

Abstract
Infections by coronaviruses such as severe acute respiratory syndrome (SARS) coronavirus (SCoV) and mouse hepatitis virus A59 (MHV-A59) result in very little type I interferon (IFN) production by host cells, which is potentially responsible for the rapid viral growth and severe immunopathology associated with SARS. However, the molecular mechanisms for the low IFN production in cells infected with coronaviruses remain unclear. Here, we provide evidence that Papain-like protease domain 2 (PLP2), a catalytic domain of the nonstructural protein 3 (nsp3) of MHV-A59, can bind to IRF3, cause its deubiquitination and prevent its nuclear translocation. As a consequence, co-expression of PLP2 strongly inhibits CARDIF-, TBK1- and IRF3-mediated IFNβ reporter activities. In addition, we show that wild-type PLP2 but not the mutant PLP2 lacking the deubiquitinase (DUB) activity can reduce IFN induction and promote viral growth in cells infected with VSV. Thus, our study uncovered a viral DUB which coronaviruses may use to escape from the host innate antiviral responses.
Supplementary information
The online version of this article (doi:10.1038/cr.2008.294) contains supplementary material, which is available to authorized users.
Keywords: MHV-A59, PLP2, deubiquitination, IRF3, type I interferons
Introduction
The coronavirus family has been extensively studied since the severe acute respiratory syndrome (SARS) broke out in 2003 1. SARS coronavirus (SCoV) belongs to the group II coronavirus and is phylogenetically related to the mouse hepatitis virus A59 (MHV-A59) 2. Studies showed that type I interferons (IFNs) including IFNα and IFNβ could inhibit replication of SCoV, and IFNβ was more effective 3, 4. However, clinical studies revealed that SCoV infection induced very low levels of type I IFNs 5, 6, 7, which likely contributed to the rampant viral replication and high mortality associated with the disease. Interestingly, a similar type I IFN response was also found in MHV-A59 infection 8, 9. It was reported recently that certain viral components of SCoV, such as nsp1, nsp3 (nonstructural protein 3), ORF3b, ORF6 and N protein, might play some roles in the inhibition of type I IFN induction 10, 11. However, the underlying molecular mechanisms remain to be elucidated.
Type I IFNs (mainly IFNβ and multiple IFNα subtypes) are believed to be the most important cytokines in innate immunity against viral infections. Depending upon the nature of invading viruses and the infected host cell types, Toll-like receptors (TLRs) such as TLR3, 7 and 9, as well as cytosolic RNA helicases, such as retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA-5), play important roles of sentinel to recognize viral genomes and trigger the type I IFN signaling through adaptor proteins TRIF, MyD88 or CARDIF 12, 13, 14, 15. Moreover, recent studies indicate that these multiple TLR-dependent and -independent pathways may converge on a common TRAF3 adaptor complex, leading to the activation of two critical serine/threonine kinases, TBK1 or IKKε 16, 17. Activated TBK1 or IKKε then phosphorylates the transcription factor IRF3, which translocates into the nucleus to activate the transcription of type I IFN genes. Secreted IFNs, via autocrine or paracrine circuit, activate the JAK-STAT pathway through the common type I interferon receptor (IFNAR) and induce the expression of an array of anti-viral related genes and cytokines 18, 19.
Recent research also indicates that host cells possess intrinsic ubiquitination and deubiquitination programs that are critically involved in the type I IFN induction pathway 20, 21. Interestingly, the nonstructural protein of SCoV, nsp3, a known protease involved in the processing of replicase polyproteins, has recently been shown to carry a conserved deubiquitinase (DUB) motif within its papain-like protease (PLpro) domain 22. In the present study, we provide evidence showing that nsp3 of MHV-A59, a group II coronavirus phylogenetically related with SCoV, can efficiently inhibit IRF3 ubiquitination and IFN induction. This novel finding therefore sheds new light into why host cells produce very low levels of type I IFNs characteristic for innate immunity evasion by SCoV and MHV 5, 6, 7, 8, 9. These results also suggest a novel target for the development of drugs against SARS.
Results
MHV-A59 could not activate type I IFN signal pathway
Using Sendai virus (SeV) as a control, we assessed whether MHV-A59 induced IFNβ expression in mouse embryonic fibroblasts (MEFs). Quantitative RT-PCR analyses showed rapid and strong IFNβ production in response to SeV infection of MEF cells (6 h post-infection), whereas MHV-A59 infection resulted in no detectable IFNβ induction along the time course, with trivial increase after 24 h of infection (Figure 1A). MHV-A59 replicated well in MEFs as revealed by the RT-PCR assay showing the expression of nucleocapsid genes and the plaque assay (Supplementary information, Figure S1). IFNβ binds to IFNAR to activate the phosphorylation of STAT1. The inability of MHV-A59 to effectively induce IFNβ gene expression correlated well with undetectable STAT1 phosphorylation, as compared with strong STAT1 phosphorylation after SeV infection (Figure 1B). IRF3 is a nucleo-cytoplasm shuttling factor that requires phosphorylation and dimerization for transcriptional activation. In contrast to SeV, MHV-A59 infection of MEFs could induce neither dimerization (Figure 1C) nor nuclear translocation (Figure 1D) of IRF3. These results therefore suggest that host cells lack the ability to produce type I IFN in response to MHV-A59 infection.
Figure 1.
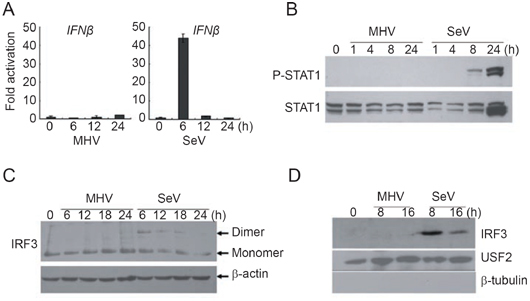
MHV-A59 infection could not activate IRF3 to induce IFNβ in MEFs. (A) MEFs produced barely detectable IFNβ mRNA upon MHV-A59 infection. MEFs were infected with MHV-A59 (MOI=5) or SeV (HA titer of 1:25 was used). At the indicated time points post-infection, endogenous IFNβ transcription levels were determined by quantitative real-time PCR. The data were normalized to that of 18s RNA and averaged as fold activation over that by mock infection (mean±SD). (B) STAT1 was not phosphorylated after MHV-A59 infection. Whole cell lysates (15-20 μg) after virus infections at the indicated time points were subject to 10% SDS-PAGE and phosphorylated STAT1 was immunoblotted. Total STAT1 was also detected as a loading control. (C) IRF3 was not activated by MHV-A59. Whole cell lysates (15-20 μg) after virus infections for the indicated time points were subject to 10% native PAGE and IRF3 dimerization was immunoblotted with anti-IRF3 antibody. β-Actin was immunoblotted as a loading control. (D) IRF3 did not translocate to the nucleus. Nuclear extracts (15 μg) from the infected MEFs were resolved in 10% SDS-PAGE and the presence of IRF3 was immunoblotted similarly to (C). USF2 and β-tubulin were immunoblotted as a loading control or to exclude contamination of the extracts, respectively. The data represented at least 3 independent experiments.
nsp3 of MHV-A59 inhibited type I IFN induction
Because ubiquitination plays an important role in the RIG-I-mediated IFN induction cascade 20, and because PLpro domain 2 (PLP2) domain of MHV-A59 contains a conserved DUB motif 22, we hypothesized that the DUB activity of PLP2 domain might block the IFNβ response during MHV-A59 infection. As the first step to identify the potential target of PLP2 DUB in the type I IFN signaling pathway, we tested whether IFN reporter activities driven by overexpressed CARDIF, TBK1 or IRF3 were affected by co-expressed PLP2. As shown in Figure 2A-C, PLP2 efficiently inhibited IFNβ reporter activities in a dose-dependent manner (data not shown for expression of various exogenous proteins). As mentioned above, PLP2 is the main active domain of nsp3; to understand whether full-length nsp3 also contains such activity we tried to clone the full-length nsp3 of MHV-A59. However, the full-length sequence appears very unstable, always resulting in smaller inserts in our constructs. We were able to obtain a construct (MHV NSP3 4.0) with an about 4.0-kb insert which comprised 2/3 of the full-length nsp3 from its N terminus and contained the intact PLP2 domain. We found that this construct could produce a 150-kDa protein and could greatly inhibit the type I IFN signaling pathway (Supplementary information, Figure S2). At the same time, we also tested the effect of full-length nsp3 of ScoV, which is a close relative of MHV-A59. Expression of this construct could produce several proteins with different sizes and could also inhibit the type I IFN signaling pathway (Supplementary information, Figure S3). Because PLP2 exhibited apparent inhibition of IRF3-driven reporter activity, and IRF3 activation is the very last step in the type I IFN signaling pathway, we suspected that inhibition of IRF3 ubiquitination by PLP2 DUB would account for the inactivation of IRF3 and reduced IFNβ production. While overexpressed IRF3 became poly-ubiquitinated, such poly-ubiquitin modification of IRF3 was effectively inhibited by co-expressed PLP2 DUB (Figure 3A). Furthermore, PLP2 could deconjugate both Lys63- and Lys48-linked polyubiquitin chains as shown with the mutant Ubi-K48 or Ubi-K63 (described in Materials and methods), respectively (Figure 3B), suggesting its rather promiscuous spectra of deubiquitination. This result was also in good agreement with its general hydrolyzing activity towards cellular polyubiquitinated proteins (data not shown). Furthermore, Flag-PLP2 and Myc-IRF3 were reciprocally co-immunoprecipitated with each other, indicating that they could form a complex (Figure 3C). All these data therefore suggest that PLP2 likely inhibited IFN induction through binding to and deubiquitinating IRF3.
Figure 2.
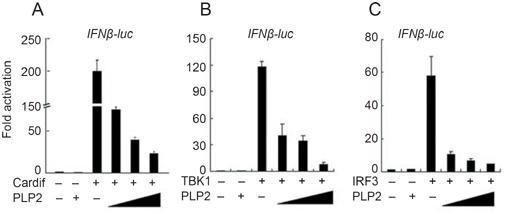
MHV-A59 PLP2 interfered with the type I interferon induction pathway. Plasmids expressing Cardif (A, 100 ng pcDNA3.1-CARDIF), TBK1 (B, 100 ng pEBB-HA-TBK1) or IRF3 (C, 200 ng pFLAG-CMV-2-IRF3) were transiently co-transfected without or with increasing amounts of pCMV-Myc-PLP2 (50-200 ng) into HEK293T cells (in 24 wells). IFNβ-Luc reporter plasmid (30 ng) together with Renilla luciferase internal control plasmid (15 ng) were co-transfected for 24 h, the luciferase activities were determined and normalized based on Renilla luciferase activity. Data were expressed as fold activation for each sample by dividing the luciferase activity by that observed in the sample containing only empty vector. Data represented the average of three independent experiments (mean±SD).
Figure 3.
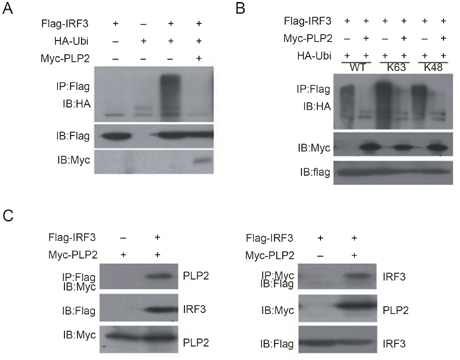
MHV-A59 PLP2 inhibited ubiquitination of IRF3. (A) IRF3 was poly-ubiquitinated. Whole cell lysates of HEK293T cells (in 35 mm plates) transiently transfected with pFLAG-CMV-2-IRF3 (1.2 μg), pRK5-HA-Ubi (0.8 μg) or pCMV-myc-PLP2 (1.5 μg) (after 24 h) were immunoprecipitated with anti-FLAG antibody (2 μg) and resolved precipitates were immunoblotted with anti-HA antibody (top panel). Expression of the exogenous epitope-tagged proteins was verified with the indicated antibodies (bottom two panels). (B) PLP2 DUB deconjugated both K63- and K48-linked polyubiquitin. HEK293T cells transiently expressing Flag-IRF3, Myc-PLP2 and HA-Ubi, or its mutant derivatives, HA-Ubi-K63 or HA-Ubi-K48, were lysed and subject to co-immunoprecipitation as that in (A). (C) IRF3 directly interacted with PLP2 DUB. FLAG-IRF3 and Myc-PLP2 transiently expressed in HEK293T were co-immunoprecipitated by anti-FLAG (left panel) or anti-myc (right panel) antibody and the precipitants were immunoblotted as indicated. Expression levels of tagged proteins were measured by indicated antibodies.
DUB activity was essential for PLP2 to block type I IFN induction
The Cys residue of the catalytic triad “Cys-His-Asp” is essential for the DUB activity of SCoV PLpro domain 22. When we mutated the conserved Cys106 to Ala, the resultant mutant (PLP2-C106A) could no longer deubiquitinate IRF3 (Figure 4A). In the following experiments we found that the C106A mutant showed reduced ability to inhibit IFNβ promoter activation driven by CARDIF, TBK1 and IRF3 (Figure 4B-4D). We also produced another mutant, PLP2-D277A (Asp277 to Ala), which changes one of the catalytic triad, Asp277. Unexpectedly, the D277A mutant still showed strong DUB activity (Figure 4E) even though its expression level was lower. Consequently, D277A could still inhibit the activation of IFNβ promoter (Figure 4F). Therefore, our data indicate that the DUB activity of PLP2 was required for the inhibition of type I IFN induction.
Figure 4.
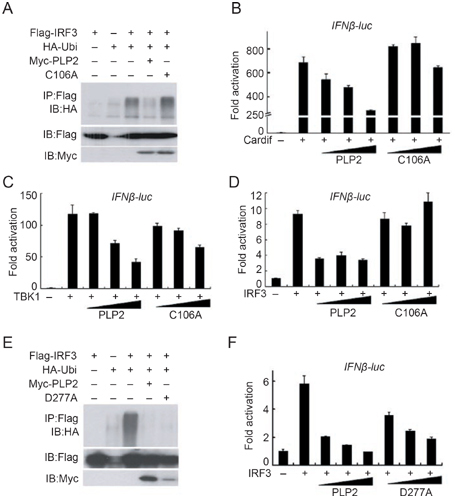
Deubiquitinase activity was required for PLP2 to block the type I interferon signaling. (A) Enzymatic dead mutant of PLP2 DUB failed to deubiquitinate IRF3. HEK293T cell lysates overexpressing FLAG-IRF3, HA-Ubi and myc-PLP2 or myc-PLP2-C106A were immunoprecipitated with anti-FLAG antibody and ubiquitin conjugation of IRF3 was verified by immunoblotting with anti-HA antibody. The input tagged proteins were verified with indicated antibodies (bottom two panels). (B-D) PLP2-C106A became inefficient to inhibit IFNβ-luc reporter activities. IFNβ-Luc reporter assays were performed as those in Figure 2, except that pCMV-myc-PLP2-C106A was used in parallel to pCMV-myc-PLP2 (10-50 ng). Data represented the average of three independent experiments (mean±SD). (E) D277 mutation could not block the DUB activity of PLP2. Procedure was the same as in (A) except the use of pCMV-myc-PLP2-D277A. (F) PLP2-D277A could still inhibit the activation of IFNβ promoter. Procedure was the same as (B-D). Data represented the average of three independent experiments (mean±SD).
PLP2 inhibited type I IFN signaling activation by viruses
IRF3 activation has been shown to be critically involved in type I IFN induction by paromyxoviruses such as SeV and VSV 23, 24. In order to substantiate our hypothesis that PLP2 DUB may be a general blocker of the type I IFN signaling pathway, we set to determine whether PLP2 inhibits IFN production induced by these viruses. Overexpression of wild-type PLP2 could greatly inhibit the activation of IFNβ promoter induced by SeV infection of HEK293T cells, while PLP2-C106A failed to do so (Figure 5A). The ability of PLP2 to inhibit IFNβ induction by SeV (Figure 5A) was due to inactivation of IRF3, as wild-type PLP2 but not the PLP2-C106A mutant inhibited IRF3 nuclear translocation (Figure 5B). Similarly, wild-type PLP2 but not PLP2-C106A could effectively inhibit VSV-mediated induction of endogenous IFNβ mRNA (Figure 5C) and, consequently, accelerate virus replication in HEK293T cells (Figure 5D). Overall, it is tempting to speculate that group II coronaviruses, including MHV-A59 and SCoV, might utilize the immediate-early gene product, nsp3, to subvert host anti-viral innate immune response by preventing ubiquitination and activation of IRF3, a critical transcription factor in the type I IFN signaling pathway.
Figure 5.
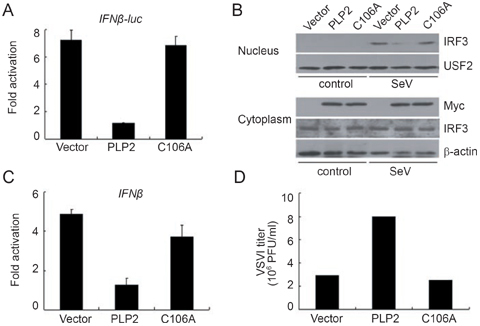
PLP2 DUB was responsible for inhibition of type I interferon pathway and acceleration of virus replication. (A) Inactivated PLP2 DUB failed to inhibit SeV-induced IFNβ-luc reporter activity. IFNβ-Luc reporter plasmid was co-transfected into HEK293T cells with pCMV-myc-PLP2 or pCMV-myc-PLP2-C106A for 24 h before SeV infection for an additional 8 h. Luciferase activities were assessed as those in Figure 2. Data were expressed as the fold activation for each sample by dividing the luciferase activity by that observed in uninfected sample. Data represented the average of three independent experiments (mean±SD). (B) PLP2 blocked SeV-induced IRF3 nuclear translocation. Nuclear translocation of endogenous IRF3 after SeV infection of HEK293T cells transiently expressing PLP2 or PLP2-C106A was monitored as that in Figure 1D, and expression level of constructs and IRF3 were also measured in cytosolic extracts. (C) Inactivated PLP2 DUB failed to inhibit VSV-induced IFNβ production. HEK293T cells transiently transfected with PLP2 or PLP2-C106A were infected with VSV (MOI=0.1) for 5 h and mRNA levels of endogenous IFNβ were measured by quantitative real-time PCR as those in Figure 1A, except that PCR primers specific to human IFNβ gene were used and GAPDH expression was used for normalization. Data were expressed as the fold activation for each sample by dividing the luciferase activity by that observed in the uninfected sample (mean±SD). (D) PLP2 DUB inhibited VSV replication. The supernatants from cells as treated in (C) were collected for plaque assays to determine the virus titer. The data represented at least 3 independent experiments.
Discussion
Coronaviruses are able to infect a broad range of vertebrates to evoke systemic immunopathological dysfunction. Lack of type I IFN production in host cells to effectively control viral replication likely plays an important role in SARS manifestation and progression. By using a mouse-specific strain of group II coronavirus, MHV-A59, we provide evidence here that a DUB activity conserved among coronavirus nsp3 proteins might specifically inhibit IRF3 activation, and thereby is responsible for viral inhibition of cellular type I IFN production.
Most mammalian cells including immune and non-immune cells can produce type I IFNs in response to various viral infections. On the other hand, as a result of host-pathogen co-evolution, viruses have developed various strategies to escape from type I IFN response. For example, VP35 of Ebola virus could inhibit the phosphorylation of IRF3 25; NS1 of influenza virus could bind dsRNA and inhibit the subsequent activation of IRF3 26. NS34A, a cysteine protease of HCV, could cleave CARDIF to block RIG-I-mediated IFN induction 27. In immune response to SARS-CoV and other coronaviruses, such as HCoV-OC43, there is an unusual lack of the antiviral IFN response 5, 6, 7, 8. The finding that MHV-A59 PLP2 potently inhibits IRF3 ubiquitination (Figure 3) and its transcriptional activation of the IFNβ gene (Figures 2, 4 and 5) provides novel insight into how the viral gene expression program targets the host innate immune system. During our study, another group reported a similar phenomenon that SCoV PLpro could block IRF3-mediated type I IFN induction 28. However, in their study, they concluded that the DUB activity of PLpro was not involved in the inhibition of type I IFN signaling pathway because the Asp mutant (D1826A), which had been shown to lose >99% of DUB activity 22, could still inhibit type I IFN induction 28. In our present study, we found that the Asp mutant of PLP2 could still strongly block the ubiquitination of IRF3 (Figure 4B). In fact, from the data of the previous study 22, one could clearly see that the D1826A mutant could cleave di-ubiquitin to produce mono-ubiquitin, even though its activity was lower than that of WT. Considering the situation of over-expression, the remaining DUB activity might be sufficient to affect the ubiquitination process of the cells. Therefore, we propose that the DUB activity of PLP2 is closely related to its function to block the type I IFN induction pathway.
A growing body of evidence indicates that ubiquitination is critically involved in the type I IFN signaling pathway. Ubiquitination of RIG-I by the E3 ubiquitin ligase TRIM25 is necessary and sufficient to trigger the downstream signaling cascade to produce type I IFNs 20. Ubiquitination of IRF7 by the E3 ligase TRAF6 has been implicated in type I IFN induction in TLR7, 8 and 9 pathways 29, 30. Studies have shown that activated IRF3 could be polyubiquitinated and degraded 31, 32, yet one could not exclude the possibility of IRF3 activation by ubiquitination just like what is observed with RIG-I 20. In the present study we found that IRF3 could be ubiquitinated by both K48 and K63 Ubi (Figure 3B). Notably, K48 ubiquitination is involved in degradation while K63 ubiquitination is usually regarded as an active form of ubiquitin-targeted proteins 33, 34. The fact that deubiquitinated IRF3 fails to translocate to the nucleus (Figure 5B) also supports the possibility that IRF3 activation may require ubiquitination. Although further studies are needed to elucidate the relationship between the activation and ubiquitination of IRF3, our present study and the recent report by others 28 clearly show that IRF3 is a target of the coronavirus PLpro. Interestingly, the newly identified cellular DUB, DUBA, inhibits the ubiquitination of TRAF3, an adaptor of RIG-I signaling, and negatively regulates the type I IFN signaling pathway 21. Coronaviruses may have picked up this mechanism from the host during their co-evolution, and deubiquitination of IRF3 by nsp3 seems to provide a general suppression of anti-viral IFN production to facilitate productive infection. This strategy is not unique to viruses because the bacterial virulence factor YopJ also functions as a DUB that inhibits TRAFs-mediated innate immune response 35.
It would be ideal to provide definitive evidence that nsp3PLP2 encodes a functionally important DUB by either reverse genetics to introduce point mutations (e.g., C106A) into the MHV-A59 genome 36 or small interference RNA against nsp3 to rescue IFN response in the infected cells. However, the nsp3 protein and the catalytic cysteine site are required for the processing of replicase polyprotein of coronaviruses 22, 37; therefore targeting of nsp3 would affect viral replication per se. Nevertheless, ectopic expression of the PLP2 domain inhibits both SeV- and VSV-induced IFN response and facilitates viral replication (Figure 5), suggesting that MHV-A59 PLP2 provides a general inactivation mechanism of the host anti-viral response through deubiquitination of important innate immunity regulatory factor(s), such as IRF3.
Taken together, the results of our study and others suggest that inactivation of IRF3 by nsp3 may contribute to the weak IFN response in coronavirus infections. Ultimate understanding of the overall interactions between coronaviruses and the host innate immune system will provide further insight into the pathogenesis of viruses in this class, and open a new avenue of therapeutic target exploration against coronavirus infections.
Materials and methods
Cells and plasmids
Wild-type MEF cells were in C57/BL6 background. The cell lines L929, HEK293T and HeLa were routinely maintained in DMEM (Hyclone, UT, USA) supplemented with 10% FCS (PAA, Pasching, Austria) and 1% penicillin and streptomycin (Hyclone, UT). Cell line 17Cl-1 is a kind gift from Dr K Holmes (University of Colorado Health Sciences Center, USA). The constructs pEBB-HA-TBK1 and pcDNA3.1-CARDIF were described previously 17, 38. Wild-type ubiquitin expression vector pRK5-HA-Ubi and the mutant derivatives Ubi-K63 and Ubi-K48 were from Dr K Lim (National Neuroscience Institute, Singapore); the K63 or K48 mutant contains arginine substitutions on all of the lysine residues except the one at position 63 or 48, respectively 39. Primer pairs specific to MHV-A59 N protein gene were forward: 5′-CAA AGA AAA GGG CGT AGA CAG G-3′ and reverse: 5′-CGC CAT CAT CAA GGA TCT GAG-3′. Full-length human IRF3 sequence was subcloned into the vector pFLAG-CMV-2 between EcoRI and SalI sites. MHV-A59 PLP2 domain (nt 1-1 080) was RT-PCR amplified from MHV-A59-infected cells and subcloned into pCMV-Myc plasmid between EcoRI and XhoI sites. PCR primers were derived from the MHV-A59 genome sequence (NCBI NC_001846, nt 5 040-6 119): forward 5′-CGG AAT TCG GGT TGA TGT CTT GTG TAC TG-3′ and reverse 5′-CCG CTC GAG TTA CAA TTT AAA GTT GGT ATA GAC-3′. Point mutagenesis was performed according to the described protocol 40. The primer pairs for generating PLP2-C106A were forward: 5′-GTC AAA TAA TAA Tgc gTA TAT AAA TGT GGC ATG TTT AAT GCT GCA ACA C-3′ and reverse: 5′-CAC ATT TAT ATA cgc ATT ATT ATT TGA CTG CTT GAA AGC AAA ATA ATT GC-3′. The primer pairs for PLP2-D277A were forward: 5′-CAG CTT TAT GcT GCT TGT AAT GTT AAT AAG GTT TCG G-3′ and reverse: 5′-CAT TAC AAG CAg CAT AAA GCT GGT ACT TGG GTT TAC ATT TC-3′ . The partial fragment (NCBI NC_001846, nt 2 706-6 617) of MHV-A59nsp3 was cloned into the vector pCMV-Myc between Sal I and Not I sites and was termed as MHV NSP3 4.0. All the clones were verified by DNA sequencing. The construct pCDβA-SCoVNSP3 expressing the Flag tagged full-length nsp3 of SCoV was kindly donated by Dr M Wathelet (University of Cincinnati College of Medicine, USA) 10.
Reagents
Antibodies specific to STAT1, IRF3 and USF2 were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA). Monoclonal antibodies to Flag and β-actin were purchased from Sigma (St Louis, MO), β-tubulin from Chemicon International Inc. (Temecula, CA), and anti-Myc and anti-HA antibodies were from Shanghai Genomics, Inc. (Shanghai, China). Antibodies specific to the phosphorylated STAT1 were obtained from Cell Signaling Technologies (Beverly, MA).
Viruses and infection
MHV-A59 kindly provided by Dr Lishan Su (University of North Carolina, USA) was expanded in 17Cl-1 cell and purified as described previously 41. The titer of the virus was determined by plaque assay in 17Cl-1 cells exactly as described 42 and MOI = 5 was used for the infection.
SeV was from the Wuhan Inst Virol, CAS, China and expanded in day 10 embryonate eggs. The allantoic fluid was harvested 72 h after inoculation. The viral titer was measured for hemagglutination (HA) with chicken erythrocytes using the pattern method 43. The HA titer was expressed as the highest dilution of the allantoic fluid showing complete HA. The HA titer of SeV stock was 1:256, and for infection 1:25 was used.
Vesicular stomatitis virus (VSV) was provided by Dr G Gao (Institute of Biophysics, CAS, China). The virus was expanded in L929 cells and titrated with plaque assay in HeLa cells as that of MHV. MOI = 0.1 was used for infection.
RT-PCR
Total RNA extraction by Trizol (Invitrogen, CA, USA) and quantitative real-time PCR reactions in a MyiQ cycler (Bio-Rad, CA, USA) using SYBR Green I (Molecular Probes, OR, USA) were performed exactly as described previously 44. The primer pairs for mouse IFNβ and 18s RNA were derived from previous reports 44, 45. The primer pairs for human IFNβ and GAPDH were IFNβ forward: 5′-AAA CTC ATG AGC AGT CTG CA-3′; IFNβ reverse: 5′-AGG AGA TCT TCA GTT TCG GAG G-3′; GAPDH forward: 5′-AAG CTC ACT GGC ATG GCC TT-3′; GAPDH reverse: 5′-AGG AGA CCA CCT GGT GCT CAG-3′. The mRNA levels of indicated genes were normalized to that of 18s RNA or GAPDH mRNA. The data represented the average of at least three independent experiments (mean±SD).
Transfection, immunoprecipitation and immunoblotting
Transient transfection of HEK293T cells with indicated plasmids was performed routinely with calcium phosphate method. Co-immunoprecipitation 46 and nuclear Western blotting 44 were performed exactly as described previously.
Analysis of IRF-3 dimerization by native PAGE
IRF-3 activation was assessed by its ability to homodimerize using native PAGE precisely as described previously 47.
Luciferase assays
IFNβ luciferase reporter plasmid described previously 48 was co-transfected with the indicated constructs into HEK293T cells. Renilla luciferase plasmid was co-transfected as internal control of transfection efficiency. Luciferase activities were measured and normalized as per instructions by the manufacturer (Dual-Luciferase reporter assay system, Promega, WI). Luciferase activities were measured 24 h after transfection or after cells were infected by SeV for an additional 8 h.
(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Proliferation of MHV-A59 in MEFs. WT MEFs were infected with MHV-A59 (MOI = 1). (PDF 42 kb)
Partial fragment of MHV NSP3 can inhibit type I interferon signal pathway. (PDF 54 kb)
SARS coronavirus NSP3 can inhibit type I interferon signal pathway. (PDF 97 kb)
Acknowledgements
We thank Drs S Vaidya and E Chow (University of California Los Angeles, USA) for their help in setting up critical experimental systems. We greatly thank Dr K Holmes (University of Colorado Health Sciences Center, USA) for sharing with us 17Cl-1 cell line and helping to optimize the protocol to produce high titered MHV-A59 virus stock. We also thank Drs R Baric and L Su (University of North Carolina, USA) for the gift of MHV-A59 and guidance of virus infection. We thank Dr K Lim (National Neuroscience Institute, Singapore) for the gift of Ubi plasmids. We thank Dr M Wathelet (University of Cincinnati College of Medicine, USA) for sharing the nsp3 construct. Also we thank Dr G Gao (Institute of Biophysics, CAS) for providing us with VSV. This research was partly supported by grants from the National Natural Science Foundation of China (30728006) to Genhong Cheng and the National Basic Research Program of MOST (2004BA519A61, 2006CB504300, 2007DFC30190) to Hong Tang.
Contributor Information
Genhong Cheng, Phone: +1-310-825-8896, FAX: +1-310-206-5553, Email: gcheng@mednet.ucla.edu.
Hong Tang, Phone: +86-10-64888438, FAX: +86-10-64848357, Email: tanghong@moon.ibp.ac.cn.
References
- 1.Oxford JS, Bossuyt S, Lambkin R. A new infectious disease challenge: Urbani severe acute respiratory syndrome (SARS) associated coronavirus. Immunology. 2003;109:326–328. doi: 10.1046/j.1365-2567.2003.01684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snijder EJ, Bredenbeek PJ, Dobbe JC. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cinatl J, Morgenstern B, Bauer G. Treatment of SARS with human interferons. Lancet. 2003;362:293–294. doi: 10.1016/S0140-6736(03)13973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng B, He ML, Wong KL. Potent inhibition of SARS-associated coronavirus (SCOV) infection and replication by type I interferons (IFN-alpha/beta) but not by type II interferon (IFN-gamma) J Interferon Cytokine Res. 2004;24:388–390. doi: 10.1089/1079990041535610. [DOI] [PubMed] [Google Scholar]
- 5.Reghunathan R, Jayapal M, Hsu LY. Expression profile of immune response genes in patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005;6:2. doi: 10.1186/1471-2172-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Subbarao K. The immunobiology of SARS*. Annu Rev Immunol. 2007;25:443–472. doi: 10.1146/annurev.immunol.25.022106.141706. [DOI] [PubMed] [Google Scholar]
- 7.Frieman M, Heise M, Baric R. SARS coronavirus and innate immunity. Virus Res. 2007;133:101–112. doi: 10.1016/j.virusres.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H, Perlman S. Mouse hepatitis virus does not induce Beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J Virol. 2007;81:568–574. doi: 10.1128/JVI.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Versteeg GA, Bredenbeek PJ, van den Worm SH, Spaan WJ. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361:18–26. doi: 10.1016/j.virol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med. 2006;84:712–725. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 13.Perry AK, Chen G, Zheng D, Tang H, Cheng G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005;15:407–422. doi: 10.1038/sj.cr.7290309. [DOI] [PubMed] [Google Scholar]
- 14.Yoneyama M, Kikuchi M, Natsukawa T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 15.Andrejeva J, Childs KS, Young DF. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA. 2004;101:17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oganesyan G, Saha SK, Guo B. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 17.Saha SK, Pietras EM, He JQ. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. Embo J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiscott J, Pitha P, Genin P. Triggering the interferon response: the role of IRF-3 transcription factor. J Interferon Cytokine Res. 1999;19:1–13. doi: 10.1089/107999099314360. [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 20.Gack MU, Shin YC, Joo CH. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 21.Kayagaki N, Phung Q, Chan S. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 22.Barretto N, Jukneliene D, Ratia K. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato H, Sato S, Yoneyama M. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 24.Kato H, Takeuchi O, Sato S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 25.Basler CF, Mikulasova A, Martinez-Sobrido L. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talon J, Horvath CM, Polley R. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol. 2000;74:7989–7996. doi: 10.1128/JVI.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meylan E, Curran J, Hofmann K. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 28.Devaraj SG, Wang N, Chen Z. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawai T, Sato S, Ishii KJ. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 30.Huye LE, Ning S, Kelliher M, Pagano JS. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol Cell Biol. 2007;27:2910–2918. doi: 10.1128/MCB.02256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saitoh T, Tun-Kyi A, Ryo A. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 2006;7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- 32.Bibeau-Poirier A, Gravel SP, Clement JF. Involvement of the IkappaB kinase (IKK)-related kinases tank-binding kinase 1/IKKi and cullin-based ubiquitin ligases in IFN regulatory factor-3 degradation. J Immunol. 2006;177:5059–5067. doi: 10.4049/jimmunol.177.8.5059. [DOI] [PubMed] [Google Scholar]
- 33.Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 34.Wang M, Cheng D, Peng J, Pickart CM. Molecular determinants of polyubiquitin linkage selection by an HECT ubiquitin ligase. Embo J. 2006;25:1710–1719. doi: 10.1038/sj.emboj.7601061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sweet CR, Conlon J, Golenbock DT, Goguen J, Silverman N. YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell Microbiol. 2007;9:2700–2715. doi: 10.1111/j.1462-5822.2007.00990.x. [DOI] [PubMed] [Google Scholar]
- 36.Yount B, Denison MR, Weiss SR, Baric RS. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J Virol. 2002;76:11065–11078. doi: 10.1128/JVI.76.21.11065-11078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanjanahaluethai A, Baker SC. Identification of mouse hepatitis virus papain-like proteinase 2 activity. J Virol. 2000;74:7911–7921. doi: 10.1128/JVI.74.17.7911-7921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry AK, Chow EK, Goodenough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim KL, Chew KC, Tan JM. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sturman LS, Holmes KV, Behnke J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J Virol. 1980;33:449–462. doi: 10.1128/jvi.33.1.449-462.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baric RS, Sullivan E, Hensley L, Yount B, Chen W. Persistent infection promotes cross-species transmissibility of mouse hepatitis virus. J Virol. 1999;73:638–649. doi: 10.1128/jvi.73.1.638-649.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salk JE. A Simplified Procedure for Titrating Hemagglutinating Capacity of Influenza-Virus and the Corresponding Antibody. J Immunol. 1944;49:87–98. [Google Scholar]
- 44.Doyle S, Vaidya S, O'Connell R. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/S1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 45.Doyle SE, O'Connell R, Vaidya SA. Toll-like receptor 3 mediates a more potent antiviral response than Toll-like receptor 4. J Immunol. 2003;170:3565–3571. doi: 10.4049/jimmunol.170.7.3565. [DOI] [PubMed] [Google Scholar]
- 46.Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J Biol Chem. 2005;280:10491–10500. doi: 10.1074/jbc.M412819200. [DOI] [PubMed] [Google Scholar]
- 47.Lin R, Yang L, Nakhaei P. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 2006;281:2095–2103. doi: 10.1074/jbc.M510326200. [DOI] [PubMed] [Google Scholar]
- 48.Guo B, Cheng G. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem. 2007;282:11817–11826. doi: 10.1074/jbc.M700017200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proliferation of MHV-A59 in MEFs. WT MEFs were infected with MHV-A59 (MOI = 1). (PDF 42 kb)
Partial fragment of MHV NSP3 can inhibit type I interferon signal pathway. (PDF 54 kb)
SARS coronavirus NSP3 can inhibit type I interferon signal pathway. (PDF 97 kb)