known neurotoxic effect of the exposure to toluene results from toluene-induced TLR4 activation [93]. Toluene also induces O&NS pathways and O&NS-induced damage, including lipid peroxidation, in different organs, e.g., liver, kidney, lung, brain, etc. [94].
TLR Memory and Neuro-immune Interactions at the TLR Level
It is important to note that exposure to TLR agonists causes sensitization of inflammatory responses [76]; thus, priming TLR4 with various agonists before the basic TLR4 stimulation enhanced inflammatory responses during the test. The sensitivity and, therefore, the grade of the TLR4 response are dependent upon TLR4 activation during the last 24 h. Therefore, it is important to recognize that TLR4 responsivity is determined by interactions between many different agonists and that hypersensitivity to one TLR agonist may result from priming with another agonist. By inference, priming together with enhanced gene expression of TLR4, MyD88, and NF-κB following TLR activation may explain the development of sensitization or even hypersensitivity of TLR responses.
Additional effects may result from psychological stressors, which also upregulate TLR4 [18]. Social stress activates TLR expression on splenic macrophages [95]. Stress responses may augment the DAMP-induced TLR activation [96]. TLR4 plays a role in the development of brain inflammation and oxidative processes following immobilization stress [97].
Moreover, TLR functions are partly determined by neuro-immune interactions while TLRs play a role in neuro-immune processes. For example, there are several cross-linkages between the nervous system and the immune system, one of the most striking findings being that hippocampal stem cell proliferation is partly regulated by TLRs. Thus, Rolls et al. [98] reported on the processes by which TLR2 and TLR4 modulate adult hippocampal neurogenesis via MyD88-related mechanisms. On the other hand, the nervous system may modulate TLR functions. For example, treatment of human mast cells with substance P, a neuropeptide, induces an upregulated expression of TLR2, TLR4, TLR8, and TLR9 [99].
ROS and Neutrophil Cytosolic Factor 1
Several diseases with a high mortality rate, including H5N1 avian flu, acute respiratory syndrome (SARS), and Yersinia pestis, anthrax, and pox infection cause a dramatic elevated production of IL-6 and ROS [4]. ROS can oxidize phospholipids, which in their oxidized form (OxPL) function as agonist of TLR4 [4]. Neutrophil cytosolic factor 1 (Ncf1), a protein of the NADPH oxidase complex, is required for the oxidative burst and formation of ROS [100]. Ncf1 mutant mice and TLR4 minus mutant mice showed a strongly attenuated reaction to, for example, infection with H5N1 avian flu or SARS [4]. Therefore, the authors conclude that modulation of ROS synthesis may protect individuals infected with H5N1 avian influenza, SARS coronavirus, or anthrax from severe lung failure [4].
OxPAPC
Oxidized 1-palmitoyl-2-arachidonyl-sn-3-glycerophosphorylcholine (OxPAPC) was shown to specifically activate TLR2. While administration of OxPAPC provoked inflammation in wild-type mice, no such reaction was found in TLR2-deficient mice [101]. The TLR2 mutant mice also were protected against tissue damage induced by carbon tetrachloride, which causes accumulation of oxidative phospholipids in the liver [101].
New Putative Treatments of TLR4-Mediated Inflammation
Activation of TLR4 by LPS is a complex process since many molecules are involved, including LPS, CD14, MD-2, and LPS-binding protein (LPB). The TLR4 signal pathway offers, therefore, several opportunities for pharmacological interventions. Basically, three different approaches are available: (1) anti-LPS strategies that aim to neutralize LPS; (2) TLR4 antagonism, including the MyD88 signaling pathway; and (3) targeting inflammation and ROS/RNS.
Neutralizing LPS
Inactivation of LPS is a promising approach to fight sepsis. Several synthetic anti-LPS peptides (SALPs) are developed, which bind LPS, thus preventing activation of TLR4 complex [102]. In a mouse model of lethal sepsis, SALPs are able to neutralize bacterial endotoxins even at very low concentrations and protect the animals from endotoxic shock [102]. In fact, this treatment approach could be applied to many more inflammatory diseases. For example, SALPs could be helpful in clinical depression or CFS related to increased bacterial translocation and in many infectious diseases mediated by LPS. Another putative treatment is competing with the interaction of human LBP with LPS, thus neutralizing endotoxicity, e.g., using purified recombinant factor C (rFC), a protein from the horseshoe crab [103]. This rFC protein does not show acute cytotoxicity in cell cultures.
Several TLR antagonists are commercially available for research purposes. For example, the company InvivoGen offers OxPAPC as an inhibitor for TLR2 and TLR4, which should act through competing with accessory proteins like CD14, LBP, and MD-2. At least for TLR2, this is surprising since this is contradictory to a report [102], describing OxPAPC as a TLR2 agonist. Further, InvivoGen offers LPS-RS, which is a LPS from Rhodobactersphaeroides, a purple bacteria. The effectiveness of LPS-RS as TLR4 antagonist is shown in mice [104]. The administration of LPS-RS also prevents the development of a mechanical hypersensitivity in a model of serum-transferred arthritis [105].
TLR4 Antagonism
Eritoran is a synthetic lipid A which binds to MD-2 and prevents LPS activation of TLR4 and is therefore useful in the treatment of different inflammatory diseases [106]. Eritoran is developed by Eisai Co. and is subject to different pharmacological studies, e.g., phase III studies in sepsis. Eritoran is quite similar to the LPS lipid A structure and, therefore, functions as a TLR4 antagonist [106]. Macagno et al. [107] characterized a LPS-like molecule named cyanobacterial product (CyP) which is extracted from the cyanobacterium Oscillatoria Planktothrix. CyP does not activate TLR, but binds effectively MD-2. Since MD-2 is essential for TLR4 activation by LPS, this lipid indirectly prevents LPS-mediated inflammatory responses by occupying the co-factor MD-2. There are many other TLR4–MD-2 antagonists, e.g., E5531 and E5564 [108]. Another possibility to inhibit the activation of TLRs is the use of specific monoclonal antibodies, e.g., the anti-hTLR4-IgG (InvivoGen, Cat. Code mabg-htlr4), which may neutralize human TLR4-induced cellular activation.
Many plant compounds may be used as antagonists to target the TLR4 complex [1]. Interestingly, many herbs used in Traditional Chinese medicine (TCM) and Ayurveda medicine seem to interact with the TLR4 complex. These comprise green tea, Glycyrrhiza uralensis, better known as licorice, Magnolia officinalis, ginger (Zingiber officinale), Salvia miltiorrhiza (red sage), and curcumin. The polyphenol component epigallocatechin-3-gallate (EGCG) in green tea is an inhibitor of kinase TBK1 and can suppress the MyD88 signal and the MyD88-independent signal pathway [109]. Kuang et al. [110] were able to prevent the development of neuropathic pain in an animal model by intrathecal injections of the TLR4 inhibitor EGCG from green tea. In these EGCG-treated animals, the expression of TLR4, HMGB1, NF-κB, TNF-α, and IL-1β was significantly reduced in comparison with the control animals after chronic constriction injury of the sciatic nerve [110]. Ginger comprises 6-shoagol as an active component. The working mechanism of 6-shoagol is based on inhibiting inhibitor-κB kinase [111]. Red sage is another important plant in TCM. The effective component in red sage is Sal B [112]. S. miltiorrhiza is widely used in the context of cerebrovascular disorders and shows protective effects, e.g., in H2O2-induced apoptosis, which may be ascribed to inhibition of the TLR4–NF-κB–TNF-α pathway [112, 113]. Curcumin, which antagonizes NF-κB activation, also inhibits the dimerization of TLR4 [114].
Another important target of the innate immune system is MyD88. Humans possess nine TLRs, i.e., TLR1 through TLR9, all of which (except TLR3) use the MyD88-dependent pathway. Therefore, it is possible that deactivation of MyD88 may attenuate the TLR-mediated cytokine pathway. High doses of cinnamon extract can suppress the induced overexpression of MyD88 in vivo and in vitro [20]. The authors showed that cinnamon extract may prevent alcohol-induced liver damage (steatosis) in mice in association with inhibition of the expression of MyD88 mRNA, inducible nitric oxide synthase (iNOS), and plasminogen activator inhibitor 1. Moreover, in vitro experiments showed that cinnamon extract suppresses not only LPS-induced MyD88 and iNOS, but also TNF-α expression and nitric oxide (NO) synthesis [20]. EM-163 is a synthetic BB-mimetic of MyD88 (i.e., targeting the BB loop region of the TIR domain), which prevents downstream MyD88 signaling and staphylococcal enterotoxin B shock-induced death [115]. EM-163 inhibits the production of pro-inflammatory cytokines in human primary cells, such as IL-1, IL-6, TNF-α, and IFNγ. These findings show that EM-163 may have a therapeutic efficacy against staphylococcal enterotoxin B intoxication and, in fact, may have a much broader potential, including the treatment of many chronic inflammatory processes. Surprisingly, also propofol, an anesthetic, has the ability to reduce ROS formation, suppress NF-κB expression, and reduce IL-6 [116]. The exact mechanism how propofol acts seems to be unclear so far, but it may act upstream of NF-κB [117]. Similar results were obtained for another anesthetic, i.e., ketamine. This drug reduces the release of pro-inflammatory cytokines and TLR4 expression, while influencing the phosphorylation of NF-κB [118].
Blocking ROS and RNS and TLR4 Signaling
A third approach is to eliminate ROS and RNS together with attenuating the activated TLR4 or TLR2 complexes. All TLR-mediated inflammatory reactions are accompanied by ROS and NOS. Radicals are a double-edged sword in the body. While ROS released during inflammatory responses inactivate bacteria, viruses, and fungi, ROS may damage its own tissues during sterile inflammation where no pathogens are present. NO, on the one hand, is an essential radical gas with several biological functions, while ROS, on the other hand, contributes to aberrations in cardiac functions, vascular tone, and endothelial function and may be a key factor in hypertension [119].
There are many different substances that may be used to eliminate ROS and RNS and that at the same time may modulate the TLR complex. Examples are synthetic substances, such as N-acetylcysteine (NAC), and natural occurring substances, such as alfa-lipoic acid, quercitine, pycnogenol, and some flavonoids (polyphenolic compounds present in dietary plants), such as micronized purified flavonoids (e.g., diosmin and hesperidin) and oxerutins as well as baicalein and catechins. NAC is the most commonly used expectorant that also displays antidepressant effects [120, 121]. NAC is a very potent radical scavenger which is frequently used in research to eliminate ROS [122]. In animal models, NAC administration attenuates the adverse effects of LPS and prevents the increases of NF-κB protein and TLR4 mRNA [123]. Alfa-lipoic acid (ALA) is an organosulfur compound, which is derived from octanoic acid. It occurs in almost all food, but is usually covalently bound, rare, and not directly available from dietary sources. For supplemental purpose, the strong antioxidant ALA is chemically synthesized. Experimental administration of ALA can prevent the development of NAFLD in rats. ALA-treated rats showed lesser TLR4, HMGB1, and inflammation markers than the control group [124]. In obese mice, ALA can attenuate innate immune infiltration and modulate the visceral adipose inflammation, which play an important role in diet-induced obesity and insulin resistance [125]. ALA acts through the TLR4 and NF-κB pathways and abrogates the effects of LPS challenge [125]. Cotreatment with ALA prevents the LPS-induced expression of TLR4, IL-6, and TNF-α [126]. Two substances glycyrrhizin (GL) and isoliquiritigenin (ILG) from the plant G. uralensis (Chinese licorice) suppress LPS-induced TLR4 signaling in different ways [127]. While GL affects the formation of the LPS–TLR4/MD-2 complex, which leads to an inhibition of homodimerization of TLR4, ILG does not affect LPS binding to TLR4/MD-2, but inhibits LPS-induced TLR4 homodimerization [127].
Generally flavonoids, including resveratrol and baicalin, are known as radical scavengers. In a placebo-controlled study, Ghanim et al. [128] showed that the combination of resveratrol and muscadine grape polyphenols significantly improved redox status. The supplementation not only induces the expression of antioxidant genes, but also suppresses enhanced TLR4, CD14, and IL-1β expression [128]. Baicalin is a flavonoid extracted from Scutellaria baicalensis, an herb used in TCM. Kim and Lee [129] found that baicalin attenuates ischemia/reperfusion stress-induced expression of TLR4 and MyD88, the nuclear translocation of NF-κB, and increased TNF-α and IL-6 mRNA levels in animal models. Baicalin administration to the genital tract of Chlamydia-infected mice significantly reduced Chlamydia trachomatis loading in association with the expression of TLR2, TLR4, and NF-κB, while in cervical tissue, iNOS and cyclooxgenase-2 activities were decreased [130]. In another model, baicalin attenuates the effects of cerebral ischemia in association with the expression of TLR2, TLR4, NF-κB, iNOS, and COX-2 [131].
Molecular Hydrogen
Another substance recently gaining scientific attention is molecular hydrogen. Hydrogen could be either inhaled or consumed as hydrogen-enriched water. Hydrogen is the lightest of all molecules and has the ability to react with several free radicals. Recently, Ohno et al. [132] reviewed that hydrogen may have a positive effect in 63 different diseases. The majority of the experiments were carried out in animal models, including Alzheimer’s and Parkinson’s disease models. Results are reviewed in human diseases/conditions, including infarction, metabolic syndrome, diabetes mellitus type 2, inflammatory and mitochondrial myopathies, and radiation-induced adverse effects [132]. For example, hydrogen-enriched water has been shown to have a clinical efficacy in treating mitochondrial dysfunctions in inflammatory and mitochondrial myopathies [133]. Part of the clinical efficacy of molecular hydrogen is attributed to its anti-inflammatory, antioxidative, and neuroprotective effects [134]. Moreover, hydrogen gas inhibits LPS-induced NF-κB production and inflammatory pathways in the lung, including the production of pro-inflammatory cytokines and chemokines, and additionally prevents lung cell apoptosis [135].
Critical Notes
Experimental studies of TLRs are, however, accompanied by several obstacles. In the past, some papers had to be retracted due to artifacts by LPS contaminations. Bacteria are ubiquitous and, therefore, cell culture experiments are error-prone to LPS contamination [136]. One potential source of LPS is bovine serum albumin used in eukaryotic cell cultures. Another problem revolves around the extreme conserved DNA sequences and structures of TLR receptor proteins. It appears to be difficult to raise specific monoclonal antibodies, which reliably recognize TLRs [137]. Nevertheless, new methodologies are available to develop sensitive antibodies [137]. Another problem is that the overall expression of TLR receptors is not very high. Other authors describe difficulties when measuring TLR4 proteins on the surface of cells [138]. Perhaps, the most reliable method for the measurement of TLR is quantitative reverse PCR. The abovementioned methodological difficulties have undoubtedly led to erroneous conclusions in the past literature. Nevertheless, the literature on TLR2/TLR4 functions and dysfunctions is consistent, and new data based on appropriate methods have been published.
Conclusions
This paper reviews that activation of the TLR4 complex may underpin chronic inflammatory diseases. Many environmental factors may activate or interfere with the TLR4 pathways and therefore induce inflammatory and O&NS pathways, e.g., ozone, atmospheric particulate matter, long-lived reactive oxygen intermediates, nanoparticles, metals, organophosphate pesticides, dioxin, pentachlorophenol, ionizing radiation, toluene, and heat shock protein 70. Figure 2 shows the environmental factors that may activate the TLR4 complex mainly through the effects of ROS/RNS-induced DAMPs. Chronic inflammation may be caused by a single high exposure or chronic subacute exposure to the abovementioned radical-inducing substances.
Fig. 2.
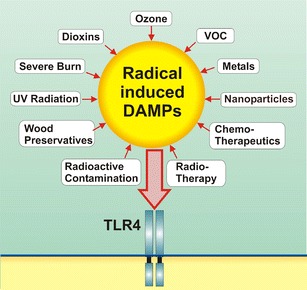
Different sources of radical-induced damage-associated molecular patterns (DAMPs). Very different chemical substances and ionizing radiation share that they can generate free radicals in tissue. These distinct substances directly or indirectly cause activation of TLR4 signaling. One common mechanism is that they all induce DAMPs, which activate TLR4 and subsequently cause inflammatory and oxidative and nitrosative stress responses. Activation of the TLR with one agonist causes an enhanced response to a second TLR agonist. Psychological stressors may further augment the responsivity of TLR signaling
As shown in Fig. 3, a vicious cycle between the TLR4 complex, production of ROS/RNS stimulating the TLR4 complex could explain the maintenance of chronic inflammatory state. We propose to term this vicious cycle the “TLR Radical Cycle.”
Fig. 3.

The TLR Radical Cycle. The initial event could be any TLR activation, e.g., an infection, chemotherapy or radiation therapy, exposure to radical inducing environmental substances or in some cases adjuvant agents, either continuous low exposure or a short time high exposure. Key is the release of reactive oxygen species (ROS), which can degrade hyaluronic acid, oxidize phospholipids and can activate high-mobility group protein B1. All these products are damage-associated molecular patterns (DAMPs) that activate the TLR4 pathway resulting in increased production of ROS, reactive nitrogen species, and inflammatory mediators. As a consequence, a vicious cycle may result between TLR activation and ROS production which is independent of the initial event. Blocking this TLR radical cycle offers opportunities to treat many TLR-related diseases. TLR4 antagonist, interference with the signal pathway, and ROS eliminating pharmaceuticals provide possibilities to treat TLR4-induced chronic inflammatory responses
The inflammatory and O&NS effects of the abovementioned TLR4 agonists may be blocked or attenuated using anti-LPS strategies that neutralize LPS (synthetic anti-LPS peptides and recombinant factor C), TLR4/MyD88 antagonists (eritoran, CyP, EM-163, licorice, M. officinalis, ginger, curcumin, and cinnamon) and antioxidants. By inference, these compounds targeting the TLR4 complex may be new “drugs” to treat TLR4-mediated inflammatory disorders. It is concluded that targeting the TLR radical cycle, i.e., suppressing the TLR4 signaling pathway in conjunction with antioxidative effects, may alleviate the suffering of millions of people affected with TLR-induced inflammatory disorders.
Acknowledgments
Conflict of interest
Kurt Lucas has filed two relevant patent applications, i.e., “Cinnamon extract for the treatment of diseases caused by induced “mismanagement” of the innate immune system (October 2011) and “Compositions for the preparation of hydrogen enriched water” (September 2012). MM does not report any conflict of interest.
Glossary
List of Abbreviations
- ACE
Acephate (organophosphate insecticide)
- AHR
Airway hyperresponsiveness
- AhR
Aryl hydrocarbon receptor
- ALA
Alfa-lipoic acid
- CD
Crohn’s disease
- CD14
Cluster of differentiation 14
- CFS
Chronic fatigue syndrome
- COPD
Chronic obstructive pulmonary disease
- COX-2
Cyclooxygenase-2 (prostaglandin-endoperoxide synthase 2)
- CyP
Cyanobacterial product
- DAMPs
Damage-associated molecular patterns
- ECM
Extracellular matrix
- EGCG
Epigallocatechin-3-gallate
- EM-163
EM-163 is a synthetic BB-mimetic of MyD88
- GL
Glycyrrhizin
- HA
Hyaluronic acid
- HMGB1
High-mobility group protein B1
- HSPs
Heat shock proteins
- IBD
Inflammatory bowel disease
- IFNγ
Interferon gamma
- IL-1β
Interleukin-1β
- IL-6
Interleukin-6
- IL-8
Interleukin-8
- IL-12
Interleukin-12
- ILG
Isoliquiritigenin
- iNOS
Inducible nitric oxide synthase
- LL37
Cathelicidin
- LPB
LPS-binding protein
- LPS
Lipopolysaccharide
- LPS-RS
LPS from Rhodobactersphaeroides
- MD-2
Myeloid differential protein-2
- MyD88
Myeloid differentiation primary response gene 88
- NAC
N-acetylcysteine
- NAFLD
Nonalcoholic fatty liver disease
- Ncf1
Neutrophil cytosolic factor 1
- NF-κB
Nuclear factor-κB
- NO
Nitric oxide
- O&NS
Oxidative and nitrosative stress
- OxLDL
Oxidized low-density lipoprotein
- OxPAPC
1-Palmitoyl-2-arachidonyl-sn-3-glycerophosphorylcholine
- OxPL
Oxidized phospholipids
- PAMPs
Pathogen-associated molecular patterns
- PCP
Pentachlorophenol
- PM
Particular matter
- PM 2.5
is the PM fraction of airborne nanoparticles with a diameter <2.5 μm
- PRRs
Pattern recognition receptors
- rFC
Recombinant factor C
- RNS
Reactive nitrogen species
- ROIs
Reactive oxygen intermediates
- ROS
Reactive oxygen species
- Sal B
Salvianolic acid B
- SALPs
Synthetic anti-LPS peptides
- SARS
Acute respiratory syndrome
- siRNA
Small interfering RNA
- sLP
Synthetic lipopeptide
- TBK1
TANK-binding kinase 1
- TCM
Traditional Chinese medicine
- TIRAP
Toll–interleukin 1 receptor (TIR) domain containing adaptor protein
- TLR1
Toll-like receptor 1
- TLR2
Toll-like receptor 2
- TLR3
Toll-like receptor 3
- TLR4
Toll-like receptor 4
- TLR6
Toll-like receptor 6
- TLR9
Toll-like receptor 9
- TLRs
Toll-like receptors
- TNF-α
Tumor necrosis factor-α
- VOC
Volatile organic compounds
References
- 1.Jeong E, Lee JY. Intrinsic and extrinsic regulation of innate immune receptors. Yonsei Med J. 2011;52(3):379–92. doi: 10.3349/ymj.2011.52.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mu HH, Hasebe A, Van Schelt A, Cole BC. Novel interactions of a microbial superantigen with TLR2 and TLR4 differentially regulate IL-17 and Th17-associated cytokines. Cell Microbiol. 2011;13(3):374–87. doi: 10.1111/j.1462-5822.2010.01540.x. [DOI] [PubMed] [Google Scholar]
- 3.Oliveira-Nascimento L, Massari P, Wetzler LM. The role of TLR2 in infection and immunity. Front Immunol. 2012;3:79. doi: 10.3389/fimmu.2012.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–49. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre JS, Lévesque T, Picard S, Paré G, Gravel A, Flamand L, Borgeat P. Extra domain A of fibronectin primes leukotriene biosynthesis and stimulates neutrophil migration through activation of Toll-like receptor 4. Arthritis Rheum. 2011;63(6):1527–33. doi: 10.1002/art.30308. [DOI] [PubMed] [Google Scholar]
- 6.Sirisinha S. Insight into the mechanisms regulating immune homeostasis in health and disease. Asian Pac J Allergy Immunol. 2011;29(1):1–14. [PubMed] [Google Scholar]
- 7.Kerkhof M, Postma DS, Brunekreef B, Reijmerink NE, Wijga AH, de Jongste JC, Gehring U, Koppelman GH. Toll-like receptor 2 and 4 genes influence susceptibility to adverse effects of traffic-related air pollution on childhood asthma. Thorax. 2010;65(8):690–7. doi: 10.1136/thx.2009.119636. [DOI] [PubMed] [Google Scholar]
- 8.Budulac SE, Boezen HM, Hiemstra PS, Lapperre TS, Vonk JM, Timens W, Postma DS, GLUCOLD study group Toll-like receptor (TLR2 and TLR4) polymorphisms and chronic obstructive pulmonary disease. PLoS One. 2012;7(8):e43124. doi: 10.1371/journal.pone.0043124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kashiwagi M, Imanishi T, Ozaki Y, Satogami K, Masuno T, Wada T, Nakatani Y, Ishibashi K, Komukai K, Tanimoto T, Ino Y, Kitabata H, Akasaka T. Differential expression of Toll-like receptor 4 and human monocyte subsets in acute myocardial infarction. Atherosclerosis. 2012;221(1):249–53. doi: 10.1016/j.atherosclerosis.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 10.Spirig R, Tsui J, Shaw S. The emerging role of TLR and innate immunity in cardiovascular disease. Cardiol Res Pract. 2012;2012:181394. doi: 10.1155/2012/181394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11(2):155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Downes CE, Crack PJ. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? Br J Pharmacol. 2010;160(8):1872–88. doi: 10.1111/j.1476-5381.2010.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pimentel-Nunes P, Teixeira AL, Pereira C, Gomes M, Brandão C, Rodrigues C, Gonçalves N, Boal-Carvalho I, Roncon-Albuquerque R, Jr, Moreira-Dias L, Leite-Moreira AF, Medeiros R, Dinis-Ribeiro M. Functional polymorphisms of Toll-like receptors 2 and 4 alter the risk for colorectal carcinoma in Europeans. Dig Liver Dis. 2013;45(1):63–69. doi: 10.1016/j.dld.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 14.Ladefoged M, Buschard K, Hansen AM (2013) Increased expression of toll-like receptor 4 and inflammatory cytokines, interleukin-6 in particular, in islets from a mouse model of obesity and type 2 diabetes. APMIS doi:. doi:10.1111/apm.12018 [DOI] [PubMed]
- 15.Amyot J, Semache M, Ferdaoussi M, Fontés G, Poitout V. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via Toll-like receptor-4 and NF-κB signalling. PLoS One. 2012;7(4):e36200. doi: 10.1371/journal.pone.0036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287(3):356–9. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 17.Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased toll-like receptor activity in patients with metabolic syndrome. Diabetes Care. 2012;35(4):900–4. doi: 10.2337/dc11-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKernan DP, Dennison U, Gaszner G, Cryan JF, Dinan TG. Enhanced peripheral toll-like receptor responses in psychosis: further evidence of a pro-inflammatory phenotype. Transl Psychiatry. 2011;1:e36. doi: 10.1038/tp.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Enstrom AM, Onore CE, Van de Water JA, Ashwood P Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain BehavImmun. 2010;24(1):64–71. doi: 10.1016/j.bbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanuri G, Weber S, Volynets V, Spruss A, Bischoff SC, Bergheim I. Cinnamon extract protects against acute alcohol-induced liver steatosis in mice. J Nutr. 2009;139(3):482–7. doi: 10.3945/jn.108.100495. [DOI] [PubMed] [Google Scholar]
- 21.Maes M, Kubera M, Leunis JC. The gut–brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol Lett. 2008;29(1):117–24. [PubMed] [Google Scholar]
- 22.Maes M, Mihaylova I, Leunis JC. Increased serum IgA and IgM against LPS of enterobacteria in chronic fatigue syndrome (CFS): indication for the involvement of gram-negative enterobacteria in the etiology of CFS and for the presence of an increased gut-intestinal permeability. J Affect Disord. 2007;99(1–3):237–40. doi: 10.1016/j.jad.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Maes M, Kubera M, Leunis JC, Berk M, Geffard M, Bosmans E (2013) In depression, bacterial translocation may drive inflammatory responses, oxidative and nitrosative stress (O&NS), and autoimmune responses directed against O&NS-damaged neoepitopes. Acta Psychiatr Scand. doi:10.1111/j.1600-0447.2012.01908.x [DOI] [PubMed]
- 24.Lim SG, Menzies IS, Lee CA, Johnson MA, Pounder RE. Intestinal permeability and function in patients infected with human immunodeficiency virus. A comparison with coeliac disease. Scand J Gastroenterol. 1993;28(7):573–80. doi: 10.3109/00365529309096090. [DOI] [PubMed] [Google Scholar]
- 25.Sundqvist T, Lindström F, Magnusson KE, Sköldstam L, Stjernström I, Tagesson C. Influence of fasting on intestinal permeability and disease activity in patients with rheumatoid arthritis. Scand J Rheumatol. 1982;11(1):33–8. doi: 10.3109/03009748209098111. [DOI] [PubMed] [Google Scholar]
- 26.Kamer AR, Dasanayake AP, Craig RG, Glodzik-Sobanska L, Bry M, de Leon MJ. Alzheimer’s disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J Alzheimers Dis. 2008;13(4):437–49. doi: 10.3233/jad-2008-13408. [DOI] [PubMed] [Google Scholar]
- 27.Gabrielli M, Bonazzi P, Scarpellini E, Bendia E, Lauritano EC, Fasano A, Ceravolo MG, Capecci M, Rita Bentivoglio A, Provinciali L, Tonali PA, Gasbarrini A. Prevalence of small intestinal bacterial overgrowth in Parkinson's disease. Mov Disord. 2011;26(5):889–92. doi: 10.1002/mds.23566. [DOI] [PubMed] [Google Scholar]
- 28.Ochoa-Repáraz J, Mielcarz DW, Begum-Haque S, Kasper LH. Gut, bugs, and brain: role of commensal bacteria in the control of central nervous system disease. Ann Neurol. 2011;69(2):240–7. doi: 10.1002/ana.22344. [DOI] [PubMed] [Google Scholar]
- 29.Geffard M, Bodet D, Martinet Y, Dabadie M-P. Detection of the specific IgM and IgA circulating in sera of multiple sclerosis patients: interest and perspectives. Immuno-Analyse and Biol Spec. 2002;17:302–310. [Google Scholar]
- 30.Shanahan F. Current concepts of the pathogenesis of inflammatory bowel disease. Ir J Med Sci. 1994;163(12):544–9. doi: 10.1007/BF02943022. [DOI] [PubMed] [Google Scholar]
- 31.Krack A, Sharma R, Figulla HR, Anker SD. The importance of the gastrointestinal system in the pathogenesis of heart failure. Eur Heart J. 2005;26(22):2368–74. doi: 10.1093/eurheartj/ehi389. [DOI] [PubMed] [Google Scholar]
- 32.Sandek A, Bauditz J, Swidsinski A, Buhner S, Weber-Eibel J, von Haehling S, Schroedl W, Karhausen T, Doehner W, Rauchhaus M, Poole-Wilson P, Volk HD, Lochs H, Anker SD. Altered intestinal function in patients with chronic heart failure. J Am CollCardiol. 2007;50(16):1561–9. doi: 10.1016/j.jacc.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 33.Sandek A, Rauchhaus M, Anker SD, von Haehling S. The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care. 2008;11(5):632–9. doi: 10.1097/MCO.0b013e32830a4c6e. [DOI] [PubMed] [Google Scholar]
- 34.Charalambous BM, Stephens RC, Feavers IM, Montgomery HE. Role of bacterial endotoxin in chronic heart failure: the gut of the matter. Shock. 2007;28(1):15–23. doi: 10.1097/shk.0b013e318033ebc5. [DOI] [PubMed] [Google Scholar]
- 35.Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H. Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1beta, and expression of caspase-11 in mice. J BiolChem. 2004;279(49):51647–53. doi: 10.1074/jbc.M407328200. [DOI] [PubMed] [Google Scholar]
- 36.Gyurcsovics K, Bertók L. Pathophysiology of psoriasis: coping endotoxins with bile acid therapy. Pathophysiology. 2003;10(1):57–61. doi: 10.1016/j.pathophys.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Hollingsworth JW, Kleeberger SR, Foster WM. Ozone and pulmonary innate immunity. Proc Am Thorac Soc. 2007;4(3):240–6. doi: 10.1513/pats.200701-023AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams AS, Leung SY, Nath P, Khorasani NM, Bhavsar P, Issa R, Mitchell JA, Adcock IM, Chung KF. Role of TLR2, TLR4, and MyD88 in murine ozone-induced airway hyperresponsiveness and neutrophilia. J Appl Physiol. 2007;103(4):1189–95. doi: 10.1152/japplphysiol.00172.2007. [DOI] [PubMed] [Google Scholar]
- 39.Oakes JL, O'Connor BP, Warg LA, Burton R, Hock A, Loader J, Laflamme D, Jing J, Hui L, Schwartz DA, Yang IV. Ozone enhances pulmonary innate immune response to a TLR2 agonist. Am J Respir Cell Mol Biol. 2013;48(1):27–34. doi: 10.1165/rcmb.2012-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connor AJ, Laskin JD, Laskin DL. Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4. Exp Mol Pathol. 2012;92(2):229–35. doi: 10.1016/j.yexmp.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garantziotis S, Li Z, Potts EN, Lindsey JY, Stober VP, Polosukhin VV, Blackwell TS, Schwartz DA, Foster WM, Hollingsworth JW. TLR4 is necessary for hyaluronan-mediated airway hyperresponsiveness after ozone inhalation. Am J RespirCrit Care Med. 2010;181(7):666–75. doi: 10.1164/rccm.200903-0381OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 42.Li Z, Potts-Kant EN, Garantziotis S, Foster WM, Hollingsworth JW. Hyaluronan signaling during ozone-induced lung injury requires TLR4, MyD88, and TIRAP. PLoS One. 2011;6(11):e27137. doi: 10.1371/journal.pone.0027137. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 43.Iuliano L. Pathways of cholesterol oxidation via non-enzymatic mechanisms. ChemPhys Lipids. 2011;164(6):457–68. doi: 10.1016/j.chemphyslip.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Lee C-W, Hsu D-J. Measurements of fine and ultrafine particles formation in photocopy centers in Taiwan. Atmospheric Environment. 2007;41(3):6598–09. doi: 10.1016/j.atmosenv.2007.04.016. [DOI] [Google Scholar]
- 45.Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis JA, Xu X, Kherada N, Brook RD, Reddy KM, Padture NP, Parthasarathy S, Chen LC, Moffatt-Bruce S, Sun Q, Morawietz H, Rajagopalan S. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res. 2011;108(6):716–26. doi: 10.1161/CIRCRESAHA.110.237560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.US Environmental Protection Agency (2012) National Ambient Air Quality Standards (NAAQS), PM2.5 NAAQS Implementation http://www.epa.gov/ttn/naaqs/pm/pm25_index.html
- 47.Miyata R, van Eeden SF. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol Appl Pharmacol. 2011;257(2):209–26. doi: 10.1016/j.taap.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 48.Shiraiwa M, Sosedova Y, Rouvière A, Yang H, Zhang Y, Abbatt JP, Ammann M, Pöschl U. The role of long-lived reactive oxygen intermediates in the reaction of ozone with aerosol particles. Nat Chem. 2011;3(4):291–5. doi: 10.1038/nchem.988. [DOI] [PubMed] [Google Scholar]
- 49.Gruijthuijsen YK, Grieshuber I, Stöcklinger A, Tischler U, Fehrenbach T, Weller MG, Vogel L, Vieths S, Pöschl U, Duschl A. Nitration enhances the allergenic potential of proteins. Int Arch Allergy Immunol. 2006;141(3):265–75. doi: 10.1159/000095296. [DOI] [PubMed] [Google Scholar]
- 50.Bermudez E, Mangum JB, Wong BA, Asgharian B, Hext PM, Warheit DB, Everitt JI. Pulmonary responses of mice, rats, and hamsters to subchronic inhalation of ultrafine titanium dioxide particles. Toxicol Sci. 2004;77(2):347–57. doi: 10.1093/toxsci/kfh019. [DOI] [PubMed] [Google Scholar]
- 51.Chen P, Migita S, Kanehira K, Sonezaki S, Taniguchi A. Development of sensor cells using NF-κB pathway activation for detection of nanoparticle-induced inflammation. Sensors (Basel) 2011;11(7):7219–30. doi: 10.3390/s110707219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui Y, Liu H, Zhou M, Duan Y, Li N, Gong X, Hu R, Hong M, Hong F. Signaling pathway of inflammatory responses in the mouse liver caused by TiO2 nanoparticles. J Biomed Mater Res A. 2011;96(1):221–9. doi: 10.1002/jbm.a.32976. [DOI] [PubMed] [Google Scholar]
- 53.Dick CA, Brown DM, Donaldson K, Stone V. The role of free radicals in the toxic and inflammatory effects of four different ultrafine particle types. Inhal Toxicol. 2003;15(1):39–52. doi: 10.1080/08958370304454. [DOI] [PubMed] [Google Scholar]
- 54.Reeves JF, Davies SJ, Dodd NJ, Jha AN. Hydroxyl radicals (*OH) are associated with titanium dioxide (TiO(2)) nanoparticle-induced cytotoxicity and oxidative DNA damage in fish cells. Mutat Res. 2008;640(1–2):113–22. doi: 10.1016/j.mrfmmm.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 55.Ionita P, Conte M, Gilbert BC, Chechik V. Gold nanoparticle-initiated free radical oxidations and halogen abstractions. Org Biomol Chem. 2007;5(21):3504–9. doi: 10.1039/b711573c. [DOI] [PubMed] [Google Scholar]
- 56.Xiong D, Fang T, Yu L, Sima X, Zhu W. Effects of nano-scale TiO2, ZnO and their bulk counterparts on zebrafish: acute toxicity, oxidative stress and oxidative damage. Sci Total Environ. 2011;409(8):1444–52. doi: 10.1016/j.scitotenv.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 57.Long TC, Saleh N, Tilton RD, Lowry GV, Veronesi B. Titanium dioxide (P25) produces reactive oxygen species in immortalized brain microglia (BV2): implications for nanoparticle neurotoxicity. Environ Sci Technol. 2006;40(14):4346–52. doi: 10.1021/es060589n. [DOI] [PubMed] [Google Scholar]
- 58.Hu R, Gong X, Duan Y, Li N, Che Y, Cui Y, Zhou M, Liu C, Wang H, Hong F. Neurotoxicological effects and the impairment of spatial recognition memory in mice caused by exposure to TiO2 nanoparticles. Biomaterials. 2010;31(31):8043–50. doi: 10.1016/j.biomaterials.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 59.Wang J, Chen C, Liu Y, Jiao F, Li W, Lao F, Li Y, Li B, Ge C, Zhou G, Gao Y, Zhao Y, Chai Z. Potential neurological lesion after nasal instillation of TiO(2) nanoparticles in the anatase and rutile crystal phases. Toxicol Lett. 2008;183:72–80. doi: 10.1016/j.toxlet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 60.Yang EJ, Kim S, Kim JS, Choi IH. Inflammasome formation and IL-1β release by human blood monocytes in response to silver nanoparticles. Biomaterials. 2012;33(28):6858–67. doi: 10.1016/j.biomaterials.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 61.Kim AS, Chae CH, Kim J, Choi JY, Kim SG, Băciut G. Silver nanoparticles induce apoptosis through the Toll-like receptor 2 pathway. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(6):789–98. doi: 10.1016/j.oooo.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 62.Jang S, Park JW, Cha HR, Jung SY, Lee JE, Jung SS, Kim JO, Kim SY, Lee CS, Park HS. Silver nanoparticles modify VEGF signaling pathway and mucus hypersecretion in allergic airway inflammation. Int J Nanomedicine. 2012;7:1329–43. doi: 10.2147/IJN.S27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scarino A, Noël A, Renzi PM, Cloutier Y, Vincent R, Truchon G, Tardif R, Charbonneau M. Impact of emerging pollutants on pulmonary inflammation in asthmatic rats: ethanol vapors and agglomerated TiO2 nanoparticles. Inhal Toxicol. 2012;24(8):528–38. doi: 10.3109/08958378.2012.696741. [DOI] [PubMed] [Google Scholar]
- 64.Chen EY, Garnica M, Wang YC, Chen CS, Chin WC. Mucin secretion induced by titanium dioxide nanoparticles. PLoS One. 2011;6(1):e16198. doi: 10.1371/journal.pone.0016198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt M, Raghavan B, Müller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen PJ, Galanos C, Roth J, Skerra A, Martin SF, Freudenberg MA, Goebeler M. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol. 2012;11(9):814–9. doi: 10.1038/ni.1919. [DOI] [PubMed] [Google Scholar]
- 66.Raghavan B, Martin SF, Esser PR, Goebeler M, Schmidt M. Metal allergens nickel and cobalt facilitate TLR4 homodimerization independently of MD2. EMBO Rep. 2012;13(12):1109–15. doi: 10.1038/embor.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takahashi H, Kinbara M, Sato N, Sasaki K, Sugawara S, Endo Y. Nickel allergy-promoting effects of microbial or inflammatory substances at the sensitization step in mice. Int Immunopharmacol. 2011;11(10):1534–40. doi: 10.1016/j.intimp.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 68.Lerner A. Aluminum as an adjuvant in Crohn’s disease induction. Lupus. 2012;21(2):231–8. doi: 10.1177/0961203311430090. [DOI] [PubMed] [Google Scholar]
- 69.Koedrith P, Seo YR. Advances in carcinogenic metal toxicity and potential molecular markers. Int J Mol Sci. 2011;12(12):9576–95. doi: 10.3390/ijms12129576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reiter RJ, Manchester LC, Tan DX. Neurotoxins: free radical mechanisms and melatonin protection. Curr Neuropharmacol. 2010;8(3):194–210. doi: 10.2174/157015910792246236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178(8):5271–6. doi: 10.4049/jimmunol.178.8.5271. [DOI] [PubMed] [Google Scholar]
- 72.Didierlaurent AM, Morel S, Lockman L, Giannini SL, Bisteau M, Carlsen H, Kielland A, Vosters O, Vanderheyde N, Schiavetti F, Larocque D, Van Mechelen M, Garçon N. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J Immunol. 2009;183(10):6186–97. doi: 10.4049/jimmunol.0901474. [DOI] [PubMed] [Google Scholar]
- 73.Poovala VS, Huang H, Salahudeen AK. Role of reactive oxygen metabolites in organophosphate-bidrin-induced renal tubular cytotoxicity. J Am Soc Nephrol. 1999;10(8):1746–52. doi: 10.1681/ASN.V1081746. [DOI] [PubMed] [Google Scholar]
- 74.Alluwaimi AM, Hussein Y. Diazinon immunotoxicity in mice: modulation of cytokines level and their gene expression. Toxicology. 2007;236(1–2):123–31. doi: 10.1016/j.tox.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 75.Singh AK, Jiang Y. Lipopolysaccharide (LPS) induced activation of the immune system in control rats and rats chronically exposed to a low level of the organothiophosphate insecticide, acephate. Toxicol Ind Health. 2003;19(2–6):93–108. doi: 10.1191/0748233703th181oa. [DOI] [PubMed] [Google Scholar]
- 76.Pestka J, Zhou HR. Toll-like receptor priming sensitizes macrophages to proinflammatory cytokine gene induction by deoxynivalenol and other toxicants. Toxicol Sci. 2006;92(2):445–55. doi: 10.1093/toxsci/kfl012. [DOI] [PubMed] [Google Scholar]
- 77.Bohonowych JE, Zhao B, Timme-Laragy A, Jung D, Di Giulio RT, Denison MS. Newspapers and newspaper ink contain agonists for the ah receptor. Toxicol Sci. 2008;102(2):278–90. doi: 10.1093/toxsci/kfn011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Masuda K, Kimura A, Hanieh H, Nguyen NT, Nakahama T, Chinen I, Otoyo Y, Murotani T, Yamatodani A, Kishimoto T. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int Immunol. 2011;23(10):637–45. doi: 10.1093/intimm/dxr072. [DOI] [PubMed] [Google Scholar]
- 79.Zhu BZ, Shan GQ. Potential mechanism for pentachlorophenol-induced carcinogenicity: a novel mechanism for metal-independent production of hydroxyl radicals. Chem Res Toxicol. 2009;22(6):969–77. doi: 10.1021/tx900030v. [DOI] [PubMed] [Google Scholar]
- 80.Ohnishi T, Yoshida T, Igarashi A, Muroi M, Tanamoto K. Effects of possible endocrine disruptors on MyD88-independent TLR4 signaling. FEMS Immunol Med Microbiol. 2008;52(2):293–5. doi: 10.1111/j.1574-695X.2007.00355.x. [DOI] [PubMed] [Google Scholar]
- 81.Sethi G, Sodhi A. In vitro activation of murine peritoneal macrophages by ultraviolet B radiation: upregulation of CD18, production of NO, proinflammatory cytokines and a signal transduction pathway. Mol Immunol. 2004;40(18):1315–23. doi: 10.1016/j.molimm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 82.Yamamoto T, Kimura T, Ueta E, Tatemoto Y, Osaki T. Characteristic cytokine generation patterns in cancer cells and infiltrating lymphocytes in oral squamous cell carcinomas and the influence of chemoradiation combined with immunotherapy on these patterns. Oncology. 2003;64(4):407–15. doi: 10.1159/000070300. [DOI] [PubMed] [Google Scholar]
- 83.Shan YX, Jin SZ, Liu XD, Liu Y, Liu SZ. Ionizing radiation stimulates secretion of pro-inflammatory cytokines: dose–response relationship, mechanisms and implications. Radiat Environ Biophys. 2007;46(1):21–9. doi: 10.1007/s00411-006-0076-x. [DOI] [PubMed] [Google Scholar]
- 84.Hayashi T, Morishita Y, Khattree R, Misumi M, Sasaki K, Hayashi I, Yoshida K, Kajimura J, Kyoizumi S, Imai K, Kusunoki Y, Nakachi K. Evaluation of systemic markers of inflammation in atomic-bomb survivors with special reference to radiation and age effects. FASEB J. 2012;26(11):4765–73. doi: 10.1096/fj.12-215228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heyman SN, Rosen S, Khamaisi M, Idée JM, Rosenberger C. Reactive oxygen species and the pathogenesis of radiocontrast-induced nephropathy. Invest Radiol. 2010;45(4):188–95. doi: 10.1097/RLI.0b013e3181d2eed8. [DOI] [PubMed] [Google Scholar]
- 86.Marquette C, Linard C, Galonnier M, Van Uye A, Mathieu J, Gourmelon P, Clarençon D. IL-1beta, TNFalpha and IL-6 induction in the rat brain after partial-body irradiation: role of vagal afferents. Int J Radiat Biol. 2003;79(10):777–85. doi: 10.1080/09553000310001610998. [DOI] [PubMed] [Google Scholar]
- 87.Taylor AG, Goehler LE, Galper DI, Innes KE, Bourguignon C. Top-down and bottom-up mechanisms in mind-body medicine: development of an integrative framework for psychophysiological research. Explore (NY) 2010;6(1):29–41. doi: 10.1016/j.explore.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goedendorp MM, Gielissen MF, Verhagen CA, Bleijenberg G (2009) Psychosocial interventions for reducing fatigue during cancer treatment in adults. Cochrane Database Syst Rev(1):CD006953. doi:10.1002/14651858.CD006953.pub2 [DOI] [PMC free article] [PubMed]
- 89.Morris G, Maes M (2012) A neuro-immune model of myalgic encephalomyelitis/chronic fatigue syndrome. Metab Brain Dis doi:10.1007/s11011-012-9324-8 [DOI] [PubMed]
- 90.Shakhov AN, Singh VK, Bone F, Cheney A, Kononov Y, Krasnov P, Bratanova-Toshkova TK, Shakhova VV, Young J, Weil MM, Panoskaltsis-Mortari A, Orschell CM, Baker PS, Gudkov A, Feinstein E. Prevention and mitigation of acute radiation syndrome in mice by synthetic lipopeptide agonists of Toll-like receptor 2 (TLR2) PLoS One. 2012;7(3):e33044. doi: 10.1371/journal.pone.0033044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Riehl TE, Foster L, Stenson WF. Hyaluronic acid is radioprotective in the intestine through a TLR4 and COX-2-mediated mechanism. Am J Physiol Gastrointest Liver Physiol. 2011;302(3):G309–16. doi: 10.1152/ajpgi.00248.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Borne J, Riascos R, Cuellar H, Vargas D, Rojas R. Neuroimaging in drug and substance abuse part II: opioids and solvents. Top Magn Reson Imaging. 2005;16(3):239–45. doi: 10.1097/01.rmr.0000192154.34563.6b. [DOI] [PubMed] [Google Scholar]
- 93.Win-Shwe TT, Kunugita N, Yoshida Y, Fujimaki H. Role of hippocampal TLR4 in neurotoxicity in mice following toluene exposure. Neurotoxicol Teratol. 2011;33(5):598–602. doi: 10.1016/j.ntt.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 94.Martínez-Alfaro M, Cárabez-Trejo A, Gallegos-Corona MA, Pedraza-Aboytes G, Hernández-Chan NG, Leo-Amador GE. Thinner inhalation effects on oxidative stress and DNA repair in a rat model of abuse. J Appl Toxicol. 2010;30(3):226–32. doi: 10.1002/jat.1488. [DOI] [PubMed] [Google Scholar]
- 95.Bailey MT, Engler H, Powell ND, Padgett DA, Sheridan JF. Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. Am J Physiol Regul Integr Comp Physiol. 2007;293(3):R1180–90. doi: 10.1152/ajpregu.00307.2007. [DOI] [PubMed] [Google Scholar]
- 96.García-Bueno B, Madrigal JL, Pérez-Nievas BG, Leza JC. Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology. 2008;149(4):1969–78. doi: 10.1210/en.2007-0482. [DOI] [PubMed] [Google Scholar]
- 97.Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke. 2008;39(4):1314–20. doi: 10.1161/STROKEAHA.107.498212. [DOI] [PubMed] [Google Scholar]
- 98.Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, Schwartz M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9(9):1081–8. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 99.Tancowny BP, Karpov V, Schleimer RP, Kulka M. Substance P primes lipoteichoic acid- and Pam3CysSerLys4-mediated activation of human mast cells by up-regulating Toll-like receptor 2. Immunology. 2010;131(2):220–30. doi: 10.1111/j.1365-2567.2010.03296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hultqvist M, Olofsson P, Holmberg J, Bäckström BT, Tordsson J, Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci U S A. 2004;101(34):12646–51. doi: 10.1073/pnas.0403831101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kadl A, Sharma PR, Chen W, Agrawal R, Meher AK, Rudraiah S, Grubbs N, Sharma R, Leitinger N. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic Biol Med. 2011;51(10):1903–9. doi: 10.1016/j.freeradbiomed.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaconis Y, Kowalski I, Howe J, Brauser A, Richter W, Razquin-Olazarán I, Iñigo-Pestaña M, Garidel P, Rössle M, Martinez de Tejada G, Gutsmann T, Brandenburg K. Biophysical mechanisms of endotoxin neutralization by cationic amphiphilic peptides. Biophys J. 2011;100(11):2652–61. doi: 10.1016/j.bpj.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li P, Ho B, Ding JL. Recombinant factor C competes against LBP to bind lipopolysaccharide and neutralizes the endotoxicity. J Endotoxin Res. 2007;13(3):150–7. doi: 10.1177/0968051907079573. [DOI] [PubMed] [Google Scholar]
- 104.Lu Z, Zhang X, Li Y, Jin J, Huang Y. TLR4 antagonist reduces early-stage atherosclerosis in diabetic apolipoprotein E-deficient mice. J Endocrinol. 2013;216(1):61–71. doi: 10.1530/JOE-12-0338. [DOI] [PubMed] [Google Scholar]
- 105.Christianson CA, Dumlao DS, Stokes JA, Dennis EA, Svensson CI, Corr M, Yaksh TL. Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain. 2011;152(12):2881–91. doi: 10.1016/j.pain.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, Wheeler J, Gogate J, Opal SM; Eritoran Sepsis Study Group Phase 2 trial of eritorantetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med. 2010;38(1):72–83. doi: 10.1097/CCM.0b013e3181b07b78. [DOI] [PubMed] [Google Scholar]
- 107.Macagno A, Molteni M, Rinaldi A, Bertoni F, Lanzavecchia A, Rossetti C, Sallusto F. A cyanobacterial LPS antagonist prevents endotoxin shock and blocks sustained TLR4 stimulation required for cytokine expression. J Exp Med. 2006;203(6):1481–92. doi: 10.1084/jem.20060136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peri F, Piazza M. Therapeutic targeting of innate immunity with Toll-like receptor 4 (TLR4) antagonists. Biotechnol Adv. 2012;30(1):251–60. doi: 10.1016/j.biotechadv.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 109.Youn HS, Lee JY, Saitoh SI, Miyake K, Kang KW, Choi YJ, Hwang DH. Suppression of MyD88- and TRIF-dependent signaling pathways of Toll-like receptor by (−)-epigallocatechin-3-gallate, a polyphenol component of green tea. Biochem Pharmacol. 2006;72(7):850–9. doi: 10.1016/j.bcp.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 110.Kuang X, Huang Y, Gu HF, Zu XY, Zou WY, Song ZB, Guo QL. Effects of intrathecalepigallocatechingallate, an inhibitor of Toll-like receptor 4, on chronic neuropathic pain in rats. Eur J Pharmacol. 2012;676(1–3):51–6. doi: 10.1016/j.ejphar.2011.11.037. [DOI] [PubMed] [Google Scholar]
- 111.Park SJ, Lee MY, Son BS, Youn HS. TBK1-targeted suppression of TRIF-dependent signaling pathway of Toll-like receptors by 6-shogaol, an active component of ginger. Biosci Biotechnol Biochem. 2009;73(7):1474–8. doi: 10.1271/bbb.80738. [DOI] [PubMed] [Google Scholar]
- 112.Liu CL, Xie LX, Li M, Durairajan SS, Goto S, Huang JD. Salvianolic acid B inhibits hydrogen peroxide-induced endothelial cell apoptosis through regulating PI3K/Akt signaling. PLoS One. 2007;2(12):e1321. doi: 10.1371/journal.pone.0001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang X, Wang Y, Jiang M, Zhu Y, Hu L, Fan G, Wang Y, Li X, Gao X. Differential cardioprotective effects of salvianolic acid and tanshinone on acute myocardial infarction are mediated by unique signaling pathways. J Ethnopharmacol. 2011;135(3):662–71. doi: 10.1016/j.jep.2011.03.070. [DOI] [PubMed] [Google Scholar]
- 114.Youn HS, Saitoh SI, Miyake K, Hwang DH. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem Pharmacol. 2006;72(1):62–9. doi: 10.1016/j.bcp.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 115.Kissner TL, Ruthel G, Alam S, Mann E, Ajami D, Rebek M, Larkin E, Fernandez S, Ulrich RG, Ping S, Waugh DS, Rebek J, Jr, Saikh KU. Therapeutic inhibition of pro-inflammatory signaling and toxicity to staphylococcal enterotoxin B by a synthetic dimeric BB-loop mimetic of MyD88. PLoS One. 2012;7(7):e40773. doi: 10.1371/journal.pone.0040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hsing CH, Lin MC, Choi PC, Huang WC, Kai JI, Tsai CC, Cheng YL, Hsieh CY, Wang CY, Chang YP, Chen YH, Chen CL, Lin CF. Anesthetic propofol reduces endotoxic inflammation by inhibiting reactive oxygen species-regulated Akt/IKKβ/NF-κB signaling. PLoS One. 2011;6(3):e17598. doi: 10.1371/journal.pone.0017598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chiu WT, Lin YL, Chou CW, Chen RM. Propofol inhibits lipoteichoic acid-induced iNOS gene expression in macrophages possibly through downregulation of toll-like receptor 2-mediated activation of Raf-MEK1/2-ERK1/2-IKK-NFkappaB. Chem Biol Interact. 2009;181(3):430–9. doi: 10.1016/j.cbi.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 118.Wu Y, Li W, Zhou C, Lu F, Gao T, Liu Y, Cao J, Zhang Y, Zhang Y, Zhou C. Ketamine inhibits lipopolysaccharide-induced astrocytes activation by suppressing TLR4/NF-ĸB pathway. Cell Physiol Biochem. 2012;30(3):609–17. doi: 10.1159/000341442. [DOI] [PubMed] [Google Scholar]
- 119.Montezano AC, Touyz RM. Molecular mechanisms of hypertension-reactive oxygen species and antioxidants: a basic science update for the clinician. Can J Cardiol. 2012;28(3):288–95. doi: 10.1016/j.cjca.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 120.Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Bush AI. N-acetyl cysteine for depressive symptoms in bipolar disorder—a double-blind randomized placebo-controlled trial. Biol Psychiatry. 2008;64(6):468–75. doi: 10.1016/j.biopsych.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 121.Magalhães PV, Dean OM, Bush AI, Copolov DL, Malhi GS, Kohlmann K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Berk M. N-acetylcysteine for major depressive episodes in bipolar disorder. Rev Bras Psiquiatr. 2011;33(4):374–8. doi: 10.1590/S1516-44462011000400011. [DOI] [PubMed] [Google Scholar]
- 122.Smaga I, Pomierny B, Krzyóanowska W, Pomierny-Chamioło L, Miszkiel J, Niedzielska E, Ogórka A, Filip M. N-acetylcysteine possesses antidepressant-like activity through reduction of oxidative stress: behavioral and biochemical analyses in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):280–7. doi: 10.1016/j.pnpbp.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 123.Hou Y, Wang L, Yi D, Ding B, Yang Z, Li J, Chen X, Qiu Y, Wu G (2013) N-acetylcysteine reduces inflammation in the small intestine by regulating redox, EGF and TLR4 signaling. Amino Acids (in press) [DOI] [PubMed]
- 124.Jung TS, Kim SK, Shin HJ, Jeon BT, Hahm JR, Roh GS. α-Lipoic acid prevents non-alcoholic fatty liver disease in OLETF rats. Liver Int. 2012;32(10):1565–73. doi: 10.1111/j.1478-3231.2012.02857.x. [DOI] [PubMed] [Google Scholar]
- 125.Deiuliis JA, Kampfrath T, Ying Z, Maiseyeu A, Rajagopalan S. Lipoic acid attenuates innate immune infiltration and activation in the visceral adipose tissue of obese insulin resistant mice. Lipids. 2011;46(11):1021–1032. doi: 10.1007/s11745-011-3603-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tian YF, Hsieh CH, Hsieh YJ, Chen YT, Peng YJ, Hsieh PS. α-Lipoic acid prevents mild portal endotoxaemia-induced hepatic inflammation and β cell dysfunction. Eur J Clin Invest. 2012;42(6):637–48. doi: 10.1111/j.1365-2362.2011.02630.x. [DOI] [PubMed] [Google Scholar]
- 127.Honda H, Nagai Y, Matsunaga T, Saitoh S, Akashi-Takamura S, Hayashi H, Fujii I, Miyake K, Muraguchi A, Takatsu K. Glycyrrhizin and isoliquiritigenin suppress the LPS sensor toll-like receptor 4/MD-2 complex signaling in a different manner. J Leukoc Biol. 2012;91(6):967–76. doi: 10.1189/jlb.0112038. [DOI] [PubMed] [Google Scholar]
- 128.Ghanim H, Sia CL, Korzeniewski K, Lohano T, Abuaysheh S, Marumganti A, Chaudhuri A, Dandona P. A resveratrol and polyphenol preparation suppresses oxidative and inflammatory stress response to a high-fat, high-carbohydrate meal. J Clin Endocrinol Metab. 2011;96(5):1409–14. doi: 10.1210/jc.2010-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim SJ, Lee SM. Effect of baicalin on toll-like receptor 4-mediated ischemia/reperfusion inflammatory responses in alcoholic fatty liver condition. Toxicol Appl Pharmacol. 2012;258(1):43–50. doi: 10.1016/j.taap.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 130.Hao H, Gufu H, Lei F, Dang L, Zhongliang Y. Baicalin suppresses expression of TLR2/4 and NF-κB in Chlamydia trachomatis-infected mice. Immunopharmacol Immunotoxicol. 2012;34(1):89–94. doi: 10.3109/08923973.2011.580756. [DOI] [PubMed] [Google Scholar]
- 131.Tu XK, Yang WZ, Shi SS, Chen Y, Wang CH, Chen CM, Chen Z. Baicalin inhibits TLR2/4 signaling pathway in rat brain following permanent cerebral ischemia. Inflammation. 2011;34(5):463–70. doi: 10.1007/s10753-010-9254-8. [DOI] [PubMed] [Google Scholar]
- 132.Ohno K, Ito M, Ichihara M, Ito M. Molecular hydrogen as an emerging therapeutic medical gas for neurodegenerative and other diseases. Oxid Med Cell Longev. 2012;2012:353152. doi: 10.1155/2012/353152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ito M, Ibi T, Sahashi K, Ichihara M, Ito M, Ohno K. Open-label trial and randomized, double-blind, placebo-controlled, crossover trial of hydrogen-enriched water for mitochondrial and inflammatory myopathies. Med Gas Res. 2011;1(1):24. doi: 10.1186/2045-9912-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li J, Dong Y, Chen H, Han H, Yu Y, Wang G, Zeng Y, Xie K. Protective effects of hydrogen-rich saline in a rat model of permanent focal cerebral ischemia via reducing oxidative stress and inflammatory cytokines. Brain Res. 2012;1486:103–11. doi: 10.1016/j.brainres.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 135.Xie K, Yu Y, Huang Y, Zheng L, Li J, Chen H, Han H, Hou L, Gong G, Wang G. Molecular hydrogen ameliorates lipopolysaccharide-induced acute lung injury in mice through reducing inflammation and apoptosis. Shock. 2012;37(5):548–55. doi: 10.1097/SHK.0b013e31824ddc81. [DOI] [PubMed] [Google Scholar]
- 136.Taylor KE, Giddings JC, van den Berg CW. C-reactive protein-induced in vitro endothelial cell activation is an artefact caused by azide and lipopolysaccharide. Arterioscler Thromb Vasc Biol. 2005;25(6):1225–30. doi: 10.1161/01.ATV.0000164623.41250.28. [DOI] [PubMed] [Google Scholar]
- 137.Kikkert R (2009) Toll-like receptors: tools, assays, and implications for in-vitro pyrogen tests. Thesis, University of Amsterdam, 29 September 2009
- 138.Józefowski S, Czerkies M, Sobota A, Kwiatkowska K. Determination of cell surface expression of Toll-like receptor 4 by cellular enzyme-linked immunosorbent assay and radiolabeling. Anal Biochem. 2011;413(2):185–91. doi: 10.1016/j.ab.2011.02.031. [DOI] [PubMed] [Google Scholar]