

Summary:
Juvenile myelomonocytic leukemia (JMML) is a childhood leukemia for which allogeneic BMT is the only curative therapy. At our pediatric stem cell transplantation unit, we performed 26 BMTs in 23 children (age 0.5–12.7 years). Conditioning was CY/TBI based (1980–1996, n=14) or BU/CY/melphalan based (1996–2001, n=9). Donors were HLA-identical siblings (n=11), unrelated volunteers (n=9) or mismatched family members (n=3). A total of 10 patients survive in CR (median follow-up 6.8 years, range 3.1–22.2 years). Relapse or persistent disease was observed in eight and two patients, respectively. Nine of these patients died, one achieved a second remission following acute nonlymphatic leukemia chemotherapy (duration to date 5.3 years). Transplant-related mortality occurred in four patients. Overall survival at 5 and 10 years was 43.5%. Using T-cell-depleted, one-antigen mismatched unrelated donors was the only significant adverse factor associated with relapse in multivariate analysis (P=0.039, hazard ratio 4.9). Together with a trend towards less relapse in patients with graft-versus-host-disease and in patients transplanted with matched unrelated donors, this suggests a graft-versus-leukemia effect of allogeneic BMT in JMML.
Keywords: juvenile myelomonocytic leukemia, graft-versus-leukemia
Main
Juvenile myelomonocytic leukemia (JMML) is a rare myeloproliferative disorder in young children, formerly grouped within the FAB classification of myelodysplastic syndromes (MDS).1 It has also been described as juvenile chronic myeloid leukemia or chronic myelomonocytic leukemia.2 In a recently proposed pediatric morphological classification,3 JMML is separated from the other subtypes of MDS, that is, RA, RAEB and RAEB-t because of different characteristics. Molecular biological investigations have shown three major, mutually exclusive abnormalities in JMML patients, that is, the presence of NF1 gene mutation,4 mutations in the Ras-signalling pathway5 and mutations in the PTPN11 region.6,7 Without treatment, the 10-year survival of JMML is 6%;8 and there are only sporadic definitive responses to chemotherapy.9,10 Allogeneic BMT has been shown to improve outcome: retrospective evaluations of series including 15 patients or more reported an overall survival (OS) of 39–58%.2,11,12,13,14,15 Woods10 found a similar OS at 6 years of 31% for 13 patients treated on a prospective study (CCG protocol 2891). The major causes of failure in all series were a high relapse rate and a high transplant-related mortality (TRM). This report describes our single center experience over a 20-year period from June 1980 until January 2001 on 23 children undergoing transplantation for JMML.
Patients and methods
Patients
From June 1980 until January 2001, 26 transplants in 23 consecutive children with JMML were undertaken in the pediatric BMT unit of the Leiden University Medical Center in Leiden, The Netherlands. Preliminary results of the first two transplants were described in a conference report16 and five other patients were included in a registry report of the European Working Group on Myelodysplastic Syndromes (EWOG-MDS)8 (Table 1). The current group of 23 consecutive patients consisted of eight female and 15 male subjects (age 6 months–12 years 8 months, median 4 years 3 months, at the time of transplant). Diagnosis of JMML was made upon clinical and laboratory signs and symptoms. In retrospect, all patients qualified for the diagnosis of JMML based on fulfilment of the three major and at least two of the five minor criteria of the International JMML Working Group.14
Table 1.
Patient data at diagnosis, pretransplant treatment and interval to BMT
| UPN | Age | Leukocytes (10 9 /l) | Monocytes (10 9 /l) | Platelets (10 9 /l) | HbF (%) | Cytogenetics | Pretransplant treatment | Interval to BMT (months) |
|---|---|---|---|---|---|---|---|---|
| 031a | 4.9 | 185 | 16.7 | 24 | 15 | nb | BU, Leukeran, Vincristin | 8.48 |
| 036a | 0.4 | 8.3 | 1.3 | 16 | NTc | n | Ara-C, Vincristin, Etoposide | 5.82 |
| 107 | 1.5 | 40 | 4.0 | 45 | 14.4 | n | IFN, CY | 4.41 |
| 140.1d | 5.3 | 12.2 | 3.4 | 174 | 2.9 | −7 | Inductione ANLL | 4.14 |
| 177 | 5.9 | 3.1 | 1.1 | 23 | NT | −7 | Induction+consolidatione ANLL | 6.58 |
| 194d | 0.3 | 79.9 | 32.0 | 26 | 11.7 | n | 6TG, Ara-C, induction ANLL | 2.24 |
| 196d | 1.0 | 41 | 4.1 | 42 | 15 | n | Induction ANLL | 2.30 |
| 210.1d | 1.8 | 69.8 | 9.1 | 26 | 24 | −6,−8,+mar2 | Induction, consolidation, intensificatione ANLL | 9.21 |
| 226 | 1.1 | 30.7 | 5.8 | 11 | NT | −7 | Splenectomy, induction ANLL | 11.31 |
| 240 | 10.5 | 162 | 8.1 | 44 | 20.9 | n | Induction ANLL | 2.27 |
| 272 | 10.8 | 32.5 | 7.8 | 153 | NT | −7 | Induction ANLL | 10.98 |
| 273 | 11.9 | 4.1 | 1.4 | 8 | NT | −7, +21 | None | 8.78 |
| 298 | 6.9 | 32.8 | 2.6 | 87 | 33.3 | n | Isotretinoide, splenectomy, Vincristin | 10.55 |
| 329 | 1.3 | 40.6 | 8.1 | 218 | 40 | n | 6MP, Vincristin, Ara-C, Etoposide | 5.98 |
| 339 | 7.6 | 3.8 | 1.4 | 56 | 3.5 | −7 | None | 5.79 |
| 355d | 0.8 | 74.6 | 16.4 | 181 | 5.9 | n | 6MP, Ara-C | 47.90 |
| 369 | 3.7 | 74.8 | 13.5 | 29 | NT | dup (3)(q21q29) | IFN | 8.28 |
| 372 | 3.7 | 89.7 | 14.4 | Not known | 13.0 | n | Splenectomy, IFN, Ara-C | 9.21 |
| 412 | 1.0 | 79.3 | 9.5 | 22 | 4.3 | n | 6MP, Ara-C | 5.92 |
| 419 | 0.3 | 62 | 12.4 | 23 | NT | n | 6MP | 9.86 |
| 430 | 2.6 | 36.4 | 5.5 | 29 | 41.2 | n | 6MP, Ara-C, induction ANLL | 14.17 |
| 445 | 1.6 | 25 | 1.3 | 346 | NT | n | None | 8.88 |
| 450.1 | 0.9 | 33 | 8.6 | 6 | NT | −7, +21 | 6MP | 4.93 |
Clinical and laboratory characteristics at diagnosis
Characteristics of the patients are given in Table 1. In all, 22 patients showed hepatosplenomegaly, 10 had petechiae or hematomas. One of the patients had Noonan syndrome (UPN 419). Three patients had a clinical diagnosis of neurofibromatosis (NF; UPN 140, 329 and 355). The median leukocyte count was 36.4 × 109/l (range, 3.1–185.0; n=23), the median platelet count was 29.0 × 109/l (range, 6–364; n=22) and the median HbF was 15.0% (range, 2.9–41.2; n=14). The karyotype was abnormal in nine cases.
Pretransplant therapy
Early on, patients were treated with intensive chemotherapy before BMT in an attempt to lower the blast cell count in the peripheral blood. This policy was largely omitted from 1994 onwards. A total of 11 children thus received intensive chemotherapy according to the acute nonlymphatic leukemia (ANLL) protocols of the Dutch Childhood Leukemia Study Group (DCLSG), consisting of AraC, daunorubicine and etoposide (induction), of prednisone, 6-thioguanine, vincristine, adriamycine, AraC and cyclophosphamide (consolidation) and of HD-AraC and etoposide (intensification). Nine children received nonintensive treatment, for example, with 6-mercaptopurine or low-dose Ara-C, while three children did not receive chemotherapy before transplantation. At the start of the conditioning for BMT, intensively pretreated patients were not different from nonpretreated or nonintensively pretreated patients with regard to age at BMT (split 4 years), hepatomegaly (yes or no), splenomegaly (yes or no/splenectomized), percentage bone marrow blasts (split 5%), cytogenetic abnormalities (yes or no). The leukocyte and monocyte counts were significantly higher in nonpretreated or nonintensively pretreated patients than in intensively pretreated patients. Taken together, we consider the two groups comparable.
Donor choice
Until 1990, only matched sibling donors (MSD) were used. Subsequently, unrelated donors (UD) and mismatched related donors (MMRD) were also considered suitable. HLA typing was performed by both serological microcytotoxic techniques and DNA techniques (which have replaced the serological typing for class II in 1992 and for class I in 1999). HLA-A, B, Cw, DR, DQ and DP loci were taken into consideration for donor matching. In case of an unrelated donor, additional tests (MLC and CTLp) were used in the donor selection procedure. Retrograde high-resolution DNA typing for all loci on UD and MMRD patient/donor pairs confirmed the previous serological typings. The patient/donor characteristics are given in Table 2. A total of 11 patients received an allogeneic BMT from an MSD, nine patients from a UD (matched n=4; one-antigen mismatched n=5: class I mismatch n=3, class II mismatch n=2). The remaining three patients received bone marrow from a MMRD (phenotypically identical aunt (n=1), 11/12 identical sister (n=2)).
Table 2.
Conditioning regimen, donor, T-cell depletion, cell dose and GvHD prophylaxis
| UPN | Conditioning | Donor a | TCD | NC × 10 8 /kg b | GvHD prophylaxis |
|---|---|---|---|---|---|
| 031 | CY, BU, Hydroxyurea, TBI | MSD | No | 4.0 | MTX |
| 036 | CY, TBI | MSD | No | 4.0 | MTX |
| 107 | CY, TBI | MSD | No | 3.0 | CyA/MTX |
| 140.1 | CY, TBI | MSD | No | 3.5 | CyA/MTX |
| 177 | Ara-C, CY, TBI | MSD | No | 2.5 | CyA/MTX |
| 194 | Ara-C, Etoposide, BU, CY | MSD | No | 2.8 | CyA/MTX |
| 196 | Etoposide, BU, CY | MSD | No | 4.0 | CyA/MTX |
| 210.1 | Ara-C, CY, Campath, anti-LFA1, TBI | MMUD | Yes | 4.0 | CyA/MTX |
| 226 | Ara-C, CY, Campath, anti-LFA1, TBI | MMUD | Yes | 4.3 | None |
| 240 | Ara-C, CY, TBI | MSD | No | 4.6 | CyA/MTX |
| 272 | Ara-C, CY, Campath, anti-LFA1, TBI | MMUD | Yes | 1.0 | MTX |
| 273 | Ara-C, CY, Campath, anti-LFA1, TBI | MMUD | Yes | 2.5 | MTX |
| 298 | Ara-C, Etoposide, CY, Campath, TBI | MUD | No | 5.3 | CyA/MTX |
| 329 | BU, CY | MMRD | No | 4.8 | CyA/MTX |
| 339 | BU, CY, Melphalan | MSD | No | 1.5 | CyA/MTX |
| 355 | BU, CY, Melphalan | MSD | No | 2.8 | CyA/MTX |
| 369 | Ara-C, CY, TBI | MMRD | No | 2.3 | CyA/MTX |
| 372 | Ara-C, CY, TBI | MMRD | No | 3.2 | CyA/MTX |
| 412 | BU, CY, Melphalan, Campath | MUD | No | 11.0 | CyA/MTX |
| 419 | BU, CY, Melphalan, Campath | MUD | No | 2.2 | CyA/MTX |
| 430 | CY, Melphalan, Campath, anti-LFA1, TBI | MMUD | Yes | 0.8 | CyA |
| 445 | BU, CY, Melphalan, Campath | MUD | No | 3.2 | CyA/MTX |
| 450.1 | BU, CY, Melphalan | MSD | No | 5.3 | CyA/MTX |
aMSD=matched sibling donor; MUD=matched unrelated donor; MMUD=mismatched unrelated donor; MMRD=mismatched related donor.
bNumber of nucleated cells in bone marrow harvest.
Conditioning regimens
The conditioning regimens (Table 2) varied with time. From 1980 until December 1996, the scheme was CY/TBI based (according to the national regimen for ANLL conditioning) except for three children below the age of 2 years at transplant, who received BU instead of TBI. The BU, CY and melphalan (Mel) regimen, used from December 1996 onwards, was proposed by Locatelli et al.17 Three patients received the CY/TBI-based regimen in this period because of pre-existent hepatic dysfunction. TBI was given as a single fraction of 7–7.5 Gy to patients younger than 10 years, and in two fractions of 6 Gy each on two consecutive days to children aged ⩾10 years. CY 60 mg/kg was administered intravenously once daily for 2 days with uromitexan prophylaxis. BU 1 mg/kg was administered orally four times daily for 4 days. MeI 140 mg/m2 was given intravenously once. Ara-C 1 g/m2 was administered intravenously twice daily for 2 days. Etoposide 300 mg/m2 was administered intravenously once daily for 3 days. In those children with a mismatched, unrelated donor, Campath-1G (0.2 mg/kg/day for 5 days, starting at day −7) and anti-LFA-1 (AntilfaR, 0.2 mg/kg/day for 12 days after 0.4 mg/kg the first day, ie at day −2) were additionally administered, whereas in the case of a matched unrelated donor only Campath-1G was added to the regimen.
Marrow manipulation and infusion
In the five cases of one-antigen mismatched unrelated donors, T-cell depletion (TCD; Table 2) of the marrow was performed by an immunorosetting technique resulting in a mean depletion of 2.5 log (range, 2.2–3.0).18 Red cell depletion was undertaken for ABO incompatibility in seven donor/recipient combinations. Bone marrow was infused either 36 h after the last infusion of CY or 24 h after infusion of Mel or at a minimum of 6 h after TBI. The median total nucleated cell dose in the harvest (in case of TCD, number before depletion) was 3.2 × 108/kg recipient weight (range 0.86–11.0 × 108/kg). Quantitation of CD34+ cells in the graft was done in nine recent transplantations only (median number 4.4 × 106/kg, range 2.2–10.8 × 106/kg).
Graft-versus-host disease (GvHD) prophylaxis and treatment (Table 2)
CyA plus short-course MTX were used for 17 BMTs for prophylaxis of GvHD. CyA alone was used once and MTX long course alone for 4 BMTs. In one case, only Campath-1G and anti-LFA-1 were given. CyA starting at day −1 was given intravenously (2 mg/kg/day) until oral medication (6 mg/kg/day) was tolerated with dose adjustment according to the results of regular blood sampling. In general, this was continued for 6 months and tapered thereafter until discontinuation at 9 months. MTX prophylaxis consisted of 10 mg/m2 once daily intravenously on day +1, 3, 6 and 11. Long course MTX, that is, continuation of 10 mg/m2 once weekly until day 102 was given without CyA addition.
Acute and chronic GvHD were graded according to the Seattle criteria.19,20 Acute GvHD>grade 1 was treated with methylprednisolone 2 mg/kg/day i.v., followed by tapering of the dose according to clinical response.
Supportive treatment
All patients received the same supportive care. They were nursed in strict protective isolation using sterilized food and beverages and received total gastrointestinal decontamination using nonreabsorbable antimicrobials.21 Microbiological control of the suppression of the potentially pathogenic microflora was carried out in samples of the throat and stool once or twice weekly.21 No systemic antimicrobial prophylaxis was given, except for Pneumocystis pneumonia prophylaxis with cotrimoxazole from day +20 onwards.
Statistical analysis
Event-free survival (EFS) and OS were calculated by the Kaplan–Meier method and the difference was tested with the log-rank test. The Cox proportional hazards model was employed to assess the independent effects of risk factors on OS, where the selection of variables was stepwise. All calculations were performed using SPSS® (version 10.0.7, Chicago, USA).
Results
Transplant outcome (Table 3)
Engraftment, defined as a sustained count of leukocytes>1.0 × 109/l (Table 3), occurred in 20/23 cases (at a median of 24 days, range 14–47; neutrophils>0.5 × 109/l at a median of 30 days, range 18–55; platelets>50 × 109/l at a median of 36 days, range 23–78). Acute GvHD was seen in five cases; two of these developed extensive chronic GvHD. Nine patients are in continuous complete remission (CCR) with full donor chimerism, two showed recipient cells emerging at +2 and +4 weeks, with JMML morphology (persisting disease), eight relapsed and four died of transplant-related causes (3/4 with viral infection or reactivation). The median follow-up of the nine patients in CCR is 6.0 years (range 3.1–22.2 years) after BMT. The probability of relapse-free survival of the total cohort is 48±12% (Figure 1). In one patient who relapsed after BMT with a T-cell-depleted UD, relapse was diagnosed 3.3 years after BMT. Intensive ANLL treatment resulted in a second remission of presently 5.3 years duration and full donor chimerism. In the seven other relapsed patients relapse occurred between 1 and 11 months after BMT. All nine patients with persisting disease or relapse in the first year after BMT died. Three of them died after a second BMT using different conditioning regimens and the same original donor; one patient additionally received leukocyte-activated killer cells.
Table 3.
Engraftment, GvHD, relapse and outcome
| UPN | Engraftment (day) | aGvHD (grade) | cGvHD | Relapse | Complications | Outcome |
|---|---|---|---|---|---|---|
| 031 | NEa | NE | NE | No | DICb | Died day 16 (pulmonary bleeding) |
| 036 | 21 | 0 | Absent | No | None | Alive and well (22 years after BMT) |
| 107 | 35 | 0 | Absent | Yes | None | Died day 192 (relapse) |
| 140.1 | 47 | 0 | Absent | Yes | None | Died day 233 (Asp. inf. after 2nd BMT) |
| 177 | 27 | 0 | Absent | Yes | None | Died day 514 (relapse) |
| 194 | 41 | 0 | Absent | No | VODc | Alive and well (11.3 years after BMT) |
| 196 | 19 | 0 | Absent | Yes | VOD | Died day 413 (relapse, Candida sepsis, ARDS) |
| 210.1 | No | 0 | NE | Yes | Convulsions | Died day 186 (persistent disease, relapse after 2nd BMT) |
| 226 | No | 0 | Absent | Yes | None | Died day 133 (persistent disease) |
| 240 | 24 | 0 | Absent | No | None | Alive and well (9.6 years after BMT) |
| 272 | 36 | 1 | Limited | Yes | None | Alive and well (8.6 years after BMT, 5.3 years after relapse) |
| 273 | 37 | 0 | Absent | No | EBV-LPDd | Died day 75 (EBV-LPD) |
| 298 | 18 | 1 | Extensive | No | None | Alive and well (7.6 years after BMT) |
| 329 | 14 | 1 | Limited | Yes | None | Died day 254 (relapse) |
| 339 | 23 | 0 | NE | No | HSVe pneumonia, Stevens–Johnson syndrome | Died day 39 (toxic epidermal necrolysis) |
| 355 | 28 | 0 | Absent | No | None | Alive and well (6 years after BMT) |
| 369 | 24 | 4 | Absent | No | Multiple infections | Died day 97 (adenovirus infection) |
| 372 | 27 | 1 | Extensive | No | None | Alive and well (5.5 years after BMT) |
| 412 | 17 | 0 | Absent | No | None | Alive and well (4.2 years after BMT) |
| 419 | 24 | 0 | Absent | No | None | Alive and well (4 years after BMT) |
| 430 | 41 | 0 | Absent | Yes | None | Died day 149 (relapse) |
| 445 | 21 | 0 | Absent | No | Severe mucositis | Alive and well (3 years after BMT) |
| 450.1 | 17 | 0 | Absent | Yes | None | Died day 651 (relapse after 2nd BMT) |
aNE=not evaluable.
bDIC=diffuse intravascular coagulation.
cVOD=veno-occlusive disease.
dEBV-LPD=EBV-lymphoproliferative disorder.
eHSV=Herpes simplex virus.
Figure 1.
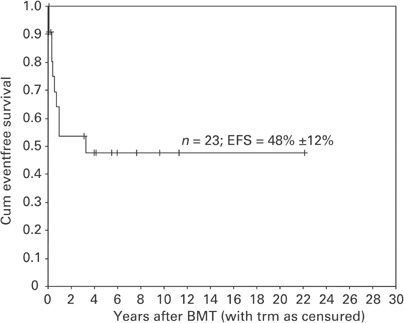
Kaplan–Meier estimates of event-free survival for all patients.
Factors associated with relapse
To assess the factors possibly predictive for relapse, the 10 children with progressive disease/relapse were compared to the 13 children without relapse. Karyotype (abnormal vs normal), treatment before BMT (intensive ANLL treatment vs none/nonintensive treatment), interval from diagnosis to BMT (split 6 months), age at BMT (split 4 years), percentage of blast cells in the bone marrow at the start of conditioning, conditioning regimen (CY/TBI vs BU/CY/Mel), donor type (MSD, UD, MMRD), graft manipulation (TCD vs no TCD) and occurrence of acute and/or chronic GvHD (yes or no; number of evaluable patients 22 and 20, respectively) were analyzed with (time to) relapse as event. Univariate analysis of the data showed a significant negative influence on EFS of intensive treatment before BMT (P=0.04, Figure 2) and of using one-Ag mismatched T-cell-depleted UD marrow (P=0.004, Figure 3). No significance was reached for the other variables tested, mainly due to limited power because of small patient numbers. Multivariate analysis showed a significant unfavorable effect of using one-Ag mismatched T-cell-depleted UD marrows corrected for the effect of TBI by keeping TBI in the multivariate model (estimated hazard ratio 4.946; 95% CI: 1.1–22.6). Of the nine UD cases (five TCD and four non-TCD), persisting disease (n=2) and relapse (n=2) were only seen in recipients of a TCD graft. There was a positive tendency for nonintensive/no pre-BMT therapy in the Cox's model when corrected for the effect of TBI (estimated hazard ratio 0.264, P=0.108).
Figure 2.

Kaplan–Meier estimates of event-free survival by pre-BMT treatment: nonintensive/no therapy vs intensive therapy.
Figure 3.
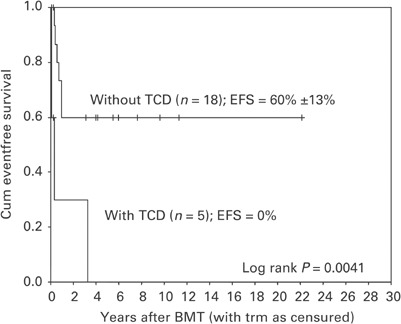
Kaplan–Meier estimates of event-free survival by TCD of the marrow: without TCD vs with TCD.
Discussion
Although the only curative approach for JMML is to perform an allogeneic BMT,2,11,12,13 this procedure is accompanied by a high relapse rate and a high TRM, resulting in an OS of 31–58% as reported in the recent literature.8,10,11,14,15 In our series, OS is 43.5±10% at 5 and 10 years; the probability of relapse-free survival (TRM censured) is 48±12%. Rapid sustained engraftment in 20 of 22 evaluable children proved that engraftment was not a problem, similar to results in other studies.14,15,17,22
Factors possibly predicting a relapse, as found by us and by others, relate to karyotype,14 intensive pre-BMT chemotherapy, conditioning regimen,11,22 donor type,11 TCD and GvHD.15 Manabe et al14 found in a multivariate analysis on the outcome of 27 JMML patients an abnormal karyotype to be the only significant risk factor for decreased OS.14 In our series, statistical significance was not reached due to limited power, but a trend towards worse outcome having an abnormal karyotype was noted. This observation needs to be confirmed with larger numbers. Intensive chemotherapy given before the start of the conditioning for BMT had a significant adverse effect (P=0.04 in univariate analysis) in our study on the relapse rate after BMT. This finding is not in agreement with the findings of other authors11,14,22 and needs further study. The influence of TBI in the conditioning regimen for JMML was studied by Matthes-Martin et al22 in a single center evaluation of 11 transplants and a review of the literature on single center reports. The relapse rate was significantly higher in patients conditioned with a TBI-containing regimen. In other series,14,15 the outcome was comparable for both types of conditioning. Locatelli et al11 found better EFS of 62% with the non-TBI regimen vs 11% with the TBI regimen for children with JMML given a BMT from an identical sibling or one-antigen-disparate relative. In our series, there is a trend towards a negative influence on EFS of the TBI-containing regimen. These combined data support the inferiority of the TBI regimen, but further study is needed to validate this finding. With regard to donor type, Locatelli et al11 found EFS for all UD transplants of 22% vs EFS of 38% for all transplants with identical sibling or one-antigen mismatched family donors (P<0.05). Our results with transplants from fully matched unrelated donors (n=4) are as good as those from sibling donors, similar to another reported series.14 Smith et al15 found significantly more relapses in using one-Ag mismatched unrelated marrows, but from his study this finding could not be associated with TCD. Using unrelated marrows, which were T-cell depleted because of one-Ag HLA disparity between donor and recipient, was the only factor in both univariate and multivariate analysis with a significant negative effect on EFS in our study. This suggests a possible graft-versus-leukemia (GvL) effect of allogeneic BMT in this disease. However, the 95% CI is rather large due to small patient numbers.
Matthes-Martin et al22 found no difference in relapse rate in patients with or without acute GvHD. In a recent multicenter overview from the National Marrow Donor Program of 46 JMML patients transplanted with unrelated donor marrow, a statistically significant relapse preventing effect of chronic GvHD was found. In our study, acute GvHD occurred only in five of our 22 evaluable patients (22.7%). Of these patients, one relapsed in the chronic GvHD phase, while of the 15 patients without GvHD, seven patients relapsed (not statistically significant).
Apart from relapse, another major problem were lethal viral infections. In recently published series,15,22 infectious complications also accounted for the largest number of transplant-related deaths. In the future, it may be possible to reduce the frequency of this complication by pre-emptive antiviral therapy following RT-PCR surveillance of viral nucleic acids in blood samples post-BMT.
Given the trend noted in this study that there may be a deleterious effect of intensive pretransplant chemotherapy, in the absence of evidence in the literature suggesting benefit from such therapy, we recommend to proceed to transplant once a donor is identified. Our experience and the literature show no benefit from TBI, therefore, we also recommend the use of a non-TBI conditioning regimen. Furthermore, we have demonstrated that using one-Ag mismatched T-cell-depleted unrelated donor marrows is associated with inferior outcome. Trials to enhance a GvL effect post BMT should be undertaken in a prospective multicenter setting.
Acknowledgements
We thank Mrs JDJ Bakker-Steeneveld for expert data managing, Dr R Wolterbeek for help with statistical analysis, Dutch pediatricians for referring patients for BMT, and the Dutch Childhood Leukemia Study Group for ANLL-chemotherapy protocols.
References
- 1.Bennett JM, Catovsky D, Daniel MT. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189–199. doi: 10.1111/j.1365-2141.1982.tb08475.x. [DOI] [PubMed] [Google Scholar]
- 2.Aricò M, Biondi A, Pui CH. Juvenile myelomonocytic leukemia. Blood. 1997;90:479–488. [PubMed] [Google Scholar]
- 3.Hasle H, Niemeyer CM, Chessels JM. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277–282. doi: 10.1038/sj.leu.2402765. [DOI] [PubMed] [Google Scholar]
- 4.Side LE, Emanuel PD, Taylor B. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood. 1998;92:267–272. [PubMed] [Google Scholar]
- 5.Flotho C, Valcamonica S, Mach-Pascual S. RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML) Leukemia. 1999;13:32–37. doi: 10.1038/sj.leu.2401240. [DOI] [PubMed] [Google Scholar]
- 6.Niemeyer CM, Tartaglia M, Büchner J. Clinical characteristics of children with JMML and germline or somatic PTPN11 mutations, RAS mutations or neurofibromatosis type 1. Leuk Res. 2003;27(Suppl 1. add):3. [Google Scholar]
- 7.Tartaglia M, Niemeyer CM, Fragale A. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 8.Niemeyer CM, Arico M, Basso G. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood. 1997;89:3534–3543. [PubMed] [Google Scholar]
- 9.Chan HSL, Estrov Z, Weitzmann SS, Freedman MH. The value of intensive combination chemotherapy for juvenile chronic myelogenous leukemia. J Clin Oncol. 1987;5:1960–1967. doi: 10.1200/JCO.1987.5.12.1960. [DOI] [PubMed] [Google Scholar]
- 10.Woods WG, Barnard DR, Alonzo TA. Prospective study of 90 children requiring treatment for juvenile myelomonocytic leukemia or myelodysplastic syndrome: a report from the Children's Cancer Group. J Clin Oncol. 2002;20:434–440. doi: 10.1200/JCO.2002.20.2.434. [DOI] [PubMed] [Google Scholar]
- 11.Locatelli F, Niemeyer C, Angelucci E. Allogeneic bone marrow transplantation for chronic myelomonocytic leukemia in childhood: a report from the European Working Group on Myelodysplastic Syndrome in Childhood. J Clin Oncol. 1997;15:566–573. doi: 10.1200/JCO.1997.15.2.566. [DOI] [PubMed] [Google Scholar]
- 12.Novitzky N. Myelodysplastic syndromes in children. A critical review of the clinical manifestations and management. Am J Hematol. 2000;63:212–222. doi: 10.1002/(SICI)1096-8652(200004)63:4<212::AID-AJH9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 13.Maguire AM, Vowels MR, Russell S. Allogeneic bone marrow transplant improves outcome for juvenile myelomonocytic leukaemia. J Paediatr Child Health. 2002;38:166–169. doi: 10.1046/j.1440-1754.2002.00764.x. [DOI] [PubMed] [Google Scholar]
- 14.Manabe A, Okamura J, Yumura-Yagi K. MDS Committee of the Japanese Society of Pediatric Hematology. Allogeneic hematopoietic stem cell transplantation for 27 children with juvenile myelomonocytic leukemia diagnosed based on the criteria of the International JMML Working Group. Leukemia. 2002;16:645–649. doi: 10.1038/sj.leu.2402407. [DOI] [PubMed] [Google Scholar]
- 15.Smith FO, King R, Nelson G. Unrelated donor bone marrow transplantation for children with juvenile myelomonocytic leukaemia. Br J Haematol. 2002;116:716–724. doi: 10.1046/j.0007-1048.2001.03333.x. [DOI] [PubMed] [Google Scholar]
- 16.Van't Veer-Korthof ET. Myelodysplastic Syndromes. 1992. Myelodysplastic syndromes in childhood: description of 11 cases; pp. 38–42. [Google Scholar]
- 17.Locatelli F, Pession A, Comoli P. Role of allogeneic bone marrow transplantation from an HLA-identical sibling or a matched unrelated donor in the treatment of children with juvenile chronic myeloid leukaemia. Br J Haematol. 1996;92:49–54. doi: 10.1046/j.1365-2141.1996.00276.x. [DOI] [PubMed] [Google Scholar]
- 18.Slaper-Cortenbach ICM, Wijngaarden-du Bois MJGJ, Vries-van Rossen Ade. The depletion of T cells from haematopoietic stem cell transplants. Rheumatology. 1999;38:751–754. doi: 10.1093/rheumatology/38.8.751. [DOI] [PubMed] [Google Scholar]
- 19.Glucksberg H, Storb R, Fefer A. Clinical manifestation of graft-versus-host disease in human recipients of marrow from HLA-matched sibling donors. Transplantation. 1974;18:295–304. doi: 10.1097/00007890-197410000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Shulman HM, Sullivan KM, Weiden PL. Chronic graft-versus-host syndrome in man. Am J Med. 1980;69:204–217. doi: 10.1016/0002-9343(80)90380-0. [DOI] [PubMed] [Google Scholar]
- 21.Vossen JM, Heidt PJ, Berg Hvanden. Prevention of infection and graft-versus-host disease by suppression of intestinal microflora in children treated with allogeneic bone marrow transplantation. Eur J Clin Microbiol Infect Dis. 1990;9:14–22. doi: 10.1007/BF01969527. [DOI] [PubMed] [Google Scholar]
- 22.Matthes-Martin S, Mann G, Peters C. Allogeneic bone marrow transplantation for juvenile myelomonocytic leukaemia: a single centre experience and review of the literature. Bone Marrow Transplant. 2000;26:377–382. doi: 10.1038/sj.bmt.1702522. [DOI] [PubMed] [Google Scholar]