

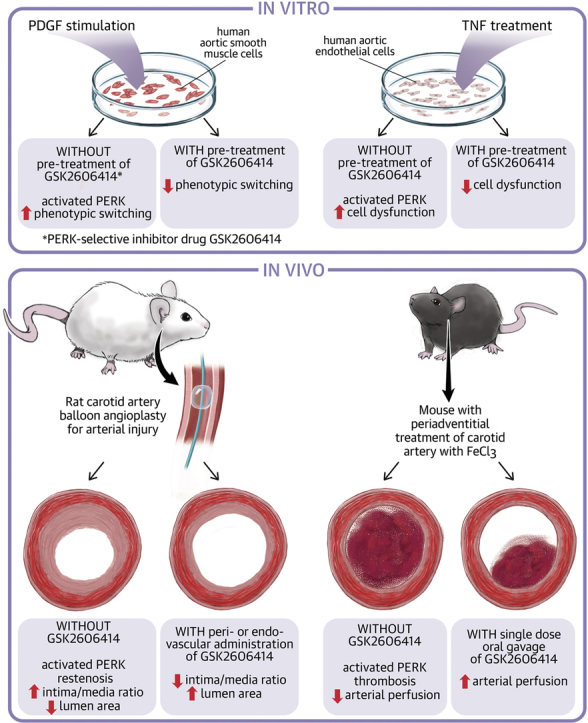
Visual Abstract
Key Words: endothelial cells, PERK, restenosis, smooth muscle cells, thrombosis
Abbreviations and Acronyms: Ad, adenovirus; ATF, activating transcription factor; CHOP, CCAAT-enhancer-binding protein homologous protein; DES, drug-eluting stents; DMSO, dimethyl sulfoxide; EC, endothelial cell; eIF2, eukaryotic translation initiation factor 2; ER, endoplasmic reticulum; FBS, fetal bovine serum; GFP, green fluorescent protein; HA, hemagglutinin; IEL, internal elastic lamina; IH, intimal hyperplasia; I/M, intima to media; IRE1, inositol-requiring kinase 1; MRTF-A, myocardin related transcription factor A; PDGF, platelet-derived growth factor; PDGF-BB, platelet-derived growth factor with 2 B subunits; PERK, protein kinase RNA-like endoplasmic reticulum kinase; siRNA, small interfering ribonucleic acid; SMA, smooth muscle actin; SMC, smooth muscle cell; SRF, serum response factor; STAT3, signal transducer and activator of transcription 3; TNF, tumor necrosis factor
Highlights
-
•
Drug-eluting stents impede neointimal smooth muscle cell hyperplasia but exacerbate endothelial cell dysfunction and thrombogenicity. It has been a challenge to identify a common target to inhibit both. Findings in this study suggest PERK as such a target.
-
•
A PERK inhibitor administered either via an endovascular (in biomimetic nanocarriers) or perivascular (in hydrogel) route effectively mitigated neointimal hyperplasia in rats.
-
•
Oral gavage of the PERK inhibitor partially preserved the normal blood flow in a mouse model of induced thrombosis.
-
•
Dampening PERK activity inhibited STAT3 while activating SRF in smooth muscle cells, and also reduced prothrombogenic tissue factor and growth impairment of endothelial cells.
Summary
Developing endothelial-protective, nonthrombogenic antirestenotic treatments has been a challenge. A major hurdle to this has been the identification of a common molecular target in both smooth muscle cells and endothelial cells, inhibition of which blocks dysfunction of both cell types. The authors’ findings suggest that the PERK kinase could be such a target. Importantly, PERK inhibition mitigated both restenosis and thrombosis in preclinical models, implicating a low-thrombogenic antirestenotic paradigm.
Cardiovascular disease is the number 1 cause of death in developed countries. Angioplasty with stenting is the most commonly performed procedure to reconstruct occluded vessels, accounting for ∼1 million cases worldwide and at a $11.18 billion expenditure in the United States alone (1). Unfortunately, reconstructed vessels, particularly those in the periphery, become renarrowed; this recurrent disease is referred to as restenosis. Restenosis occurs primarily due to the formation of neointimal lesions in the vessel wall, a process termed intimal hyperplasia (IH). Its central etiology is smooth muscle cell (SMC) phenotypic switching and endothelial cell (EC) dysfunction (2,3). These 2 cell types are vital because they compose the vasculature tunica media and endothelial inner lining. Angioplasty damages SMCs and the endothelial lining, exposing SMCs to a myriad of stimuli such as growth factors and cytokines. In response, SMCs undergo phenotypic changes and lose normal functions. Specifically, phenotypically switched SMCs dedifferentiate, becoming contraction deficient and proliferative/migratory cells that form neointima (4,5). Thus, an ideal antirestenotic treatment should not only block SMC’s phenotypic switching but also be EC-protective (6,7).
Drug-eluting stents (DES) are commonly deployed to reduce post-angioplasty restenosis. However, they do not completely prevent IH and, in fact, worsen thrombogenic risks (6). In-stent restenosis occurs in ∼5% to 10% of coronary DES applications (8) and >50% peripheral DES applications (9). Moreover, stent thrombosis has become a major concern, with up to 40% sudden death among those patients (10). Increasing evidence reveals that DES mitigate the disease phenotypes of SMCs but not that of ECs; rather, they elicit EC dysfunction (11,12). Dysfunctional ECs become growth-impaired and apoptotic, and produce thrombogenic factors (e.g., tissue factor) while releasing proinflammatory signal molecules that enhance SMC phenotypic switching thereby promoting IH and thrombosis (7,13). Therefore, the ultimate outcome of DES implantation for restenosis prevention, paradoxically, can be the persistence of in-stent restenosis and exacerbated thrombogenic risks in many patients (6). Anticoagulant therapies cannot completely prevent stent thrombosis, and long-term application incurs a significant risk of hemorrhage (14). To tackle the downsides of DES, alternative strategies such as drug-eluting balloons and biodegradable stents have been clinically tested. However, their applications still rely on EC-toxic drugs, and recent studies have demonstrated increased mortality associated with their usage (15). Therefore, there is a clear clinical need for an endothelium-protective and stent-free antirestenotic therapy (16). Currently, basic and translational research to address this need has been limited (11,17). In order to rescue the endothelium thereby reducing thrombogenic risk while mitigating IH, it is essential to identify and inactivate a common target in both SMCs and ECs that is responsible for their disease phenotypes.
Recently, endoplasmic reticulum (ER) stress response pathways have emerged as novel interventional targets for cardiovascular diseases. Three unique pathways, represented by inositol-requiring kinase 1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase RNA-like endoplasmic reticulum kinase (PERK), are selectively or simultaneously activated depending on the ER stress context (18,19). Of note, the IRE1 and ATF6 pathways have been reported to play a significant role in restenosis and other vascular diseases (18,19). By contrast, the role of PERK in restenosis remained unclear. In this pathway, PERK serves as the sensor for ER stress; its autophosphorylated form activates elongation initiation factor (eIF)-2α. Activated eIF2α suppresses global protein translation while paradoxically activating the transcription of subsets of genes in a context-dependent fashion, for example, ATF4, a key counter-stress transcription factor (20). In addition, activated PERK can also regulate other signaling pathways via its kinase activity (21). Therefore, global translational repression, together with selective activation of a subset of genes as well as crosstalk with other pathways, all elicited by PERK activation, may define specific outcomes in a context (e.g., cell type) dependent manner (22).
Context dependence of PERK-mediated responses and implications from the other 2 ER-stress response pathways led us to explore a potential involvement of PERK in restenosis. We found that PERK inhibition was robustly effective in stymying restenosis in a rat carotid artery angioplasty model. More significantly, we further uncovered that PERK inhibition was also antithrombotic in a FeCl3-induced mouse model. This finding was unexpected given that an antithrombotic effect has not been shown in the interventions of the other 2 pathways. Selective inhibitors for PERK have been well developed, and the first-in-class GSK2606414 has demonstrated outstanding therapeutic efficacy in multiple disease models, including prion infection, Alzheimer’s disease, diabetes, hemorrhagic stroke, and cancers (23). In the current study, we explored the potential of PERK inhibition as an antirestenotic option featuring low thrombogenic risks.
Methods
Ethics statement
All animal studies conform to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication No. 85–23, 1996 revision) and protocols approved by the institutional animal care and use committee at the University of Wisconsin and The Ohio State University.
Reagents and materials
Human aortic SMCs and ECs and their respective media were purchased from Lonza (Walkersville, Maryland). BrdU (5-bromo-2′-deoxyuridine) enzyme-linked immunosorbent assay colorimetric kit was purchased from Roche-Sigma Aldrich (Indianapolis, Indiana). Human recombinant platelet-derived growth factor (PDGF) with 2 B subunits (PDGF-BB) and tumor necrosis factor (TNF)-α were purchased from R&D Systems (Minneapolis, Minnesota). TRIzol, High Capacity cDNA synthesis kit, SYBR Green PCR Master Mix, and small interfering ribonucleic acids (siRNAs) (scrambled control and PERK-targeting siRNA) were purchased from Thermo Fisher (Waltham, Massachusetts). GSK2606414 was purchased from Apexbio Technology (Houston, Texas).
Primary vascular SMC and EC cultures
Human aortic SMCs and ECs were cultured in SmGM-2 complete medium containing 5% fetal bovine serum (FBS) and EGM-2 complete medium containing 2% FBS, respectively. Cells between passages 5 and 7 were used for all experiments and maintained at 37°C with 5% CO2. For cell culture expansion, 0.25% Trypsin was used for detachment of human SMCs, whereas Accutase (Thermo Fisher Scientific) was used for human ECs.
For siRNA-mediated PERK knockdown, small interfering ribonucleic acids (siRNAs) for PERK (Eif2ak3) were ordered and tested for knockdown efficiency. Two validated siRNAs (4390824, Silencer Select, and AM51331, Silencer; Thermo Fisher Scientific) were transfected into SMCs by using RNAiMax Reagent (13778-075, Thermo Fisher Scientific) following the manufacturer’s protocol at a concentration of 50 nmol/l.
For in vitro PERK overexpression, adenoviral vectors expressing green fluorescent protein (GFP) (Ad-GFP) and human PERK (Ad-PERK) were constructed using a 1-step Adeno-X adenoviral system (632267 and 632266, Takara Bio, Mountain View, California) following the manufacturer’s instructions. Virus were amplified, purified, and then titrated as previously described (1). SMCs were infected for 6 h with adenoviruses (3 × 104 particles/cell) in SmBm2 basal medium containing 2% FBS, and recovered for 20 h with 10% FBS, and then starved with 0.5% FBS for 24 h followed by treatments as illustrated in each experiment.
Preparation of delivery tools for PERK inhibitor in vivo application
Platelet membrane-coated nanoclusters were synthesized as previously described (16). Briefly, PAMAM-PVL-OH was reacted with succinic anhydride with presence of 4-dimethylaminopyridine at a weight ratio of 100:16.4:25 to generate PAMAM-PVL-COOH for nanocluster synthesis. PAMAM-PVL-COOH was then dialyzed against distilled water to remove impurities, and then lyophilized for long-term storage. For nanocluster drug loading, PAMAM-PVL-COOH and PERK inhibitor GSK2606414 were dissolved in dimethyl sulfoxide (DMSO) at a weight ratio of 5:2. Distilled water was then added drop-wise into the aforementioned solution over the course of 30 min. The unloaded drugs and DMSO were subsequently removed by dialysis. Drug-loaded nanoclusters were then fused with platelet membrane vesicles under sonication for 2 min (100 μg of PAMAM to 20 μl of platelet membrane solution).
Perivascular triblock gel was synthesized as previously described (24). Briefly, heat-dried OH–PEG–OH was reacted with LA and GA at a weight ratio of 2.4:5:1.2 and then dried under vacuum for 30 min at 70°C. Upon complete melting, the mixture was then added with Sn(Oct)2 as catalyst for initiation of the polymerization process, at a catalyst: (LA:GA) molar ratio of 1:500. After 8 h of reaction at 150°C, the mixture was dissolved in 4°C cold water and then reheated to 80°C to precipitate the copolymers and remove other impurities; this process was repeated 3 times. The final product was lyophilized for long-term storage, and the triblock gel working solution was prepared by dissolving the polymer in water at 23% by weight.
Cell proliferation assay
BrdU enzyme-linked immunosorbent assay cell proliferation assay (Abcam, Cambridge, Massachusetts) was performed following the manufacturer’s instructions. For SMCs: SMCs were seeded in 96-well plates at a density of 4,000 cells per well with a final volume of 200 μl, and starved for 24 h in SmBM-2 basal medium containing 0.5% FBS. Cells were then pre-treated with 1 μmol/l GSK2606414 or an equal volume of vehicle control (DMSO) for 2 h in fresh starvation medium before PDGF-BB mitogenic stimulation (final 10 ng/ml). For ECs: ECs were seeded in 96-well plates at a density of 8,000 cells per well with a final volume of 200 μl of EBM-2 medium containing 10% FBS. Cells were then pre-treated with 1 μmol/l GSK2606414 or an equal volume of vehicle control (DMSO) for 2 h in fresh EBM-2 medium before TNF-α stimulation (final 20 ng/ml). At 22 h after stimulation, cells were labeled with BrdU by a 2 h incubation at 37°C, and then fixed with a FixDenat solution for 30 min, followed by a 90-min incubation at room temperature with an anti-BrdU-POD antibody (1:100 dilution). After washing with phosphate-buffered saline for 3 times, substrate was added. Plates were incubated at room temperature for 30 min, and then colorimetric signals were measured on a FlexStation 3 Benchtop Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, California) at 370 nm with a reference wavelength of 492 nm.
Real-time quantitative polymerase chain reaction
mRNA was isolated from cultured primary cells or rat carotid arteries snap-frozen in liquid nitrogen, using TRIzol following the manufacturer's instructions. Purified mRNA (1 μg) was used for the first-strand complementary DNA synthesis, and quantitative real-time polymerase chain reaction was performed using the 7500 Fast and QuantStudio 3 Real-Time PCR Systems (Applied Biosystems, Carlsbad, California). Each cDNA template was amplified in triplicates using SYBR Green PCR Master Mix.
Cellular and tissue protein extraction and western blotting
SMCs and ECs were lysed in radioimmunoprecipitation assay buffer containing protease inhibitors (50 mmol/l Tris, 150 mmol/l NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, and 10 μg/ml aprotinin). For tissue protein extraction, freshly harvested tissues were immediately snap-frozen in liquid nitrogen and completely pulverized with a plastic mini-pestle in a liquid nitrogen bath, followed by addition of radioimmunoprecipitation assay buffer. Protein concentrations of cell or tissue lysates were determined using a Bio-Rad DC Protein Assay kit (Bio-Rad Laboratories, Hercules, California). Approximately 50 μg of proteins from each sample were separated on 4% to 20% Mini-PROTEAN TGX precast gels (Bio-Rad Laboratories) and transferred to a polyvinylidene fluoride membrane. Proteins of interest were detected by immunoblotting using the following primary antibodies and dilution ratios (Supplemental Table S1): Rabbit anti-phospho-PERK (1:100) from Santa Cruz Biotechnology (sc-32577; Santa Cruz Biotechnology, Dallas, Texas), rabbit anti-phospho-eIF2α (1:1000) from Cell Signaling Technology (3398; Cell Signaling Technology, Danvers, Massachusetts), mouse anti–phospho-signal transducer and activator of transcription 3 (STAT3) (1:1,000) from Cell Signaling Technology (4113), rabbit anti–phospho-p65 (1:1,000) from Cell Signaling Technology (3033), rabbit anti-myocardin related transcription factor A (MRTF-A) (1:1,000) from Cell Signaling Technology (14760), rabbit anti-CD142/Tissue Factor (1:500) from Thermo Fisher (PA5-27278), rabbit anti–CAAT-enhancer-binding protein homologous protein (CHOP) (1:100) from Santa Cruz Biotechnology (sc-8327), rabbit anti-ATF4 (1:1,000) from (ab184909, Abcam, Cambridge, Massachusetts), rabbit anti-cyclin D1 (1:1,000) from Abcam (ab134175), mouse anti-smooth muscle α-actin (α-SMA) (1:5,000) from Sigma-Aldrich (St. Louis, Missouri), and mouse anti–β-actin (1:5,000) from Sigma-Aldrich. After incubation of the blots with horseradish peroxidase–conjugated secondary antibodies (1:3,000 for goat anti-rabbit or 1:10,000 for goat anti-mouse, Bio-Rad Laboratories), specific protein bands on the blots were visualized by applying enhanced chemiluminescence reagent Clarity Max Western ECL Substrate (Bio-Rad Laboratories) and then recorded with a LAS-4000 Mini imager (GE, Piscataway, New Jersey) and an Azure C600 imager (Azure Biosystems, Dublin, California). Band intensity was quantified using ImageJ software, version 1.49 (NIH, Bethesda, Maryland).
Rat carotid artery balloon angioplasty
Carotid artery balloon angioplasty was performed in male Sprague–Dawley rats (300 to 350 g, Charles River Laboratories, Wilmington, Massachusetts) as previously described (16). Briefly, rats were anesthetized with isoflurane (5% for inducing and 2.5% for maintaining anesthesia). A longitudinal incision was made in the neck, and the carotid arteries were exposed. A 2-F balloon catheter (Edwards Lifesciences, Irvine, California) was inserted through an arteriotomy on the left external carotid artery. To produce arterial injury, the balloon was inflated at a pressure of 1.5 atm and withdrawn to the carotid bifurcation; this action was repeated 3 times. The external carotid artery was then permanently ligated, and blood flow was resumed. Throughout the surgery, the animal was kept anesthetized via isoflurane inhaling at a flow rate of 2 l/min. Carprofen (5 mg/kg) and bupivacaine (0.25% at the incision site) were injected subcutaneously. Sample size was determined using power analysis (G*Power software, version 3.1.7, Heinrich Heine University, Dusseldorf, Germany), based on our existing data and pilot study. Two weeks after balloon angioplasty, common carotid arteries were collected from anesthetized animals (under 2.5% isoflurane) following perfusion fixation at a physiological pressure of 100 mm Hg. Animals were euthanized in a chamber gradually filled with CO2. Sample size was determined using power analysis (G*Power software), on the basis of our existing data and pilot study.
In vivo overexpression of PERK in angioplastied carotid arteries was performed as we have previously described with modifications (25). Briefly, immediately after angioplasty, an intravenous catheter (24GA, Insyte Autoguard; Becton Dickinson, Franklin Lakes, New Jersey) was inserted through the previously created arteriotomy on the external carotid artery and advanced past the bifurcation while hemostats were still in place for the common and internal carotid arteries. The catheter and external carotid artery were then tightly fastened using ligatures to prevent leaking. A syringe containing adenovirus solution was connected to the catheter, and its position was adjusted and secured using a flexible magnetic mount arm platform (Quadhands, Charleston, South Carolina). A total of 150 μl of adenoviral solution (>1 × 109 infectious units/ml) was then gradually infused into the common carotid artery to create local adenoviral infection, and incubated for 25 min. Saline-soaked gauge or Kimwipe were applied to keep the exposed carotid arteries and surrounding tissues from drying. Upon the completion of adenovirus solution infusion and incubation, the catheter was then withdrawn, and the common carotid artery lumen was then flushed repeatedly with saline containing 20 U/ml heparin. Heparin was administered perioperatively to prevent thrombosis during the procedure, especially in the temporarily ligated internal carotid artery. Back flushing/bleeding from both common and internal carotid arteries was confirmed. In addition to carprofen and bupivacaine, buprenorphine was also subcutaneously injected (0.05 mg/kg) for this experiment. Sample size was determined using power analysis (G*Power software), on the basis of our existing data and pilot study.
Morphometric analysis of neointima
Paraffin sections (5 μm thick) were excised from carotid arteries at equally spaced intervals and then Verhoeff-van Gieson stained for morphometric analysis, as described in our previous reports (16). Planimetric parameters as follows were measured on the sections and calculated using Image J software: area inside external elastic lamina, area inside internal elastic lamina (IEL) area, lumen area, intima area (intima area = IEL area − lumen area), and media area (media area = external elastic lamina area − IEL area). Intimal hyperplasia was quantified as a ratio of intima area versus media area. Measurements were performed by a researcher blinded to the experimental conditions using 3 to 6 sections from each of animal. The data from all sections were pooled to generate the mean for each animal. The means from all the animals in each treatment group were then averaged, and the standard error of the mean (SEM) was calculated.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described. Briefly, paraffin sections of rat carotid arteries were deparafinized, followed by antigen retrieval using a decloaking chamber (Decloaking Chamber NxGen, DC2012, Biocare Medical, Pacheco, California). The slides were incubated without (negative staining) or with primary antibodies for 12 h and rinsed at least 3 times. The sections were then incubated with an anti-rabbit/mouse secondary antibody conjugated with Alexa Fluor 594 (A-11037/A-21203, Invitrogen, Carlsbad, California) or anti-mouse secondary antibody conjugated with Alexa Fluor 488 (ab150117, Abcam) for 1.5 h and rinsed. The specific antigen was then visualized with EVOS FL Auto 2 Imaging System (Thermo Fisher Scientific). Detailed information of antibodies is included in Supplemental Table S1. For quantification, 3 to 4 immunostained sections from each animal were used. Fluorescence intensity in each image field was quantified by using ImageJ software and normalized to the number of DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei in the media and neointima layers. The values from all sections were pooled to generate the mean for each animal.
Luciferase assay for SRF transcriptional activity
Luciferase assay was performed as previously described (26). Primary human aortic SMCs were first transfected with a luciferase reporter vector, and these transfected SMCs were then split for infection with Ad-GFP and Ad-PERK so as to rule out variation in luciferase vector transfection efficiency between these 2 groups. Specifically, human primary aortic SMCs were transfected with pGL4.34 Vector plasmids (E1350, Promega, Madison, Wisconsin) using Effectene Transfection Reagent (301425, Qiagen, Germantown, Maryland). After screening with Hygromycin B, SMCs were then recovered for another passage and then seeded in 24-well plates at a density of 20,000 cells/well in complete SmGm2 medium after being detached using Accutase. Cells were then infected with adenovirus as aforementioned and cultured for an additional 24 h before lysis in Bright-Glo (2610, Promega). Luminescence signals were measured on a FlexStation 3 Benchtop Multi-Mode Microplate Reader (Molecular Devices), and CellTiter-Glo luminescence readings from duplicate wells were used for normalization.
Coimmunoprecipitation assay
All co-immunoprecipitation assays were performed in MOVAS SMC cell lines (CRL-2797, ATCC, Manassas, Virginia) as previously described (26). Briefly, cells were stably transfected with either hemagglutinin (HA) or HA-STAT3 vectors, followed by selection with ampicillin and subsequent treatment with PDGF-BB and GSK2606414. At the end of each treatment, cells were collected by using an immunoprecipitation lysis buffer (87787, Thermo Fisher Scientific) containing a protease inhibitor cocktail (78430, Thermo Fisher Scientific), and kept on ice for 30 min to ensure complete lysis. Cell lysates of equal protein amounts were incubated overnight at 4°C (constant rotation) with 2 μg of an anti-HA antibody or immunoglobulin G control (sc2027, Santa Cruz Biotechnology). Coimmunoprecipitation of STAT3 with associated proteins was then performed by using Protein A/G Agarose beads included in the Pierce Classic IP Kit (26146, Thermo Fisher Scientific) followed by immunoblotting to detect immunoprecipitated HA/HA-STAT3 or coimmunoprecipitated PERK and MRTF-A.
FeCl3-induced arterial thrombosis model
Adult C57BL/6J wild-type mice were administered either GSK2606414 at the dosage of 150 mg/kg or vehicle control as a single oral gavage. Compound was mixed in 0.5% carboxymethyl cellulose and 0.05% Tween. Four hours after treatment, mice were anesthetized with ketamine (55 mg/kg) plus xylazine (15 mg/kg), and rectal temperatures were maintained at 37°C by a thermo-regulated heating pad (Physitemp TCAT-2DF Controller, Physitemp, Clifton, New Jersey). Noninvasive blood pressure by tail cuff measurement was recorded at baseline before injury and 60 min after injury (CODA, Kent Scientific, Torrington, Connecticut). The common carotid artery was gently dissected; and a pulsed Doppler flow probe (MA0.5PSB, Transonic Systems, Ithaca, New York) placed to record blood flow velocity. Electrocardiography leads were placed to allow monitoring of electrocardiograms in addition to continuous heart rate recording throughout the entire procedure (LabChart, ADInstruments, Sydney, Australia). Carotid artery injury was induced by application of Whatman filter paper (1 mm2) saturated with 10% FeCl3 solution on the adventitial surface proximal to the flow probe for 3 min. The flow as a percentage of baseline and the time to thrombotic occlusion (blood flow, 0 ml/min) was measured from the placement of the FeCl3-saturated filter paper. Animals were reanesthetized as needed with ketamine to maintain deep anesthesia throughout the procedure and sacrificed 60 min after initiation of FeCl3 application. Both FeCl3-applied carotid arteries and the contralateral uninjured carotid arteries were collected for histology purposes. Sample size was determined using power analysis (G*Power), based on our existing data and pilot study.
Ex vivo culture of aortic rings
Male Sprague–Dawley rats (300 to 350 g, Charles River Laboratories) were sacrificed and perfused with cold heparinized Hanks balanced salt solution. Thoracic aortas were collected, with all branches carefully dissected and removed in heparinized Hanks balanced salt solution. Aortas were cut into rings of ∼5 mm in length, and cultured in Dulbecco's Modified Eagle Medium supplemented with 1% FBS in 24-well plates. Rings were evenly distributed in each well, and were pre-treated with 1 μmol/l GSK2606414 at least 2 h before stimulation with TNF-α at a final concentration of 100 ng/ml. At 4 h post-stimulation, aortic rings were rinsed and snap-frozen in liquid nitrogen for protein extraction.
Statistical analysis
Statistical analyses were conducted using either a nonparametric test (Mann-Whitney test for unpaired 2-group) or parametric test (Student’s t-test or 1-way analysis of variance followed by post hoc Bonferroni), on the basis of the normality of data determined by the Shapiro-Wilk normality test. Data are presented as mean ± SEM or median with interquartile range accordingly, as specifically stated in figure legends. Statistical significance was set at p < 0.05. GraphPad Prism software, version 7.04 (GraphPad Software, San Diego, California) was used for all statistical analyses.
Results
Endovascular delivery of PERK inhibitor via a biomimetic nano approach markedly mitigates neointima lesion and restenosis in a rat balloon angioplasty model
Recently, we have developed a novel biomimetic platform that could achieve stent-free, targeted delivery to vascular lesions (16). Here, we applied this technology to the preclinical therapeutic evaluation of PERK inhibition in the context of antirestenotic intervention, as a concerted effort to provide a safer and effective alternative to DES. In our previous study, platelet membrane-coated (biomimetic) nanoclusters proved to be nonimmunogenic and able to home to the injured artery wall following intravenous injection in the rat balloon angioplasty model (16). To evaluate the effect of PERK inhibition on IH, we injected these biomimetic nanoclusters loaded with GSK2606414 (dosage of 2.5 mg/kg) into the tail vein immediately after balloon angioplasty. The second injection followed at day 5 post-angioplasty (Figure 1A). At day 14, arteries were collected for histological analysis (Figure 1B). An inhibitory effect of GSK2606414 on PERK activation was confirmed by reduced phospho-PERK immunofluorescence (Supplemental Figure S1). Morphometric analysis showed a profound antirestenotic effect of GSK2606414 compared with vehicle control, with a >70% reduction in the intima to media (I/M) ratio and ∼30% increase in lumen area (Figure 1C). Of note, this 2.5 mg/kg dose is approximately one-fifth of that used in a perivascular hydrogel delivery route (see later in the text) (Figure 2A). More significantly, it is only about 1/250 to 1/800 of that in a delivery regimen of consecutive 14 days (50 to 150 mg/kg, twice daily by mouth) commonly used in the published reports (23,27). To the best of our knowledge, we have provided herein the first in vivo evidence showing the antirestenotic efficacy of PERK inhibition. In addition, delivery of PERK inhibitor via an injectable biomimetic nanoplatform may cater to the clinical need for stent-free antirestenotic therapies.
Figure 1.

Endovascular Targeted Delivery of PERK Inhibitor via a Biomimetic Nano Approach Mitigates IH in the Rat Model of Carotid Artery Angioplasty
Drug delivery via biomimetic (platelet membrane-coated) nanoclusters that home to balloon-injured sites of rat carotid arteries was established in our recent report (16). We used this targeted delivery method in the same rat angioplasty model for administration of vehicle (Veh) (dimethyl sulfoxide control) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) inhibitor (GSK2606414 [GSK], 2.5 mg/kg). Biomimetic nanoclusters were tail-vein injected twice (immediately and 96 h after angioplasty). Arteries were collected 14 days after angioplasty for morphometric analysis. (A) Schematic illustration of tail-vein administration of PERK inhibitor carried in biomimetic nanoclusters. (B) Representative Verhoeff-van Gieson (VVG)-stained sections. Neointima lesion is indicated between arrowheads. Scale bar: 100 μm for 10×, 50 μm for 40×. (C) Quantification of IH (the intima/media [I/M] area ratio) and lumen area. Mean ± SEM, n ≥ 4 animals; *p < 0.05, **p < 0.01, ***p < 0.001 1-way analysis of variance with Bonferroni post hoc test. IH = intimal hyperplasia; IV = intravenous; r.u. = relative units.
Figure 2.

Perivascular Local Delivery of PERK Inhibitor Mitigates IH
Vehicle or PERK inhibitor (GSK2606414, 25 mg/kg) carried in a slow-release triblock gel was perivascularly applied immediately after balloon angioplasty. Arteries were collected 14 days after angioplasty for morphometric analysis. (A) Schematic illustration of periadventitial (common carotid artery) administration of PERK inhibitor carried in a triblock hydrogel. (B) Representative VVG-stained sections. Neointima lesion is indicated between arrowheads. Scale bar: 100 μm for 10×, 50 μm for 40×. (C) Quantification of I/M area ratio. (D) Quantification of lumen area. Mean ± SEM, n = 4 to 5 rats; *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student’s t-test. Abbreviations as in Figure 1.
Perivascular local delivery of PERK inhibitor effectively ameliorates neointima lesion and restenosis
To draw a solid conclusion on the antirestenotic efficacy of PERK inhibition and to broaden its potential therapeutic application, we next tested GSK2606414 in a perivascular delivery route in the same rat balloon angioplasty model. To this end, we used a PLGA-PEG-PLGA thermosensitive triblock gel (liquid on ice and semisolid at body temperature), which in our previous studies exhibited excellent stability and a steady sustained (∼4-week) drug release profile (24). Using this gel system, we applied GSK2606414 (25 mg/kg, 1-time delivery) to the periadventitial space immediately after angioplasty (Figure 2A). Morphometric analysis (Figure 2B) of arteries collected at day 14 indicated an ∼80% reduction of the I/M ratio (Figure 2C) and an ∼50% increase of lumen area (Figure 2D) in comparison to vehicle control. These results confirmed a robust antirestenotic effect of the PERK inhibitor. Moreover, because this perivascular delivery method is in line with a prospective antirestenotic management of open surgeries (e.g., bypass vein grafts) (28), PERK inhibition has a potential to be broadly applied.
The PERK pathway is activated in balloon angioplasty-injured arteries; its overexpression increases neointima
Given the antirestenotic efficacy of PERK inhibitor, we next determined a possible injury-induced activation of the PERK pathway. Uninjured and injured rat common carotid arteries at day 3, day 7, and day 14 post-angioplasty were harvested, and their homogenates were used for Western blot analysis of PERK pathway proteins. The data showed that dramatic PERK activation (a 4-fold increase of phospho-PERK) occurred in the artery 3 days after injury and persisted throughout the 14-day time course (Figures 3A and 3B). This time-course result was confirmed by immunostained P-PERK on artery cross sections (Figure 3C, Supplemental Figure S2). Although immunofluorescence showed P-PERK in the media layer at day 3 post-injury, as indicated by the staining of SMC marker α-SMA, its distribution shifted at day 7 and day 14 to the neointima layer, where SMCs are typically dedifferentiated as evidenced by reduced α-SMA. In accordance, key players downstream of PERK were also up-regulated, with P-eIF2α and CHOP peaking at day 3, and ATF4 most expressed at day 7 (Supplemental Figure S2). eIF2α as a central effector kinase is one of the direct substrates of PERK (29). Another central player in ER stress responses, ATF4, is a transcription factor downstream of eIF2α. CHOP is a downstream transcription factor of the extended PERK pathway and is often associated with cell death when cellular stress is too severe (30). It is noteworthy that the up-regulation of these players did not last throughout post-injury day 14 as P-PERK did. On the one hand, PERK has multiple alternative substrates such as NRF2 (31). On the other hand, the P-eIF2α-ATF4-CHOP axis is also regulated by kinases other than PERK, for example, GCN2 and PKR (32). Therefore, we infer that given a 14-day time span, cross-talks of different pathways in the injured artery may have rendered the upregulation of the PERK downstream players temporally variable. Nonetheless, these results collectively indicate that the PERK pathway was strongly activated due to arterial injury, consistent with a role in neointima development and its targetability by an inhibitor as evidenced above (Figures 1 and 2).
Figure 3.

Injury-Induced Activation of the PERK Pathway and Exacerbated IH in Balloon-Injured Rat Carotid Arteries Overexpressing PERK
(A) Activation of the PERK pathway in injured arteries. Following angioplasty, rat common carotid arteries were harvested at days 3, 7, and 14 (3 to 14 days) and homogenized for immunoblotting analysis. (B) Quantitation of PERK phosphorylation level following rat balloon angioplasty. Mean ± SEM, n = 3 rats; *p < 0.05, 1-way analysis of variance with Bonferroni post hoc test. (C) Increase of P-PERK on cross sections of injured arteries. Immunofluorescent staining of P-PERK and α-SMA was performed as described in Methods. Shown are representative images, and quantified data are presented in Supplemental Figure S2. Neointima lesion is indicated between arrowheads. (D) In vivo PERK gain-of-function. Adenovirus (Ad) GFP (Ad-GFP) or Ad-PERK was intraluminally infused with the injured artery for 25 min immediately after balloon angioplasty. Shown are representative VVG-stained sections. Scale bar: 100 μm for 10×, 50 μm for 40×. (E and F) Quantification of I/M area ratio and lumen area. Mean ± SEM, n = 6 rats; *p < 0.05, unpaired Student’s t-test. ATF = activating transcription factor; CHOP = CCAAT-enhancer-binding protein homologous protein; GFP = green fluorescent protein; SMA = smooth muscle actin; other abbreviations as in Figure 1.
We then further verified PERK’s specific role in promoting neointima, via its overexpression using adenovirus intraluminally infused into the artery wall following endothelium-denuding angioplasty. Effective transduction of PERK-expressing adenovirus was confirmed by Western blot analysis of artery homogenates (Supplemental Figure S3A). Morphometric analysis of post-angioplasty 14-day artery sections indicates a significantly higher I/M ratio in arteries transduced with PERK-expressing adenovirus compared with the Adeno-GFP empty vector control (Figures 3D to 3F). Additionally, using tissue mRNA samples from a portion of the injured carotid artery, we also observed hallmarks of phenotypic switching, including reduced expression of α-SMA (Supplemental Figure S3B). Therefore, these gain-of-function results support a specific role for PERK in promoting neointima formation.
PERK activation promotes SMC phenotype switching
Phenotypic switching of SMCs is the core cellular mechanism underlying the development of neointima lesion and consequential restenosis (5). We therefore determined the specific function of PERK in this process via loss- and gain-of-function experiments. We used PDGF-BB, a widely used potent inducer, to stimulate SMC phenotypic switching which was measured as enhanced de-differentiation (decreased expression of SMC markers) and proliferation (BrdU incorporation and cyclin D1 expression). As shown in Figure 4A, whereas treatment of human primary aortic SMCs with PDGF-BB stimulated PERK activation (Figures 4A, 4E, and 4I) and cell proliferation (Figure 4C, 4G, and 4K), and reduced α-SMA by 2- to 3-fold (Figures 4D, 4H, and 4L), pre-treatment with the PERK inhibitor GSK2606414 (1 μmol/l) abrogated these PDGF-induced changes (Figures 4A to 4D). We then confirmed this result via gene knockdown using a PERK-specific siRNA. The data indicate that PERK silencing versus scrambled siRNA control closely phenocopied the effects of GSK2606414 on SMC phenotypic switching (Figures 4E to 4H). Importantly, consistent evidence was also derived from the gain-of-function experiments. As shown in Figures 4I to 4L, PDGF-BB stimulated SMC proliferation and cyclin D1 expression and reduced α-SMA, this effect was enhanced in cells transduced with PERK-expressing adenovirus compared with empty-vector control. We also determined 2 other authentic SMC markers, calponin (CNN1) and myosin heavy chain (MYH11), and found a pattern of change similar to that of α-SMA (Supplemental Figure S4). In vivo, whereas PERK overexpression in injured arteries reduced α-SMA mRNA levels (Supplemental Figure S3), PERK inhibitor versus vehicle (through either endo- or perivascular delivery) prominently increased α-SMA while reducing P-PERK immunofluorescence (Supplemental Figure S1). Combined, these loss- and gain-of-function results support a specific role of PERK in promoting SMC phenotypic switching in vitro and in vivo.
Figure 4.

PERK Loss- and Gain-of-Function Respectively Abrogate and Exacerbate SMC Phenotypic Switching
To determine the specific role of PERK in smooth muscle cell (SMC) phenotypic switching (reduced α-SMA and increased proliferation), human primary aortic SMCs were cultured, starved, and pretreated with PERK inhibitor (A to D), small interfering ribonucleic acid (siRNA) (E to H), or adenovirus (I to L), and then treated with solvent or platelet-derived growth factor (PDGF)-BB (100 ng/ml). Cells were harvested 24 h after PDGF stimulation for Western blot analysis of PERK pathway proteins and BrdU proliferation assay. (A to D) Pharmacological blockade of PERK kinase activity. Cells were pre-treated with vehicle (dimethyl sulfoxide) or 1 μmol/l GSK2606414 for 2 h before adding PDGF-BB. (A) Representative immunoblots of PERK pathway (P-PERK and P-eIF2α) and SMC phenotypic switching (α-SMA and CyclinD1). (B and D) Quantitation of PERK phosphorylation level and α-SMA expression in SMCs stimulated with PDGF-BB and PERK inhibitor pre-treatment. Protein band densitometry was normalized to β-actin; n = 3. (C) Proliferation of SMCs were determined using BrdU incorporation enzyme-linked immunosorbent assay; n = 3. (E to H) PERK genetic silencing. Starved SMCs were transfected with 50 nmol/l PERK-specific siRNA for 48 h, and then starved for 24 h before PDGF stimulation. Similar to A to D, immunoblots of PERK pathway and SMC phenotypic switching markers are presented (E), together with proliferation assay (G; n = 4) and quantitation of PERK phosphorylation level (F; n = 3) and α-SMA expression (H; n = 3). (I to L) Adenovirus-mediated PERK gain-of-function. Cells were transduced with Ad-GFP or Ad-PERK for 6 h, recovered for 24 h and then starved for 24 h before PDGF stimulation. Similar to A to D and E to H, immunoblots of PERK pathway and SMC phenotypic switching markers are presented (I), together with proliferation assay (K; n = 4) and quantitation of PERK phosphorylation level (J; n = 3) and α-SMA expression (L; n = 3). All data are presented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, 1-way analysis of variance with Bonferroni post hoc test. eIF = eukaryotic translation initiation factor; IB = immunoblot; other abbreviations as in Figures 1 and 3.
PERK activation is paralleled by STAT3 activation and SRF inhibition upon SMC phenotypic switching
To investigate the molecular mechanism underlying PERK’s functional role in SMC phenotypic switching, we explored its possible regulation of the determinants of SMC phenotypes. Serum response factor (SRF) is a well-established master transcription factor that governs α-SMA expression and SMC differentiation; myocardin family proteins such as MRTF-A act as cofactors by activating SRF (33). It is well-documented that suppressed MRTF/SRF signaling leads to SMC phenotypic switching (34). In addition, recent studies have shown that STAT3 is highly sensitive to a range of cytokines, and it acts as a major mediator of SMC phenotypic switching by binding myocardin family proteins, hence sequestering them from activating SRF (35). Interestingly, we observed that PERK inhibition with either GSK2606414 or siRNA nearly abolished PDGF-induced STAT3 activation (Figures 5A and 5B). Further, coimmunoprecipitation experiments showed that HA-STAT3 specifically pulled down endogenous PERK in a PDGF-dependent manner, and this STAT3/PERK association was blocked by pre-treatment with the PERK inhibitor (Figure 5C). Moreover, the PDGF-induced and PERK inhibitor-blocked coimmunoprecipitation was also observed between HA-STAT3 and MRTF-A, similar to the published evidence (36). We were thus prompted to determine whether PERK regulates the SRF transcriptional activity. Indeed, PERK overexpression (vs. GFP control) markedly inhibited the SRF activity in SMCs transfected with a luciferase vector containing the SRF-binding motif (CArG box) (Figure 5D).
Figure 5.

PERK Regulates the Activation of Transcription Factors STAT3 and SRF
SMCs were cultured, starved, and then pre-treated with PERK inhibitor (A) or siRNA (B) and then treated with solvent or PDGF-BB (100 ng/ml) for 24 h as described previously. (A and B) Immunoblots of STAT3 phosphorylation in SMCs. Shown are representative blots from 1 of 2 similar experiments. (C) Coimmunoprecipitation. HA-STAT3 or HA vector control was transfected into MOVAS before pre-treatment with PERK inhibitor and treatment with PDGF-BB. Cell lysates were enriched using HA antibody, and then probed for PERK and MRTFA. (D) Luciferase assay for SRF transcriptional activity. Human aortic SMCs were transfected with a vector containing SRF response elements. Putative SRF activity was compared between 2 conditions: adenoviral overexpression of PERK (Ad-PERK) or GFP control (Ad-GFP), n = 5; mean ± SEM, ***p < 0.001, paired Student’s t-test. HA = hemagglutinin; MRTF-A = myocardin related transcription factor A; STAT3 = signal transducer and activator of transcription 3; other abbreviations as in Figures 1, 3, 4, and 5.
Up to this point, our data revealed a novel role for PERK in exacerbating neointima formation in injured arteries, and coherently, in mediating PDGF-induced SMC phenotypic switching. This PERK function in SMCs was at least partially accounted for by its positive regulation of STAT3 activation that has been previously implicated as inhibitory for the MRTF/SRF axis (35).
PERK inhibition rescues TNFα-induced human primary aortic EC dysfunction
Bearing in mind the EC-toxic effect of DES—the clinical standard for management of restenosis—we evaluated the impact of the PERK-targeting strategy on EC health. ECs are extremely susceptible to inflammatory challenges; a cytokine storm following endovascular injury is known to be notoriously detrimental to the homeostasis of endothelium. We used TNF-α as a cytokine-storm mimic to induce EC dysfunction, as broadly performed in previous studies (37). EC dysfunction manifests mainly as growth impairment, and elevated production and secretion of tissue factor, which is a potent proinflammatory factor and thrombosis initiator (11,12). As shown in Figure 6, treatment of human primary aortic ECs with TNF-α activated PERK (Figures 6A and 6B) and dramatically up-regulated tissue factor by 40-fold (Figure 6C) and hampered EC proliferation (BrdU assay) (Figure 6D); pre-treatment with the PERK inhibitor almost completely averted these changes. Importantly, TNF-α–induced up-regulation of PERK and tissue factor, as well as its abrogation by PERK inhibitor, was replicated ex vivo using rat aortic artery explants (Figure 6F). At the transcription level, necrosis factor-kappa B (NFκB) is known as a master transcription factor that dictates EC homeostasis or dysfunction if overactivated in a cytokine environment (38). Consistently, we observed TNF-α–stimulated p65 activation, which was attenuated by PERK inhibition (Figure 6E). Taken together, these results support a positive role for PERK in EC dysfunction in addition to that in SMC phenotypic switching. This finding is significant because PERK-mediated EC dysfunction (in particular tissue factor up-regulation) has not been previously reported. Moreover, restoration of EC homeostasis via PERK inhibition implicates a potential approach to lowering thrombogenic risks.
Figure 6.

PERK Inhibition Rescues EC Dysfunction and Mitigates Thrombosis
(A to E) Human primary aortic endothelial cells (ECs) were pre-treated with vehicle or 1 μmol/l GSK2606414 for 2 h before TNF-α stimulation (20 ng/ml). Cells were harvested at 6 h and 24 h after TNF-α treatment for immunoblotting and BrdU assays, respectively. p65 phosphorylation was determined 15 min after TNF-α stimulation. (A) Representative immunoblots of PERK pathway (P-PERK and P-eIF2α) and EC dysfunction marker (thrombogenic marker tissue factor [TF]). (B and C) Quantitation of PERK phosphorylation level and TF expression in ECs stimulated with PDGF-BB and PERK inhibitor pre-treatment. Protein band densitometry was normalized to β-actin; n = 3. (D) Proliferation of ECs was determined using BrdU incorporation enzyme-linked immunosorbent assay; n = 3. Data in B to D are presented as mean ± SEM, *p < 0.05, **p < 0.01, 1-way analysis of variance with Bonferroni post hoc test. (E) Representative immunoblots of NFκB pathway (p65 phosphorylation). Shown are representative blots from 2 similar experiments. (F) Ex vivo PERK pathway activation by TNF-α and its blockade by pretreatment with GSK2606414. Cultured rat aortic rings were pre-treated with vehicle or 1 μmol/l GSK2606414 for 2 h before a 6-h stimulation with TNF-α (20 ng/ml). Shown are representative blots from 2 similar experiments. (G and H) Mice were pre-treated with either vehicle or GSK2606414 (150 mg/kg) via oral gavage for 4 h, and then subjected to FeCl3 topical application to induce carotid artery thrombosis. (G) Real-time recording of blood velocity in FeCl3-injured carotid arteries. (H) Scatter plot of arterial flow at the end of the 60-min recording. A 0% flow indicates complete occlusion due to thrombosis. Median with interquartile range, n = 15 to 16 mice. *p < 0.05, Mann-Whitney nonparametric test. Abbreviations as in Figure 1.
Systemic application of PERK inhibitor mitigates thrombosis in a FeCl3-induced murine model
Inspired by effective repression of tissue factor in vitro and ex vivo via PERK inhibition, we finally tested an antithrombotic strategy in vivo. We used a widely recognized FeCl3-induced thrombosis mouse model that has been established in our previous studies (39). GSK2606414 was administered via oral gavage 4 h before FeCl3 application. On the basis of the published information regarding the pharmacokinetics and pharmacodynamics of GSK2606414 and its derivatives, we chose a regimen of 150 mg/kg, which was estimated to result in an effective local level at the endothelium. After treating the animals with the PERK inhibitor, we topically applied FeCl3 around the carotid artery to initiate acute endothelium damage and thrombosis. Interestingly, despite a slight decrease in carotid artery flow in the first 20 min, pre-treatment of the mice with GSK2606414 maintained a higher arterial perfusion as evidenced by higher blood velocity and patency (compared with vehicle control) that began ∼40 min after injury and became significantly improved at 56 min (Figures 6G and 6H, Supplemental Figure S5).
Discussion
The standard of care for occlusive cardiovascular diseases using DES is plagued by in-stent restenosis and thrombogenic risks (6,11). So far, efforts toward replacement of DES have not led to long-term clinical success. Here, we provide evidence for an alternative antirestenotic drug target; in other words, a potential PERK-targeting therapeutic paradigm that may circumvent the drawbacks of DES. Specifically, we found that treatment with a selective PERK inhibitor mitigated both IH and thrombosis in preclinical models. These highly desirable in vivo outcomes are rationalized by the in vitro dual beneficial effects of PERK inhibition, that is, not only abrogation of SMC dedifferentiation/proliferation but also rescue of EC dysfunction. By contrast, increasing reports demonstrate that the antiproliferative drugs used on DES (rapamycin and paclitaxel) exacerbate EC dysfunction (6,11,40).
In the present study, PERK inhibition proved effective in containing IH/restenosis in 2 drastically different (peri- and endovascular) drug-delivery routes. Although the former was in line with a prospective antirestenotic method suited for open surgeries (e.g., coronary artery bypass grafting), the latter using a biomimetic nanoplatform was conceived for a stent-free paradigm to prevent post-angioplasty restenosis, both of which we have described in our recent publications (16,24). Bypass surgery and angioplasty account for the majority of revascularization procedures. Of particular interest, endovascular administration of the PERK inhibitor GSK2606414 carried in lesion-homing biomimetic nanoclusters reduced IH by >70% with a drug dose ∼250- to 800-fold lower than those used for direct intraperitoneal or systemic intravenous injection in different disease models (23,27). Consistently, in our recent study, endovascular delivery using this biomimetic nanoplatform reduced the antirestenotic effective dose of an epigenetic modulator (JQ1) by 70-fold compared with delivery of naked drug (16). Whereas early studies using PERK inhibitors have noted systemic toxicity (23,27), this barrier in therapeutic translation may be resolved using our lesion-targeting endovascular delivery modality because it allows for the use of lower doses.
Lower effective dosages lend strong support to a target (PERK) specificity of the observed antirestenotic efficacy of GSK2606414. This is an important feature, especially in view of the use of pharmacological approaches to investigate the role for PERK in neointima formation. Arguing against possible off-target effects, the GSK2606414 dose in biomimetic nanoclusters is much lower than that in published studies that demonstrated a potent inhibition of PERK in several GSK2606414-treated tissues (23,27). These studies have confirmed that in a dose range of 50 to 150 mg/kg administered twice daily, GSK2606414 is an inhibitor highly selective for PERK over other ER stress response kinases. Chances of GSK2606414 off-target effects were also reduced in our second route of delivery, considering that the drug applied outside of the artery was not expected to enter circulation directly and rapidly (28). Furthermore, we observed a 4-fold up-regulation of activated PERK in injured versus uninjured arteries at day 3 and day 7 post-angioplasty, which should have substantially enhanced its targetability for GSK2606414. Moreover, strongly supporting a PERK target specificity, transgenically elevating PERK in the injured artery wall significantly enhanced IH. Taken together, these in vivo data of loss-of-function (PERK inhibitor) and gain-of-function (PERK up-regulation and overexpression) provide compelling evidence in support of a positive role for PERK in neointima development.
PERK’s pro-IH function is also supported by our in vitro results from loss-/gain-of-function experiments. Indicating a specific role of PERK, its silencing with an siRNA and overexpression with an adenoviral vector markedly inhibited and enhanced SMC phenotypic switching, respectively, as measured by cell proliferation and levels of SMC markers including α-SMA, calponin, and smooth muscle myosin heavy chain. It is well documented that SMC phenotypic switching is key to the development of IH (4,5). Furthermore, revealing the underlying PERK-mediated molecular mechanisms, PERK silencing and overexpression respectively reduced and increased cyclin D1, a cell division factor key to SMC proliferation. Although a pro-proliferative role of PERK has been reported in cell types other than SMCs such as cancer cells (41), PERK-specific inhibition of SMC contractile gene expression, the hallmark of a differentiated SMC state, has not been previously reported. More detailed investigation uncovered that the PERK protein complexed with STAT3, a transcription factor known to promote SMC dedifferentiation (35), and also MRTF-A, which activates SRF—the master transcription factor governing SMC contractile gene expression and SMC differentiation (42). Although it is a plausible scenario that the PERK/MRTF-A interaction may prevent MRTF-A from binding to SRF, whether disrupted PERK interactions with STAT3 and MRTF-A can explain the decreased proliferation and increased contractile gene expression observed in SMCs treated with the PERK inhibitor remains an open question.
Another important novel finding from this study is that PERK exacerbates EC dysfunction, particularly regarding the increased expression of tissue factor, a well-known potent thrombogenic factor (12). Although treatment with TNF-α inhibited human aortic primary EC proliferation and dramatically up-regulated tissue factor protein by 40-fold, pre-treatment with GSK2606414 abrogated this effect. Coincident with this result, in our parallel study of high throughput screening for drugs that inhibit SMC proliferation with lesser impact on EC viability, 3 PERK inhibitors that are GSK2606414 derivatives were among the top hits (Supplemental Figure S6). Additional data in the current study indicated that the observed PERK function was probably mediated by NFκB (p65), the master transcription factor in ECs known to direct the expression of tissue factor (43). Furthermore, the inhibitory effect of GSK2606414 on tissue factor expression was replicated in rat aorta samples in ex vivo culture, implicating an antithrombotic effect in vivo. Indeed, our in vivo experiments using the FeCl3-induced thrombosis model demonstrated that pre-treatment with GSK2606414 completely prevented arterial occlusion in 10 of 16 mice, whereas untreated animals incurred nearly complete flow blockage within 60 min after induction with FeCl3. Consistent with this result, histology data (Supplemental Figure S5) confirmed much less thrombus inside of the arteries of GSK2606414-treated (vs. untreated) mice.
The finding of the antithrombotic efficacy of the PERK inhibitor is intriguing and somewhat serendipitous, especially taking into account the antirestenotic effect of the same PERK inhibitor first observed in this study. All the available therapeutics in the clinic have proven to be damaging to the fragile endothelium, a fact primarily underlying the increase of late and very late thrombosis incidence observed in patients receiving these therapies with DES (44). On the other hand, hitherto reports on molecular targets that enable an antithrombotic and antirestenotic dual beneficial intervention have been rare. Early studies identified various agents that inhibit SMC, but not EC, proliferation (40). However, in most cases, their specific molecular targets remained unknown or unproven. Several recent reports pointed to potential interventional targets, such as ADAMTS7, and CTP synthase 1, for differential inhibition of SMC (vs. EC) proliferation (17,45). However, these studies were conducted in global knockout animals, and hence their SMC- and EC-specific roles in vivo remained unclear. Moreover, currently, there are no potent and selective small molecule inhibitors available for these proteins, limiting their potential for interventional targeting. Of particular interest, it was recently discovered that PIK75, a preclinical selective inhibitor of PI3K/p110α, exhibited both antirestenotic and endothelium-protective effects in mouse models (11). The PIK75 effects were similar to that of overexpressing PI3K/p85α (11,46), which is the inhibitory subunit keeping the catalytic subunit PI3K/p110α inactive. Although encouraging, achieving an antirestenotic/antithrombotic dual beneficial clinical outcome by targeting PI3K/p110α or PI3K/p85α would require an extraordinary drug selectivity in vivo. This appears challenging, given a total of 4 classes of PI3K heterodimers, each formed combinatorially with 1 of multiple regulatory subunits and 1 of multiple catalytic subunits that are functionally diverse or opposite (47,48).
Although to our knowledge we are the first to provide evidence for the dual beneficial role of PERK inhibition in mitigating both IH and thrombosis, other ER stress response pathways have been previously reported to be significant regulators of SMC pathophysiology in vascular diseases. For example, ATF4, 1 of the downstream effectors in the extended PERK pathway, was implicated in the development of restenosis, particularly in the acute phase (49). Our data showed a relatively delayed peak of ATF4 expression post-angioplasty. This discrepancy is possibly relevant to a different experimental setting or signaling cascade. Interestingly, another major ER stress response pathway, IRE1/XBP1, was shown to be actively involved in SMC differentiation; its dysregulation led to SMC phenotypic switching and neointima formation (19). Published evidence also implicates an involvement of ER stress response (or related) pathways in atherosclerosis and aneurysm (50,51). In aggregate, other reports and our current study underscore the importance of these pathways in SMC and EC pathophysiology and associated vascular diseases.
Study limitations
Our study using rodent models revealed a potentially favorable role of PERK inhibition in stymying neointima and a potential antithrombogenic effect following vascular injury. This PERK-targeting paradigm implicates an improved strategy to deal with neointima and thrombogenicity, 2 issues persistent with DES. However, in the clinical setting, these 2 issues occur with the same injured artery where DES is implanted. Therefore, in future studies, the beneficial outcomes of PERK inhibition need to be further examined with models that better recapitulate human arterial pathophysiology (e.g., hypercholesterolemic rabbit aortic stenting model). Along this line, insufficient re-endothelialization after DES implantation has been noted to heighten thrombogenic risks and also exacerbate restenosis (6). Although we observed that PERK inhibition rescued EC dysfunction in vitro, a potentially beneficial effect on re-endothelialization remains to be determined by optimizing conditions such as drug dosage, delivery frequency, and homing efficiency of the biomimetic drug carrier (Figure 1). Another limitation of this study is that our data could not differentiate the relative contributions of PERK inhibition in SMCs, ECs, and other cell types (e.g., platelets) to the observed therapeutic effects on restenosis and thrombosis. Future studies with SMC- or EC-specific PERK knockout should help delineate these details. In addition, the exact molecular machinery underlying PERK’s involvement in SMC phenotypic switching and EC dysfunction warrant further in-depth determination. For example, because our proposal of the PERK-STAT3-MRTFA signaling axis is partly based on findings from the proteins exogenously expressed in immortalized SMCs, this hypothesis needs to be corroborated with evidence of endogenous interactions in primary cells.
Conclusions
This study provides evidence for an interventional paradigm of PERK targeting to achieve dual inhibition of SMC phenotypic switching and EC dysfunction. That is, in vitro, PERK-specific silencing blocks SMC dedifferentiation and proliferation, and also rescues impaired EC growth; in vivo, treatment with a PERK inhibitor mitigates both restenosis and thrombosis.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: DES cannot eliminate in-stent restenosis yet can exacerbate thrombogenic risks—a major medical problem that remains unsolved. We found that targeting PERK thwarts both prorestenotic SMC phenotypic change and prothrombogenic EC dysfunction; this strategy proved effective in mitigating both restenosis and thrombosis in preclinical models. Given that PERK inhibitors are in human tests for other disease conditions, PERK targeting coupled with local-delivery technologies such as lesion-homing nanoplatforms may potentially translate into next-generation antirestenotic therapies with low or no thrombogenicity.
TRANSLATIONAL OUTLOOK 1: In this study, we used an acute thrombosis model. Future studies will be designed for testing a long-term (e.g., 3 months) therapeutic effect in a stenting model.
TRANSLATIONAL OUTLOOK 2: To devise optimized interventions for an antirestenotic, antithrombotic, and re-endothelialization-promoting synergistic effect with minimal toxicity, studies are warranted to determine different regimens of PERK inhibitor(s) and formulations of lesion-homing biomimetic drug nanocarriers.
Footnotes
This work was supported by National Institutes of Health grants R01HL143469, R01HL129785 (to Drs. Gong, Guo, and Kent), R01HL133665 (to Dr. Guo), K25CA166178 (to Dr. Gong), and American Heart Association pre-doctoral awards 17PRE33670865 (to Dr. Zhang) and 16PRE30160010 (to Dr. Wang). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Appendix
For supplemental figures and table, please see the online version of this paper.
Contributor Information
Lian-Wang Guo, Email: lianwang.guo@osumc.edu.
K. Craig Kent, Email: kc.kent@osumc.edu.
Appendix
References
- 1.Benjamin E.J., Muntner P., Alonso A. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e66. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Lafont A., Durand E., Samuel J.L. Endothelial dysfunction and collagen accumulation: two independent factors for restenosis and constrictive remodeling after experimental angioplasty. Circulation. 1999;100:1109–1115. doi: 10.1161/01.cir.100.10.1109. [DOI] [PubMed] [Google Scholar]
- 3.Marx S.O., Totary-Jain H., Marks A.R. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4:104–111. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomez D., Owens G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rzucidlo E.M., Martin K.A., Powell R.J. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45(Suppl A):A25–A32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Inoue T., Croce K., Morooka T., Sakuma M., Node K., Simon D.I. Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis. J Am Coll Cardiol Intv. 2011;4:1057–1066. doi: 10.1016/j.jcin.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kipshidze N., Dangas G., Tsapenko M. Role of the endothelium in modulating neointimal formation: vasculoprotective approaches to attenuate restenosis after percutaneous coronary interventions. J Am Coll Cardiol. 2004;44:733–739. doi: 10.1016/j.jacc.2004.04.048. [DOI] [PubMed] [Google Scholar]
- 8.Byrne R.A., Joner M., Kastrati A. Stent thrombosis and restenosis: what have we learned and where are we going? The Andreas Gruntzig Lecture ESC 2014. Eur Heart J. 2015;36:3320–3331. doi: 10.1093/eurheartj/ehv511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zen K., Takahara M., Iida O. Drug-eluting stenting for femoropopliteal lesions, followed by cilostazol treatment, reduces stent restenosis in patients with symptomatic peripheral artery disease. J Vasc Surg. 2017;65:720–725. doi: 10.1016/j.jvs.2016.10.098. [DOI] [PubMed] [Google Scholar]
- 10.Kirtane A.J., Stone G.W. How to minimize stent thrombosis. Circulation. 2011;124:1283–1287. doi: 10.1161/CIRCULATIONAHA.110.976829. [DOI] [PubMed] [Google Scholar]
- 11.Holy E.W., Jakob P., Eickner T. PI3K/p110alpha inhibition selectively interferes with arterial thrombosis and neointima formation, but not re-endothelialization: potential implications for drug-eluting stent design. Eur Heart J. 2014;35:808–820. doi: 10.1093/eurheartj/eht496. [DOI] [PubMed] [Google Scholar]
- 12.Steffel J., Latini R.A., Akhmedov A. Rapamycin, but not FK-506, increases endothelial tissue factor expression: implications for drug-eluting stent design. Circulation. 2005;112:2002–2011. doi: 10.1161/CIRCULATIONAHA.105.569129. [DOI] [PubMed] [Google Scholar]
- 13.Durand E., Scoazec A., Lafont A. In vivo induction of endothelial apoptosis leads to vessel thrombosis and endothelial denudation: a clue to the understanding of the mechanisms of thrombotic plaque erosion. Circulation. 2004;109:2503–2506. doi: 10.1161/01.CIR.0000130172.62481.90. [DOI] [PubMed] [Google Scholar]
- 14.Capodanno D., Angiolillo D.J. Management of antiplatelet therapy in patients with coronary artery disease requiring cardiac and noncardiac surgery. Circulation. 2013;128:2785–2798. doi: 10.1161/CIRCULATIONAHA.113.003675. [DOI] [PubMed] [Google Scholar]
- 15.Katsanos K., Spiliopoulos S., Kitrou P., Krokidis M., Karnabatidis D. Risk of death following application of paclitaxel-coated balloons and stents in the femoropopliteal artery of the leg: a systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc. 2018;7 doi: 10.1161/JAHA.118.011245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang B., Chen G., Urabe G. A paradigm of endothelium-protective and stent-free anti-restenotic therapy using biomimetic nanoclusters. Biomaterials. 2018;178:293–301. doi: 10.1016/j.biomaterials.2018.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang R., Cui X.B., Wang J.N., Chen S.Y. CTP synthase 1, a smooth muscle-sensitive therapeutic target for effective vascular repair. Arterioscler Thromb Vasc Biol. 2013;33:2336–2344. doi: 10.1161/ATVBAHA.113.301561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin J.K., Blackwood E.A., Azizi K. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. 2017;120:862–875. doi: 10.1161/CIRCRESAHA.116.310266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng L., Li Y., Yang J. XBP 1-deficiency abrogates neointimal lesion of injured vessels via cross talk with the PDGF signaling. Arterioscler Thromb Vasc Biol. 2015;35:2134–2144. doi: 10.1161/ATVBAHA.115.305420. [DOI] [PubMed] [Google Scholar]
- 20.Baird T.D., Wek R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr. 2012;3:307–321. doi: 10.3945/an.112.002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tam A.B., Mercado E.L., Hoffmann A., Niwa M. ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS One. 2012;7 doi: 10.1371/journal.pone.0045078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maas N.L., Diehl J.A. Molecular pathways: the PERKs and pitfalls of targeting the unfolded protein response in cancer. Clin Cancer Res. 2015;21:675–679. doi: 10.1158/1078-0432.CCR-13-3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreno J.A., Halliday M., Molloy C. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5 doi: 10.1126/scitranslmed.3006767. 206ra138. [DOI] [PubMed] [Google Scholar]
- 24.Chen G., Shi X., Wang B. Unimolecular micelle-based hybrid system for perivascular drug delivery produces long-term efficacy for neointima attenuation in rats. Biomacromolecules. 2017;18:2205–2213. doi: 10.1021/acs.biomac.7b00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kundi R., Hollenbeck S.T., Yamanouchi D. Arterial gene transfer of the TGF-beta signalling protein Smad3 induces adaptive remodelling following angioplasty: a role for CTGF. Cardiovasc Res. 2009;84:326–335. doi: 10.1093/cvr/cvp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang M., Urabe G., Little C. HDAC6 regulates the MRTF-A/SRF axis and vascular smooth muscle cell plasticity. J Am Coll Cardiol Basic Trans Science. 2018;3:782–795. doi: 10.1016/j.jacbts.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H.J., Raphael A.R., LaDow E.S. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaudhary M.A., Guo L.W., Shi X. Periadventitial drug delivery for the prevention of intimal hyperplasia following open surgery. J Control Release. 2016;233:174–180. doi: 10.1016/j.jconrel.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebeau J., Saunders J.M., Moraes V.W.R. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep. 2018;22:2827–2836. doi: 10.1016/j.celrep.2018.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sano R., Reed J.C. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Starck S.R., Tsai J.C., Chen K. Translation from the 5' untranslated region shapes the integrated stress response. Science. 2016;351:aad3867. doi: 10.1126/science.aad3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamanouchi D., Kato K., Ryer E.J., Zhang F., Liu B. Protein kinase C delta mediates arterial injury responses through regulation of vascular smooth muscle cell apoptosis. Cardiovasc Res. 2010;85:434–443. doi: 10.1093/cvr/cvp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mack C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee M., San Martín A., Valdivia A. Redox-sensitive regulation of myocardin-related transcription factor (MRTF-A) phosphorylation via palladin in vascular smooth muscle cell differentiation marker gene expression. PLoS One. 2016;11 doi: 10.1371/journal.pone.0153199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao X.H., Wang N., Zhao D.W. STAT3 protein regulates vascular smooth muscle cell phenotypic switch by interaction with myocardin. J Biol Chem. 2015;290:19641–19652. doi: 10.1074/jbc.M114.630111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meares G.P., Liu Y., Rajbhandari R. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol. 2014;34:3911–3925. doi: 10.1128/MCB.00980-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang B., Zhang M., Takayama T. BET bromodomain blockade mitigates intimal hyperplasia in rat carotid arteries. EBioMedicine. 2015;2:1650–1661. doi: 10.1016/j.ebiom.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown J.D., Lin C.Y., Duan Q. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. doi: 10.1016/j.molcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huttinger A.L., Wheeler D.G., Gnyawali S. Ferric chloride-induced canine carotid artery thrombosis: a large animal model of vascular injury. J Vis Exp. 2018 doi: 10.3791/57981. (139):57981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goel S.A., Guo L.W., Wang B. High-throughput screening identifies idarubicin as a preferential inhibitor of smooth muscle versus endothelial cell proliferation. PLoS One. 2014;9 doi: 10.1371/journal.pone.0089349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jamison S., Lin Y., Lin W. Pancreatic endoplasmic reticulum kinase activation promotes medulloblastoma cell migration and invasion through induction of vascular endothelial growth factor A. PLoS One. 2015;10 doi: 10.1371/journal.pone.0120252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du K.L., Ip H.S., Li J. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23:2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parry G.C., Mackman N. Transcriptional regulation of tissue factor expression in human endothelial cells. Arterioscler Thromb Vasc Biol. 1995;15:612–621. doi: 10.1161/01.atv.15.5.612. [DOI] [PubMed] [Google Scholar]
- 44.Ertas G., van Beusekom H.M., van der Giessen W.J. Late stent thrombosis, endothelialisation and drug-eluting stents. Neth Heart J. 2009;17:177–180. doi: 10.1007/BF03086242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kessler T., Zhang L., Liu Z. ADAMTS-7 inhibits re-endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin-1. Circulation. 2015;131:1191–1201. doi: 10.1161/CIRCULATIONAHA.114.014072. [DOI] [PubMed] [Google Scholar]
- 46.Torella D., Gasparri C., Ellison G.M. Differential regulation of vascular smooth muscle and endothelial cell proliferation in vitro and in vivo by cAMP/PKA-activated p85alphaPI3K. Am J Physiol Heart Circ Physiol. 2009;297:H2015–H2025. doi: 10.1152/ajpheart.00738.2009. [DOI] [PubMed] [Google Scholar]
- 47.Geering B., Cutillas P.R., Nock G., Gharbi S.I., Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci U S A. 2007;104:7809–7814. doi: 10.1073/pnas.0700373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsolakos N., Durrant T.N., Chessa T. Quantitation of class IA PI3Ks in mice reveals p110-free-p85s and isoform-selective subunit associations and recruitment to receptors. Proc Natl Acad Sci U S A. 2018;115:12176–12181. doi: 10.1073/pnas.1803446115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malabanan K.P., Kanellakis P., Bobik A., Khachigian L.M. Activation transcription factor-4 induced by fibroblast growth factor-2 regulates vascular endothelial growth factor-A transcription in vascular smooth muscle cells and mediates intimal thickening in rat arteries following balloon injury. Circ Res. 2008;103:378–387. doi: 10.1161/CIRCRESAHA.107.168682. [DOI] [PubMed] [Google Scholar]
- 50.Hotamisligil G.S. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia L.X., Zhang W.M., Zhang H.J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J Pathol. 2015;236:373–383. doi: 10.1002/path.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.