

Abstract
Methods to improve plasmid-mediated transgene expression are needed for gene medicine and gene vaccination applications. To maintain a low risk of insertional mutagenesis-mediated gene activation, expression-augmenting sequences would ideally function to improve transgene expression from transiently transfected intact plasmid, but not from spurious genomically integrated vectors. We report herein the development of potent minimal, antibiotic-free, high-manufacturing-yield mammalian expression vectors incorporating rationally designed additive combinations of expression enhancers. The SV40 72 bp enhancer incorporated upstream of the cytomegalovirus (CMV) enhancer selectively improved extrachromosomal transgene expression. The human T-lymphotropic virus type I (HTLV-I) R region, incorporated downstream of the CMV promoter, dramatically increased mRNA translation efficiency, but not overall mRNA levels, after transient transfection. A similar mRNA translation efficiency increase was observed with plasmid vectors incorporating and expressing the protein kinase R-inhibiting adenoviral viral associated (VA)1 RNA. Strikingly, HTLV-I R and VA1 did not increase transgene expression or mRNA translation efficiency from plasmid DNA after genomic integration. The vector platform, when combined with electroporation delivery, further increased transgene expression and improved HIV-1 gp120 DNA vaccine-induced neutralizing antibody titers in rabbits. These antibiotic-free vectors incorporating transient expression enhancers are safer, more potent alternatives to improve transgene expression for DNA therapy or vaccination.
Supplementary information
The online version of this article (doi:10.1038/gt.2010.149) contains supplementary material, which is available to authorized users.
Keywords: non-viral vector, plasmid, antibiotic-free, DNA vaccination, HIV
Subject terms: Genetic vectors, Gene expression
Introduction
Gene therapy using transiently expressed non-integrative plasmid vectors is the ideal solution for many public health applications. For example, no current therapy for diabetic foot ulcers directly improves the biology of wound healing as would gene therapy using the hypoxia-inducible factor 1α (HIF-1α) master regulator of wound healing genes.1 A topically delivered gene-based drug is ideal for such growth factor replacement therapies, as plasmid has a far longer duration of expression compared with rapidly degraded recombinant peptides.
Extensive research to improve plasmid delivery has resulted in advanced methods such as electroporation (EP)-facilitated uptake, which dramatically improves gene transfer compared with passive DNA uptake. Plasmid-directed gene expression is now reaching the efficacy barrier that, to date, has prevented commercialization of gene-based plasmids for human application.2
Although plasmid delivery has been the focus of gene transfer research, complementary vector design innovations may also be applied to improve transgene expression. We previously reported the development of potent minimalized antibiotic-free (AF) mammalian expression vectors.3 AF vectors are derivatives of the pDNAVACCUltra expression vectors,4 which incorporate and express a 150 bp RNA-OUT antisense RNA that represses expression of a host strain chromosome-encoded counter-selectable marker (SacB-encoded levansucrase) that is toxic in the presence of sucrose (Figure 1a). These sucrose selectable vectors combine AF selection with highly productive fermentation manufacturing (>1 g l−1 plasmid DNA yields) and improved transgene expression levels compared with existing vectors.3, 5, 6, 7, 8 These plasmids were designed to remove non-essential extraneous sequences (Figure 1) and are minimalized FDA (Food and Drug Administration)-compliant9 vectors for gene therapy or genetic immunization applications. These AF vectors are also compliant with European regulatory agency recommendations to eliminate antibiotic resistance markers from plasmid therapy vectors.10
Figure 1.

AF selection vectors: (a) RNA-OUT-mediated AF selection. Left: In a plasmid-free cell, levansucrase was expressed from a chromosomally integrated SacB gene, leading to cell death in the presence of sucrose. Right: RNA-OUT from the plasmid repressed translation of the SacB gene, achieving plasmid selection; (b) NTC8385 EGFP AF vector with RNA-OUT selectable marker and HTLV-I R transient expression enhancer; (c) NTC8485 EGFP AF vector, with transient expression enhancers HTLV-I R, VA1 and SV40 enhancer. Optimization of the SV40-CMV boundary (BE deletion) resulted in a vector, NTC8685, with further improved expression; (d) gWIZ EGFP kanR vector with locations of non-essential spacer and junk DNA (TN903 inverted repeat, polyC, polyG and ampR promoter), annotated. The basepairs 1–245 pUC19 region functioned to maintain an optimal junction between the CMV promoter and the prokaryotic backbone. This sequence was retained in the equivalent location (in the NTC8385 vector-UP) and was replaced by the SV40 enhancer in the NTC8485 and NTC8685 vectors. In these vectors, the 1–245 bp pUC19 region was moved and added as an extension to the pUC origin to add back a leading strand primosomal assembly site (PAS-BH) present in pBR322. This site was deleted when the pUC vector was created by imprecise deletion of the repressor of primer (ROP) gene.6 NTC8485 and NTC8685 PAS-BH vectors had higher plasmid copy number and manufacturing yields than did NTC8385 or gWIZ.6
Although individual expression-augmenting sequences have been identified, they have not been used combinatorially to improve vector performance. We report herein the incorporation of rationally designed additive combinations of expression enhancers into optimized minimalistic AF vectors to effect improved transgene expression. The resultant high-production-yield, minimal, AF mammalian expression vectors incorporate novel vector backbone functionalities that further improve plasmid-directed transgene expression after transient transfection (transient expression enhancers; TEE platform: Figures 1b and c). The viral human T-lymphotropic virus type I (HTLV-I) R, adenoviral viral associated (VA) RNAI (VA1) and SV40 enhancers used in this study were derived from non-coding regions of the respective viruses and did not have significant sequence homology to the human genome. These studies demonstrate that dramatic increases in vector-directed transgene expression can be obtained through innovations in vector design.
Results
Vector design criteria
To reduce chances in chromosomal integration, sequences added to a plasmid to increase transgene expression should contain no significant homology to the human genome. This may be determined by BLAST search, specifying to search for short, nearly exact matches against the human genome.5, 11
Regions encoding antigenic peptides should also not be present in vector backbones. These include cryptic open reading frames (ORFs) in bacterial or eukaryotic sequences that may be expressed in eukaryotic cells to generate unwanted and potentially detrimental cytotoxic T-cell12, 13, 14 or humoral responses. The removal of spacer and junk sequences and the use of RNA-based selectable markers to eliminate the kanamycin-resistant (kanR) ORF reduce this risk.
Minimalized vector design
The NTC8385, NTC8485 (Figures 1b and c) and NTC8685 vectors (NTC8485 incorporating the Boundary Element deletion; Figure 1c) described herein are minimalized vectors that do not contain extraneous spacer or junk sequences. These vectors incorporate a short 140 bp RNA-based selection marker rather than an antibiotic resistance marker (Figure 1a). This resulted in much higher vector potency through elimination of approximately 2 kb of DNA compared with the gWIZ (Genlantis, San Diego, CA, USA) vector, which includes the kanR gene and associated extraneous DNA including the transposon TN903 inverted repeat, the ampicillin resistance marker promoter and polyC and polyG tailing site cloning footprints (Figure 1d).
3′ untranslated region (UTR): 3′ UTR ORFs are translated
Many DNA vaccine vectors contain a single copy of the human or woodchuck hepatitis B virus posttranscriptional regulatory element (PRE or WPRE) immediately downstream of the stop codon before the transcriptional terminator. This element is commonly used in retroviral and lentiviral vectors as an alternative to the lentiviral Rev-response element to direct export of unspliced full-length viral genomic mRNA. PRE elements are included in DNA vaccines to increase spliced mRNA nuclear export and transgene expression. However, hepatitis genomes are highly compacted such that this enhancer also encodes a 178 amino-acid fragment of the hepatitis C virus polymerase gene.15 The PRE, by design, is within the eukaryotic expression cassette; this type of design is known to generate T cells reactive to major histocompatibility complex-I epitopes in a cryptic ORF.13 Such antigenic regions might significantly alter immune responses in individuals with previous exposure and memory T cells reactive to hepatitis.
To assess this risk, we quantified the translation of a coding region inserted, in different reading frames, downstream of the transgene stop codon in a cytomegalovirus (CMV) promoter expression vector. A neomycin resistance gene (NeoR) without an upstream Kozak sequence was cloned downstream of an enhanced green fluorescent protein (EGFP) transgene in different configurations similar to that used with PREs. Quantifiable neoR translation products were present in all tested configurations, as was biologically active neoR protein after plasmid transfection into both HEK293 and CHO cell lines (Supplementary Table S1). These results demonstrated that expression enhancers should ideally be non-coding regions and that cryptic ORFs present downstream of the transgene within the exported eukaryotic mRNA can be translated, even when the ORF is not in the same reading frame as the upstream transgene.
5′ UTR: HTLV-I R transient expression enhancer
Although a number of 5′ UTR sequences have been identified that can be used to increase transgene expression (for example, see16), the HTLV-I R element is very potent. When cloned downstream of the transcription start site, HTLV-I R improved expression through a variety of promoters, including SV4017 and CMV,18 and improved transgene expression in non-human primates.18 CMV-HTLV-I R promoter plasmid vectors were found to be safe for human vaccination in NIH Vaccine Research Center-sponsored clinical trials of HIV, Ebola and severe acute respiratory syndrome (SARS) DNA vaccines. The CMV-HTLV-I R promoter has HTLV-I R incorporated as part of exon 1 and intron 1 downstream of the CMV promoter (Figure 1b). This configuration improved DNA vaccine expression and immunogenicity.3, 18 Incorporation of HTLV-I R into NTC8385 (Figure 1b) dramatically increased transgene expression in HEK293 total cellular extracts (Figure 2a) and fluorescence-activated cell sorting (FACS) sorted cells (Figure 2b) after transient transfection. This was due to increased mRNA translation efficiency (that is, HTLV-I R increased the amount of EGFP transgene produced per pg cytoplasmic EGFP mRNA; Figure 2c), but not due to increased overall cytoplasmic mRNA levels. By contrast, no effect on integrated expression (either total mean fluorescence or fluorescence per integrated transgene copy) was observed in HEK293 (Figure 2d) or CHO-K1 (Supplementary Figure S1) cells. Consistent with this, the cytoplasmic mRNA translational efficiency improvement observed with HTLV-I R from transiently transfected plasmid did not occur with cytoplasmic RNA produced from genomically integrated plasmid (Figure 2c). Interestingly, the overall amount of EGFP transgene produced per pg of cytoplasmic EGFP mRNA was dramatically higher than that from genomically integrated plasmid compared with transiently transfected plasmid for both CMV and CMV-HTLV-I R promoter plasmids (Figure 2c).
Figure 2.
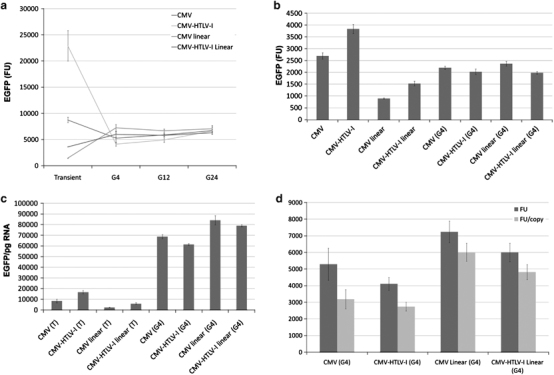
HTLV-I R is a transient expression enhancer. HEK293 cells were transfected in triplicate with CMV or CMV-HTLV-I R NTC8385 backbone vectors (supercoiled or linearized with DraIII; see Figure 1b) expressing an EGFP-IRES-neo transgene (Supplementary Table S1). At T=48 h post transfection, cells for FACS, cytoplasmic RNA for RT-PCR and extracts for EGFP fluorescence were prepared. Transfected cells were replated and stable integrants were selected by 10-day treatment with Geneticin. Samples for EGFP fluorescence were taken after 4, 12 and 24 generations propagation post selection (G4, G12, G24). Samples for (1) FACS; (2) genomic DNA isolation for RT-PCR-integrated plasmid copy number determination; and (3) cytoplasmic RNA for RT-PCR were taken at G4: (a) HTLV-I R increased transgene expression in transient transfection, but did not increase integrated gene expression. Total mean±s.d. fluorescence (FU) of EGFP extracts from the three transfections at the indicated time points is shown; (b) HTLV-I R affected transgene expression in individual cells rather than transfection efficiency. Total mean±s.d. fluorescence (FU) of transient and G4 cells after FACS is shown. The percentage of total cells gated (percentage of expressing cells) was equivalent between the CMV and CMV-HTLV-I R groups (transient: supercoiled 75–79%, linear 61–65%; G4: supercoiled 79–86%, linear 84–87%); (c) HTLV-I R increased mRNA translation after transient transfection. The amount of EGFP protein produced per unit EGFP cytoplasmic mRNA (EGFP FU/ (pg EGFP RNA/ng cytoplasmic mRNA)) pre- (T) and post integration (G4) is shown. (d) HTLV-I R did not increase transgene expression after genomic integration. Total mean±s.d. fluorescence of EGFP extracts from G4 integrated cell lines (FU) and FU per integrated transgene copy number per genome (FU/copy) are shown. Integrated copy number was determined using RT-PCR quantification of the EGFP transgene in isolated quantified genomic DNA.
The in vivo effect of the HTLV-I R region on plasmid vaccination was determined in rabbits. Immunization of rabbits with a CMV-HTLV-I R promoter HIV-1 gp120 DNA vaccine vector combined with EP delivery induced up to ½ log higher titers of HIV-1 virus-neutralizing antibodies (IC50) than a comparator CMV promoter vector (Figure 3). Improved IC50 titers were observed with both neutralization-sensitive (NL4-3 and SF162) and -resistant (6535 and JRCSF) viruses. This is likely attributable to a higher gp120 expression with the CMV-HTLV-I R plasmid. These results demonstrate that this modified 5′ UTR vector design improved vector performance in vivo when combined with an optimal DNA delivery platform (EP).
Figure 3.
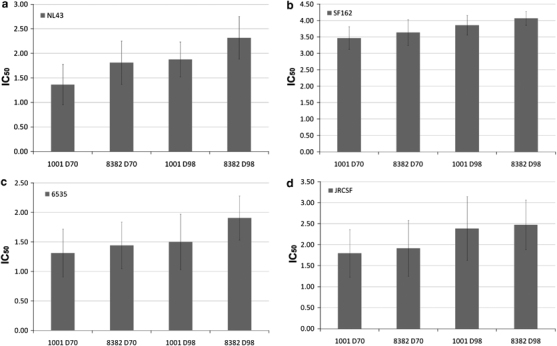
Comparison of CMV and CMV-HTLV-I R promoter HIV-1 gp120 DNA vaccine vectors in inducing neutralizing antibodies. The gp120 viral envelope protein was expressed by either CMV promoter vector pMAmp (giving plasmid 1001) or AF, CMV-HTLV-I R promoter NTC8382 (8382). Neutralization activity on days 70 (2 weeks after the last of three DNA injections) and 98 (2 weeks after a gp120 protein boost) to four HIV-1 strains was determined. The y axis shows the log10 transformation of the half-maximal inhibitory concentration (IC50) of neutralizing antibody titers (higher numbers represent stronger neutralization). Neutralization was tested on two neutralization-sensitive viruses (a) NL4-3 and (b) SF162 and on two resistant viruses (c) 6535 and (d) JRCSF.
Protein kinase regulated by RNA (PKR) inhibitor: VA1 transient expression enhancer
Activation of PKR inhibits mRNA translation through eukaryotic initiation factor-2α phosphorylation.19, 20 PKR is activated either by its ligand, double-stranded RNA or by the PKR-associated activator protein (PACT, and in murine cells by the RAX ortholog) in response to diverse non-nucleic acid-based cell stresses including endoplasmic reticulum stress (the unfolded protein response).21 Cell stress is induced by certain plasmid deliveries (for example, liposomal or calcium phosphate formulations) and/or by expression of aggregation-prone proteins (for example, hydrophobic membrane-domain-containing proteins, polyepitope synthetic proteins). Therefore, inclusion of a PKR inhibitor in the vector backbone may improve plasmid-mediated expression under stress conditions.
The adenovirus serotype 5 VA RNAI (VA1) and the Epstein–Barr Virus EBER1 RNAs are non-coding RNA Pol III-expressed small RNA-based PKR inhibitors. VA RNAI was included in the pAdVAntage Vector (Promega, Madison, WI, USA) plasmid backbone to increase target gene expression of co-transfected plasmids.22 For human vaccination, VA RNAI is preferable to EBER1, as EBER1 binds multiple cellular proteins and has documented cell growth-stimulating properties. These adverse properties have not been attributed to VA1, and, as a natural adenovirus gene, VA1 is present in multiple adenoviral vectors evaluated in human clinical trials.
CMV-HTLV-I R promoter plasmids encoding VA1 (for example, NTC8385-VA1-EGFP; Figure 1b) had increased transgene expression compared with the parent CMV-HTLV-I R promoter vector after transient expression of human HEK293 cells (Figure 4b). CMV-HTLV-I R- and CMV promoter-directed transgene expression levels were not affected by addition of RNA Pol III inhibitor to the transfection (RNA Pol III independent; Figure 4a). In contrast, as expected, VA1-mediated increased transgene expression was RNA Pol III dependent, as it was inhibited at all tested RNA Pol III inhibitor concentrations (Figure 4b). VA1 increased transiently transfected mRNA translation efficiency (EGFP transgene produced per pg of cytoplasmic EGFP mRNA; Supplementary Figure S2), but had no effect on translation efficiency after forced integration in HEK293 cells (EGFP protein produced per integrated transgene; Figure 4d). VA1- and HTLV-I R-directed transient expression enhancement was also observed in human A549 and murine NIH-3T3 and L929 cell lines (Supplementary Figure S3). These results demonstrated that both HTLV-I R and VA1 increased translation of transiently expressed mRNAs and that their effects were not redundant. The variability in the activity of these elements between these cell lines probably reflected differences in levels of cellular cofactors required for VA1 and HTLV-I R activity.
Figure 4.
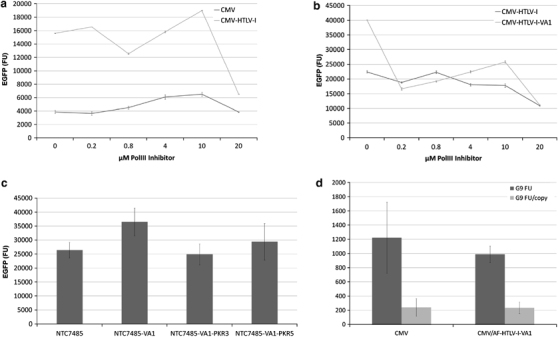
VA1 is a transient expression enhancer: (a) Pol III transcription is dispensable for HTLV-I R-mediated expression enhancement. HEK293 cells were plated and pretreated 10 h with RNA Polymerase III inhibitor (Pol III inhibitor; EMD Biosciences Gibbstown, NJ, USA) at the indicated concentration, then transfected (Lipofectamine 2000) with EGFP plasmids containing the CMV or CMV-HTLV-I R promoter in the presence of Pol III inhibitor. Cell extract EGFP fluorescence (FU) was determined 40 h post transfection. Cell growth was inhibited with 20 μM of Pol III inhibitor, which accounts for the observed reduced FU at this concentration; (b) Pol III transcription was required for VA1 expression enhancement effect. The assay was in the same format as in (a), except for the fact that Pol III inhibitor-treated cells were transfected with EGFP plasmids containing the CMV-HTLV-I R promoter with or without VA1; (c) Inhibition of PKR, not of adenosine deaminase acting on RNA (ADAR) or RNA interference (RNAI), was required for VA1 expression enhancement effect. HEK293 cells were transfected with NTC7485-EGFP (same backbone as NTC8485, except for kanR instead of RNA-OUT) and NTC7485-EGFP- modified to contain: VA1 (PKR+RNAI+ADAR+inhibitor); VA1-PKR3 (PKR−RNAI+ADAR?inhibitor), in which VA1 incorporated the L3 point mutation,26 (the designation ‘ADAR?’ was used, as the effect of L3 on ADAR inhibition has not been established); or VA1-PKR5 (PKR−RNAI+ADAR+inhibitor) in which VA1 incorporated a 1 bp change (pm91) in the central domain.27 (d) VA1 did not increase transgene expression after genomic integration. Postintegration expression of (1) kanR gWIZ (CMV) compared with (2) AF gWIZ derivative containing the CMV-HTLV-I R promoter (instead of CMV) and the VA1 gene (CMV/AF-HTLV-I-VA1). CHO cells were transfected with EGFP-IRES-neo transgene versions of both plasmids using lipofectamine LTX in quadruplicate (using four different ratios of plasmid to LTX). Integrants were selected with Geneticin for 10 days. Results presented are after nine generations post selection (G9). Total mean±s.d. fluorescence of EGFP extracts from four G9 integrated cell lines per vector (FU) and FU per integrated transgene copy number per genome (FU/copy) are shown. Integrated copy number was determined using RT-PCR quantification of the EGFP transgene in isolated quantified genomic DNA.
VA1-mediated PKR inhibition may account for increased transgene expression. Alternatively, PKR-independent stimulation of gene expression may be through VA1 inhibition of adenosine deaminase acting on RNA23 or through RNA interference.24, 25 This was determined by testing the effects of function-specific point mutations in the vector-encoded VA1 gene (Figure 4c) on EGFP expression. VA1-PKR3 incorporated the L3 mutation. This is a 2 bp change in the central domain that has been shown to be inactive for PKR inhibition,26 yet is transcribed at high level in cell culture and maintains the VA RNAI-like RNA secondary structure necessary for RNA interference inhibition. VA1-PKR5 incorporated a 1 bp change (pm91) in the central domain that is strongly reduced for PKR inhibition,27 but retained adenosine deaminase acting on RNA-inhibiting activity23 and the VA RNAI-like RNA secondary structure required for RNA interference inhibition. The results (Figure 4c) suggest that PKR inhibition, and not adenosine deaminase acting on RNA or RNA interference inhibition, was critical for VA1-mediated transgene expression enhancement, as this effect was lost with both PKR3 and PKR5 point mutants.
Nuclear membrane transit: SV40 enhancer transient expression enhancer
Sequences that, when incorporated into a vector backbone, improve plasmid nuclear localization will also enhance non-viral gene expression, as DNA transport to the nucleus is a limiting factor in non-dividing cells (reviewed in Lam and Dean,28 and Wagstaff and Jans29). Such sequences would be true transient expression enhancers, as they function to enhance plasmid nuclear entry, not mRNA production or translation, which could also affect chromosomal gene expression. Mechanisms for sequence-specific plasmid DNA nuclear import are being delineated,30 and a number of DNA transcription factor binding sites have been identified that increase plasmid nuclear import when added to a plasmid vector backbone.28 Several of these were incorporated into the NTC8385-VA1 vector backbone and evaluated for improved expression.
SV40 enhancer
Inclusion of the SV40 72 bp enhancer upstream or downstream of the expression cassette in a DNA vaccine vector backbone increased in vivo expression up to 20-fold using EP delivery in muscle tissue.31, 32 This enhancement was not observed in vitro.31 The NTC8485 vector (Figure 1c) contains the SV40 enhancer (two copies of the 72 and the 21 bp repeats) (Figure 5a). This enhancer region, as part of the SV40 promoter, has not caused safety concerns in human clinical trials of SV40 promoter-containing vectors. NTC8485 had improved manufacturing yields because of improved copy number mediated by the SV40 enhancer and the PAS-BH replication origin.6 This configuration of the SV40 enhancer with the CMV promoter was also shown to improve transgene expression after EP-mediated delivery to murine muscle.33 As expected, in vitro expression of NTC8485 was similar to the NTC8385 vector (Figure 5b). Treatment with tumor necrosis factor-α, which induces nuclear localization of nuclear factor-κB (NF-κB), thereby increasing transgene expression by activation of multiple NF-κB sites in the CMV promoter, resulted in a higher expression than NTC8385 (Figure 5b), perhaps through activation of the two NF-κB sites in the SV40 enhancer (Figure 5a). Addition of three NF-κB binding sites to NTC8485 (NTC8485-κB) further improved expression after tumor necrosis factor-α activation (Figure 5b).
Figure 5.
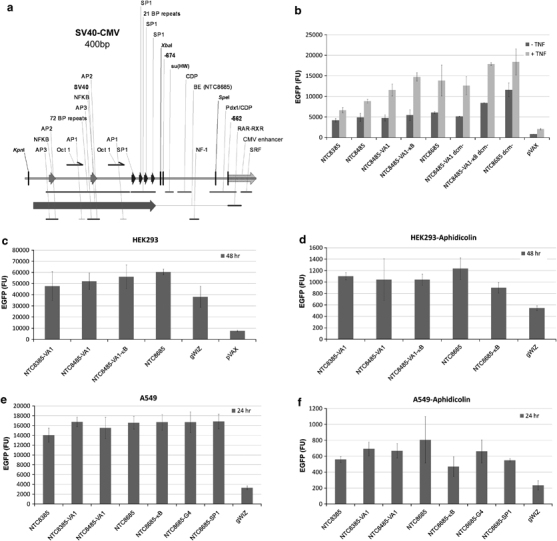
SV40 enhancer-containing NTC8485 and NTC8685 vectors: (a) annotation of the SV40 enhancer (two copies of both the 72 and 21 bp repeats) in NTC8485. The NTC8685 deletion of the CMV upstream region (boundary element, BE) between the CMV enhancer and the SV40 enhancer is shown (BE (NTC8685)), as well as the location of a variety of transcription factor binding sites in the SV40 enhancer and CMV upstream region and enhancer.34, 35, 36 (b) Average±s.d. of total fluorescence (EGFP expression) in the HEK293 cell line of CMV-HTLV-I R promoter vectors, with or without VA1 and SV40 enhancer (NTC8485 and NTC8685), compared with CMV promoter vector pVAX1. Cells were transfected for 3 h, and then incubated with tumor necrosis factor (TNF)-α, versus no TNFα control, for 3 h before addition of fresh medium overnight. EGFP expression was determined 21 h post transfection. (c) HEK293 cells were transfected in triplicate with the indicated EGFP plasmid (dcm−) and total cell EGFP fluorescence was determined 48 h post transfection. Average±s.d. of total fluorescence is shown; (d) HEK293 cells were transfected with dcm− plasmid as in (c), except that cells were growth arrested with aphidicolin before and during the transfection. Results without TNFα stimulation are shown; similar results were obtained with TNFα treatment post transfection. (e) A549 cells were transfected in triplicate with the indicated EGFP plasmid (dcm−) and total cell EGFP fluorescence was determined 24 h post transfection. Average±s.d. of total fluorescence is shown. Similar results were obtained with TNFα treatment for 3 h post transfection. (f) A549 cells were transfected as in (e), except that cells were growth arrested with aphidicolin before and during transfection. Results without TNFα stimulation are shown; similar results were obtained with 3 h TNFα treatment post transfection.
NTC8485 contains the AT-rich unique region upstream of the CMV enhancer. The unique region contains a boundary domain34 and several binding sites for cellular repressor proteins PDX135 and CDP (Figure 5a).36 These sequences may interfere with SV40 enhancer interactions with the CMV promoter. Consistent with this, deletion of the unique region between the SV40 and CMV enhancer junction (to create NTC8685) increased transgene expression in dividing HEK293 cells compared with the NTC8485 parent vector (Figures 5b and c). As the SV40 enhancer did not generally improve expression in vitro,31 the unique region deletion in NTC8685 may allow SV40 enhancer-mediated activation of the CMV promoter. This optimized configuration had higher expression than NTC8485-κB, with and without tumor necrosis factor-α, and approximately 10-fold higher expression than the kanR pVAX1 (Invitrogen, Carlsbad, CA, USA) CMV promoter vector (Figure 5b).
The SV40 enhancer NF-κB sites are substrates for the E coli dcm methylase that methylates the internal cytosine residues in the recognition sequence 5′-CC*AGG-3′ or 5′-CC*TGG-3′ to 5-methyl-cytosine (5mC). Methylation of these sites may reduce NF-κB binding and SV40 enhancer-mediated activation of the CMV promoter in the optimized NTC8685 vector. Consistent with this, NTC8685 plasmid produced in a dcm− host strain had much higher expression than dcm+plasmid (Figure 5b).
Expression levels of NTC8385, NTC8485 and NTC8685 after in vitro EP were determined in human dendritic cells (DC) versus a CMV promoter comparator (Supplementary Figure S4). All three NTC CMV-HTLV-I R promoter vectors had improved expression compared with a CMV promoter vector. In this system, in which isolated dividing cells are subjected to in vitro EP, a strong effect of the NTC8485 and NTC8685 encoding VA1 RNA or SV40 enhancer was not observed, although NTC8685 was the highest expressing vector in all three donors. This would indicate that cell stress was not induced by these in vitro conditions, and cell division eliminated the need for SV40-mediated nuclear entry (as reported in Li et al.31).
Other nuclear import binding proteins
NF-κB and NM23-H2 nuclear shuttle protein binding sites increase plasmid nuclear localization when added to base vectors.37, 38 The SP1 protein is speculated to be a chromatin modulator that functions to maintain expression-competent euchromatin through association with histone acetyl transferases.39 The SP1 and NM23-H2 sites also form a G-quadruplex (G4) structure. This is a local single-stranded DNA region, with a single-stranded three-loop structure formed by interactions with four 3-guanine motifs and three linking loops. G4 structures exclude nucleosomes and are recognized by a variety of DNA binding enzymes40 that may function as nuclear shuttle proteins.
Binding sites for these proteins were integrated into the NTC8685 backbone. NTC8685 expression in dividing or non-dividing (aphidicolin treated) A549 was not improved by inclusion of DNA binding sequences for nuclear localizing proteins NF-κB (κβ), NM23-H2 (G4) or SP1 (Figures 5e and f). A549 is known to express NM23-H2.40 However, expression of NTC8385, NTC8485 and NTC8685 base vectors in aphidicolin-treated non-dividing A549 (Figure 5f) and non-dividing HEK293 (Figure 5d) cells was much higher than that of the gWIZ comparator. All these vectors were produced in dcm− hosts; hence, differences in expression levels could not be attributed to effects of plasmid dcm methylation. This implies that nuclear localization was optimal in these NTC backbones, and that inclusion of additional shuttle protein binding sites was redundant to existing nuclear localization signals present in these vectors.
Discussion
We report the development of regulatory-agency-compliant minimal AF DNA vaccine/gene therapy plasmid vectors that combined >1 g l−1 fermentation yields41 of high-quality plasmid with improved transgene expression by incorporation of TEE: (1) HTLV-I R; (2) adenoviral VA RNAI (VA1); and/or (3) SV40 72 and 21 bp repeats. These transient expression enhancers were non-coding sequences with no homology to the human genome. The expression-enhancing effects were limited to transiently transfected plasmids; no transgene expression enhancement was observed from integrated vector. Unlike matrix-attachment site vectors, TEE sequences did not increase the frequency of genome integration (Williams et al. manuscript in preparation). A vector family, incorporating various TEE combinations, is now available, allowing researchers to select the optimal combination for their application. TEE vectors could also be adapted to improve expression in specific cell types, through swapping of an alternative tissue-specific cellular promoter for the ubiquitous CMV promoter.
Three transient expression-augmenting sequences were identified and incorporated into minimal AF vectors in order to increase the in vivo expression of encoded transgenes. The HTLV-I R region, inserted downstream of the CMV promoter, dramatically increased transgene expression by enhancing mRNA translation efficiency. A similar and additive mRNA translation efficiency increase was observed with plasmid vectors further incorporating and expressing the protein kinase R (PKR)-inhibiting adenoviral VA RNAI (VA1). Finally, the SV40 enhancer, cloned upstream of the CMV enhancer, increased transgene expression in non-dividing cells. Importantly, these vectors did not increase transgene expression from plasmid DNA after forced genomic integration.
This phenomenon of translation stimulation restriction to extrachromosomal vector-derived mRNAs has been previously observed with VA1,42 and was attributed to localized activation of PKR leading to translation inhibition through eukaryotic initiation factor-2α phosphorylation.42, 43 The similar cis-acting effect of HTLV-I R to enhance transient but not integrated plasmid mRNA translation is mechanistically distinct, because HTLV-I R and VA1 effects were additive. A critical difference between transient and genomically produced RNA is that transient mRNA is inefficiently translated (Figure 2c). This may be due to aberrant mRNA splicing, differential cytoplasmic mRNA localization, altered stability of ribosome bound-RNA or differences in ribosome loading/composition/stability with transient compared with genomic source mRNA. The fragment of HTLV-I R incorporated into the 5′ UTR may alleviate these transient RNA-restricted effects.
Incorporation of DNA binding sites for nuclear shuttling proteins NF-κB and NM23-H2 into the NTC8685 vector backbone did not result in improved nuclear localization. Previous studies with these factors were with less-optimal vectors.37, 38, 44 Nuclear localization capacity may be maximized with the extensively optimized minimalized NTC8685 vector.
The vector platform described herein, when combined with advanced delivery methods such as EP, further increased gene expression. This led to improved HIV-1 gp120 neutralizing antibody titers in rabbits after EP-mediated delivery of an HTLV-I R TEE vector. The reduced size compared with alternative vectors may be critical to improving gene transfer with large transgenes such as dystrophin, as large vectors have reduced transfection efficiency with EP delivery.45 TEE vectors therefore have application to generally improve DNA vaccination or gene therapy. These improvements may be critical to enable future gene medicine licensure for public health applications.
Materials and methods
Strains and plasmids
Vectors were constructed using standard molecular biology methods.46 Plasmids NTC8385, NTC8485 and NTC8685 contain a 140 bp DraIII-KpnI RNA-based sucrose selectable marker (RNA-OUT). NTC8485 and NTC8685 incorporate the high copy number PAS-BH-SV40 backbone from the kanR vector NTC7485.6 NTC8485-κB and NTC8685-κB contain three copies of an optimized NF-κB binding site (underlined44) inserted into the NotI site (bold) (Figure 1c). NTC8685-κB 5′-GCGGCCGGGACTTTCCAGCTGGGGACTTTCCAGCTGGGGACTTTCCGCGGCCGC-3′. NTC8685-2x SP1 contains two copies of an optimized SP1 transcription factor binding site (underlined47) inserted between the NotI and NheI sites (bold) of the vector:2x SP1: 5′-GCGGCCGCTAGACGGGGCGGGGGCTCGACGGGGCGGGGGCTAGC-3′. NTC8685-G4 contains one copy of the C-myc NM23-H2 transcription factor binding site (underlined38) inserted between the NotI and NheI sites (bolded) of the vector: G4: 5′-GCGGCCGCTAGGGGAGGGTGGGGAGGGTGGGGAAGGTGGGGAGCTAGC-3′.
kanR plasmids were grown in either Escherichia coli strain coli DH5α (F-φ80dlacZΔM15 Δ(lacZYA -argF) U169 recA1 endA1 hsdR17 (rK−, mK+) phoA supE44 λ-thi-1 gyrA96 relA1) or dcm− version NTC48107 DH5α dcm, whereas AF plasmids were grown in either dcm+ NTC4862 (DH5α attλ::P5/6 6/6-RNA-IN-SacB, CmR) or dcm− NTC48165 (DH5α dcm attλ::P5/6 6/6-RNA-IN- SacB, CmR). For cell culture and immunization testing, low endotoxin (<100 EU mg−1) plasmid DNA was purified using Nucleobond AX 2000 or AX 10 000 columns (Macherey Nagel, Düren, Germany). Where indicated, plasmid DNA was linearized by restriction enzyme digestion before transfection. Linearized plasmid DNA was purified by phenol/chloroform extraction and ethanol precipitation before being resuspended in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0) for transfection.
Cell culture
Adherent HEK293 (human embryonic kidney), A549 (human lung carcinoma), NIH3T3 (murine embryonic fibroblast), L929 (murine areolar fibroblast) and CHO-K1 (Chinese Hamster Ovary) cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). Cell lines were propagated in Dulbecco's modified Eagle's medium/F12 containing 10% fetal bovine serum and split (0.25% trypsin-EDTA) using Invitrogen (Carlsbad, CA, USA) reagents and conventional methodologies. For transfections, cells were plated on either 6- or 24-well tissue culture dishes. Supercoiled or linear plasmids were transfected into cell lines using Lipofectamine 2000 (or Lipofectamine LTX for CHO-K1) following the manufacturer's instructions (Invitrogen) and analyzed for duration and variability of expression in integrated (EGFP-IRES-neo transgene) and transient (EGFP transgene) expression systems as indicated. Plasmid integrants were selected using media containing 500 μg ml−1 Geneticin (Invitrogen) over 10 days. Total cellular lysates for EGFP determination were prepared by resuspending cells in cell lysis buffer (BD Biosciences Pharmingen, San Diego, CA, USA), lysing cells by incubating for 30 min at 37 °C, followed by a freeze–thaw cycle at −80 °C. Lysed cells were clarified by centrifugation and the supernatants assayed for EGFP by FLX800 microplate fluorescence reader (Bio-Tek, Winooski, VT, USA). Single cell suspensions for FACS flow cytometry were prepared by dissociating cells with trypsin treatment, and washing with media to remove trypsin and then phosphate-buffered saline for FACS analysis using the BD (Franklin Lakes, NJ, USA) FACSCalibur dual-laser cytometer.
Transfection of non-dividing cells was as described in Monkonge et al.38 Briefly, HEK293 or A549 cells were pretreated overnight with 5 μg ml−1 aphidicolin (Sigma, St Louis, MO, USA), which inhibits cell division and synchronizes cells to early S phase. Transfection of EGFP plasmids and outgrowth were then performed in the presence of aphidicolin. Cell outgrowths were incubated with or without 25 ng ml−1 tumor necrosis factor-α (Invitrogen) for 3 h to induce NF-κB nuclear shuttling. Fresh medium was then added and total cell extracts were prepared for EGFP fluorescence determination 24 h (A549) or 48 h (HEK293) post transfection.
Real time (RT)-PCR (genomic DNA)
Genomic DNA was isolated from integrated cell lines using the DNeasy blood and tissue kit (Qiagen Sciences, Germantown, MD, USA) and total DNA was quantified using the FL × 800 microplate fluorescence reader (Bio-Tek) by determining picogreen (Invitrogen) fluorescence of samples versus a linearized vector standard curve. RT-PCR to quantify vector copies in genomic DNA was performed used a TaqMan EGFP transgene 6FAM-ACAGCCACAACTCT-MGBNFQ probe and flanking primers (5′-GGGCACAAGCTGGAGTACAAC-3′; 5′-TCTGCTTGTCGGCCATGATA-3′) in a TaqMan gene expression assay using Applied Biosystems (Foster City, CA, USA) TaqMan reagents and the Step One Real Time PCR System (Applied Biosystems, Carlsbad, CA, USA). Linearized vector was used for the RT-PCR standard curve.
Reverse transcriptase (RT)-PCR (cytoplasmic RNA)
Cytoplasmic RNA was isolated from transfected HEK293 cells using the protein and RNA isolation system (PARIS kit, Ambion, Austin TX, USA) and quantified by A260. Samples were DNase treated (DNA-free DNase; Ambion) before reverse transcriptase RT-PCR using the Agpath-ID One step RT-PCR kit (Ambion) and the EGFP transgene specific probe. DNase treatment was confirmed by the lack of detectable signal in control RT-PCR reactions run without the initial 48 °C reverse transcriptase step using heat-treated 25 × PCR buffer. Heat treatment inactivates heat-labile reverse transcriptase but not Taq DNA polymerase. Assay linearity was verified using sample dilutions.
Rabbit immunization
The HIV-1 gp120 envelope glycoprotein gene from the JRCSF strain was cloned into the pMAmp and AF NTC8382 backbones. The pMAmp plasmid has been described previously48 and contains the CMV promoter and intron 1 for gene expression and the bovine growth hormone polyadenylation site. NTC8382 is the NTC8385 vector modified to secrete transgene using a tissue plasminogen activator secretion leader.4 Both vectors express the identical tissue plasminogen activator–gp120 fusion protein. Each plasmid was injected into rabbits (six per group) on days 0, 28 and 56, followed by EP with the MedPulser electroporation system (Inovio Biomedical, San Diego, CA, USA). All rabbits were bled on days 42 and 70. On day 84, the rabbits received a booster injection of alum-adjuvanted gp120 protein. Rabbits were bled on day 98 and the study was terminated. Sera were prepared from the day 70 and 98 blood samples for measurement of the levels of HIV-1 neutralization activity. Neutralization was tested on two neutralization-sensitive viruses (SF162 and NL4-3) and two resistant viruses (6535 and JRCSF). Rabbit EP and virus neutralization assays were as described.48 This animal study was conducted at Aldevron (Fargo, ND, USA) and was approved by Aldevron's Institutional Review Board.
Human DC EP
DCs from three independent donors were generated as previously described49 and nucleofected 24 h after maturation using the Amaxa DC nucleofection kit (Amaxa, Koeln, Germany). A total of 0.5–1 × 106 DCs were prepared as per manufacturer's instructions, nucleofected with 2 μg of plasmid DNA and subsequently resuspended in complete RPMI media (10% fetal bovine serum, 2 mmol l−1 GlutaMAX and 1 mmol l−1 sodium pyruvate (Sigma)), supplemented with the cytokine maturation cocktail containing 10 ng ml−1 interleukin-1β, 10 ng ml−1 tumor necrosis factor-α, 100 ng ml−1 interleukin-6 (R&D Systems, Minneapolis, MN, USA), 1 μg ml−1 prostaglandin E2 (Sigma), 800 U ml−1 granulocyte macrophage colony-stimulating factor (Sargramostim Leukine; Immunex, Seattle, WA, USA) and 1000 U ml−1 interleukin-4 (R&D Systems). Nucleofection efficiency and EGFP transgene expression were assessed 18 h after nucleofection using the BD FACSCalibur flow cytometer. The data were analyzed using Cell Quest software (BD).
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Marni England-Hill (Aldevron) and Jennifer Bath (Concordia College) for oversight of the rabbit study at Aldevron, and Danielle Shea (University of Nebraska, Lincoln) for performing flow cytometry. We also thank Kim Hanson (Nature Technology) for purifying the plasmid DNA used in this study and Sheryl Anderson (Nature Technology) for linear vector preparation. This paper described work supported by NIH grants R44 GM072141-03 and R43 GM080768-01 to JAW. AML is supported by a Specialized Centers for Cell-based Therapy Grant NIH-NHLBI 1 U54 HL081007 and an Amy Strelzer Manasevit Scholar Award. UG is supported by an Asbmt Young Investigator Award and a Leukemia and Lymphoma Society Special Fellow in Clinical Research Award.
Competing interests
JML, JMV, CPH and JAW have an equity interest in Nature Technology Corporation. SXD and RGW have an equity interest in AltraVax.
Footnotes
Supplementary Information accompanies the paper on Gene Therapy website
References
- 1.Liu L, Marti GP, Wei X, Zhang X, Zhang H, Liu YV. Age-dependent impairment of HIF-1α expression in diabetic mice: correction with electroporation-facilitated gene therapy increases wound healing, angiogenesis, and circulating angiogenic cells. J Cell Physiol. 2008;217:319–327. doi: 10.1002/jcp.21503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodles-Brakhop AM, Heller R, Draghia-Akli R. Electroporation for the delivery of DNA-based vaccines and immunotherapeutics: current clinical developments. Mol Ther. 2009;17:585–592. doi: 10.1038/mt.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luke J, Carnes AE, Hodgson CP, Williams JA. Improved antibiotic-free DNA vaccine vectors utilizing a novel RNA based plasmid selection system. Vaccine. 2009;27:6454–6459. doi: 10.1016/j.vaccine.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams JA, Luke J, Johnson L, Hodgson CP. pDNAVACCultra vector family: high throughput intracellular targeting DNA vaccine plasmids. Vaccine. 2006;24:4671–4676. doi: 10.1016/j.vaccine.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 5.Williams JA, Carnes AE, Hodgson CP. Plasmid DNA vector design; impact on efficacy, safety and upstream production. Biotechnol Adv. 2009;27:353–370. doi: 10.1016/j.biotechadv.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams JA, Luke J, Langtry S, Anderson S, Hodgson CP, Carnes AE. Generic plasmid DNA production platform incorporating low metabolic burden seed-stock and fed-batch fermentation processes. Biotechnol Bioeng. 2009;103:1129–1143. doi: 10.1002/bit.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carnes AE, Williams JA. Low metabolic burden plasmid production. Genetic Eng Biotech News. 2009;29:56–57. [Google Scholar]
- 8.Carnes AE, Hodgson CP, Luke J, Vincent J, Williams JA. Plasmid DNA production combining antibiotic-free selection, inducible high yield fermentation, and novel autolytic purification. Biotechnol Bioeng. 2009;104:505–515. doi: 10.1002/bit.22415. [DOI] [PubMed] [Google Scholar]
- 9.FDA. Guidance for Industry: Considerations for Plasmid DNA Vaccines for Infectious Disease Indications 2007: US Food and Drug Administration: Rockville, MD, USA.
- 10.EMA. Non-clinical Studies Required before First Clinical use of Gene Therapy Medicinal Products 2008: European Medicines Agency: London, England.
- 11.Wang Z, Troilo PJ, Wang X, Griffiths TG, Pacchione SJ, Barnum AB. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004;11:711–721. doi: 10.1038/sj.gt.3302213. [DOI] [PubMed] [Google Scholar]
- 12.van Hall T, van de Rhee NE, Schoenberger SP, Vierboom MP, Verreck FA, Melief CJ. Cryptic open reading frames in plasmid vector backbone sequences can provide highly immunogenic cytotoxic T-lymphocyte epitopes. Cancer Res. 1998;58:3087–3093. [PubMed] [Google Scholar]
- 13.Schirmbeck R, Riedl P, Fissolo N, Lemonnier FA, Bertoleti A, Reimann J. Translation from cryptic reading frames of DNA vaccines generates an extended repertoire of immunogenic, MHC class I-restricted epitopes. J Immunol. 2005;174:4647–4656. doi: 10.4049/jimmunol.174.8.4647. [DOI] [PubMed] [Google Scholar]
- 14.Maness NJ, Wilson NA, Reed JS, Piaskowski SM, Sacha JB, Walsh AD. Robust, vaccine-induced CD8+ T lymphocyte response against an out-of-frame epitope. J Immunol. 2010;184:67–72. doi: 10.4049/jimmunol.0903118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuh CH, Ting LP. The genome of hepatitis B virus contains a second enhancer: cooperation of two elements within this enhancer is required for its function. J Virol. 1990;64:4281–4287. doi: 10.1128/jvi.64.9.4281-4287.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mariati Ho SC, Yap MG, Yang Y. Evaluating post-transcriptional regulatory elements for enhancing transient gene expression levels in CHO K1 and HEK293. Protein Expr Purif. 2010;69:9–15. doi: 10.1016/j.pep.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Takebe Y, Seiki M, Fujisawa J, Hoy P, Yokata K, Arai K. SR alpha promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol Cell Biol. 1988;8:466–472. doi: 10.1128/MCB.8.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM. A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J Virol. 2005;79:8828–8834. doi: 10.1128/JVI.79.14.8828-8834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams BR. PKR; a sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- 20.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee ES, Yoon CH, Kim YS, Bae YS. The double-strand RNA-dependent protein kinase PKR plays a significant role in a sustained ER stress-induced apoptosis. FEBS Lett. 2007;581:4325–4332. doi: 10.1016/j.febslet.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Groskreutz D, Schenborn E. Increased gene expression in mammalian cell lines using pAdVAntage DNA as a cotransfectant. Promega Notes. 1994;48:8–13. [Google Scholar]
- 23.Lei M, Liu Y, Samuel CE. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology. 1998;245:188–196. doi: 10.1006/viro.1998.9162. [DOI] [PubMed] [Google Scholar]
- 24.Andersson MG, Haasnoot PCJ, Xu N, Berenjian S, Berkhout B, Akusjarvi G. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol. 2005;79:9556–9565. doi: 10.1128/JVI.79.15.9556-9565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu S, Cullen BR. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and microRNA biogenesis. J Virol. 2004;78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma Y, Mathews MB. Secondary and tertiary structure in the central domain of adenovirus type 2 VA RNA I. RNA. 1996;2:937–951. [PMC free article] [PubMed] [Google Scholar]
- 27.Rahman A, Malhotra P, Shar R, Kewalramani T, Thimmapaya B. Effect of single-base substitutions in the central domain of virus-associated RNA I on its function. J Virol. 1995;69:4299–4307. doi: 10.1128/jvi.69.7.4299-4307.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam AP, Dean DA. Progress and prospects: nuclear import of nonviral vectors. Gene Ther. 2010;61:603–613. doi: 10.1038/gt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagstaff KM, Jans DA. Nucleocytoplasmic transport of DNA: enhancing non-viral gene transfer. Biochem J. 2007;406:185–202. doi: 10.1042/BJ20070505. [DOI] [PubMed] [Google Scholar]
- 30.Miller AM, Munkonge FM, Alton EW, Dean DA. Identification of protein cofactors necessary for sequence-specific plasmid DNA nuclear import. Mol Ther. 2009;17:1897–1903. doi: 10.1038/mt.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, MacLaughlin FC, Fewell JG, Gondo M, Wang J, Nicol F. Muscle-specific enhancement of gene expression by incorporation of SV40 enhancer in the expression plasmid. Gene Ther. 2001;8:494–497. doi: 10.1038/sj.gt.3301419. [DOI] [PubMed] [Google Scholar]
- 32.Blomberg P, Eskandarpour M, Xia S, Sylven C, Islam KB. Electroporation in combination with a plasmid vector containing SV40 enhancer elements results in increased and persistent gene expression in mouse muscle. Biochem Biophys Res Commun. 2002;298:505–510. doi: 10.1016/S0006-291X(02)02486-5. [DOI] [PubMed] [Google Scholar]
- 33.Williams JA . Vectors and methods for genetic immunization. World Patent Application 2006; WO2006078979.
- 34.Angulo A, Kerry D, Huang H, Borst EM, Razinsky A, Wu J. Identification of a boundary domain adjacent to the potent human cytomegalovirus enhancer that represses transcription from the divergent UL127 promoter. J Virol. 2000;74:2826–2839. doi: 10.1128/JVI.74.6.2826-2839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chao SH, Harada JN, Hyndman F, Gao X, Nelson CG, Chanda SK. PDX1, a cellular homeoprotein, binds to and regulates the activity of human cytomegalovirus immediate early promoter. J Biol Chem. 2004;279:16111–16120. doi: 10.1074/jbc.M312304200. [DOI] [PubMed] [Google Scholar]
- 36.Lee J, Klase Z, Gao X, Caldwell JS, Stinski MJ, Kashanchi F. Cellular homeoproteins, SATB1 and CDP, bind to the unique region between the human cytomegalovirus UL127 and major immediate-early genes. Virology. 2007;366:117–125. doi: 10.1016/j.virol.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 37.Mesika A, Grigoreva I, Zohar M, Reich Z. A regulated NFkB-assisted import of plasmid DNA into mammalian cell nuclei. Mol Ther. 2001;3:653–657. doi: 10.1006/mthe.2001.0312. [DOI] [PubMed] [Google Scholar]
- 38.Munkonge FM, Amin V, Hyde SC, Green AM, Pringle IA, Gill DR. Identification and functional characterization of cytoplasmic determinants of plasmid DNA nuclear import. J Biol Chem. 2009;284:26978–26987. doi: 10.1074/jbc.M109.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hilton TL, Li Y, Dunphy EL, Wang EH. TAF1 histone acetyltransferase activity in Sp1 activation of the cyclin D1 promoter. Mol Cell Biol. 2005;25:4321–4332. doi: 10.1128/MCB.25.10.4321-4332.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thakur RK, Kumar P, Halder K, Verma A, Kar A, Parent JL. Metastases suppressor NM23-H2 interaction with G-quadruplex DNA within c-MYC promoter nuclease hypersensitive element induces c-MYC expression. Nucleic Acids Res. 2009;37:172–183. doi: 10.1093/nar/gkn919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carnes AE, Luke JM, Vincent JM, Anderson S, Schukar A, Hodgson CP. Critical design criteria for minimal antibiotic-free plasmid vectors necessary to combine robust RNA Pol II and Pol III-mediated eukaryotic expression with high bacterial production yields. J Gene Med. 2010;12:818–831. doi: 10.1002/jgm.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufman RJ, Murtha P. Translational control mediated by eukaryotic initiation factor-2 is restricted to specific mRNAs in transfected cells. Mol Cell Biol. 1987;7:1568–1571. doi: 10.1128/MCB.7.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terenzi F, deVeer MJ, Ying H, Restifo NP, Williams BR, Silverman RH. The antiviral enzymes PKR and RNase L suppress gene expression from viral and non-viral based vectors. Nucleic Acids Res. 1999;27:4369–4375. doi: 10.1093/nar/27.22.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breuzard G, Tertil M, Goncalves C, Cheradame H, Geguan P, Pichon C. Nuclear delivery of NFB-assisted DNA/polymer complexes: plasmid DNA quantification by confocal laser scanning microscopy and evidence of nuclear polyplexes by FRET imaging. Nucleic Acids Res. 2008;36:e71. doi: 10.1093/nar/gkn287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Molnar MJ, Gilbert R, Lu Y, Liu AB, Guo A, Larochelle N. Factors influencing the efficacy, longevity, and safety of electroporation-assisted plasmid-based gene transfer into mouse muscles. Mol Ther. 2004;10:447–455. doi: 10.1016/j.ymthe.2004.06.642. [DOI] [PubMed] [Google Scholar]
- 46.Williams JA . Vectors and methods for genetic immunization. World Patent Application 2008; WO2008153733.
- 47.Lednicky J, Folk WR. Two synthetic Sp1-binding sites functionally substitute for the 21-base-pair repeat region to activate simian virus 40 growth in CV-1 cells. J Virol. 1992;66:6379–6390. doi: 10.1128/jvi.66.11.6379-6390.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Du SX, Idiart RJ, Mariano EB, Chen H, Jiang P, Xu L. Effect of trimerization motifs on quaternary structure, antigenicity, and immunogenicity of a noncleavable HIV-1 gp120 envelope glycoprotein. Virology. 2009;395:33–44. doi: 10.1016/j.virol.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerdemann U, Christin AS, Vera JF, Ramos CA, Fujita Y, Liu H. Nucleofection of DCs to generate multivirus-specific T cells for prevention or treatment of viral infections in the immunocompromised host. Mol Ther. 2009;17:1616–1625. doi: 10.1038/mt.2009.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.