

Abstract
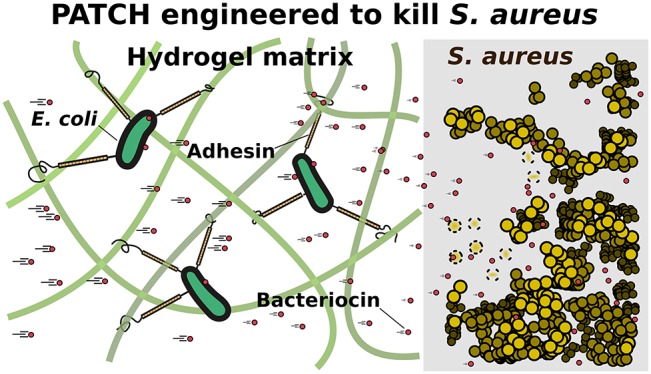
Engineered living materials have the potential for wide-ranging applications such as biosensing and treatment of diseases. Programmable cells provide the functional basis for living materials; however, their release into the environment raises numerous biosafety concerns. Current designs that limit the release of genetically engineered cells typically involve the fabrication of multilayer hybrid materials with submicrometer porous matrices. Nevertheless the stringent physical barriers limit the diffusion of macromolecules and therefore the repertoire of molecules available for actuation in response to communication signals between cells and their environment. Here, we engineer a novel living material entitled “Platform for Adhesin-mediated Trapping of Cells in Hydrogels” (PATCH). This technology is based on engineered E. coli that displays an adhesion protein derived from an Antarctic bacterium with a high affinity for glucose. The adhesin stably anchors E. coli in dextran-based hydrogels with large pore diameters (10–100 μm) and reduces the leakage of bacteria into the environment by up to 100-fold. As an application of PATCH, we engineered E. coli to secrete the bacteriocin lysostaphin which specifically kills Staphyloccocus aureus with low probability of raising antibiotic resistance. We demonstrated that living materials containing this lysostaphin-secreting E. coli inhibit the growth of S. aureus, including the strain resistant to methicillin (MRSA). Our tunable platform allows stable integration of programmable cells in dextran-based hydrogels without compromising free diffusion of macromolecules and could have potential applications in biotechnology and biomedicine.
Synthetic biology aims to design programmable cells that combine sensing and molecular computing operations with on-demand production of therapeutic relevant proteins.1−4 Engineered living materials (ELMs) integrate genetically engineered cells with free-standing materials and represent a new class of environmentally responsive living devices with designer physicochemical and material properties.5−8 Ideally, ELMs provide mechanical robustness to engineered cells, prevent their migration to the environment, and allow cells to be viable for extended periods of time. As it has been difficult to efficiently immobilize genetically modified microorganisms (GMMs) in ELMs, their release into the environments poses a major challenge for future synthetic biology applications.9,10 To date, strategies for containing GMMs inside a living device are based on the physical confinement by multilayer materials.11−13 Hybrid micropatterned devices that combine layers of elastomer and microporous hydrogel have enabled the exchange of information with the surrounding environment via diffusion of chemical inducers and their sensing by GMMs while displaying high mechanical resilience.11 Nevertheless, the low porosity of the physical barriers mitigating bacterial escape also significantly hampers the diffusion of macromolecules, reducing the repertoire of synthetic biology applications to biosensing and release of small therapeutic molecules.
Programmable interactions between GMMs and their surroundings have recently gained interest as the design of multicellular structures such as engineered biofilms enables a higher control over shape and functionality of ELMs.14,15 Inducible secretion of fibrous proteins drives the emergence of molecular architectures supporting the artificial biofilms. Interestingly, such biofilms are self-regenerating and are able to perform advanced tasks such as multistep enzymatic bioremediation, and are viable over weeks.14−17
Inspired by this strategy, we propose to leverage the formation of programmable interactions between surface-displayed proteins on genetically engineered cells and hydrogel constituents to ensure cell containment within a living material while simultaneously allowing production and delivery of large therapeutic molecules.The “Platform for Adhesin-mediated Trapping of Cells in Hydrogels” (PATCH) is based on engineered E. coli expressing a recombinant adhesion protein that stably anchors the cells to a hydrogel matrix, avoiding entrapment by stringent physical barriers (Figure 1a). Bacterial adhesins are modular cell-surface proteins that bind different ligands in a highly specific manner. While strategies for programmable cell–cell interactions via adhesins have been reported,18,19 methods that allow adhesin-mediated cell-material interactions have not been described. We engineer E. coli bacteria to surface-display a Ca2+-dependent glucose-binding adhesin which has strong interactions with dextran-based hydrogels (Figure 1b,c for details). The engineered adhesin, referred to as MpA, is derived from the Marinomonas primoryensis ice-binding protein (MpIBP) which helps these bacteria to assemble into multispecies biofilms on ice.20−22 Compared to conventional living material designs, our hydrogel-based material has large pores with diameters ranging from 10 to 100 μM, which substantially improves the diffusion of macromolecules through the matrix. Furthermore, our results reveal efficient containment of adhesin displaying bacteria with a retention factor of up to 100-fold higher compared to bacteria that lack the adhesin. PATCH may help simplify the design of ELMs and could reduce the use of multimaterial or multilayer devices aimed at immobilizing GMMs.
Figure 1.
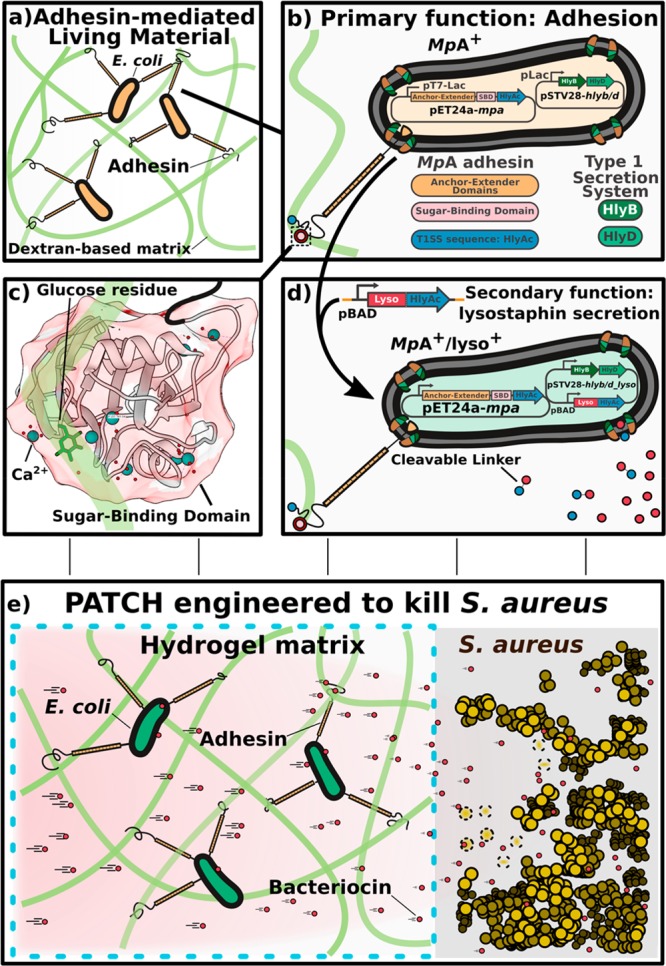
Overview of the design elements for PATCH. (a) Escherichia coli (E. coli, orange ovals with black outlines) expressing a cell-surface adhesin (orange-black sticks) with a high affinity for glucose are retained inside dextran-based hydrogels. (b) Cell surface display of the MpA adhesin is regulated by cotransforming E. coli with two expression vectors (MpA+; yellow). MpA was cloned into a pET24a vector (pET24a-mpa) under the control of a T7 promoter (pT7-Lac) inducible by IPTG. The engineered adhesin contains the membrane anchor (orange), extender (orange), and sugar-binding domains (SBD, pink circle) derived from MpIBP of the Antarctic bacterium, Marinomonas primoryensis. The 23-kDa C-terminal fragment of hemolysin protein A (HlyAc, dark blue circle) functions as a Type I Secretion Pathway (T1SS) sequence specific to E. coli, which was grafted to the end of MpA to promote its cell-surface-display. The other two E. coli T 1SS components HlyB (dark green) and HlyD (light green) were expressed from a pSTV28 vector (pSTV28-hlyb/d) under the control of a lac promoter (pLac) inducible by IPTG. The T1SS outer-membrane component (TolC) constitutive to E. coli BL21 cells is colored brown. (c) X-ray crystal structure of the MpA sugar-binding domain (SBD), which is responsible for binding to the dextran component of the hydrogel matrix. Ca2+ are illustrated as large blue-green spheres, waters are indicated by small red spheres. The sugar-binding site is occupied by a glucose molecule (green backbone; stick representation). The figure was rendered using UCSF Chimera.52 (d) E. coli (green; MpA+/lyso+) is re-engineered to surface-display the MpA adhesin and secrete the bacteriocin lysostaphin via the T1SS. HlyAc (small dark-blue circles) was added to the C-terminus of lysostaphin (small red circles) via a cleavable linker. The lysostaphin-HlyAc fusion construct was under the control of a pBAD promoter (inducible by arabinose) on the pSTV28 vector that also contains the pLac-HlyB/D. The modified vector is named pSTV28-hlyb/d_lyso.e) Schematics of the designed living material with anti-S. aureus activity. Left: Surface-expression of MpA allows E. coli to bind to the dextran matrix, thereby retaining the bacteria in the hydrogel (boundary marked by blue dashed lines). The engineered E. coli inside the hydrogel can secrete lysostaphin (small red circles), which diffuses freely to the exterior environment to inhibit the growth of S. aureus (gold spheres), including a methicillin-resistant strain (MRSA). Gold spheres with dashed boundaries indicate dead S. aureus cells killed by lysostaphin.
Hydrogels with antimicrobial activity have become a major focus in biomedical research due to their unique qualities such as high biocompatibility and ease of drug loading and release.23,24 As an application of PATCH, we anchor engineered E. coli that produce and secrete an antimicrobial enzyme called lysostaphin inside a dextran-based hydrogel (Figure 1d,e) and show successful inhibition of S. aureus growth including a methicillin-resistant (MRSA) strain. Due to the rising incidence of resistance, MRSA is becoming increasingly difficult to treat by conventional antibiotics.25 In the United States alone, MRSA causes approximately 11 000 deaths annually.25 Since lysostaphin functions by cleaving the pentaglycine components of the cell wall of S. aureus including MRSA,26 it is highly specific in killing the bacterium. Importantly, lysostaphin is an enzyme that differs from small-molecule antibiotics, and is effective against antibiotic resistant infections.27 Taken together, our study demonstrates PATCH as a programmable living material foundry with potential applications in biosensing and controlled delivery of therapeutic proteins.
Results and Discussion
Synthesis and Characterization of Dextran-Based Hydrogels
Dextran-based hydrogels are relatively simple to synthesize, robust in various shapes and highly biocompatible.28−30 The glucose-based nature of the biopolymer makes dextran an ideal material for anchoring E. coli expressing MpA. The dextran (500 kDa) polymer was functionalized by methacrylation to yield methacrylated dextran (Dex-MA) (Figure S1.1–1.2), which was subsequently cross-linked into a covalent hydrogel via radical polymerization.31 The resulting hydrogel is easy to handle (i.e., not brittle or very viscous), and can be adapted into various shapes tailored for different applications. Further analysis of the wash solution of these hydrogels using NMR and FT-IR spectroscopy demonstrated that negligible concentrations of unreacted monomers are released from the hydrogel (Figure S1.3Figure S2), which makes it suitable for keeping bacteria viable and likely safe for various medical applications. Scanning electron microscope (SEM) analysis showed that the dextran-based hydrogel has an overall microporous structure with pore diameters ranging from 10 to 100 μm (Figure S3). These large pores would allow unrestricted diffusion of macromolecules, as well as the propagation of native E. coli cells (2–5 μm), resulting in the migration of bacteria to the outside of hydrogel (vide infra).
Cell-Surface Display of MpA
To stably anchor E. coli to dextran-based hydrogel, we designed a new adhesion protein, MpA, that binds glucose with high affinity and can be surface-displayed on E. coli. MpA is derived from the giant adhesin MpIBP (1.5 MDa) (Figure 2a) found on the cell surface of the Antarctic Gram-negative bacterium, Marinomonas primoryensis. MpIBP binds its bacterium to ice and facilitates the formation of symbiotic biofilms with other microorganisms.18MpIBP has a C-terminal signal sequence enabling its secretion by the Type I Secretion System (T1SS) specific to M. primoryensis (Figure 2a).21 At the N terminus, MpIBP contains an “anchor module” that helps retain the protein to the cell surface by plugging the T1SS outer-membrane pore.21,22,32−34 The protein further consists of 120 identical 104-aa tandem repeats that serve to project a set of ligand-binding modules away from the cell surface to interact with their target molecules, including various carbohydrates, proteins, and ice. The sugar-binding domain (SBD) of MpIBP binds to glucose in a Ca2+-dependent manner,21,22,35 and is used in this study to allow engineered bacteria to bind to dextran-based hydrogels.
Figure 2.
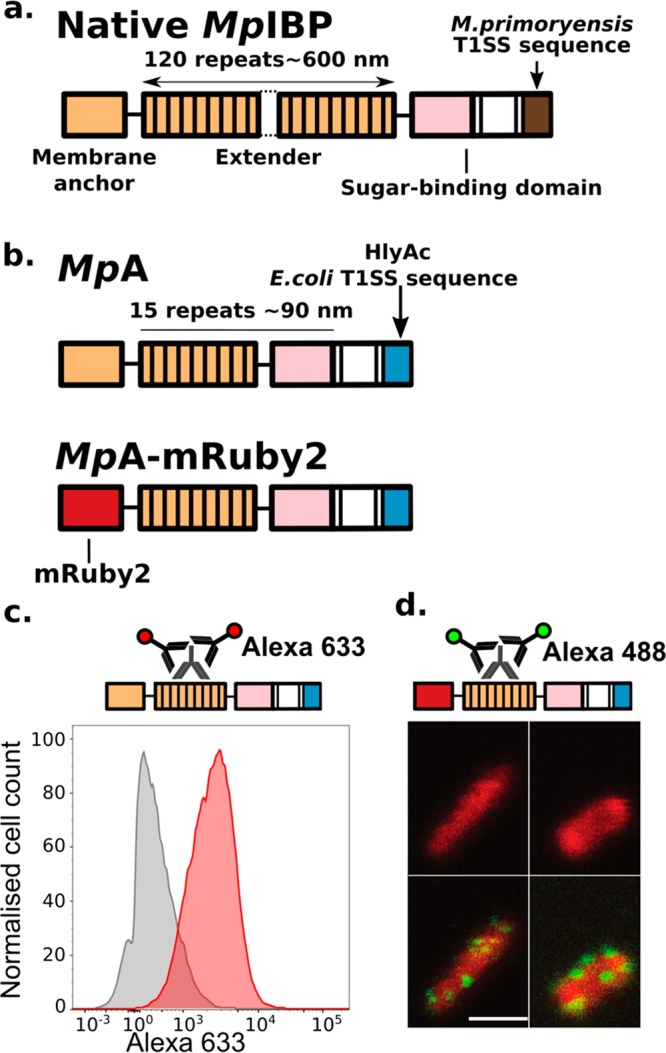
Construct design of the engineered MpA adhesins and its localization to E. coli cell surface. (a) Linear domain map of the native 1.5-MDa MpIBP with four of its functional regions illustrated as membrane anchor (orange), central extenders (orange repeats), sugar-binding domain (pink), and M. primoryensis T1SS sequence (brown). Dotted lines indicate the ∼100 central repeats omitted in the figure. (b) Linear domain maps of engineered constructs of MpA expressed and surface-displayed by E. coli. (c) Immuno-detection of MpA by flow cytometry. Top schematic shows immunodetection of MpA through an antibody raised against its central extender domains. Histograms (bottom panel) illustrating fluorescence distribution of the immuno-detection experiments done with MpA–/lyso+ (gray) and MpA+/lyso+ (red) cells. (d) Fluorescence localization after immunostaining (green) of MpA-mRuby2 (red) obtained using confocal microscopy. Top schematic shows the immunodetection of MpA through an antibody raised against its central extender domains. Bottom panels show images obtained from confocal microscopy. The MpA-mRuby2+ cells are displayed in the top two panels, while the detection of MpA on E. coli outer surface by immuno-staining with fluorescently labeled secondary antibodies (Alexa 488, green patches on the cells) are shown in the bottom two panels. Scale bar indicates 2 μm, and is representative for all confocal images in this panel.
The extremely large molecular weight of MpIBP would likely prevent its use as a modular protein for cell-surface display in E. coli. We therefore introduced several modifications resulting in an engineered MpIBP-based variant, MpA, optimized for cell-surface display. First, we reduced the number of central MpIBP repeats from 120 to 15, lowering the molecular weight by approximately 5-fold (to 330 kDa; Figure 2b). These 15 tandem extender domains can project the sugar-binding domain (SBD) approximately 90 nm away from the cell surface.18 Next, the C-terminal domain (181 aa) of MpIBP was replaced by a 23 kDa C-terminal sequence of the E. coli hemolysin protein A (HlyAc) (Figure 2b), which enables translocation of the protein to the cell surface via the Type I secretion pathway (T1SS) specific to E. coli.36−39 The engineered mpa genes were placed in a pET24a vector. Since the genome of E. coli BLR (DE3) does not contain the T1SS machinery genes hlyb and hlyd required for secretion (pSTV28-hlyb/d; Figure 1b),40 a pSTV28 vector carrying these two genes was cotransformed with the adhesin-pET24a vector (Figure 1b). We refer to bacterial cells cotransformed with these plasmids as MpA+ cells and will use this notation throughout this article for other variants. Additional variants were created by replacing the N-terminal domains of MpA with a fluorescent protein (MpA-mRuby2+), which was used to validate the localization of the adhesin by confocal microscopy (Figure 2b). It has been reported that fluorescent proteins fold rapidly in the bacterial cytosol before being secreted by T1SS, and their large size prevents full passage through the outer-membrane pore of the secretion system.38,39 Thus, mRuby2 serves as an “anchor” to hold MpA to the cell surface, while the remaining parts of the protein protrude into the extracellular environments. As a control, we prepared MpA– cells by cotransformation with a pET24a plasmid that only encodes for HlyAc (pET24a-hlyac) and the pSTV28_hlyb/d vector. As detailed later in the paper, we also created MpA+ cells capable of secreting the bacteriocin lysostaphin to target Staphyloccocus aureus. The resulting variant is referred to as MpA+/lyso+.
Next, a combination of flow cytometry and confocal microscopy was used to validate the expression of MpA on the outer surface of E. coli. MpA+/lyso+ (cells capable of expressing both MpA and lysostaphin proteins, Figure 1d, details below) and MpA–/lyso+ control cells were incubated with rabbit antisera raised against the central repeats of MpA. After unbound primary antibodies were removed by washing, cells were further incubated with a fluorescently labeled (Alexa 488) secondary antibody (Figure 2c, top schematics). As expected, MpA+/lyso+ cells (Figure 2c, red histogram) showed a median fluorescence level approximately 5-fold higher than that of the MpA–/lyso+ cells (Figure 2c, gray histogram), validating the surface localization of the adhesin. Confocal microscopy studies using the MpA-mRuby2+ variant revealed successful overexpression of the protein as evident by the evenly distributed red fluorescence inside of the cells. The surface display of this protein is demonstrated by the green immuno-fluorescence on the exterior of the cells. As the primary antibody targets the MpA extender region consisting of 15 identical 104-aa repeats, it is likely that multiple antibodies bind to an individual adhesin protruding from the T1SS outer-membrane pore. This results in amplification of green fluorescence from the Alexa488 secondary antibodies, which appears as green clusters on the cell surface (Figure 2d). These data are in agreement with the flow cytometry data. In summary, we designed a novel MpIBP-based protein for cell-surface display in E. coli and characterized the expression and localization of this protein using flow cytometry and confocal microscopy.
MpA Efficiently Retains E. coli inside Dextran-Based Hydrogels
To validate if the surface expression of MpA retains E. coli inside dextran-based hydrogels, we designed two experiments to quantify the migration of bacteria through the hydrogel matrix into surrounding liquid or solid media. MpA+ and MpA– cells were loaded into cubic-shaped hydrogels (Figure S4), which were subsequently partially submerged in LB medium (Figure 3a). To estimate bacterial migration from the gel to the surrounding medium, samples of the medium were harvested at different time points and plated for quantification by counting the colony forming units (CFUs). Samples taken immediately after loading of bacteria showed similar numbers (∼102) of MpA+ and MpA– cells leaking into the medium. We hypothesize that this initial leakage originates from the large pore size of the hydrogel matrix resulting in a portion of loaded bacteria directly flowing through the gel without binding to dextran. After unbound cells were removed by refreshing the medium, very few MpA+ bacteria were present in the medium outside of the hydrogel in the next 24 h (approx. 30 CFUs). In sharp contrast, up to 100-fold MpA– cells were present in the medium (approx. 2 ×103 CFUs) (Figure 3b). SEM analysis was performed to gain visual insight into how bacteria interacted with the hydrogel. We observed that MpA+ bacteria formed thin layers of microcolonies on the lamina of the hydrogel due to their strong interaction with the gel matrix (Figure 3c,d). In contrast, MpA– bacteria showed a significantly sparser distribution (Figure S5a–d), indicating a weaker interaction with the hydrogel, which was consistent with the results obtained from the leakage experiments. In a different experiment we assessed the capability of MpA in retaining the E. coli to the dextran-based matrix, by comparing the relative affinity of MpA+ and MpA– cells to small hydrogel cylinders (∼100 mm3). Hydrogels were preincubated with either MpA+ or MpA– overnight, and then extensively washed to remove unbound bacteria. MpA+ or MpA– that remained bound to the hydrogels were released into a solution by homogenization and were quantified by CFU counting. As expected, hydrogels retained roughly 4-fold more MpA+ bacteria compared to those that contained MpA– (Figure S5e), demonstrating the significantly stronger interaction between the bacteria and the hydrogel caused by MpA expression.
Figure 3.
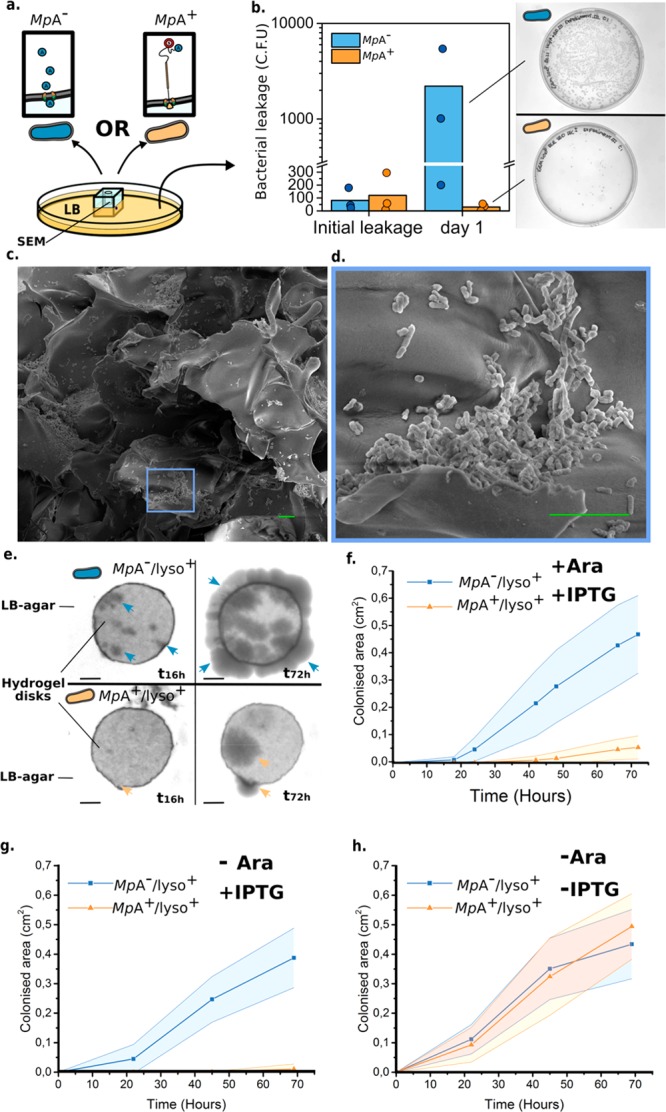
Characterization of the MpA adhesin mediated retention of E. coli inside a dextran-based hydrogel matrix. (a) Experimental design to quantify bacterial migration from the hydrogel matrix into liquid medium. Leakage of MpA+ (orange) from a hydrogel cube was assessed by counting the colony forming units (CFUs) present in the LB medium surrounding the hydrogel, and compared with the leakage obtained from MpA– cells (blue, negative control). (b) Bacterial migration was quantified immediately after bacterial loading (initial leakage) and over the following day. Right panel: representative images of plates at the same dilution are shown to illustrate the difference in migration between MpA+ and MpA– samples after 24 h (Day 1). Dots on the bar graph represent the results of individual experiments. (c) Scanning electron microscopy images obtained from the dextran-based hydrogel inoculated with the MpA+E. coli inside of the bacterial loading chamber (cross-section). (d) Zoomed-in view of an area of panel c, showing a bacterial microcolony of MpA+ formed on the lamina of the hydrogel matrix. Scale bars (green) in panels c and d indicate 10 μm. (e) Representative images showing the migration of E. coli through the matrix of a disk-shaped hydrogel, which was assessed by estimating the surface area colonized by the bacteria (MpA+/lyso+ or MpA–/lyso+) on the outside of the hydrogel disk over time. Arrowheads indicate the presence of nascent colonies of E. coli (MpA–/lyso in blue) and (MpA+/lyso+ in orange). Scale bar represents 2 mm. (f,g,h) Quantification of the time-dependent migration of engineered E. coli (MpA+/lyso+ in blue and MpA–/lyso+ in orange) through the hydrogel matrix. Average area and standard deviation of the bacteria-colonized regions outside of the hydrogel were calculated from replicates of that in panel e over 72 h (n = 4). (f) Experiments performed in the presence of both inducing agents, IPTG for adhesin surface display, and arabinose for lysostaphin expression (Ara). (g) Experiments performed in the presence of IPTG, but in the absence of Ara (n = 3). (h) Experiments performed in the absence of IPTG or Ara (n = 3).
Next, we tested whether surface display of MpA also inhibits migration of bacteria into a surrounding solid medium. Small disk-shaped dextran hydrogels (thickness, 0.75 mm; diameter, 8 mm) were fabricated. The hydrogel disk was then placed on a LB-agar plate, and MpA+/lyso+ cells or MpA–/lyso+ control (1 μL) was loaded to the center of the disk. Bacterial migration from the center of the hydrogel matrix was monitored for up to 72 h (Figure 3e). We quantified the average colonized area outside the hydrogel disk based on four replicates. We observed that after roughly 10 h, MpA–/lyso+ cells started to migrate to the outside of the hydrogel, whereas MpA+/lyso+ cells were retained for at least another 25 h (Figure 3f,g). After 72 h MpA–/lyso+ cells colonized a surface 8-fold larger compared to MpA+/lyso+ cells (Figure 3f,g). Furthermore, uninduced MpA+/lyso+ cells did not show significant differences in migratory behavior compared to either induced or uninduced MpA–/lyso+ control (-IPTG, Figure 3f–h). The expression and secretion of an additional protein, lysostaphin, which was induced by arabinose (details below, Figure 1d), did not appear to hamper MpA-mediated retention of the engineered E. coli to the hydrogel matrix (Figure 3f). These results further consolidated our findings that MpA surface display can substantially enhance the retention of bacteria inside dextran-based hydrogels through its sugar-binding capability.
Application of PATCH - Living Material Engineered to Kill MRSA
Having developed PATCH, we next assessed whether MpA+ bacteria can be used to deliver functional proteins in situ. We focused on engineering E. coli to secrete lysostaphin, a bacteriocin produced by Staphylococcus simulans to kill S. aureus.26 The full-length lysostaphin is a 50-kDa preproenzyme with a modular domain architecture including a 36-aa N-terminal signal peptide followed by a region comprising 15 tandem repeats of 13-aa, and the peptidase and cell-wall-targeting (CWT) domains at the C terminus.41 After its secretion from S. simulans, the signal peptide and the repetitive region of prepro-lysostaphin are sequentially cleaved to reveal its active form, with the CWT domain binding to its substrate on S. aureus cell surface and the peptidase domain responsible for the catalytic cleavage of the glycil–glycine bonds.
As the functional construct of lysostaphin was engineered for secretion via the T1SS by E. coli, the N-terminal S. simulans signal peptides and the tandem repeats of the preprolysostaphin were deemed redundant and therefore not included in the design (Figure 1b). Instead, the HlyAc sequence required by E. coli T1SS was fused to the C-terminus of lysostaphin via a linker consisting of a thrombin-cleavage site flanked by tetraglycine motifs. Although the lysostaphin would in theory compete with the MpA adhesin for the T1SS ducts, our data reveal sufficient secretion of functional lysostaphin (vide infra). The design of the linker motifs was intended such that the HlyAc domain can be selectively removed to release the catalytic form of lysostaphin. In addition, a 6X Histidine-tag was fused to the C-terminal end of the lysostaphin-HlyAc fusion protein for purification and detection purposes. The lysostaphin-HlyAc construct under the control of a pBAD promoter (inducible by arabinose) was incorporated into the pSTV28-hlyb/d, resulting in the pSTV28_hlyb/d_lyso vector (Figure 1d).42,43 The pSTV28-hlyb/d_lyso was cotransformed with pET24a_mpa vector to create MpA+/lyso+ bacteria. As a control, we also prepared cells lacking MpA expression (MpA–/lyso+), which have been transformed with the pET24a_hlyac and the pSTV28_hlyb/d_lyso vectors.
To reveal successful secretion of the 50-kDa lysostaphin-HlyAc, cell-free medium of MpA–/lyso+ was analyzed by Western blotting with antihistag primary antibodies. As expected, a band of approximately 50 kDa was detected for the intact lysostaphin-HlyAc (Figure S6). In addition, a much more intense band was observed at approximately 25 kDa, which indicated the presence of HlyAc alone. This observation suggested that lysostaphin-HlyAc construct had been cleaved after secretion. Given the capability of lysostaphin to cleave pentaglycine and the presence of two tetraglycine motifs linking lysostaphin and HlyAc, it is possible that the fusion protein underwent site-specific autolysis. Nevertheless, this would have no negative impact on the enzymatic capability of lysostaphin as it frees up its catalytic domains from HlyAc.
We quantified the efficacy of the secreted lysostaphin-HlyAc against S. aureus, using an adapted version of Kirby-Bauer (disk diffusion) test (Figure 4a).44 We applied cell-free medium of MpA–/lyso+ bacteria to Mueller-Hinton agar plates prestreaked with S. aureus. After an overnight incubation period, clear round exclusion zones formed where the medium was applied, indicating the effective inhibition of S.aureus growth (Figure 4b). In contrast, cell-free medium from the MpA– cells showed no activity against S. aureus, indicating that growth inhibition was solely caused by lysostaphin secretion. MpA–/lyso+ cells were unable to inhibit the growth of a different Gram-positive pathogen, Streptoccocus agalactiae, demonstrating the specificity of lysostaphin. To further consolidate this result, we spotted living MpA+/lyso+ cell cultures onto a Mueller-Hinton agar plate streaked with the notorious methicillin-resistant Staphylococcus aureus (MRSA), which is a major cause for hospital-acquired infections worldwide.45,46 Strikingly, we observed clear halo-like exclusion zones surrounding the MpA+/lyso+ cells, indicating the inhibition of MRSA growth (Figure 4c). These results demonstrate that sufficient amount of functional lysostaphin was secreted even though the MpA adhesin and lysostaphin compete for T1SS ducts. In sharp contrast, no such inhibition zones separated the E. coli cells without lysostaphin-producing capability (MpA– cells) and MRSA. Consistent with the results above, MpA+/lyso+ cells could not kill S. agalactiae. These results demonstrated the high specificity and promising potential of in situ lysostaphin secretion for treating antibiotic-resistant S. aureus-related infections.
Figure 4.

PATCH with Anti-S. aureus activity. (a) Schematic of the adapted Kirby-Bauer test for assessing the anti-S. aureus activity of lysostaphin-secreting E. coli as an application of PATCH. (b) Evaluation of bactericidal activity of the cell-free media collected from MpA–/lyso+, MpA–, MpA+/lyso+ and MpA+/lyso– cells against Staphylococcus aureus and Streptococcus agalactiae. White arrows indicate where the samples were applied. Black arrows indicate the target bacteria streaked on the plates. Growth inhibition zones are marked by a red arrow. (c) Evaluation of bactericidal activity of MpA–/lyso+, MpA–, MpA+/lyso+ and MpA+/lyso– (no lysostaphin-producing capability) E. coli cells against Methicillin-resistant Staphylococcus aureus (MRSA) and S. agalactiae. Images were captured after an incubation time of approximately 48 h. d) Evaluation of bactericidal activity of the living hydrogels with engineered MpA+/lyso+E. coli against MRSA. MpA+/lyso+E. coli cells were spotted to the centers of the hydrogel disks, which were placed onto Mueller-Hinton agar plates streaked with MRSA. The images were captured after an incubation period of 96 h. The adhesin- and lysostaphin-functionalities were controlled by induction by IPTG and/or Arabinose (Ara), respectively. White arrowheads point to the edge of the hydrogel disks as outlined by thin white circles.
After demonstrating that MpA+/lyso+ cells can secrete functional lysostaphin in situ, we next investigated whether PATCH could kill MRSA, without leakage of GMMs. This would be a key step toward demonstrating the feasibility of this novel platform for different applications. As anticipated, very few E. coli cells induced with MpA+/lyso+ migrated outside of the hydrogel disks into the surrounding Mueller-Hinton agar medium after 96 h (Figure 4d). Remarkably, the bacteria-loaded hydrogel disks are also effective in killing MRSA, as exclusion zones developed around the periphery of the devices. This demonstrates free diffusion of functional lysostaphin through the hydrogel matrix to the exterior environments. In contrast, when MpA expression was not induced (IPTG-), MpA+/lyso+ cells were not restrained to the hydrogel, resulting in their migration into the exterior agar medium (Figure 4d, two panels on the right). When only lysostaphin expression was induced (IPTG-, Ara+), was the inhibition zone significantly larger than when both MpA and lysostaphin were induced (IPTG+, Ara+). We reason that since the adhesin plugs a significant fraction of T1SS machineries, halting MpA production resulted in higher secretion levels of lysostaphin. Furthermore, a control experiment in which lysostaphin expression was not induced (IPTG-, Ara-) resulted in the absence of an inhibition zone, which validates the responsiveness of our system toward arabinose induction (Figure 4d). Taken together, we have created a living hydrogel hosting adhesin-mediated self-retaining E. coli. As an application of this versatile platform, we demonstrated a prototype living material with anti-MRSA activity.
Conclusions and Outlook
One challenge that impedes the deployment of living functional materials for biomedical applications outside the lab is the requirement of sustaining viable cells while restricting their escape from the device. To the best of our knowledge, this study shows the first example toward confinement of GMMs by actively implementing adhesive interactions between microorganisms and a fully synthetic material. The basis of PATCH is in contrast with conventional methods for engineering living materials, which typically rely on the presence of a physical barrier provided by submicrometer matrices of multilayer hybrid materials. Our work demonstrates one application of PATCH as a living material foundry for clearing pathogenic bacteria by in situ production and secretion of lysostaphin.
PATCH presents a versatile toolbox that can be tailored to the need of various applications such as biosensing and smart drug delivery in a deployable “Sense-kill” manner,47,48 which opens the door for a wide range of future research. Given the modularity of bacterial adhesins, PATCH can be easily tailored to other materials. Besides the SBD, another ligand-binding domain of MpA can be used to bind peptide-based materials.21,22 In addition, the E. coli pilus domain FimH has high specificity for mannose-based structures. Other constituents of PATCH such as gel porosity can be tuned to modulate the diffusion of various therapeutic compounds secreted by the bacteria.49 Identification of soluble S.aureus molecules capable of regulating the expression of lysostaphin would enhance the responsiveness and specificity of the current system. Implementing an alternative secretion system for lysostaphin might result in higher levels of in situ drug delivery. Crucially, integration of the expression systems into the genomes of the engineered microorganisms will help diminish the spreading of antibiotic resistant genes to pathogens via the plasmids. To address further safety concerns, we envision that the platform could be adapted to other microorganisms that previously demonstrated their innocuity for humans, and were used in other animal models for synthetic biology applications, such as E. coli Nissle 1917 strain (EcN)48 or Bacillus subtilis.50 As such, this work constitutes an important methodological improvement in the field of living functional materials and living material foundries, and could have implications for therapeutic applications.
Materials and Methods
Gene Synthesis of MpA
A gene encoding a smaller version of MpIBP (Genbank accession: ABL74378.1 and ABL74377.1) with only 15 central repeats was synthesized and purchased from GenScript (https://www.genscript.com). The gene was codon-optimized for expression in E. coli, and the central repeats were made as different as possible at the DNA level to reduce the chance of recombination. Next, the M.primoryensis-specific Type I Secretion System (T1SS) sequence near its C terminus (181 aa) was replaced by the E. coli T1SS sequence (last 213 aa of the Hemolysin A protein, HlyAc, also codon-optimized and synthesized by GenScript) to result in the MpA adhesin (Figure 2b). A fluorescent variant of MpA, MpA-mRuby2, was created by replacing the membrane anchor (412 aa at the N terminus) of the adhesin with mRuby2 (Figure 2b). Each MpA construct was placed between the NdeI and XhoI sites in a pET24a vector. Genes encoding for two other machinery proteins (hlyb and hlyd) of E. coli T1SS were placed between the SacI and BamHI sites in a pSTV28 plasmid as described.40 The hlyb/d genes were PCR amplified from a pLG575 plasmid kindly provided by Professor Dr. Peter Sebo (Institute of Microbiology of the Czech Academy of Sciences).
Flow Cytometry
Approximately 106 of either induced MpA+/lyso+ or MpA–/lyso+ cells were collected, blocked with 1 mg/mL bovine serum albumin (Sigma-Aldrich) in a buffer containing 20 mM Tris-HCl pH 9, 150 mM NaCl, and 5 mM CaCl2 (Buffer A), and incubated (1:100, 1 h) with rabbit antiserum raised against the central repeats of MpA. After washing with buffer A (2×), cells were incubated with a goat antirabbit secondary antibody (Alexa 633, Invitrogen; 1:100, 1 h) and then analyzed by a Fluorescence Activated Cell Sorter (Aria III, BD Sciences). The stained cells were gated based on forward and side scattering. At least 10 000 gated events were analyzed and plotted using the FlowJo software.
Confocal Microscopy
Cells were prepared in a similar way as those for flow cytometry experiments described above. MpA-mRuby2-expressing cells were collected, blocked with 1 mg/mL bovine serum albumin (Sigma-Aldrich) in Buffer A, and incubated (1:100, 1 h) with rabbit antiserum raised against the central repeats of MpA. After washing with buffer A (2×), a secondary (goat antirabbit) antibody conjugated with a fluorophore (Alexa 488, invitrogen) was used to label the cell-surface-exposed MpA-mRuby2. Samples were mounted on the stage of a confocal laser scanning microscope (Leica SP8) equipped with solid-state lasers (488 and 638 nm), a Plan APO 63× water immersion objective (NA 1.2) and hybrid detector. Fluorescent images were acquired using the Leica LAS X software.
Synthesis of Methacrylated Dextran
Dextran T500 (500 kDa, Pharmacosmos) (4 g, 24.7 mmol glucopyranose unit) was dissolved in 36 mL of dimethyl sulfoxide (DMSO) and degassed with argon for 15 min. After dissolving 800 mg of 4-dimethylaminopyridine (DMAP, Sigma-Aldrich), 2.4 mL of glycidyl methacrylate (GMA, Sigma-Aldrich) was added. The solution was stirred at room temperature for 44 h and the reaction was stopped by adding 6.6 mL of 1 M HCl to neutralize the DMAP. The reaction mixture was transferred to a SnakeSkin Dialysis bag (molecular weight cutoff: 10 kDa, Thermo scientific) and dialyzed against demineralized water at room temperature with changing water 3–4 times a day for 72 h. Next, the methacrylated dextran solution was lyophilized and stored at −30 °C before use. For NMR measurements, pharmaceutical-grade dextran 500 and methacrylated dextran samples were prepared in 10 mg/mL CDCl3 and 1 mg/mL D2O, respectively. The samples were measured on a Bruker Avance 3 HD NanoBay spectrometer (1H NMR 400 MHz) at 23 °C. The degree of substitution, which is the percentage of methacrylated glucose monomers, was calculated with the formula (x/1.04y) × 100, where x stands for the average integral of the protons of the double bond, indicated with 22′ and 22′′ in the spectrum, y stands for the integral of the anomeric proton, indicated with 4 in the spectrum, and 1.04 is the correction factor for the average 4% of α – 1, 3 linkages in dextran (Figure S1.2).
Dextran-Based Hydrogel Preparation
An aqueous solution of methacrylated dextran (5% w/v, 200 mg methacrylated dextran in 4 mL of H2O) and ammonium persulfate (4.5 mg) was degassed with argon, stirred, and cooled in an ice bath for 15 min. From here on, all preparation was done at 4 °C in a cold room. After the addition of 5 μL of tetramethylethylenediamine (TEMED), the solution was mixed vigorously for 4–5 s and poured into the mold for hydrogel formation via freezing at −20 °C.
Assessment and Quantification of Unreacted Monomers Present in Hydrogel Wash Solutions by NMR and FT-IR
Samples of the wash solution were prepared by soaking a ready-to-use hydrogel (10 × 10 mm) in 5 mL of UP-mQ (ultrapure miliQ for analytical purposes). After 72 h the gel was removed, and the UP-mQ was freeze-dried in a 50 mL falcon tube. Subsequently, 800 μL of D2O was added to the falcon tube and sonicated for 5 min. This solution was filtered through a syringe filter (0.2 μm) to remove residual dextran hydrogel that was still present in the sample. For NMR measurement, a sample (0.2% methanol as an internal standard) was measured on a Bruker Avance 3 HD NanoBay spectrometer (1H-NMR 400 MHz) with a water suppression scan. The liquid-state IR spectrum for the sample was recorded on a PerkinElmer Spectrum One spectrometer in a CaF2 cell. The NMR and FT-IR experiments were performed at 23 °C.
Mold Fabrication for the Cubic Hydrogel
The device mold was fabricated in two steps. First, a poly(methyl methacrylate) (PMMA) mold was made using a CO2 laser (see Figure S4a) and glued to a flat PMMA using an acrylic glue. Second, a PDMS prepolymer mixture was prepared in a 10:1 ratio (PDMS: curing agent) and poured over the mold. This was thermally cured in an oven at 65 °C for 2 h, and then the polymer was carefully peeled off from the mold (see Figure S4b). A side hole (θ = 2 mm) was made at the center using metallic punches. In the side holes, glass needles were inserted before the hydrogel was poured in.
Preparation of the Cubic Hydrogel for the Quantification of Bacterial Leakage
Activated methacrylated dextran solution was poured into a PDMS mold with a pin placed through the central hole from one side to the center of the cubic mold. Removal of the pin after the solidification of the pregel solution resulted in an empty chamber within the hydrogel for loading bacteria. The casted methacrylated dextran was kept frozen at −20 °C overnight, and later thawed at room temperature, washed with 20 mL of Milli-Q, and allowed to swell for 5 min. Next, the gel was washed with 20 mL of 0.05 M NaOH followed by 20 mL of Milli-Q and stored at 4 °C in a falcon tube with fresh Milli-Q.
Hydrogel Disk Preparation
Activated methacrylated dextran was casted in a mold used for fabrication of acrylamide-based gels for SDS-PAGE (Mini PAGE mold, Bio-Rad). This resulted in the fabrication of a 0.75 mm-thick homogeneous sheet. After washing, the hydrogel disks were formed by punching off the sheet with a 8 mm hole puncher, and stored in fresh Milli-Q at room temperature.
Cloning
Cloning procedures were performed following the Gibson assembly method.42,43 Reagents and competent DH5a cells used for cloning were from the NEBuilder HiFi DNA Cloning Kit (Product E5520S New England Biolabs). Q5 High-Fidelity 2X Master Mix was used for long PCRs (Product M0492SNew England Biolabs 240 County Road Ipswich, MA 01938–2732). The correct gene products were confirmed by DNA sequencing (BaseClear BV. Sylviusweg 74 2333 BE Leiden, The Netherlands, Tables S1 and S2).
Bacterial Culturing
Unless otherwise noted, all bacteria were grown at 37 °C. Cultures were grown overnight in LB supplemented with selective antibiotics from single colonies of freshly transformed cells. Overnight cultures were further diluted 100-fold in fresh LB supplemented with selective antibiotics. After 3 h incubation, cells were chemically induced (0 or 5 mM arabinose, 0 or 0.5 mM IPTG) (OD600 nm = 0.8) and further grown for 3 h. Note timing is important for ensuring optimum protein expression levels and therefore functionality of the system.
Bacterial Leakage Experiment with the Cubic Hydrogels (Liquid LB Medium)
Cubic hydrogels were soaked in LB medium supplemented with 30 μg/mL chloramphenicol, 100 μg/mL kanamycine, 5 mM CaCl2, and 0.5 mM IPTG for 1 h and transferred to an empty sterile Petri-dish by tweezers with the opening of the bacteria-loading chamber facing upward. Excess LB was pipetted out of the central chamber. Engineered E. coli (7 μL, OD600 nm is approximately 1) were pipetted into the central chamber of the cubic hydrogel placed in a sterile Petri-dish. Next, 25 mL of medium was added to the plate gradually to roughly 80% (8 mm) of the height of the cubic hydrogel incubated at room temperature. The medium was collected at two different time points after the bacterial loading (10 min and 24 h) and plated (100 μL) for the quantification of bacterial leakage by CFU counting. The LB medium and Petri-dish were refreshed after each sample collection to prevent the overgrowth of bacteria.
Experiments for Assessing Bacterial Retention to the Hydrogels
Approximately 100 μL of activated methacrylated dextran solution was poured into individual wells of 96-well plate to make hydrogel cylinders. To bind bacteria to the dextran-based hydrogel, each hydrogel cylinder was incubated with 5 mL of MpA+ or MpA– (OD = ∼1) in the presence of 0.5 mM IPTG and 2 mM CaCl2 in a shaking incubator overnight (37 °C, 250 rpm). The hydrogels were then transferred to a fresh culture tube containing 5 mL of a sterile buffer (10 mM Tris-HCl, 150 mM NaCl, and 2 mM CaCl2) with antibiotics (for MpA–, chloramphenicol only; for MpA+, both kanamycin and chloramphenicol). The hydrogels were washed in the buffer with shaking (200 rpm, 20 °C) for 1 h. The hydrogels were then transferred to fresh culture tubes and homogenized with a glass rod before resuspending in 100 μL of the wash buffer. Serial dilutions of the samples were plated onto agar plates with appropriate antibiotics, and CFUs were counted the next day for quantification of bacteria bound to the hydrogel.
Scanning Electron Microscopy
Dextran gels seeded with cells were first fixated in 4% formaldehyde for 20 min, and further kept in Milli-Q water. Samples were further sliced using a scalpel to show the cross-section of the bacterial loading chamber and sputter coated with a 9 nm-gold layer using an EMITECH K575X Peltier-cooled SOP Turbo sputter coater Dual. The gels were imaged at 5 kV in a FEI Quanta 3D field-emission SEM, equipped with an Everhart-Thornley secondary electron detector
Bacterial Leakage Experiment with the Hydrogel Disks (LB-Agar Medium)
Prior to use, dextran-based hydrogel disks were soaked in LB medium supplemented with 30 μg/mL chloramphenicol, 100 μg/mL kanamycine, and 5 mM CaCl2. Next, 100 μL of a solution containing chemical inducers (0 or 5 mM arabinose, 0 or 0.5 mM IPTG) were spread on LB-agar plates supplemented with antibiotics and CaCl2. The surface of the LB-agar substrate was allowed to dry by incubating the plates at 37 °C for 30 min. Hydrogel disks were set on the agar-LB surface and quickly dabbed dry using clean, dust-free, paper tissue, and further incubated for 10 min at 37 °C. A drop (1 μL) of engineered E. coli culture was placed in the center of hydrogel surface on the LB agar medium within Petri dishes. The migration of the engineered E. coli was tracked by imaging for 4 days. Pictures were analyzed using ImageJ/Fiji.51
Antibacterial Activity Tests (Modified Kirby-Bauer Test)
Master mix solutions (100 μL) containing various chemical inducers (0 or 5 mM arabinose, 0 or 0.5 mM IPTG, 5 mM CaCl2) were spread onto 15 mL Mueller-Hinton agar plates, which were dried by incubating them at 37 °C. Further, a protocol of a standard Kirby-Bauer test was followed. Briefly, bacterial cultures of Staphylococcus aureus (methicillin resistant) or Streptoccocus agalactiae was diluted to OD600 nm of 0.4–0.6, and streaked across the Mueller-Hinton agar plates with a cotton swab before they were tested against various antibacterial treatments. Various engineered E. coli cells including MpA+/lyso–, MpA–/lyso–, MpA+/lyso+ and MpA+/Lyso– were spotted (5–10 μL) onto the plates, and their effects on the growth of S. aureus were observed and photographed.
Cell-Free Medium Preparation
Approximately 3 h after lysostaphin induction with 5 mM arabinose, 100 mL of engineered E. coli cell culture was spun down by centrifugation (4000 rpm) for 15 min at 4 °C. The cell-free-medium was then harvested after the removal of residual cells by filtering (0.2 μm cutoff). To remove the antibiotics present in the growth medium, the cell-free-medium was buffer-exchanged into buffer A and concentrated approximately 30-fold using Millipore spin cassettes (molecular weight cutoff: 10-kDa). The cell-free medium was further tested by applying a drop of 5 μL onto the surface of the prestreaked plates (see antibacterial activity tests).
Living Hydrogel with anti-MRSA Activity
Dextran-based hydrogel disks were briefly dried using a sterile cotton swab and then placed on the Mueller-Hinton agar plates streaked with methicillin-resistant Staphylococcus aureus. Prior to their infusion into the hydrogel disks, the engineered E. coli cells were washed to 2 times in LB to ensure complete removal of antibiotics (growth medium). Next, a drop (1 μL) of the induced E. coli cells was placed at the center of the dextran-based hydrogel disk surface. Plates were further incubated at 37 °C for up to 4 days, and the effects of bacteriocin-producing living hydrogels were visualized by imaging with a DSLR camera (Canon 600D, 50 mm lens). Pictures were analyzed using ImageJ.
Acknowledgments
The authors would like to acknowledge the financial support for this research of the STW-foundation, the EPSRC-NSFC Joint Research Project (No. 51461135005), the European Union (ERC-StG No. 635928 & 677313), the Dutch Science Foundation (NWO ECHO Grant No. 712.016.002 and NWO-VIDI Grant No. 723.016.003), and the Dutch Ministry of Education, Culture and Science (Gravity Program 024.001.035). Dextran was kindly provided by Pharmacosmos A/S. We thank Ingeborg Schreur-Piet for her support for SEM imaging. We thank Prof. Peter Sebo for generously providing the plG575 plasmid for amplifying the HlyB/HlyD genes. We thank Bas Rosier for his help and valuable advice with Figure 3b. We thank Mathijs F.J. Mabesoone for his help with acquiring and interpreting NMR and IR data. We are indebted to Prof. Luc Brunsveld for his important guidance and financial support while supervising the Tue 2018 iGEM team. Molecular graphics and analyses were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.9b00404.
Quantification of unreacted monomers released from the hydrogel; characterization of dextran-based hydrogel microstructure; fabrication of cubic-dextran-based hydrogel; quantification of bacterial retention to the dextran-based hydrogel (PDF)
Author Contributions
# These authors contributed equally to this work
The authors declare no competing financial interest.
Supplementary Material
References
- Pardee K.; Slomovic S.; Nguyen P. Q.; Lee J. W.; Donghia N.; Burrill D.; Ferrante T.; McSorley F. R.; Furuta Y.; Vernet A.; Lewandowski M.; Boddy C. N.; Joshi N. S.; Collins J. J. (2016) Portable, On-Demand Biomolecular Manufacturing. Cell 167 (1), 248–259. e12. 10.1016/j.cell.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P.; Han N.; Cleto S.; Cao J.; Purcell O.; Shah K. A.; Lee K.; Ram R.; Lu T. K. (2016) Synthetic biology and microbioreactor platforms for programmable production of biologics at the point-of-care. Nat. Commun. 7, 12211. 10.1038/ncomms12211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Din M. O.; Danino T.; Prindle A.; Skalak M.; Selimkhanov J.; Allen K.; Julio E.; Atolia E.; Tsimring L. S.; Bhatia S. N.; Hasty J. (2016) Synchronized cycles of bacterial lysis for in vivo delivery. Nature 536 (7614), 81–85. 10.1038/nature18930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.; Perez-Pinera P.; Lowenhaupt K.; Wu M. R.; Purcell O.; de la Fuente-Nunez C.; Lu T. K. (2018) Versatile and on-demand biologics co-production in yeast. Nat. Commun. 9 (1), 77. 10.1038/s41467-017-02587-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. Y.; Zhong C.; Lu T. K. (2015) Engineering living functional materials. ACS Synth. Biol. 4 (1), 8–11. 10.1021/sb500113b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. Q.; Courchesne N. D.; Duraj-Thatte A.; Praveschotinunt P.; Joshi N. S. (2018) Engineered Living Materials: Prospects and Challenges for Using Biological Systems to Direct the Assembly of Smart Materials. Adv. Mater. 30 (19), e1704847 10.1002/adma.201870134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C.; Ellis T. (2019) Biological Engineered Living Materials: Growing Functional Materials with Genetically Programmable Properties. ACS Synth. Biol. 8 (1), 1–15. 10.1021/acssynbio.8b00423. [DOI] [PubMed] [Google Scholar]
- Appiah C.; Arndt C.; Siemsen K.; Heitmann A.; Staubitz A.; Selhuber-Unkel C. (2019) Living Materials Herald a New Era in Soft Robotics. Adv. Mater. 31 (36), e1807747 10.1002/adma.201807747. [DOI] [PubMed] [Google Scholar]
- Lee J. W.; Chan C. T. Y.; Slomovic S.; Collins J. J. (2018) Next-generation biocontainment systems for engineered organisms. Nat. Chem. Biol. 14 (6), 530–537. 10.1038/s41589-018-0056-x. [DOI] [PubMed] [Google Scholar]
- Schaffner M.; Ruhs P. A.; Coulter F.; Kilcher S.; Studart A. R. (2017) 3D printing of bacteria into functional complex materials. Sci. Adv. 3 (12), eaao6804 10.1126/sciadv.aao6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber L. C.; Koehler F. M.; Grass R. N.; Stark W. J. (2012) Incorporation of penicillin-producing fungi into living materials to provide chemically active and antibiotic-releasing surfaces. Angew. Chem., Int. Ed. 51 (45), 11293–6. 10.1002/anie.201204337. [DOI] [PubMed] [Google Scholar]
- Liu X.; Tang T. C.; Tham E.; Yuk H.; Lin S.; Lu T. K.; Zhao X. (2017) Stretchable living materials and devices with hydrogel-elastomer hybrids hosting programmed cells. Proc. Natl. Acad. Sci. U. S. A. 114 (9), 2200–2205. 10.1073/pnas.1618307114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpetti F.; Petrova E.; Maerkl S. J. (2017) A Microfluidic Biodisplay. ACS Synth. Biol. 6 (11), 1979–1987. 10.1021/acssynbio.7b00088. [DOI] [PubMed] [Google Scholar]
- Wang X.; Pu J.; An B.; Li Y.; Shang Y.; Ning Z.; Liu Y.; Ba F.; Zhang J.; Zhong C. (2018) Programming Cells for Dynamic Assembly of Inorganic Nano-Objects with Spatiotemporal Control. Adv. Mater. 30 (16), e1705968 10.1002/adma.201705968. [DOI] [PubMed] [Google Scholar]
- Huang J.; Liu S.; Zhang C.; Wang X.; Pu J.; Ba F.; Xue S.; Ye H.; Zhao T.; Li K.; Wang Y.; Zhang J.; Wang L.; Fan C.; Lu T. K.; Zhong C. (2019) Programmable and printable Bacillus subtilis biofilms as engineered living materials. Nat. Chem. Biol. 15 (1), 34–41. 10.1038/s41589-018-0169-2. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S.; Aubin-Tam M. E.; Meyer A. S. (2019) 3D Printing for the Fabrication of Biofilm-Based Functional Living Materials. ACS Synth. Biol. 8 (7), 1564–1567. 10.1021/acssynbio.9b00192. [DOI] [PubMed] [Google Scholar]
- Gao C.; Xu P.; Ye C.; Chen X. L.; Liu L. M. (2019) Genetic Circuit-Assisted Smart Microbial Engineering. Trends Microbiol. 27 (12), 1011–1024. 10.1016/j.tim.2019.07.005. [DOI] [PubMed] [Google Scholar]
- Pinero-Lambea C.; Bodelon G.; Fernandez-Perianez R.; Cuesta A. M.; Alvarez-Vallina L.; Fernandez L. A. (2015) Programming controlled adhesion of E. coli to target surfaces, cells, and tumors with synthetic adhesins. ACS Synth. Biol. 4 (4), 463–73. 10.1021/sb500252a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass D. S.; Riedel-Kruse I. H. (2018) A Synthetic Bacterial Cell-Cell Adhesion Toolbox for Programming Multicellular Morphologies and Patterns. Cell 174 (3), 649–658. e16. 10.1016/j.cell.2018.06.041. [DOI] [PubMed] [Google Scholar]
- Guo S.; Garnham C. P.; Whitney J. C.; Graham L. A.; Davies P. L. (2012) Re-evaluation of a bacterial antifreeze protein as an adhesin with ice-binding activity. PLoS One 7 (11), e48805 10.1371/journal.pone.0048805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.; Stevens C. A.; Vance T. D. R.; Olijve L. L. C.; Graham L. A.; Campbell R. L.; Yazdi S. R.; Escobedo C.; Bar-Dolev M.; Yashunsky V.; Braslavsky I.; Langelaan D. N.; Smith S. P.; Allingham J. S.; Voets I. K.; Davies P. L. (2017) Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci. Adv. 3 (8), e1701440 10.1126/sciadv.1701440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.; Vance T. D. R.; Stevens C. A.; Voets I. K.; Davies P. L. (2019) RTX Adhesins are Key Bacterial Surface Megaproteins in the Formation of Biofilms. Trends Microbiol. 27 (5), 470. 10.1016/j.tim.2019.02.001. [DOI] [PubMed] [Google Scholar]
- Li S. Q.; Dong S. J.; Xu W. G.; Tu S. C.; Yan L. S.; Zhao C. W.; Ding J. X.; Chen X. S. (2018) Antibacterial Hydrogels. Adv. Sci. 5 (5), 1700527 10.1002/advs.201700527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. Y.; Wang H.; Hu J. F.; Hu J. J.; Zhang S.; Yang Z.; Li Y. W.; Cheng Y. Y. (2019) Smart Hydrogels with Antibacterial Properties Built from All Natural Building Blocks. Chem. Mater. 31 (18), 7678–7685. 10.1021/acs.chemmater.9b02547. [DOI] [Google Scholar]
- Gross M. (2013) Antibiotics in crisis. Curr. Biol. 23 (24), R1063–R1065. 10.1016/j.cub.2013.11.057. [DOI] [PubMed] [Google Scholar]
- Schindler C. A.; Schuhardt V. T. (1964) Lysostaphin: A New Bacteriolytic Agent for the Staphylococcus. Proc. Natl. Acad. Sci. U. S. A. 51, 414–21. 10.1073/pnas.51.3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. T.; Wroe J. A.; Agarwal R.; Martin K. E.; Guldberg R. E.; Donlan R. M.; Westblade L. F.; Garcia A. J. (2018) Hydrogel delivery of lysostaphin eliminates orthopedic implant infection by Staphylococcus aureus and supports fracture healing. Proc. Natl. Acad. Sci. U. S. A. 115 (22), E4960–E4969. 10.1073/pnas.1801013115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque S. G.; Lim R. M.; Shoichet M. S. (2005) Macroporous interconnected dextran scaffolds of controlled porosity for tissue-engineering applications. Biomaterials 26 (35), 7436–46. 10.1016/j.biomaterials.2005.05.054. [DOI] [PubMed] [Google Scholar]
- van Dijk-Wolthuis W. N. E.; Franssen O.; Talsma H.; van Steenbergen M. J.; Kettenes-van den Bosch J. J.; Hennink W. E. (1995) Synthesis, Characterization, and Polymerization of Glycidyl Methacrylate Derivatized Dextran. Macromolecules 28 (18), 6317–6322. 10.1021/ma00122a044. [DOI] [Google Scholar]
- vanDijkWolthuis W. N. E.; KettenesvandenBosch J. J.; vanderKerkvanHoof A.; Hennink W. E. (1997) Reaction of dextran with glycidyl methacrylate: An unexpected transesterification. Macromolecules 30 (11), 3411–3413. 10.1021/ma961764v. [DOI] [Google Scholar]
- Szafulera K.; Wach R.ła. A.; Olejnik A. K.; Rosiak J. M.; Ulanski P. (2018) Radiation synthesis of biocompatible hydrogels of dextran methacrylate. Radiat. Phys. Chem. 142, 115–120. 10.1016/j.radphyschem.2017.01.004. [DOI] [Google Scholar]
- Guo S.; Langelaan D. N.; Phippen S. W.; Smith S. P.; Voets I. K.; Davies P. L. (2018) Conserved structural features anchor biofilm-associated RTX-adhesins to the outer membrane of bacteria. FEBS J. 285 (10), 1812–1826. 10.1111/febs.14441. [DOI] [PubMed] [Google Scholar]
- Smith T. J.; Font M. E.; Kelly C. M.; Sondermann H.; O’Toole G. A. (2018) An N-Terminal Retention Module Anchors the Giant Adhesin LapA of Pseudomonas fluorescens at the Cell Surface: a Novel Subfamily of Type I Secretion Systems. J. Bacteriol. 200 (8), e00734-17 10.1128/JB.00734-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. J.; Sondermann H.; O’Toole G. A. (2018) Type 1 Does the Two-Step: Type 1 Secretion Substrates with a Functional Periplasmic Intermediate. J. Bacteriol. 200 (18), e00168-18 10.1128/JB.00168-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance T. D. R.; Guo S.; Assaie-Ardakany S.; Conroy B.; Davies P. L. (2019) Structure and functional analysis of a bacterial adhesin sugar-binding domain. PLoS One 14 (7), e0220045 10.1371/journal.pone.0220045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N.; Nicaud J. M.; Gray L.; Holland I. B. (1985) Identification of polypeptides required for the export of haemolysin 2001 from E. coli. Mol. Gen. Genet. 201 (3), 529–36. 10.1007/BF00331351. [DOI] [PubMed] [Google Scholar]
- Bakkes P. J.; Jenewein S.; Smits S. H.; Holland I. B.; Schmitt L. (2010) The rate of folding dictates substrate secretion by the Escherichia coli hemolysin type 1 secretion system. J. Biol. Chem. 285 (52), 40573–80. 10.1074/jbc.M110.173658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenders M. H.; Beer T.; Smits S. H.; Schmitt L. (2016) In vivo quantification of the secretion rates of the hemolysin A Type I secretion system. Sci. Rep. 6, 33275. 10.1038/srep33275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenders M. H.; Weidtkamp-Peters S.; Kleinschrodt D.; Jaeger K. E.; Smits S. H.; Schmitt L. (2015) Directionality of substrate translocation of the hemolysin A Type I secretion system. Sci. Rep. 5, 12470. 10.1038/srep12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L.; Chen S.; Yi L.; Woodard R. W.; Chen J.; Wu J. (2012) Extracellular overexpression of recombinant Thermobifidafuscacutinase by alpha-hemolysin secretion system in E. coli BL21(DE3). Microb. Cell Fact. 11, 8. 10.1186/1475-2859-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T.; Schneewind O. (1996) Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J. 15 (18), 4789–97. 10.1002/j.1460-2075.1996.tb00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G.; Smith H. O.; Hutchison C. A. 3rd; Venter J. C.; Merryman C. (2010) Chemical synthesis of the mouse mitochondrial genome. Nat. Methods 7 (11), 901–3. 10.1038/nmeth.1515. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R. Y.; Venter J. C.; Hutchison C. A. 3rd; Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6 (5), 343–5. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Bauer A. W.; Perry D. M.; Kirby W. M. (1959) Single-disk antibiotic-sensitivity testing of staphylococci; an analysis of technique and results. AMA Arch. Int. Med. 104 (2), 208–16. 10.1001/archinte.1959.00270080034004. [DOI] [PubMed] [Google Scholar]
- Sisirak M.; Zvizdic A.; Hukic M. (2010) Methicillin-resistant Staphylococcus aureus (MRSA) as a cause of nosocomial wound infections. Bosnian J. Basic Med. Sci. 10 (1), 32–7. 10.17305/bjbms.2010.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga B. T.; Sumida W. K.; Taira D. A.; Davis J. W.; Seto T. B. (2016) Hospital-Acquired Methicillin-resistant Staphylococcus aureus Bacteremia Related to Medicare Antibiotic Prescriptions: A State-Level Analysis. Hawaii J. Med. Public Health 75 (10), 303–309. [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Bram E. E.; Weiss R. (2013) Genetically programmable pathogen sense and destroy. ACS Synth. Biol. 2 (12), 715–23. 10.1021/sb4000417. [DOI] [PubMed] [Google Scholar]
- Hwang I. Y.; Koh E.; Wong A.; March J. C.; Bentley W. E.; Lee Y. S.; Chang M. W. (2017) Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 8, 15028. 10.1038/ncomms15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogfelt K. A.; Bergmans H.; Klemm P. (1990) Direct Evidence That the Fimh Protein Is the Mannose-Specific Adhesin of Escherichia-Coli Type-1 Fimbriae. Infect. Immun. 58 (6), 1995–1998. 10.1128/IAI.58.6.1995-1998.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M.; Wu F.; Hao G.; Qi Q.; Li R.; Li N.; Wei L.; Chai T. (2017) Bacillus subtilis Improves Immunity and Disease Resistance in Rabbits. Front. Immunol. 8, 354. 10.3389/fimmu.2017.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J. Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9 (7), 676–82. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25 (13), 1605–12. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.