

Abstract
As coxsackievirus B3 (CoxB3) and adenoviruses may cause acute myocarditis and inflammatory cardiomyopathy, isolation of the common coxsackievirus–adenovirus-receptor (CAR) has provided an interesting new target for molecular antiviral therapy. Whereas many viruses show high mutation rates enabling them to develop escape mutants, mutations of their cellular virus receptors are far less likely. We report on antiviral efficacies of CAR gene silencing by short hairpin (sh)RNAs in the cardiac-derived HL-1 cell line and in primary neonatal rat cardiomyocytes (PNCMs). Treatment with shRNA vectors mediating RNA interference against the CAR resulted in almost complete silencing of receptor expression both in HL-1 cells and PNCMs. Whereas CAR was silenced in HL-1 cells as early as 24 h after vector treatment, its downregulation in PNCMs did not become significant before day 6. CAR knockout resulted in inhibition of CoxB3 infections by up to 97% in HL-1 cells and up to 90% in PNCMs. Adenovirus was inhibited by only 75% in HL-1 cells, but up to 92% in PNCMs. We conclude that CAR knockout by shRNA vectors is efficient against CoxB3 and adenovirus in primary cardiac cells, but the efficacy of this approach in vivo may be influenced by cell type-specific silencing kinetics in different tissues.
Keywords: viral cardiomyopathy, coxsackievirus–adenovirus-receptor, RNA interference, gene silencing, adenovirus vector
Introduction
Initially, cardiac viral infections were documented in the clinical context of acute myocarditis. Coxsackievirus B3 (CoxB3) was the first virus detected in this condition in humans,1 and adenoviruses of serotypes 2 and 5 were later described as common agents of myocarditis in children.2 Systematic screening of patients with dilated cardiomyopathy has recently revealed that a rather broad spectrum of viruses may also chronically persist in human myocardium.3 It is therefore assumed that acute cardiac viral infections may cause not only acute illness, but also chronic heart disease if not definitely eliminated from the heart.4 The disease may progress to terminal heart failure and then require heart transplantation as a last therapeutic option.
RNA interference (RNAi) is a process of posttranscriptional gene silencing mediated by double-stranded RNA (dsRNA). The introduction of double-stranded small interfering RNAs (siRNA) or vectors expressing short hairpin RNA (shRNA) has already been successfully used to inhibit the replication of multiple viruses in vitro and in vivo including respiratory viruses,5 hepatitis B6, 7 and C viruses,8 foot-and-mouth disease virus,9 human herpesvirus-6,10 cytomegalovirus,11 SARS coronarvirus,12 HIV-113 and CoxB3.14, 15 However, the efficiency of RNAi depends on exact homology of the siRNA to the target sequence. A single mismatch can be sufficient to abrogate silencing by RNAi.16
Many RNA viruses encode polymerase enzymes that lack proofreading abilities and, as a result, have high mutation rates. Thus, there is a high probability that viruses will rapidly develop resistance to a particular siRNA during virus replication by incorporation of nucleotide mutations within the target sequence of the siRNA. Thus, it has been shown that hepatitis C virus becomes resistant against a particular siRNA after several cycles of replication by the incorporation of point mutations within the siRNA target sequence.17 Similar data have been reported for HIV-118 and poliovirus infections.19 Therefore, therapeutic targeting of non-variable virus-binding receptors on susceptible cells or other host cell molecules directly or indirectly involved in virus infections is an interesting alternative to targeting of the virus itself.15, 20, 21, 22, 23, 24, 25 One approach of this type showed that downregulation of the CD4-independent attachment receptor (DC-SIGN) significantly inhibited HIV infection of dendritic cells (DC) and prevented the transfer of infectious HIV-1 particles from DC to T cells.22
Adenoviruses of types 2 and 5 and CoxB3 use the coxsackievirus–adenovirus-receptor (CAR) to infect their target cells, either for virus attachment (adenoviruses) or virus internalization (CoxB3).26 Therefore, CAR represents an attractive target for the inhibition of cardiac CoxB3 and adenovirus infections. It has been shown recently that suppression of CAR by RNAi results in inhibition of CoxB3 infections.15, 27 These studies were carried out with cells of non-cardiac origin, and chemically synthesized siRNAs were used to inhibit CAR expression. However, the therapeutic use of synthetic siRNAs is significantly limited by their rapid degradation in target cells resulting in only transient gene silencing.28 Moreover, most cells of cardiac origin are very difficult to transfect with chemically synthesized siRNAs even in vitro. These problems are significantly aggravated in vivo. Despite intense efforts during recent years, systemic and target organ-specific delivery of siRNAs remains a major hurdle for in vivo applications of RNAi.
Therefore, we investigated the antiviral potential of CAR gene silencing in cells of cardiac origin by use of newly developed adenoviral vectors (AdVs) that generate shRNA against murine or rat CAR (shCAR). shCAR treatment of HL-1 cells and primary neonatal rat cardiomyocytes (PNCMs) led to almost complete silencing of CAR gene expression and efficient inhibition of CoxB3 infection in both cell types, whereas efficient inhibition of adenovirus infections was observed only in the primary cells. The studies thus indicate cell type-specific responses of cellular CAR to shCAR treatment, which need to be considered as important determinants of this new antiviral approach in vivo.
Results
Selection and specificity of mouse CAR shRNAs
To investigate the efficacy of CAR silencing for the inhibition of adenovirus and CoxB3 infection in cardiac HL-1 cells, we generated a total of three shRNAs (shCAR2m, shCAR3m, shCAR4m) directed against the extracellular domains of the mCAR splice variants mCAR-126 and mCAR-229 (Figure 1) following published siRNA selection criteria.30 DNA oligonucleotides encoding these shRNAs were then cloned into expression plasmids under the control of a murine RNA polymerase III U6 promotor. To test their efficacies, we co-transfected 293T cells with shCAR2m-, shCAR3m- or shCAR4m-expressing plasmid, together with plasmids expressing recombinant mCAR1 and mCAR2. As a transfection control, a plasmid expressing an irrelevant shRNA was used. To determine the extent of recombinant mCAR1- and mCAR2-mRNA downregulation, total cellular RNA was isolated 48 h after transfection and Northern blot hybridization was carried out. These experiments showed that two shRNAs were highly efficient: shCAR2m with 93% silencing and shCAR4m with 97% silencing of mCAR1- and mCAR2-mRNA. The control shRNA affected neither mCAR1- nor mCAR2-mRNA expression (Figure 2a).
Figure 1.
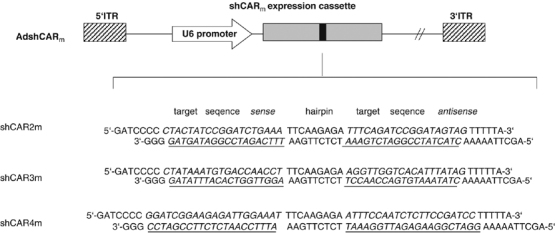
Structure of adenovectors for shRNA transcription. Schematic illustration of recombinant AdshCARm and the sequence of shCAR: the 5′-ITR represents nucleotide positions 1–342 of adenovirus type 5; the shCAR expression cassette is composed of a U6=RNA polymerase III promoter and an shCAR sequence; the individual shCAR motifs consist of 19 (shCAR2m, shCAR3m) or 21 nucleotides (shCAR4m) corresponding to the coding regions of mCAR1 and mCAR2. The two motifs that form the sense and antisense strand of the shRNA are separated by a loop of nine nucleotides. A transcriptional termination signal of five thymidines is added at the 3′ end of the inverted repeat. Whereas shCAR2m and shCAR3m are directed against the IG1 extracellular domain of CAR (shCAR2m=nucleotides 246–264; shCAR3m=nucleotides 311–329), shCAR4m is directed against the nucleotides 448–468 of the IG2 extracellular domain; 3′-ITR=right inverted terminal repeat of adenovirus 5. Note: siRNA sequence in shCAR2m matches completely to mouse and rat CAR target sequences.
Figure 2.
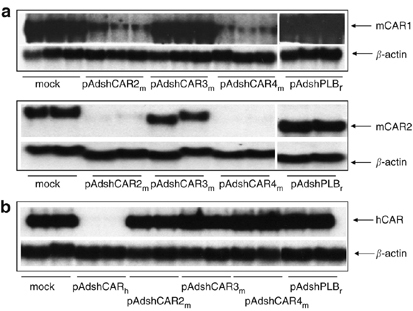
Efficiency and specificity of shCARm-mediated mCAR silencing. (a) Selection of shCARm. 293T cells were co-transfected with mCAR1 (upper panel) or mCAR2 (lower panel) plus shCAR2m, shCAR3m or shCAR4m expression plasmid. Northern blot analysis performed 48 h after transfection showed efficient downregulation of both mCAR1- and mCAR2-mRNA expression by shCAR2m and shCAR4m as compared to untransfected and control-shRNA (shPLBr)-transfected cells. Experiments are made in triplicate (upper panel) and duplicate (lower panel). (b) Specificity of shCARm. 293T cells were co-transfected with human soluble CAR (hCAR) plus shCARm or hCAR-shRNA (pAdshCARh) expression plasmids. Northern blot analysis was performed as above. None of the shCARm downregulated hCAR, whereas hCAR-shRNA led to strong downregulation of hCAR-mRNA. Determination of β-actin expression was included as loading control. Experiments are made in duplicate.
To analyze the specificities of shCAR2m and shCAR4m, both were also tested for downregulation of human CAR (hCAR) by co-transfection with a plasmid expressing recombinant soluble hCAR. None of the mouse CAR-specific shRNAs influenced hCAR-mRNA expression. In contrast, a human-specific shRNA efficiently silenced hCAR-mRNA expression (Figure 2b). Considering that shCAR2m has only one mismatch and shCAR4m three mismatches as compared to hCAR-mRNA, these experiments indicate very high species specificity of the selected mCAR-shRNAs.
Silencing of mCAR in mouse cardiac HL-1 cells
To determine the optimal AdV dose for transduction of mouse cardiac HL-1 cells, we first transduced HL-1 cells with a GFP-expressing marker AdV at a multiplicity of infection (MOI) from 5 to 1000. At an MOI from 60 to 100, GFP expression was seen in 76–80% of the cells, whereas no cytotoxic effects were observed. Further dose escalation resulted in an only slight further increase of transduction rate (Figure 3a), and at an MOI of 500 or higher, cytotoxic effects became evident (increase of apoptotic cells, reduction of cell growth). HL-1 cells express mCAR1 but no mCAR2 on their cell surface (not shown). Therefore, the silencing capacity of the new AdshCAR4m (Figure 1), which expresses the above-evaluated most efficient shCAR4m, was investigated with respect to mCAR1 expression in HL-1 cells only. To assess the amount of AdV-generated shCAR4m, HL-1 cells were transduced with AdshCAR4m at an MOI from 5 to 1000. The expression of shCAR4m was analyzed 24, 48 and 72 h later. A time- and dose-dependent transcription of shCAR4m was detected (Figure 3b). As a consequence of shRNA expression, strong downregulation of mCAR1-mRNA was observed 48 and 72 h after transduction. Near maximal reduction of 85–90% was observed at an MOI of 100. Higher doses did not result in significant further downregulation of mCAR1-mRNA (Figure 3c). These results indicate that an MOI of 100 was optimal for HL-1 transduction, as this dose allowed strong downregulation of mCAR1-mRNA in HL-1 cells, although cytotoxicity of the viral vector was still very low.
Figure 3.
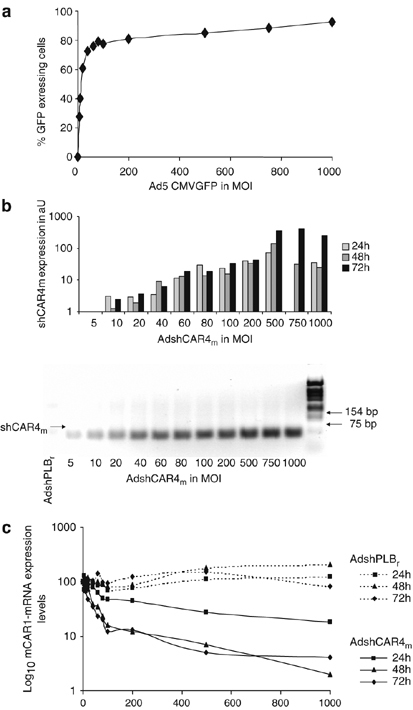
Downregulation of mCAR mRNA in HL-1 cells by AdshCAR4m. (a) AdV-mediated gene transfer efficacy. HL-1 cells were transduced with Ad5CMVGFP (MOI from 5 to 1000) and the fraction of cells expressing GFP was quantified by flow cytometry after 48 h. (b) AdV-mediated shCAR4m expression levels. HL-1 cells were transduced with AdshCAR4m (MOI from 5 to 1000) and the shCAR4m expression level (shown as arbitrary units, aU) was then measured by real-time RT-PCR (upper panel) and agarose gel electrophoresis (lower panel, 24 h after transduction) at the indicated time points. HL-1 cells transduced with the vector AdshPLBr served as controls. (c) Downregulation of mCAR-mRNA expression by AdshCAR4m. HL-1 cells were transduced with AdshCAR4m or AdshPLBr (MOI from 5 to 1000) and mCAR-mRNA expression was measured by real-time RT-PCR at the indicated time points. Normalization was for MOI=0 (100%).
We next investigated mCAR1 protein expression after transduction of HL-1 cells with AdshCAR4m at an MOI of 100. To disrupt pre-existing membrane-associated mCAR1, HL-1 cells were trypsinized 24 h after AdshCAR4m transduction and then re-seeded. This procedure led to ablation of mCAR1 protein at the cell surface 2 days after transduction (Figure 4a and b). In contrast, control cells rapidly re-expressed mCAR1 (Figure 4b). Two days after AdshCAR4m treatment, a few cells still displayed some membrane-associated mCAR1 immunoreactivity (Figure 4b), probably reflecting residual mCAR1 domains still anchored in the cell membrane after trypsinization. At days 3 and 4 after AdshCAR4m transduction, mCAR1 immunoreactivity disappeared from HL-1 cells, whereas it remained permanently detectable in the control cells (Figure 4b). Interestingly, CAR was expressed only at cell-to-cell contact sites in HL-1 cells, suggesting that the observed stability of CAR in intact monolayers is determined by homophilic or heterophilic CAR interaction at cell-to-cell contact sites. Together, the results indicate that AdshCAR4m treatment efficiently inhibits the de novo synthesis of mCAR1.
Figure 4.
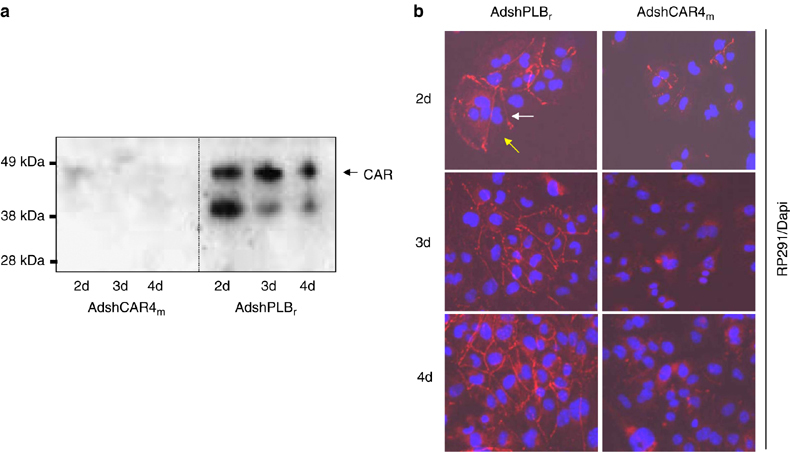
Downregulation of mCAR1 protein in HL-1 cells by AdshCAR4m. (a) Detection of mCAR1 ablation by Western blot analysis. HL-1 cells were transduced with AdshCAR4m or the control vector AdshPLBr (MOI of 100) for 24 h. Cells were then trypsinized, re-seeded and investigated using the polyclonal rabbit anti-CAR antibody H-300 directed against the extracellular domain of CAR. mCAR1 protein became undetectable as early as 2 days after transduction with AdshCAR4m, whereas in the controls mCAR1 protein was abundantly expressed. (b) Detection of mCAR1 knockdown by indirect immunofluorescence. HL-1 cells were treated as in (a). mCAR1 expression was detected by indirect immunofluorescence using the polyclonal rabbit antibody RP-29156 directed against the CAR variant with intracellular SIV tail (mCAR variant 1, accession no. NM_001025192). mCAR1 was abundantly expressed over the whole investigation period (4 days after transduction) in HL-1 cells transduced with control vector (left-hand panels). In contrast, mCAR1 was visible at low levels 2 days after transduction but became undetectable at later time points (right-hand panels). Cell nuclei are stained with 4,6-diamidino-2-phenylindole and overlaid images are shown. Notably, mCAR1 was localized only at cell–cell contact sites (white arrows), whereas cell membrane regions not in contact with other cells displayed no mCAR1 protein (yellow arrows). mCAR2 expression was not detectable in HL-1 cells by using polyclonal rabbit antibody RP-19456 directed against the CAR variant with intracellular TVV tail (mCAR variant 2, accession no. NM_009988) (not shown). We later noted that CAR protein was downregulated by AdshCAR4 with similar kinetics if the trypsinization and re-seeding step was omitted. Figures 5b and 6 show that also the virus-related functionality of CAR in HL-1 cells was abandoned irrespective of trypsinization.
Inhibition of CoxB3 replication in HL-1 cells by AdshCAR4m
The antiviral effect of mCAR1 silencing was now investigated with respect to CoxB3 for which cellular CAR constitutes the internalization receptor. For this purpose, we first investigated the CoxB3 protective efficacy of AdshCAR4m as a function of dose, at an MOI of AdshCAR4m ranging from 10 to 100, using the same trypsin-involving experimental procedure as described above. HL-1 cells were infected with CoxB3 48 h after transduction with AdshCAR4m and the amount of newly generated progeny virus was determined 6 days later. Virus plaque assays clearly demonstrated that CoxB3 replicates in HL-1 cells, but at a rather low frequency because no cytopathogenic effect was visible during the 6-day investigation period. As expected, the strongest reduction of CoxB3 progeny virus production by 97% was observed at the highest AdshCAR4m dose with an MOI of 100. Nevertheless, MOIs of 50 and 25 still led to significant inhibition of CoxB3 replication by 85 and 75%, respectively, whereas an MOI of 10 showed no significant effect (Figure 5a). To investigate whether disruption of pre-existing CAR affects CoxB3 infections, we either treated HL-1 cells with the trypsin-involving experimental procedure described above, or omitted the trypsinization step after AdshCAR4m transduction. The titers of CoxB3 progeny virus generated 5 and 6 days after CoxB3 infection were reduced strongly by about 97% in AdshCAR4m-transduced cells, irrespective of whether membrane-associated CAR was disrupted by trypsinization or not (Figure 5b). These experiments indicate that AdshCAR4m inhibits CoxB3 replication in HL-1 cells with high efficacy. Moreover, proteolysis of membrane-associated CAR apparently does not affect CoxB3 infection.
Figure 5.
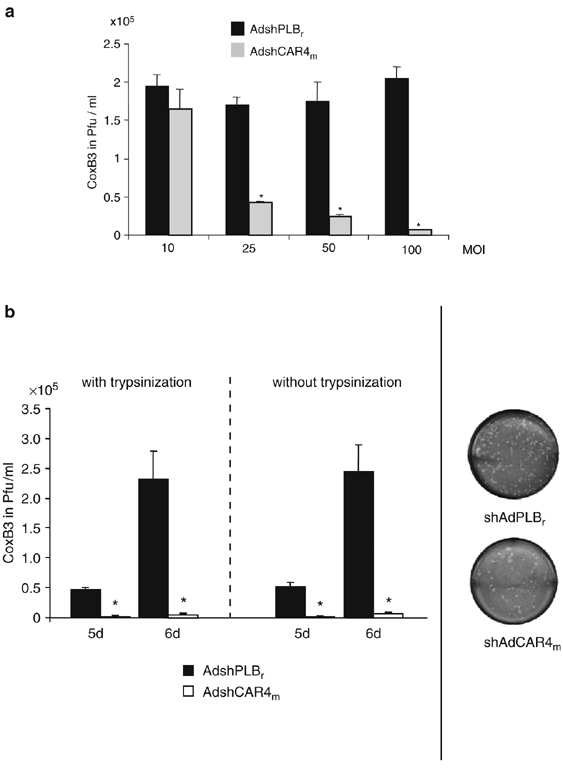
Inhibition of CoxB3 infection by mCAR silencing in HL-1 cells. (a) Inhibition of CoxB3 replication as a function of AdshCAR4m dose. HL-1 cells were transduced with AdshCAR4m or control vector at variable MOIs ranging from 10 to 100. The cells were trypsinized 24 h later, re-seeded and infected with CoxB3 (at an MOI of 1) 24 h later. Plaque assays were carried out 6 days after infection. Asterisks indicate significant differences (P<0.05). (b) Inhibition of CoxB3 replication by AdshCAR4m as a function of trypsin treatment. HL-1 cells were transduced with AdshCAR4m or control vector (MOI of 100) as in Figure 4. After 24 h, cells were trypsinized and re-seeded (with trypsinization) or the medium was changed (without trypsinization). At 48 h after transduction, the cells were infected with CoxB3 at an MOI of 1. Five and six days later, cells were lysed by three freeze/thaw cycles and virus titers were determined by plaque assay on HeLa cells (left-hand diagram). Right side: example for plaque assay on HeLa cells using a dilution of 1:500 of HL-1 cell lysate from AdshCAR4m- and control vector-transduced cells.
Inhibition of adenovirus infections of HL-1 cells by AdshCAR4m
CAR is also involved in adenovirus infection as an attachment receptor. To investigate the long-term efficacy of AdshCAR4m with respect to the inhibition of adenovirus infections and to examine whether proteolysis of pre-existing CAR affects adenovirus infections, HL-1 cells were transduced with AdshCAR4m versus control vector at an MOI of 100. After 24 h, cells were either trypsinized and re-seeded, or the medium was simply changed (treatment without trypsinization). Cells were infected with a luciferase marker adenovirus (MOI of 5) 2–5 days after transduction. The measurement of luciferase activity 24 h later revealed persistent reduction of marker gene expression of about ≈75% in AdshCAR4m-treated HL-1 cells during the 5-day investigation period. This clearly demonstrates that CAR-shRNA treatment inhibits adenovirus uptake into HL-1 cells, but markedly less efficient than for CoxB3. Inhibition of adenovirus infections was closely similar between trypsinized and non-trypsinized cells (Figure 6) indicating that proteolysis of CAR does not affect the function of CAR in adenovirus infections.
Figure 6.
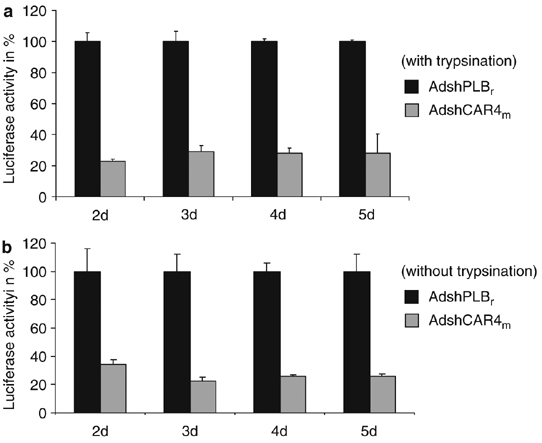
Inhibition of adenovirus infection by mCAR silencing in HL-1 cells. (a) Inhibition of adenovirus infection by AdshCAR4m (with trypsinization). HL-1 cells were treated as in Figure 4. Two to five days after transduction, the cells were infected with a luciferase marker adenovirus (MOI of 5) and luciferase expression was measured after 24 h. (b) Inhibition of adenovirus infection by AdshCAR4m (without trypsinization). HL-1 cells were transduced with AdshCAR4m or the control vector AdshPLBr (each at an MOI of 100). The medium was changed after 24 h. Two to five days after transduction, the cells were infected with a luciferase marker adenovirus (MOI of 5) and luciferase expression was measured 24 h later.
Anti-adenovirus and anti-CoxB3 effects of CAR silencing in PNCMs
As HL-1 cells is a permanent heart-derived cell line, we were also interested in investigation of the effects of CAR silencing upon viral infections of primary cardiac cells. For this reason, PNCMs were transduced with AdshCAR2m whose shRNA sequence completely matches with mouse and rat CAR sequences, or with the control vector AdshGFP, both at an MOI of 100. At this dose, nearly 80% of PNCMs were transduced with AdV (not shown), which is closely similar to the HL-1 cells. Following the treatment of PNCMs with AdshCAR2m, CAR expression was unaffected up to day 3 after transduction, but became strongly downregulated on days 6 and 8 after transduction (Figure 7a). As a consequence of this delayed CAR downregulation, challenge of AdshCAR2m-treated PNCMs with CoxB3 (MOI of 1) resulted in delayed inhibition of CoxB3 replication. In fact, significant reduction of CoxB3 replication was seen only if PNCMs were infected with CoxB3 on days 6 and 8 but not on day 3 after AdshCAR2m treatment. The degree of CoxB3 inhibition was strongly dependent on the AdshCAR2m dose. At an MOI of 100, the AdshCAR2m treatment led to reduction of CoxB3 titers by 66 and 90%, whereas at an MOI of 25, CoxB3 titers were reduced by only 16 and 34% on days 6 and 8, respectively (Figure 7b). Similar results were obtained when AdshCAR2m-transduced PNCMs (MOI of 100) were infected with adenovirus. Its uptake was reduced by 85 and 92%, respectively, on days 6 and 8 after AdshCAR2m transduction, whereas no effect was seen on day 3 (Figure 7c).
Figure 7.
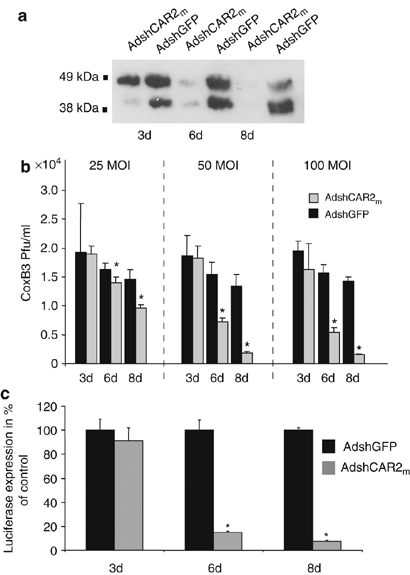
Inhibition of CoxB3 and adenovirus infection in PNCMs by CAR silencing. (a) Detection of rat CAR downregulation by Western blot analysis. PNCMs were transduced with AdshCAR2m or the control vector AdshGFP (each at an MOI of 100) for 3, 6 and 8 days and then investigated for CAR expression using the polyclonal rabbit anti-CAR antibody H-300. (b) PNCMs were transduced with AdshCAR2m or the control vector AdshGFP (at MOIs of 25, 50 and 100) for 3, 6 and 8 days and then infected with CoxB3 (MOI of 1). Cells were harvested 3 days later and CoxB3 titers were determined by plaque assay. Asterisks indicate significant differences (P<0.05). (c) PNCMs were transduced with AdshCAR2m or the control vector AdshGFP (each at an MOI of 100) for 3, 6 and 8 days and then transduced with a luciferase marker adenovirus (MOI of 5). Luciferase expression was measured 24 h later. Asterisks indicate significant differences (P<0.005).
Discussion
CoxB3 and adenoviruses are common agents of acute myocarditis and inflammatory cardiomyopathy.1, 3 Although interferon-β treatment has the potential to eliminate cardiotropic viruses and to improve heart function in patients with myocardial persistence of viral genomes,31 no specific treatment against CoxB3 and adenovirus infections is available to date. With respect to antiviral therapy, RNAi is a promising new technology. Its therapeutic potential against viral infections has been demonstrated in multiple in vitro and in vivo studies.5, 6, 7, 13, 14, 15 Despite encouraging results, however, several reports have revealed a fundamental general problem of siRNA-based antiviral therapy. Owing to the requirement for strict sequence-specificity between therapeutic siRNA and target mRNA, even single point mutations in the viral target sequence may result in the rapid selection of escape mutants during persistent viral infections.17, 18, 19
In this study, we show that inhibition of CoxB3 and adenovirus infections is possible via silencing of the CAR receptor by means of vector-generated CAR-shRNA. This strategy is an attractive alternative to siRNA-based silencing of virus-encoded gene, because it drastically reduces the probability of escape mutant development.22 In contrast to previous work,15 our investigations were carried out in myocardial cells representing the actual targets for CoxB3 and adenovirus infections in acute myocarditis and inflammatory cardiomyopathy.32 Treatment of HL-1 cells with the shCAR vector led to efficient and stable removal of mCAR protein from the cell surface, and consequently, to significant inhibition of CoxB3 and adenovirus infection of these cells. Remarkably, adenovirus internalization was reduced by only ≈75% despite the near-complete silencing of mCAR expression, whereas CoxB3 infection was inhibited by up to 97%. In contrast, CAR silencing in PNCMs resulted in strong inhibition of both adenovirus and CoxB3 infections (by 92 and 90%, respectively). Adenovirus binding to cells is not exclusively mediated by CAR. Type 2 and 5 adenoviruses use CAR to attach to the cell surface,33 whereas its internalization occurs through αvβ1, αvβ3 and αvβ5 integrin.29, 34, 35 Others have shown that heparan sulfate glycosaminoglycans and major histocompatibility complex class I molecules may also be involved in the binding of adenoviruses 2 and 5.36, 37 Moreover, the expression of receptors involved in adenovirus infections is highly variable between different cell types and tissues.38 Thus, variable receptor expression levels and interactions of adenoviruses with cell type-specifically expressed alternative receptors are the most likely explanation for the failure of complete CAR silencing to achieve efficient blockade of adenovirus infection in HL-1 cells. To enhance efficacy in inhibition of adenovirus infection via the anti-receptor approach, it seems to be necessary to simultaneously inhibit CAR and its co-receptors.
In contrast to the anti-adenovirus experiments, the anti-CAR approach was efficient with respect to CoxB3 in both HL-1 cells and PNCMs. The magnitude of inhibition of CoxB3 infection correlated well with the vector-induced ablation of mCAR from the cell surface and emphasizes the key role of CAR in CoxB3 infections of cardiac cells. The molecular reason for the differences of anti-CAR treatment in adenovirus versus CoxB3 infections may be the unique role played by CAR during CoxB3 infection as it induces conformational changes in the virus capsid that are essential for CoxB3 entry into the target cell.27 The high efficacy of the shCAR vector against this virus indicates that CAR silencing is a promising therapeutic approach against CoxB3 infections of the myocardium. Previous studies have shown a similar degree of inhibition when using siRNAs directed against CoxB3 viral genomic RNA or its RNA intermediate.14, 15, 39 Combination of direct antiviral approaches, which are prone to escape mutant development with the current anti-receptor strategy, is likely to further increase the efficacy of RNAi-based antiviral therapy.
Recently, it has been shown that silencing of CAR by chemically synthesized siRNA inhibited CoxB3 infection in human cells of non-cardiac origin.27 However, this approach decreased virus titers by only ≈60%15 which is markedly less than in the present study employing AdV-generated shCARs. This difference should be the result of high transduction efficacy of AdVs and the prolonged high-level expression of the shCAR. A bottleneck of chemically synthesized siRNAs is its transient silencing activity of ≈3 days in cardiac cells.28 Viral vectors expressing shRNAs, however, enable long-term silencing of target genes in cells of cardiac origin40 and allow efficient delivery of transgenic sequences to the heart.41, 42 Among the currently available viral vectors, pseudotyped AAV9 vectors appear to be most promising for cardiac gene therapy as they allow highly efficient targeted transduction of the heart by way of simple intravenous injection.43, 44
CAR is a cell adhesion protein mediating homotypic45, 46 and heterotypic47 intercellular interactions. It is a component of specialized intercellular junctions including epithelial tight junctions,45 neuro-muscular junctions48 and myocardial intercalated discs.49 We found it to be localized exclusively at cell-to-cell contact sites between HL-1 cells, possibly as a consequence of cell membrane-associated CAR stabilization through homotypic (CAR–CAR) or heterotypic (CAR–protein) intercellular protein interactions. In contrast to previous CAR silencing attempts using synthetic siRNAs, the newly developed viral vectors AdshCAR4m and AdshCAR2m achieved near complete and stable knockout of CAR. This was an essential prerequisite to reveal significantly different kinetics of CAR protein ablation in different cell types, in particular, an unexpectedly high stability of CAR in primary cells versus a stable cell line. These data may be explained by cell type-specific CAR protein kinetics, but differential CAR-mRNA stability or CAR-shRNA processing through the cellular microRNA/shRNA machinery also need to be taken into account. The delayed downregulation of CAR following vector-mediated CAR-shRNA delivery in primary cardiomyocytes is relevant for in vivo investigations because the time lag between vector application and CAR silencing needs to be considered in the development of antiviral therapy protocols.
An important concern associated with downregulation of a cellular receptor molecule for antiviral therapy are the possible side effects resulting from ablation of the normal cellular functions of the receptor. During organogenesis, CAR is highly expressed in the heart, but rapidly downregulated post partum.50 In a recently published genomic mouse knockout model, cardiomyocyte-specific CAR deletion resulted in severe cardiac anomalies and death between days 11.5 and 13.5 of embryonal development.51, 52 If the CAR gene became ablated late in embryonic development, however, the CAR-deficient animals survived into adulthood and had no evident cardiac anomalies.51 This finding suggests that CAR silencing by RNAi, as employed in our study, should be well tolerable over considerable periods of time, during which virus migration and spreading were efficiently inhibited. Furthermore, treatment of HL-1 cells with AdV-generated shCAR revealed no side effects as we found no changes in cell growth, cell morphology or F-actin cytoskeleton protein expression (not shown). As CAR is a component of tight junctions and may play a permanent role in tissue function of several organs, further experiments have to examine if CAR knockdown by RNAi has no side effects in vivo even over prolonged treatment times.
In summary, high efficacy of CAR gene silencing by shRNAs in cardiac-derived cells was observed against CoxB3, whereas cell type-dependent results were obtained against adenovirus. In contrast to previous silencing attempts using siRNAs or shRNA-plasmid, the newly developed shRNA vectors were able to achieve nearly complete and stable knockout of CAR transcription. This, in turn, revealed significantly different responses of cellular CAR protein to CAR-shRNA treatment, which need to be considered as important determinants of this new antiviral approach in vivo.
Materials and methods
Cell cultures
The HL-1 cell line, a cardiac muscle cell line established from an AT-1 mouse atrial cardiomyocyte tumor lineage, was a kind gift from William C Claycomb. The cells were maintained in Claycomb medium supplemented with 10% fetal calf serum (FCS) (JRH Bioscience, Lenexa, USA), 1% each of penicillin and streptomycin, 0.1 mM norepinephrine (Sigma, München, Germany) and 2 mM L-glutamine (Invitrogen, Karlsruhe, Germany). HeLa (human cervical carcinoma) cells, HEK293 (human embryonal kidney) and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco BRL, Karlsruhe, Germany) supplemented with 10% FCS and 1% penicillin/streptomycin. PNCMs were generated and maintained as described previously.40
Construction of expression plasmids
For each mCAR-shRNA, four short oligonucleotides that cover antisense, loop and sense sequence of the shRNA were synthesized (Oligoservice, Berlin, Germany). The target sequence of shCAR2m and shCAR3m encompassed 19 nucleotides, and that of shCAR4m 21 nucleotides (see Figure 1). The forward and reverse oligos were phosphorylated by T4 polynucleotide kinase (New England BioLabs, Frankfurt, Germany), annealed and cloned into HindIII/BamHI-digested pSilencerTM 2.1-U6 neo (Ambion, Austin, TX, USA). The shRNA expression cassettes were then cloned into pAdshPLBr40 via EcoRI/HindIII to yield the plasmids pAdshCAR2m, pAdshCAR3m and pAdshCAR4m.
For the generation of the mCAR1 expression plasmid pAdmCAR1, total RNA was first isolated from HL-1 cells and reverse-transcribed by the use of SuperScriptII reverse transcriptase (Invitrogen). The mCAR1-cDNA was then amplified using the primer pair MCAR105s (5′-CGGCAGCYACCATGGCGC-3′) and HCAR-1157 (5′-CTATACTATAGACCCATCCTTG-3′) in a 35-cycle PCR with Pwo polymerase (Roche, Mannheim, Germany), and the resulting fragment was then cloned in the pCR2.1-TOPO vector (Invitrogen). An EcoRI fragment containing mCAR1-cDNA was excised and inserted into the shuttle plasmid pZS2.53 For generation of the recombinant mCAR2 expression plasmid pAdmCAR2, the mCAR2 cDNA was excised from pCR3.1-mCAR2 with HindIII/PmeI and cloned into pZS2 via SmaI/HindIII. Correctness of all plasmids was proven by sequencing on an ABI 310 genetic analyzer (Applied Biosystems, Foster city CA, USA).
Development of AdVs
The adenoviral shuttle plasmids pAdshCAR4m and pAdshCAR2m were linearized with SpeI and ligated to the 5′ ‘long arm’ of the XbaI-digested E1−E3− adenovirus mutant RR5, transfected in HEK293 cells and propagated as described.54 This resulted in the AdVs AdshCAR2m, AdshCAR4m and AdshPLBr. An additional shGFP expressing AdV (AdshGFP) containing an siRNA target sequence (5′-GGTTATGTACAGGAACGCA-3′) directed against GFP was generated in the same way as AdshCAR4m and AdshCAR2m. The AdVs Ad5CMVGFP, Ad5CMVluc38 and AdshPLBr40 have been described. All AdVs were tested for possible RCA contamination by PCR as described.53 Virus titers were determined by standard plaque assays on HEK293 cells.
Plasmid transfections
Plasmid transfections were carried out by calcium phosphate precipitation. Cells were seeded in six-well plates at a density of 5 × 105 cells/well and transfected with 1 μg of each plasmid/well in 250 μl 2 × HBS buffer, pH 7.1 and 250 μl 0.25 M CaCl2 in the presence of DMEM containing 10% FCS. After 24 h, cells were washed three times with phosphate-buffered solution (PBS) and supplied with fresh growth medium.
Northern blots
Total RNA was isolated with TRIzol Reagent (Invitrogen) following the manufacturer's instruction. Ten micrograms of isolated RNA was electrophoretically separated on 1% formaldehyde agarose gels. After transfer to a Hybond N nylon membrane (Amersham, Buckinghamshire, UK), the membranes were hybridized with a single-stranded (ss) antisense mCAR1-specific DNA probe using the mCAR1-cDNA excised from pAdmCAR1 as template. Rehybridization to control for equal DNA loading was carried out with an ss-antisense β-actin-specific DNA probe as described,55 using a PCR fragment amplified from HeLa cells with the primer pair 5′-AAGGATTCCTATGTGGTCG-3′ and 5′-CTCCTTAATGTCACGCAGGA-3′. The labeling of the probes with [32P]dCTP in a PCR-like reaction was described.38 Hybridized filters were exposed to Kodak Biomax MS film (Integra Biosciences, Fernwald, Germany) and hybridization signal intensity was then determined by phosphoimaging in a Fuji Film BAS-1500 imager (Fuji Photo Film, Dusseldorf, Germany).
Flow cytometry
To determine the fraction of cells transduced by an AdV, HL-1 cells were seeded in a six-well plate at a density of 4 × 105 cells/well. After 24 h, the cells were transduced with different MOI of adenoviral GFP marker vector (Ad5CMVGFP).38 Flow cytometry was performed 48 h later on a FACS Calibur with CellQuest software (Becton Dickinson, Franklin Lakes, NJ, USA).
Quantitative real-time reverse transcription-PCR
Total RNA from HL-1 cells was isolated with RNAzol. Four micrograms of RNA was incubated with peqGOLD DNaseI (Peqlab, Erlangen, Germany) at 37°C for 30 min. The reaction was stopped by addition of 2.5 μl of 25 mM ethylenediaminetetraacetic acid (EDTA) and heating for 15 min to 65°C. RNA was reverse-transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems) according to the manufacturer's instruction. For the quantification of gene expression, quantitative PCR assay was performed on a Mastercycler ep Gradient S (Eppendorf) with TaqMan Universal Master Mix UNG (Applied Biosystems) using the standard conditions determined by the company. After AmpliTaqGold activation for 10 min at 95°C, 40 cycles were run at a denaturing temperature of 95°C (15 s) and an annealing/extension temperature of 60°C (1 min). A unique combination of forward and reverse primers and fluorescent MGB probes designed by the company was used for the mCAR1 target gene, and the HPRT and GAPDH housekeeping genes, respectively, which were used for normalization. cDNA corresponding to 50 ng of RNA was used per 20 μl reaction and each reaction was performed in triplicate. The level of expression was calculated on the basis of the PCR cycle number (Ct) at which the exponential growth in fluorescence from the probe passes a certain threshold value. Relative expression (ΔCt) was determined by the difference in the Ct values for the target gene after normalization to RNA input level, using HPRT Ct values. Relative quantification was determined by standard 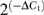 calculation in percent.
calculation in percent.
Assessment of cellular shCAR4 expression levels
Semiquantitative analysis of shCAR4m expression included two steps: an RT reaction containing 50 ng/μl total RNA samples, 50 nM shCAR4 stem–loop RT primer (5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTTCCAA-3′), 1 × RT buffer (Applied Biosystems), 0.25 mM each of dNTP, 3.33 U/μl MultiScribe reverse transcriptase (Applied Biosystems) and 0.25 U/μl RNAse inhibitor (Peqlab). The 7.5 μl reactions were incubated for 30 min at 16°C, 30 min at 42°C, 5 min at 85°C and then held at 4°C. The 20 μl PCR reactions included 1 μl of RT product, 1 × Gene Amp PCR buffer and 1.25 U AmpliTaq DNA polymerase (Roche), 0.25 mM each of dNTP and 1.5 μ
M forward primer (5′-GCGGCGGATCGGAAGAGATT-3′) and reverse primer (5′-GTGCAGGGTCCGAGGT-3′) specific for shCAR4m-cDNA. The reactions were incubated in a Gene Amp PCR System 9700 (Applied Biosystems) at 95°C for 4 min, followed by 30 cycles at 95°C for 15 s and 60°C for 1 min and analyzed by gel electrophoresis. Alternatively, shCAR4m quantification was carried out by real-time PCR. The TaqMan reactions contained 1 × TaqMan Universal PCR Master Mix (Applied Biosystems), 1 μl of RT product, 200 nM forward primer, 200 nM reverse primer and Sybr Green (Invitrogen) at 1:10 000. The reactions were incubated in a 96-well plate at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min, and run in triplicate. The threshold cycle (Ct) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold. The relative shCAR4m expression was calculated by the formula 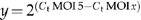 .
.
Western blots
Cells were solubilized in general lysis buffer (20 mM Tris, pH 8, 10 mM NaCl, 0.5% (v/v) Triton X-100, 5 mM EDTA, 3 mM MgCl2). After boiling of the lysate at 95°C for 5 min, proteins were separated on NuPAGE 4–12% Bis–Tris gels (Invitrogen) under denaturing and reducing conditions and transferred onto a PVDF membrane (Bio-Rad, Hercules, CA, USA). Subsequently, the membrane was incubated with anti-CAR polyclonal antibody H-300 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h in dry milk. Secondary swine–anti-rabbit antibodies conjugated with peroxidase (DAKO, Hamburg, Germany) were used at 1:10 000 dilution. Detection by chemiluminescence was achieved by using an enhanced chemiluminescence system (Amersham Biosciences).
Indirect immunofluorescence
HL-1 cells were fixed with 1 ml of Tris-buffered solution (TBS) containing 4% formaldehyde and permeabilized by 0.5% Triton X-100. After blocking in TBS buffer with 5% FCS and 0.1% Triton X-100, cells were incubated with anti-mCAR1 rabbit polyclonal antibody RP-29156 diluted 1:100 in blocking solution for 1 h. After extensive rinsing, the cells were incubated with a biotin-labeled donkey–anti-rabbit secondary antibody (Dianova, Hamburg, Germany) at a dilution of 1:400 for 1 h. Cells were subsequently incubated with Cy3-conjugated streptavidin (Dianova) (dilution 1:500) for 30 min and nuclei were stained with 4,6-diamidino-2-phenylindole (0.5 μg/ml) (Sigma). After covering with Fluoromount GTM (Southern Biotechnology, Birmingham, AL, USA), images were taken by using an Olympus BX60 immunofluorescence microscope.
Luciferase assays
Luciferase assays were performed using the luciferase reporter gene assay kit (Roche) following the manufacturer's instructions. Briefly, cells were washed twice with PBS buffer and lysed directly by adding 250 μl of cell lysis reagent and incubating for 15 min at room temperature. Cell debris was pelleted in a microfuge at maximum speed for 15 s and 10 μl of the supernatant was then transferred to a 5-ml tube (Sarstedt, Nürnberg, Germany). Luciferase substrate (50 μl) was added and the luciferase activity was immediately measured in a Lumat LB 9501 luminometer (Berthold, Bad wildbad, Germany).
Virus plaque assays
HeLa cells were cultured in six-well cell culture plates as confluent monolayers at a density of 1.2 × 106 cells/well and incubated at 37°C in Eagle's MEM (minimal essential medium) with 5% (v/v) FCS under 5% (v/v) CO2. After 24 h, cells were overlaid with 1 ml of diluted supernatant from HL-1 cells, which were treated by three freeze/thaw cycles to release intracellular CoxB3 virus. The HeLa cell culture was then incubated at 37°C for 30 min, the supernatant removed and the cells overlaid with 2 ml agar containing Eagle's MEM. Three days later, cells were stained with 0.025% (w/v) neutral red. The virus titer was determined by plaque counting 3 h after staining. Mean and standard deviation were calculated from two independent experiments, each performed in duplicate.
Statistical analysis
To test for statistical significance, Student's t-test was applied. A value of P<0.05 was considered statistically significant.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft through grants SFB-TR-19/C5 to WP and HF and SFB-TR-19/C1 to JK. We thank Denise Werk for generating the shRNA expression cassette against hCAR.
Glossary
- AdV
adenoviral vector
- CAR
coxsackievirus–adenovirus-receptor
- CoxB3
coxsackievirus B3
- PNCMs
primary neonatal rat cardiomyocytes
- RNAi
RNA interference
- shCAR
shRNA against CAR
- shRNA
short hairpin RNA
- siRNA
short interfering RNA.
Accession codes
Accessions
GenBank/EMBL/DDBJ
Footnotes
H Fechner and S Pinkert: These authors contributed equally to this work.
References
- 1.Bowles NE, Richardson PJ, Olsen EG, Archard LC. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet. 1986;1:1120–1123. doi: 10.1016/S0140-6736(86)91837-4. [DOI] [PubMed] [Google Scholar]
- 2.Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R. Detection of viruses in myocardial tissues by polymerase chain reaction. Evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. 2003;42:466–472. doi: 10.1016/S0735-1097(03)00648-X. [DOI] [PubMed] [Google Scholar]
- 3.Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005;112:1965–1970. doi: 10.1161/CIRCULATIONAHA.105.548156. [DOI] [PubMed] [Google Scholar]
- 4.Poller W, Kuhl U, Tschoepe C, Pauschinger M, Fechner H, Schultheiss HP. Genome-environment interactions in the molecular pathogenesis of dilated cardiomyopathy. J Mol Med. 2005;83:579–586. doi: 10.1007/s00109-005-0664-2. [DOI] [PubMed] [Google Scholar]
- 5.Bitko V, Musiyenko A, Shulyayeva O, Barik S. Inhibition of respiratory viruses by nasally administered siRNA. Nat Med. 2005;11:50–55. doi: 10.1038/nm1164. [DOI] [PubMed] [Google Scholar]
- 6.McCaffrey AP, Nakai H, Pandey K, Huang Z, Salazar FH, Xu H. Inhibition of hepatitis B virus in mice by RNA interference. Nat Biotechnol. 2003;21:639–644. doi: 10.1038/nbt824. [DOI] [PubMed] [Google Scholar]
- 7.Carmona S, Ely A, Crowther C, Moolla N, Salazar FH, Marion PL. Effective inhibition of HBV replication in vivo by anti-HBx short hairpin RNAs. Mol Ther. 2006;13:411–421. doi: 10.1016/j.ymthe.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Takigawa Y, Nagano-Fujii M, Deng L, Hidajat R, Tanaka M, Mizuta H. Suppression of hepatitis C virus replicon by RNA interference directed against the NS3 and NS5B regions of the viral genome. Microbiol Immunol. 2004;48:591–598. doi: 10.1111/j.1348-0421.2004.tb03556.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen W, Liu M, Jiao Y, Yan W, Wei X, Chen J. Adenovirus-mediated RNA interference against foot-and-mouth disease virus infection both in vitro and in vivo. J Virol. 2006;80:3559–3566. doi: 10.1128/JVI.80.7.3559-3566.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon JS, Kim SH, Shin MC, Hong SK, Jung YT, Khang IG. Inhibition of herpesvirus-6B RNA replication by short interference RNAs. J Biochem Mol Biol. 2004;37:383–385. doi: 10.5483/bmbrep.2004.37.3.383. [DOI] [PubMed] [Google Scholar]
- 11.Wiebusch L, Truss M, Hagemeier C. Inhibition of human cytomegalovirus replication by small interfering RNAs. J Gen Virol. 2004;85:179–184. doi: 10.1099/vir.0.19453-0. [DOI] [PubMed] [Google Scholar]
- 12.Lu A, Zhang H, Zhang X, Wang H, Hu Q, Shen L. Attenuation of SARS coronavirus by a short hairpin RNA expression plasmid targeting RNA-dependent RNA polymerase. Virology. 2004;324:84–89. doi: 10.1016/j.virol.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park WS, Hayafune M, Miyano-Kurosaki N, Takaku H. Specific HIV-1 env gene silencing by small interfering RNAs in human peripheral blood mononuclear cells. Gene Therapy. 2003;10:2046–2050. doi: 10.1038/sj.gt.3302099. [DOI] [PubMed] [Google Scholar]
- 14.Merl S, Michaelis C, Jaschke B, Vorpahl M, Seidl S, Wessely R. Targeting 2A protease by RNA interference attenuates coxsackieviral cytopathogenicity and promotes survival in highly susceptible mice. Circulation. 2005;111:1583–1592. doi: 10.1161/01.CIR.0000160360.02040.AB. [DOI] [PubMed] [Google Scholar]
- 15.Werk D, Schubert S, Lindig V, Grunert HP, Zeichhardt H, Erdmann VA. Developing an effective RNA interference strategy against a plus-strand RNA virus: silencing of coxsackievirus B3 and its cognate coxsackievirus-adenovirus receptor. Biol Chem. 2005;386:857–863. doi: 10.1515/BC.2005.100. [DOI] [PubMed] [Google Scholar]
- 16.Sabariegos R, Gimenez-Barcons M, Tapia N, Clotet B, Martinez MA. Sequence homology required by human immunodeficiency virus type 1 to escape from short interfering RNAs. J Virol. 2006;80:571–577. doi: 10.1128/JVI.80.2.571-577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson JA, Richardson CD. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J Virol. 2005;79:7050–7058. doi: 10.1128/JVI.79.11.7050-7058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gitlin L, Stone JK, Andino R. Poliovirus escape from RNA interference: short interfering RNA-target recognition and implications for therapeutic approaches. J Virol. 2005;79:1027–1035. doi: 10.1128/JVI.79.2.1027-1035.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray JL, Mavrakis M, McDonald NJ, Yilla M, Sheng J, Bellini WJ. Rab9 GTPase is required for replication of human immunodeficiency virus type 1, filoviruses, and measles virus. J Virol. 2005;79:11742–11751. doi: 10.1128/JVI.79.18.11742-11751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson J, Akkina R. HIV-1 resistance conferred by siRNA cosuppression of CXCR4 and CCR5 coreceptors by a bispecific lentiviral vector. AIDS Res Ther. 2005;2:1. doi: 10.1186/1742-6405-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arrighi JF, Pion M, Wiznerowicz M, Geijtenbeek TB, Garcia E, Abraham S. Lentivirus-mediated RNA interference of DC-SIGN expression inhibits human immunodeficiency virus transmission from dendritic cells to T cells. J Virol. 2004;78:10848–10855. doi: 10.1128/JVI.78.20.10848-10855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao X, Wang H, Sairenji T. Inhibition of Epstein–Barr virus (EBV) reactivation by short interfering RNAs targeting p38 mitogen-activated protein kinase or c-myc in EBV-positive epithelial cells. J Virol. 2004;78:11798–11806. doi: 10.1128/JVI.78.21.11798-11806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ping YH, Chu CY, Cao H, Jacque JM, Stevenson M, Rana TM. Modulating HIV-1 replication by RNA interference directed against human transcription elongation factor SPT5. Retrovirology. 2004;1:46. doi: 10.1186/1742-4690-1-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kameoka M, Nukuzuma S, Itaya A, Tanaka Y, Ota K, Ikuta K. RNA interference directed against poly(ADP-ribose) polymerase 1 efficiently suppresses human immunodeficiency virus type 1 replication in human cells. J Virol. 2004;78:8931–8934. doi: 10.1128/JVI.78.16.8931-8934.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergelson JM, Krithivas A, Celi L, Droguett G, Horwitz MS, Wickham T. The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J Virol. 1998;72:415–419. doi: 10.1128/jvi.72.1.415-419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe A, Arai M, Yamazaki M, Koitabashi N, Wuytack F, Kurabayashi M. Phospholamban ablation by RNA interference increases Ca2+ uptake into rat cardiac myocyte sarcoplasmic reticulum. J Mol Cell Cardiol. 2004;37:691–698. doi: 10.1016/j.yjmcc.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Tomko RP, Xu R, Philipson L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA. 1997;94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 31.Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107:2793–2798. doi: 10.1161/01.CIR.0000072766.67150.51. [DOI] [PubMed] [Google Scholar]
- 32.Kandolf R. Virus etiology of inflammatory cardiomyopathy. Dtsch Med Wochenschr. 2004;129:2187–2192. doi: 10.1055/s-2004-831863. [DOI] [PubMed] [Google Scholar]
- 33.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 34.Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-E. [DOI] [PubMed] [Google Scholar]
- 35.Li E, Brown SL, Stupack DG, Puente XS, Cheresh DA, Nemerow GR. Integrin alpha(v)beta1 is an adenovirus coreceptor. J Virol. 2001;75:5405–5409. doi: 10.1128/JVI.75.11.5405-5409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong SS, Karayan L, Tournier J, Curiel DT, Boulanger PA. Adenovirus type 5 fiber knob binds to MHC class I alpha2 domain at the surface of human epithelial and B lymphoblastoid cells. EMBO J. 1997;16:2294–2306. doi: 10.1093/emboj/16.9.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dechecchi MC, Tamanini A, Bonizzato A, Cabrini G. Heparan sulfate glycosaminoglycans are involved in adenovirus type 5 and 2-host cell interactions. Virology. 2000;268:382–390. doi: 10.1006/viro.1999.0171. [DOI] [PubMed] [Google Scholar]
- 38.Fechner H, Haack A, Wang H, Wang X, Eizema K, Pauschinger M. Expression of Coxsackie adenovirus receptor and alphav-integrin does not correlate with adenovector targeting in vivo indicating anatomical vector barriers. Gene Therapy. 1999;6:1520–1535. doi: 10.1038/sj.gt.3301030. [DOI] [PubMed] [Google Scholar]
- 39.Yuan J, Cheung PK, Zhang HM, Chau D, Yang D. Inhibition of coxsackievirus B3 replication by small interfering RNAs requires perfect sequence match in the central region of the viral positive strand. J Virol. 2005;79:2151–2159. doi: 10.1128/JVI.79.4.2151-2159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fechner H, Suckau L, Kurreck J, Sipo I, Wang X, Pinkert S. Highly efficient and specific modulation of cardiac calcium homeostasis by adenovector-derived short hairpin RNA targeting phospholamban. Gene Therapy. 2007;14:211–218. doi: 10.1038/sj.gt.3302872. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda Y, Gu Y, Iwanaga Y, Hoshijima M, Oh SS, Giordano FJ. Restoration of deficient membrane proteins in the cardiomyopathic hamster by in vivo cardiac gene transfer. Circulation. 2002;105:502. doi: 10.1161/hc0402.102953. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 43.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- 44.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen CJ, Shieh JTC, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci USA. 2001;98:15191–15196. doi: 10.1073/pnas.261452898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I. The coxsackievirus–adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res Mol Brain Res. 2000;77:19–28. doi: 10.1016/S0169-328X(00)00036-X. [DOI] [PubMed] [Google Scholar]
- 47.Zen K, Liu Y, McCall IC, Wu T, Lee W, Babbin BA. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol Biol Cell. 2005;16:2694–2703. doi: 10.1091/mbc.e05-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw CA, Holland PC, Sinnreich M, Allen C, Sollerbrant K, Karpati G. Isoform-specific expression of the Coxsackie and adenovirus receptor (CAR) in neuromuscular junction and cardiac intercalated discs. BMC Cell Biol. 2004;5:42. doi: 10.1186/1471-2121-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Noutsias M, Fechner H, de Jonge H, Wang X, Dekkers D, Houtsmuller AB. Human coxsackie-adenovirus receptor is colocalized with integrins alpha(v)beta(3) and alpha(v)beta(5) on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy: implications for cardiotropic viral infections. Circulation. 2001;104:275–280. doi: 10.1161/01.CIR.104.3.275. [DOI] [PubMed] [Google Scholar]
- 50.Fechner H, Noutsias M, Tschoepe C, Hinze K, Wang X, Escher F. Induction of coxsackievirus-adenovirus-receptor expression during myocardial tissue formation and remodeling: identification of a cell-to-cell contact-dependent regulatory mechanism. Circulation. 2003;107:876–882. doi: 10.1161/01.CIR.0000050150.27478.C5. [DOI] [PubMed] [Google Scholar]
- 51.Chen JW, Zhou B, Yu QC, Shin SJ, Jiao K, Schneider MD. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ Res. 2006;98:923–930. doi: 10.1161/01.RES.0000218041.41932.e3. [DOI] [PubMed] [Google Scholar]
- 52.Dorner AA, Wegmann F, Butz S, Wolburg-Buchholz K, Wolburg H, Mack A. Coxsackievirus-adenovirus receptor (CAR) is essential for early embryonic cardiac development. J Cell Sci. 2005;118:3509–3521. doi: 10.1242/jcs.02476. [DOI] [PubMed] [Google Scholar]
- 53.Fechner H, Wang X, Wang H, Jansen A, Pauschinger M, Scherubl H. Trans-complementation of vector replication versus Coxsackie–adenovirus-receptor overexpression to improve transgene expression in poorly permissive cancer cells. Gene Therapy. 2000;7:1954–1968. doi: 10.1038/sj.gt.3301321. [DOI] [PubMed] [Google Scholar]
- 54.Marienfeld U, Haack A, Thalheimer P, Schneider-Rasp S, Brackmann HH, Poller W. ‘Autoreplication’ of the vector genome in recombinant adenoviral vectors with different E1 region deletions and transgenes. Gene Therapy. 1999;6:1101–1113. doi: 10.1038/sj.gt.3300928. [DOI] [PubMed] [Google Scholar]
- 55.Sipo I, Hurtado Pico A, Wang X, Eberle J, Petersen I, Weger S. An improved Tet-On regulatable FasL-adenovirus vector system for lung cancer therapy. J Mol Med. 2006;84:215–225. doi: 10.1007/s00109-005-0009-1. [DOI] [PubMed] [Google Scholar]
- 56.Sollerbrant K, Raschperger E, Mirza M, Engstrom U, Philipson L, Ljungdahl PO. The Coxsackievirus and adenovirus receptor (CAR) forms a complex with the PDZ domain-containing protein ligand-of-numb protein-X (LNX) J Biol Chem. 2003;278:7439–7444. doi: 10.1074/jbc.M205927200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.