

Abstract
A large number of adenoviral agents are being developed for the treatment of cancer. However, the treatment-related death of a patient with ornithine transcarbamylase deficiency following adenovirus administration by hepatic artery has led to serious concerns regarding the safety of intravascular adenovirus. Both replication-incompetent (rAd.p53, e.g., SCH58500) and replication-selective (dl1520, aka Onyx-015; CG7870) oncolytic adenoviruses, by intravascular administration, are in clinical trials. We review Phases I and I/II results from these clinical trials. dl1520 and rAd.p53 were well-tolerated following hepatic artery infusion at doses of up to 2×1012 and 2.5×1013 particles, respectively. At a dose of 7.5×1013 particles, rAd.p53 was associated with dose-limiting cardiac output suppression; dl1520 dose escalation did not proceed higher than 2×1012. Intravenous (i.v.) infusions of dl1520 and CG7870 have been well tolerated by i.v. infusion at doses of 2×1013 and 6×1012, respectively, without identification of a maximally tolerated dose to date. Mild/moderate transaminitis was demonstrated in some patients on both the hepatic arterial and i.v. trials at doses ≥1012 particles. Interleukin (IL)-6 and IL-10 were induced in a dose-dependent manner in most patients, but significant interpatient and intrapatient (on repeat doses) variabilities were demonstrated. Evidence of p53 gene expression (Ad.p53) or viral replication (dl1520) was demonstrated in the majority of patients receiving ≥1012 particles. Over 100 cancer patients have been treated with intravascular adenovirus constructs to date with an acceptable toxicity profile; further clinical trial testing appears appropriate in cancer patients.
Keywords: adenovirus, oncolytic virus, gene therapy, clinical trial, cancer
Main
Adenoviruses have been developed as cancer therapeutics using two major strategies. Initially, mutants with deletions in one or more critical viral genes (e.g., E1, E4) were constructed as replication-incompetent vehicles to deliver therapeutic genes including tumor-suppressor genes, prodrug-activating enzymes, or cytokines.1,2,3 Adenoviruses expressing p53 were demonstrated to have selective antitumoral effects, including cell cycle arrest and apoptosis. Of note, replacement of p53 alone was enough to kill tumor cells with a great number of genetic alterations. Subsequently, adenoviruses were engineered to replicate selectively within cancers.4 The original approach proposed by McCormick was to delete the E1B–55 kDa gene.5 This gene product was known to bind p53 (in complex with E4orf6), leading to p53 inhibition and/or degradation. The theory was that E1B–55 kDa–deleted adenoviruses such as dl1520 (later designated Onyx-015) would not be able to inactivate p53 in normal cells and, as a result, the viral replication cycle would not be completed. In cancer cells lacking normal p53 function, however, replication was predicted to proceed. Subsequently, other gene deletion mutants were evaluated to target tumor cells lacking G1–S checkpoint control and/or loss of pRB function.6,7 A subsequent approach to emerge was construction of replication-selective adenoviruses by placing the expression of E1A under control of tumor/tissue–specific promoters.8,9,10 This approach has been used with PSA, E2F, AFP, and other promoters.
Whereas these approaches both held promise, the initial clinical trials with these agents were limited to cancers for which intratumoral (i.t.) injection was feasible in order to maximize safety. Head and neck cancers11 and localized lung3 or prostate12 cancers were evaluated extensively. Once safety and gene expression or replication was demonstrated with i.t. injection, e.g., with dl1520, intraperitoneal (i.p.) injections were performed in patients with ovarian carcinoma.13 After safety and feasibility of i.p. delivery to ovarian cancer patients were demonstrated, intravascular administration was contemplated. However, the feasibility of vascular delivery to tumors in the face of innate immunity and neutralizing antibodies was unknown. The dose-limiting toxicity in mice was typically hepatotoxicity with adenovirus vectors.14 Finally, the tragic death of a patient on an adenoviral gene therapy trial for ornithine transcarbamylase (OTC) deficiency at the University of Pennsylvania eventually raised serious concerns about the safety of intravascular adenovirus administration and led to numerous trials in the US and Europe being put on hold.15,16,17,18 We summarize publicly disclosed and/or published data on clinical trials of intravascular adenovirus in cancer patients.
Therapeutic adenoviruses
dl1520 (aka Onyx-015)
dl1520 (Onyx-015) is a first-generation replication-selective adenovirus type 2/5 chimera with a deletion in the E1B–55 kDa gene,19 as well as the E3 10.4/14.5 and 14.7 genes. The virus contains a deletion between nucleotides 2496 and 3323 in the E1B region encoding the 55-kDa protein. In addition, a C-to-T transition at position 2022 in E1B generates a stop codon at the third codon position of the protein. These alterations eliminate expression of the E1B–55 kDa gene in Onyx-015–infected cells. Because E1B–55 kDa binds to and inactivates the p53 tumor-suppressor gene product, this mutant should theoretically be unable to overcome the p53-mediated blockade of viral replication in a normal cell.20 In a tumor cell lacking p53 function, in contrast, the E1B–55 kDa protein should be expendable for p53 inhibition and replication should proceed.21 Initial studies demonstrated p53-dependent selectivity in matched cells with and without dominant-negative p53. Normal cells were subsequently shown to be relatively resistant to the effects of the virus. Publications from preclinical studies with different cell systems and/or endpoints have reported both supportive data regarding the original McCormick hypothesis and data that seemed to contradict it.20,21,22,23,24,25 At least some of the confusion can be attributed to the fact that many tumor cell lines with wild-type p53 genomes actually do not have normal p53 function.26 For example, loss of p14arf can lead to loss of p53 induction; replacement of p14arf was able to restore resistance to the virus in one cell line.
Onyx-015 has shown promise in Phases I and II clinical trials following direct i.t. injection into recurrent head and neck cancers.11,27,28 Tumor-selective viral replication and necrosis were demonstrated, and the treatment was well tolerated without dose-limiting toxicities; flu-like symptoms and injection site pain were frequently noted. Although durable responses were rare as a single agent in these advanced refractory tumors,11 a potentially synergistic interaction was subsequently discovered between Onyx-015 and chemotherapy.29,30,31,32 Further development of Onyx-015 was, therefore, indicated in combination with chemotherapy. Intravenous (i.v.) administration of Onyx-015 in nude mouse–human tumor xenograft models led to infection and growth inhibition of distant tumors in a dose-dependent fashion.14
SCH58500 (rAd.p53)
This replication-deficient type 5 adenovirus has a deletion in the critical E1A gene and encodes the entire human p53 gene under control of the human cytomegalovirus (CMV) immediate-early promoter. Preclinical studies reported dose-dependent, p53-mediated tumor suppression in various mouse tumor models.33,34 Early Phases I and II clinical trials demonstrated the safety and feasibility of delivery by i.t. injection.35
CG7870 (formerly CV787)
This replication-selective adenovirus is engineered for prostate cell selectivity in the following manner. The E1A gene was placed under control of the rat probasin promoter, whereas E1B was under control of the PSA promoter.10 Unlike other first-generation adenoviruses,36,37 the entire E3 region was reinserted into this virus. This virus was initially tested by i.t. administration into locally recurrent prostate carcinomas at doses of 1012–1013 particle units (T DeWeese, personal communication). The intraprostatic virus injections were well tolerated, although local injection site pain and bleeding were noted (T DeWeese, personal communication).
Objectives
The primary objectives of these studies were to determine the safety and maximally tolerated dose of single or repeated administrations of these agents by hepatic artery infusion (h.a.i.) or i.v. infusions, alone and/or in combination with chemotherapy. In addition, the pharmacokinetic profiles, the humoral and cytokine immune responses, the feasibility of delivery to tumors and antitumoral efficacy was assessed.
Treatment schedule: dl1520 (Onyx-015), h.a.i.
Onyx-015 administered through the hepatic artery was determined following single infusions on days 1 and 8 (cycles 1 and 2)38 (Table 1). Starting on day 22, treatment cycles were 28 days and consisted of Onyx-015 infusions followed by i.v. chemotherapy within 6 hours following virus infusion; leucovorin 20 mg/m2, i.v. was followed by 5-FU 425 mg/m2/day, i.v. bolus. After completion of cycle 4, up to four additional cycles were optional based on toxicity and tumor response. Viral doses were escalated in half-log increments from 2×109 to 2×1012 particles. Phase II dosing was at the top dose level.39
Table 1.
Trial design and toxicity data from clinical trials of oncolytic adenoviruses (dl1520/Onyx-015; CG7870) administered by intravascular infusion

Treatment schedule: dl1520 (Onyx-015), i.v.
Onyx-015 was administered i.v. on a weekly basis for three consecutive weeks followed by a 1-week rest (Table 1).40 Following two such cycles, Onyx-015 infusions were followed by i.v. paclitaxel and carboplatin on a weekly basis. Viral doses were escalated incrementally from 2×1010 to 2×1013 particles; three patients were treated at the 2×1012 dose level but only one at the 6×1012 and 2×1013 dose levels.
Treatment schedule: CG7870, i.v.
CG7870 was administered i.v. on day 1 only (Table 1). Viral doses were escalated incrementally from 1010 to 6×1012 particles; dose escalation has not yet been completed in this study.
Treatment schedule: rAd.p53 (SCH58500), h.a.i.
In the first study, the virus was administered through the right hepatic artery as a single infusion on day 1 (Table 2). Doses were escalated between patients in half-log increments (nine dose levels) at doses of viral doses were escalated in half-log increments from 7.5×109 to 7.5×1013 particles. In a subsequent trial, repeat dosing (daily ×5, repeated every 4 weeks) was explored at doses of 7.5×1011–2.5×1013 particles. Finally, a randomized Phase II trial was initiated to compare h.a.i. FUDR (days 114, repeated every 4 weeks) plus/minus h.a.i. SCH58500 (day 15). This study was put on hold for nonmedical reasons after seven patients were treated.
Table 2.
Trial design and toxicity data from clinical trials of Adp53 (SCH58500) administered by intravascular infusion
Results
Viral pharmacokinetics post-h.a.i.: peripheral and hepatic venous blood
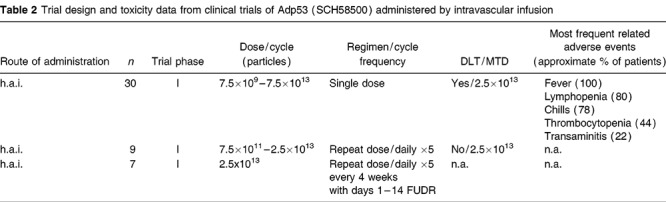
Quantitative PCR testing of the peripheral venous blood for Onyx-015 was performed at predetermined timepoints over the first 6 hours after injection on cycles 1 and 3 at the highest dose level (Fig 1). The virus was rapidly cleared from the blood over 6 hours. The pharmacokinetic parameters were nearly identical during cycles 1 and 3 (following high-level antibody titer increases): t1/2α (10 vs 14 minutes, respectively) and t1/2β (113 vs 135 minutes, respectively) were, therefore, not demonstrably affected by neutralizing antibody levels.
Figure 1.
Pharmacokinetics of Onyx-015 following h.a.i. The first five patients treated at the 2×1012 particles dose level had pharmacokinetic blood draws taken on cycle 1 (day 1) at the following timepoints after infusion: 5, 10, 30, 60, 90, 120, 180, and 360 minutes. Plasma was tested for virus by quantitative PCR.
SCH58500 levels were measured in the hepatic vein by Q-PCR during and immediately following the infusion of virus. Samples were positive only following doses of 2.5×1011 particles or higher. Peripheral venous levels were lower and more variable, but at 2×1013 particles, all patients were positive during and/or immediately following the infusion.
Onyx-015, h.a.i.: adverse events and maximum feasible dose
Dose escalation proceeded from 2×108 to 2×1012 particles without occurrence of any dose-limiting toxicities (Table 1). Specifically, no treatment-emergent clinical hepatotoxicity occurred during dose escalation, despite preexisting liver abnormalities due to intrahepatic metastases in over half of the patients at baseline. Table 1 describes the most common Onyx-015–related adverse events. Nearly all patients reported flu-like symptoms, including fever, myalgias, asthenia, and/or chills. Chills, myalgias, and flu-like symptoms were mild to moderate (grades 1 and 2) in most cases, and the duration of these symptoms was typically short (<48 hours). No patients discontinued therapy on the basis of flu-like symptoms. Transient, dose-related suppression of lymphocytes was demonstrated. These decreases resolved in less than 7 days and were not clinically significant. Transaminitis was experienced by approximately one-third of patients at the highest dose level but was generally mild–moderate and resolved within less than 10 days. Cycles 3 and above were administered with chemotherapy, and therefore, the chemotherapy-related toxicities of leukopenia and mucositis were more common during these cycles. It did not appear from this study that Onyx-015 administration worsened the toxicity associated with chemotherapy. In summary, Onyx-015 was well tolerated both as a single agent by h.a.i., and in combination with i.v. 5-FU and leucovorin, at doses of up to 2×1012 particles.
Onyx-015, i.v.: adverse events and maximum feasible dose
Dose escalation proceeded from 2×1010 to 2×1013 particles without occurrence of any dose-limiting toxicities (Table 1). All patients reported flu-like symptoms, including fever, myalgias, asthenia, and/or chills. Chills, myalgias, and flu-like symptoms were mild to moderate (grades 1 and 2) in most cases, and the duration of these symptoms was typically short (<48 hours). No patients discontinued therapy on the basis of flu-like symptoms. Transaminitis, experienced by most patients receiving doses of ≥2×1012 particles, was generally mild–moderate and resolved within 10–14 days. Cycles 3 and above were administered with carboplatin/paclitaxel chemotherapy. It did not appear from this study that Onyx-015 administration worsened the toxicity associated with chemotherapy. In summary, i.v. Onyx-015 was well tolerated both as a single agent and in combination with chemotherapy at doses of up to 2×1013 particles. However, a very small number of patients have been treated at doses ≥2×1012 particles, so additional safety data at these doses are desirable.
CG7870, i.v.: adverse events and maximum feasible dose
This study has not yet been completed. Preliminary data were presented at the American Society for Gene Therapy Conference (Boston, MA, 2002). Dose escalation proceeded from 1010 to 6×1012 particles without occurrence of any dose-limiting toxicities (n=23 to date; Table 1). Chills, myalgias, and flu-like symptoms were reportedly common. Transient, mild-to-moderate transaminitis was reported in most patients receiving doses of ≥3×1012 particles. Final data from this trial are awaited.
SCH58500, h.a.i.: adverse events and maximally tolerated dose
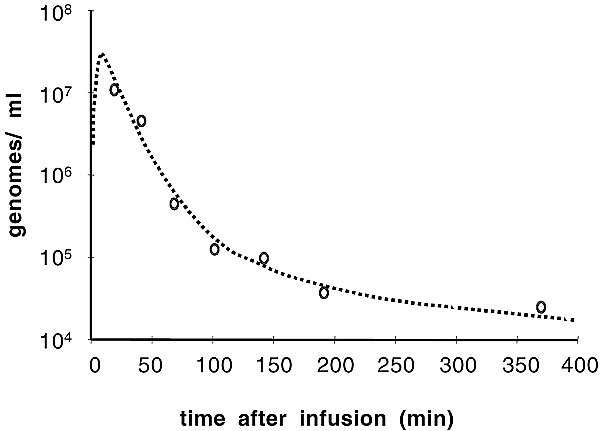
Dose escalation proceeded through 2.5×1013 particles (i.e., 10-fold higher than the top dose with Onyx-015) without occurrence of any dose-limiting toxicities. At 7.5×1013 particles, however, transient dose-limiting cardiac output suppression and hypotension occurred (Table 2); as this toxicity was seen in preclinical models, the patient had an indwelling right heart catheter in place at the time. Therefore, 2.5×1013 particles was the maximum tolerated dose (MTD). No treatment-emergent clinical hepatotoxicity occurred during dose escalation. Table 2 describes the most common SCH58500-related adverse events. These toxicities were remarkably similar to those reported with Onyx-015 and, therefore, these tolerable toxicities appear to be due to adenovirus itself rather than the transgene. Transient, dose-related suppression of lymphocytes, monocytes, eosinophils, and platelets was demonstrated (Fig 2A), generally within the first 24–48 hours. These decreases resolved in less than 7 days and were not clinically significant. Associated increases in fibrin degradation products and increases in PT/PTT were demonstrated within a similar timeframe but were not clinically significant; fibrinogen levels actually increased. Transient transaminitis was common at the highest dose levels as well (Fig 2B), resolving within 10 days. In summary, SCH58500 was well tolerated by h.a.i., both with single and repeated doses, at doses of up to 2.5×1013 particles.
Figure 2.
Laboratory changes versus dose following h.a.i. of SCH58500 rAd.p53. The maximum nadir following treatment was determined and related to baseline values for blood counts (Panel A) and transaminases (Panel B). Blood counts generally dropped within 72 hours and transaminases increased within 5 days. Laboratory abnormalities generally returned to normal within 10 days.
Immune response to intravascular adenovirus
Neutralizing antibody titers to Onyx-015 (Ad5 protein coat) were positive in approximately 50% of patients prior to treatment (antibody positivity was an enrollment criterion for the SCH58500 trials). Titers increased and/or became positive in all patients following intravascular administration with Onyx-015, regardless of viral dose; the median antibody titer after a single cycle was approximately 1:10,000. Titers continued to rise after a second cycle of treatment, as well. Similar antibody increases were seen in patients on SCH58500 trials. In neither trial did antibodies prevent biological activity (i.e., replication/shedding or gene expression).
Evidence for adenovirus replication and/or transgene expression
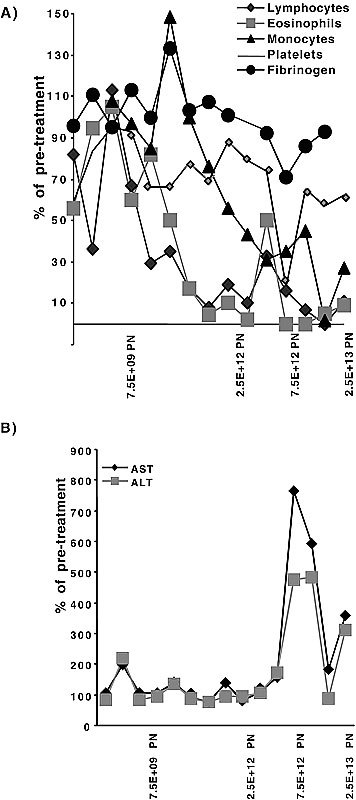
Following h.a.i. of Onyx-015, quantitative PCR of the blood was performed on day 4 (±1) to assess viral replication and shedding from tumor tissue; this was possible because input virus was rapidly cleared within 6 hours after the infusion to levels at or below the level of detection (see representative curve in Fig 3). No patients had positive blood results at doses of <2×1011 particles.38 In contrast, approximately half of all patients treated at higher doses (≥2×1011 particles) had detectable virus in day 4 blood samples following treatment (Fig 4A).38 Similar results were obtained following i.v. administration; three of four patients treated with ≥2×1011 particles had viremia on day 4 posttreatment.40
Figure 3.
Evidence for viral replication and shedding into the blood. Quantitative PCR of the blood for detection of Onyx-015 was performed on this patient daily posttreatment. Since Onyx-015 was cleared from the blood within the first 6 hours after infusion, the presence of virus at later timepoints was indicative of ongoing viral replication and shedding into the blood.
Figure 4.
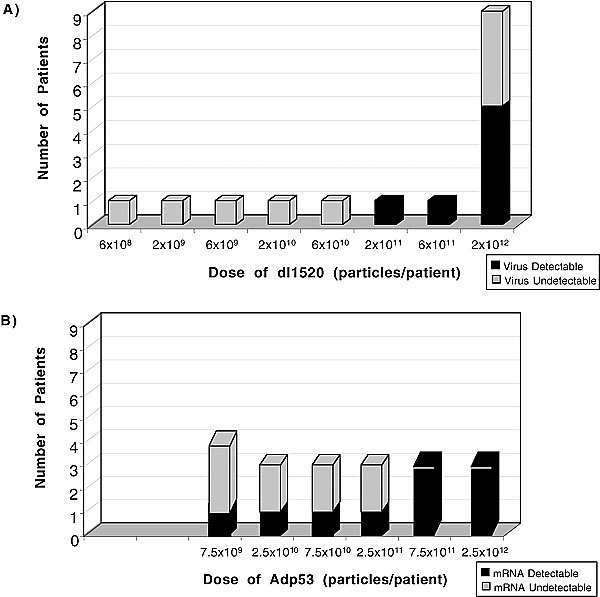
Evidence for viral infection in cancer patients: correlation with viral dose. Onyx-015 replication postinfection was measured on day 4 indirectly as described in the legend (Panel A). SCH58500 rAd.p53 gene expression was measured using RT-PCR on biopsies obtained at laparotomy 2–7 days post-HAI (Panel B). The dose at which biological activity was reproducibly seen was similar in the two trials.
Following h.a.i. of SCH58500, the majority of tumor samples taken posttreatment (days 2–7, postcycle 1) were positive for gene expression from the virus, especially at doses ≥2.5×1011 particles (Fig 4B). Therefore, a similar dose threshold was seen for both viruses, above which the majority of tumors appeared to be infected.
Summary
Both replication-incompetent (rAd.p53, aka SCH58500) and replication-selective (dl1520, aka Onyx-015; CG7870) adenoviruses are being developed for the treatment of p53-deficient cancers. Intravascular administration has either been through the i.v. route or the hepatic arterial route (h.a.i.). H.a.i. has historically been used to selectively target primary or metastatic tumors within the liver, and therefore, regional therapy with adenovirus in this setting is an attractive approach. Phases I and II trials were performed with dl1520 and rAd.p53 viruses administered by h.a.i. The MTD of rAd.p53 was 2.5×1013; cardiac output suppression was the dose-limiting toxicity at 7.5×1013 particles. No maximally tolerated dose or treatment-emergent clinical hepatotoxicity was identified following dl1520 infusion at doses up to 2×1012 (i.e., Ad.p53 was tested at 25-fold higher doses).38 Mild to moderate fever, rigors, and transaminitis were the most common adverse events.39 Evidence of p53 gene expression (Ad.p53) or viral replication (dl1520) was demonstrated in the majority of patients receiving ≥6×1011 particles. Similar safety has been seen to date in trials of dl1520 or CG7870 administered by i.v. routes to patients with metastatic carcinoma at doses up to approximately 2×1013 or 6×1012, respectively.40 Of particular note given the underlying liver pathology in hepatocellular carcinoma patients, another Phase I trial of rAd.p53 was performed in these patients at doses up to 7.5×1012 (n=8, half with viral hepatitis; Robert Warren, unpublished data). Acute inflammatory cytokine induction occurred within 3–6 hours postinfusion (e.g., interleukin (IL)-6; IFN-γ; tumor necrosis factor, TNF) in a dose-dependent fashion. IL-10 induction generally occurred after IL-6 induction (e.g., 18–24 hours). However, the levels and patterns of cytokine induction varied greatly between patients; of note, the mild and clinically insignificant consumptive coagulopathy demonstrated with rAd.p53 was similar to that seen in previous trials with TNF protein.41 Blood tests were not routinely performed during the first 48 hours post-dl1520 and, therefore, it remains unknown whether these coagulation changes occur following h.a.i. with this virus. Overall, therefore, the safety and feasibility of delivery with these two adenoviruses were very similar in these clinical trials. Neutralizing antibodies did not prevent infection of tumors in any trial. Additional studies of h.a.i. adenoviruses are indicated.
Serious concerns were raised about the safety of intravascular adenovirus following the patient death on a clinical trial for patients with OTC deficiency at the University of Pennsylvania.15,16,17,18 This patient received a dose of approximately 4×1013 particles with a replication-deficient adenovirus expressing the OTC gene. By report, in less than 24 hours, the patient experienced hyperammonemia, adult respiratory distress syndrome (ARDS), and disseminated intravascular coagulation; this was followed over the next few days by multiorgan system failure and death.15 On the trials reported here these severe complications were not reported. Several factors may account for these apparent differences in safety. One plausible explanation is that the patient populations enrolled onto these two trials differed in their sensitivity to viral infection and the ensuing systemic inflammatory response. Patients with OTC deficiency have a well documented heightened sensitivity to viral and bacterial infections compared to the general population, and that hyperammonemia, ARDS, and death can result.42 Unfortunately, the OTC patient reportedly had an elevated blood ammonia level prior to treatment on the trial. This sort of metabolic stress may not be tolerated in OTC patients. Finally, it is theoretically possible that differences in the virus preparations themselves may have contributed, although the protein coats of the viral particles were identical on each study. Based on these studies, it is clear that adenoviruses, including replication-selective adenoviruses, can be well tolerated following administration into the bloodstream at doses that result in gene transduction and/or replication.
Future directions: replication-selective virotherapy
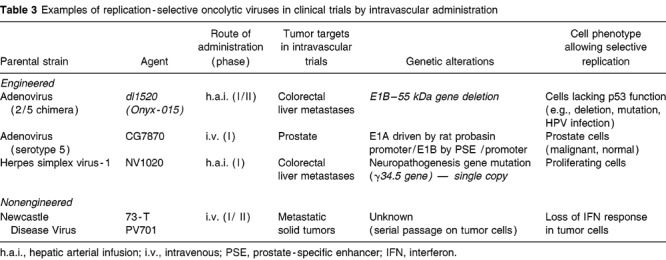
The findings from these studies have implications for the fields of viral and gene therapies. Arterial and i.v. deliveries are now being explored with several virotherapy agents in clinical trials (Table 3). This opens up the possibility of treating a wide range of tumors using this approach. The chemosensitization demonstrated by Onyx-015 on trials of head and neck cancer patients29 has now been shown with another tumor type and another route of administration.39 Other replication-selective viruses also appear to be potentially synergistic with chemotherapy.43,44 Therefore, i.v. virotherapy may be able to improve metastatic tumor response rates to chemotherapy agents. An oncolytic herpesvirus mutant, NV1020, is now being administered by h.a.i. in a Phase I clinical trial (Y Fong, R Martuza, personal communication). Reovirus has demonstrated i.v. efficacy in immunodeficient murine tumor models.45 Finally, Newcastle Disease Virus (PV701) has been tested by i.v. infusion in 79 patients.46
Table 3.
Examples of replication-selective oncolytic viruses in clinical trials by intravascular administration
The field of replication-selective virotherapy has expanded dramatically over the past 5 years.4 Viruses have evolved to dramatically alter the phenotype of the infected cell to maximize their replication and survival. The cellular changes induced by viral infection are often strikingly similar to the cellular changes acquired during carcinogenesis (e.g., p53 tumor-suppressor protein inactivation, inhibition of apoptosis). Given this genetic convergence, it is not surprising that many viruses inherently grow preferentially in tumor cells and/or that viruses can be engineered for tumor selectivity. Five general mechanistic approaches to tumor-selective replication have been described to date. These include (a) the use of viruses with inherent tumor selectivity (e.g., NDV, reovirus, VSV, autonomous parvovirus);47 (b) deletion of entire genes (e.g., HSV, adenovirus)48 or (c) functional gene regions (e.g., adenovirus, poliovirus)6 that are necessary for efficient replication and/or toxicity in normal cells but are expendable in tumor cells; (d) engineering of tumor/tissue–specific promoters into viruses to limit expression of gene(s) necessary for replication to cancer cells (e.g., adenovirus, HSV);8 and (e) modification of the viral coat to target uptake selectively to tumor cells (e.g., adenovirus, poliovirus).49,50
Several encouraging strategies are being explored to improve the potential utility of these agents. First, because replication-selective viral treatment should not lead to cross-resistance with standard therapies, combinations with radiotherapy and chemotherapy may lead to additive or synergistic efficacy;25,29 viral replication does not appear to be completely blocked by these agents. Endogenous viral gene expression can be modified to enhance antitumoral potency; examples with adenovirus include reintroduction or overexpression of the adenovirus death protein,51 deletion of the E1B–19 kDa gene, or deletion of the E1A CR2 region.6 Viral replication within tumors can lead to induction of cytokines with antitumoral and antivascular properties, as well as tumor-specific CTL. Viruses can be “armed” to express exogenous therapeutic genes including cytokines or prodrug-activating enzymes.52 Finally, strategies to immunomodulate the host are being explored. For example, antibody clearance from the blood and complement inhibition are strategies that have been used in murine tumor models with adenoviruses and HSV, respectively.
Acknowledgements
We thank the following individuals for their contributions to these clinical trials: Jim Abbruzzese, Joe Rubin, Eva Galanis, and Alan Venook.
References
- 1.McCormick F. Cancer gene therapy: fringe or cutting edge? Nat Rev Cancer. 2001;1:130–141. doi: 10.1038/35101008. [DOI] [PubMed] [Google Scholar]
- 2.Kozarsky KF, Wilson JM. Gene therapy: adenovirus vectors. Curr Opin Genet Dev. 1993;3:499–503. doi: 10.1016/0959-437X(93)90126-A. [DOI] [PubMed] [Google Scholar]
- 3.Roth J, Cristiano RJ. Gene therapy for cancer: what have we done and where are we going? J Natl Cancer Inst. 1997;89:21–39. doi: 10.1093/jnci/89.1.21. [DOI] [PubMed] [Google Scholar]
- 4.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 5.Bischoff JR, Kirn DH, Williams A. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 6.Heise C, Hermiston T, Johnson L. An adenovirus E1A mutant that demonstrates potent and selective antitumoral efficacy. Nat Med. 2000;6:1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 7.Fueyo J, Gomez-Manzano C, Alemany R. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 8.Hallenbeck PL, Chang YN, Hay C. A novel tumor-specific replication-restricted adenoviral vector for gene therapy of hepatocellular carcinoma. Hum Gene Ther. 1999;10:1721–1733. doi: 10.1089/10430349950017725. [DOI] [PubMed] [Google Scholar]
- 9.Yu D, Sakamoto G, Henderson DR. Identification of the transcriptional regulatory sequences of human kallikrein 2 and their use in the construction of calydon virus 764, an attenuated replication competent adenovirus for prostate cancer therapy. Cancer Res. 1999;59:1498–1504. [PubMed] [Google Scholar]
- 10.Yu D, Chen Y, Seng M, Dilley J, Henderson DR. The addition of adenovirus region E3 enables calydon virus 787 to eliminate distant prostate tumor xenografts. Cancer Res. 1999;59:4200–4203. [PubMed] [Google Scholar]
- 11.Nemunaitis J, Ganly I, Khuri F. Selective replication and oncolysis in p53 mutant tumors with Onyx-015, an E1B–55 kD gene–deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 2000;60:6359–6366. [PubMed] [Google Scholar]
- 12.DeWeese T, Van der Poel H, Li S. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001;61:7464–7472. [PubMed] [Google Scholar]
- 13.Vasey P, Shulman L, Gore M, Kirn D, Kaye S. Phase I trial of intraperitoneal Onyx-015 adenovirus in patients with recurrent ovarian carcinoma. Proc Am Soc Clin Oncol. 2000;19:1512. [Google Scholar]
- 14.Heise C, Williams A, Xue S, Propst M, Kirn D. Intravenous administration of ONYX-015, a selectively-replicating adenovirus, induces antitumoral efficacy. Cancer Res. 1999;59:2623–2628. [PubMed] [Google Scholar]
- 15.Marshall E. Clinical trials: gene therapy death prompts review of adenovirus vector. Science. 1999;286:2244–2245. doi: 10.1126/science.286.5448.2244. [DOI] [PubMed] [Google Scholar]
- 16.Beardsley T. Gene therapy setback. Sci Am. 2000;282:36–37. doi: 10.1038/scientificamerican0200-36. [DOI] [PubMed] [Google Scholar]
- 17.Jenks S. Gene therapy death — everyone has to share in the guilt. J Natl Cancer Inst. 2000;92:98–100. doi: 10.1093/jnci/92.2.98. [DOI] [PubMed] [Google Scholar]
- 18.Miller H. Letter to the editor. Science. 2000;287:591. doi: 10.1126/science.287.5453.591c. [DOI] [Google Scholar]
- 19.Barker DD, Berk AJ. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology. 1987;156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- 20.Bischoff JR, Kirn DH, Williams A. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells [see comments] Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 21.Heise C, Sampson JA, Williams A, McCormick F, Von HD, Kirn DH. ONYX-015, an E1B gene–attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents [see comments] Nat Med. 1997;3:639–645. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 22.Harada J, Berk A. p53-independent and -dependent requirements for E1B–55 kD in adenovirus type 5 replication. J Virol. 1999;73:5333–5344. doi: 10.1128/jvi.73.7.5333-5344.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M, zur Hausen H. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J Virol. 1998;72:9470–9478. doi: 10.1128/jvi.72.12.9470-9478.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodrum FD, Ornelles DA. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J Virol. 1998;72:9479–9490. doi: 10.1128/jvi.72.12.9479-9490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogulski K, Freytag S, Zhang K. In vivo antitumor activity of ONYX-015 is influenced by p53 status and is augmented by radiotherapy. Cancer Res. 2000;60:1193–1196. [PubMed] [Google Scholar]
- 26.Ries SJ, Brandts CH, Chung AS. Loss of p14ARF in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015) Nat Med. 2000;6:1128–1133. doi: 10.1038/80466. [DOI] [PubMed] [Google Scholar]
- 27.Kirn D, Hermiston T, McCormick F. ONYX-015: clinical data are encouraging. Nat Med. 1998;4:1341–1342. doi: 10.1038/3902. [DOI] [PubMed] [Google Scholar]
- 28.Kirn D, Nemunaitis J, Ganly I. A phase II trial of intratumoral injection with an E1B-deleted adenovirus, ONYX-015, in patients with recurrent, refractory head and neck cancer. Proc Am Soc Clin Oncol. 1998;17:391a. [Google Scholar]
- 29.Khuri F, Nemunaitis J, Ganly I. A controlled trial of Onyx-015, an E1B gene–deleted adenovirus, in combination with chemotherapy in patients with recurrent head and neck cancer. Nat Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 30.Kirn DH, Khuri F, Ganly I. A phase II trial of ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Proc Am Soc Clin Oncol. 1999;18:1505. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 31.Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene–attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents [see comments] Nat Med. 1997;3:639–645. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 32.Reid A, Galanis E, Abbruzzese J, Romel L, Rubin J, Kirn D . A phase I/II trial of ONYX-015 administered by hepatic artery infusion to patients with colorectal carcinoma EORTC-NCI-AACR Meeting on Molecular Therapeutics of Cancer 1999
- 33.Wills KN, Maneval DC, Menzel P. Development and characterization of recombinant adenoviruses encoding human p53 for gene therapy of cancer. Hum Gene Ther. 1994;5:1079–1088. doi: 10.1089/hum.1994.5.9-1079. [DOI] [PubMed] [Google Scholar]
- 34.Mujoo K, Maneval D, Anderson S, Gutterman J. Adenoviral-mediated p53 tumor suppressor gene therapy of human ovarian carcinoma. Oncogene. 1996;12:1617–1623. [PubMed] [Google Scholar]
- 35.Nemunaitis J, Swisher SG, Timmons T. Adenovirus-mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non-small-cell lung cancer. J Clin Oncol. 2000;18:609–622. doi: 10.1200/JCO.2000.18.3.609. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez R, Schuur ER, Lim HY, Henderson GA, Simons JW, Henderson DR. Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997;57:2559–2563. [PubMed] [Google Scholar]
- 37.Barker DD, Berk AJ. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology. 1987;156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- 38.Reid T, Galanis E, Abbruzzese J. Intra-arterial administration of a replication-selective adenovirus (dl1520) in patients with colorectal carcinoma metastatic to the liver: a phase I trial. Gene Ther. 2001;8:1618–1626. doi: 10.1038/sj.gt.3301512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reid T, Galanis E, Abbruzzese J. Hepatic artery infusion of Onyx-015, a replication-selective adenovirus, in combination with 5-FU/leucovorin for gastrointestinal carcinoma metastatic to the liver: a phase I/II clinical trial. Proc Am Soc Clin Oncol. 2000;19:953. [Google Scholar]
- 40.Nemunaitis J, Cunningham C, Buchanan A. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001;8:746–759. doi: 10.1038/sj.gt.3301424. [DOI] [PubMed] [Google Scholar]
- 41.Alexander HR, Bartlett DL, Libutti SK, Fraker DL, Moser T, Rosenberg SA. Isolated hepatic perfusion with tumor necrosis factor and melphalan for unresectable cancers confined to the liver. J Clin Oncol. 1998;16:1479–1489. doi: 10.1200/JCO.1998.16.4.1479. [DOI] [PubMed] [Google Scholar]
- 42.Brusilow S, Maestri N. Urea cycle disorders: diagnosis, pathophysiology and therapy. Adv Pediatr. 1996;43:127–170. [PubMed] [Google Scholar]
- 43.Yu D, Chen Y, Dilley J. Antitumor synergy of CN787, a prostate cancer–specific adenovirus, and paclitaxel and docetaxel. Cancer Res. 2001;61:517–525. [PubMed] [Google Scholar]
- 44.Chalavi A, Todo T, Martuza R, Rabkin S. Replication-competent herpes simplex vector G207/cisplatin combination therapy for head and neck squamous cell carcinoma. Neoplasia. 1999;1:162–169. doi: 10.1038/sj.neo.7900016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norman K, Lee P. Reovirus as a novel oncolytic agent. J Clin Invest. 2000;105:1035–1038. doi: 10.1172/JCI9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pecora AL, Rizvi N, Cohen GI. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol. 2002;20:2251–2266. doi: 10.1200/JCO.2002.08.042. [DOI] [PubMed] [Google Scholar]
- 47.Coffey M, Strong J, Forsyth P, Lee P. Reovirus therapy of tumors with activated ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 48.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 49.Roelvink P, Mi G, Einfeld D, Kovesdi I, Wickham T. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science. 1999;286:1568–1571. doi: 10.1126/science.286.5444.1568. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki K, Fueyo J, Krasnykh VPR, Curiel D, Alemany R. A replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7:120–126. [PubMed] [Google Scholar]
- 51.Doronin K, Toth K, Kuppuswamy M, Ward P, Tollefson A, Wold W. Tumor-specific, replication-competent adenovirus vectors overexpressing the adenovirus death protein. J Virol. 2000;74:6147–6155. doi: 10.1128/JVI.74.13.6147-6155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hermiston T. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J Clin Invest. 2000;105:1169–1172. doi: 10.1172/JCI9973. [DOI] [PMC free article] [PubMed] [Google Scholar]