

Abstract
Immune responses against multiple epitopes are required for the prevention of hepatitis C virus (HCV) infection, and the progression to phase I trials of candidates may be guided by comparative immunogenicity studies in non-human primates. Four vectors, DNA, SFV, human serotype 5 adenovirus (HuAd5) and Modified Vaccinia Ankara (MVA) poxvirus, all expressing hepatitis C virus Core, E1, E2 and NS3, were combined in three prime-boost regimen, and their ability to elicit immune responses against HCV antigens in rhesus macaques was explored and compared. All combinations induced specific T-cell immune responses, including high IFN-γ production. The group immunized with the SFV+MVA regimen elicited higher E2-specific responses as compared with the two other modalities, while animals receiving HuAd5 injections elicited lower IL-4 responses as compared with those receiving MVA. The IFN-γ responses to NS3 were remarkably similar between groups. Only the adenovirus induced envelope-specific antibody responses, but these failed to show neutralizing activity. Therefore, the two novel regimens failed to induce superior responses as compared with already existing HCV vaccine candidates. Differences were found in response to envelope proteins, but the relevance of these remain uncertain given the surprisingly poor correlation with immunogenicity data in chimpanzees, underlining the difficulty to predict efficacy from immunology studies.
Supplementary information
The online version of this article (doi:10.1038/gt.2016.55) contains supplementary material, which is available to authorized users.
Subject terms: Virology, DNA vaccines, Cell death and immune response, Hepatitis C, Genetic vectors, Vaccines
Introduction
Hepatitis C virus (HCV) vaccine candidates inducing HCV-specific immune responses may protect naive chimpanzees from heterologous experimental challenge; however, the mechanism behind protection remains elusive.1, 2, 3, 4, 5, 6 Although information regarding the immune correlates of protective immunity is incomplete, sufficient evidence points toward a pivotal role for a potent functional cellular immune response against several HCV proteins.7, 8, 9, 10 Vaccine strategies inducing such strong cellular immunity in mice, rhesus macaques and humans often combine priming of the immune system with booster immunizations using DNA and recombinant viral vectors, including adenovirus and modified virus Ankara (MVA).10, 11
Preclinical evaluation of the potential efficacy of HCV vaccine candidates is limited to the chimpanzee and humanized mouse models.8, 12, 13, 14 Several HCV vaccine candidates have been created, including viral vectors and typically encoding multiple target antigens, and relied on preclinical immunogenicity studies in non-human primates such as rhesus macaques.8, 15, 16, 17 Comparison of different vectors and regimen in non-human primates is highly valuable for HCV vaccine development, to shed light on factors specifically influencing HCV vaccine immunogenicity as well as providing technological improvement in the field of genetic vaccination, as is performed in HIV development.18 In a recent study, HCV adenovirus vaccine boosting induced robust cellular responses, HCV pseudoparticles enhanced the humoral response, and poxvirus priming induced both humoral and cellular responses. Immune responses were optimized with heterologous prime-boost regimens.16
Vectors included plasmid DNA, recombinant SFV, replication-incompetent adenovirus and poxvirus (MVA) encoding the structural HCV antigens Core (C), the two envelope glycoproteins E1 and E2, and the non-structural antigen NS3 were developed and evaluated for immunogenicity in mice, alone or in various prime-boost combinations. These vectors successfully elicited cellular and humoral immune responses including in HLA-A2 mice.19 Moreover, a DNA-DNA-MVA-MVA vaccine regimen induced robust immune responses associated with an early control of heterologous HCV infection in chimpanzees.20 In the present study, we explored the B- and T-cell immune responses induced by this vaccine in rhesus macaques and investigated whether two different immunization regimens, not investigated to date, have the potential to improve the HCV-specific immune responses: DNA followed by adenovirus and SFV followed by MVA. We characterized the immune responses to each of the four vaccine antigens induced by the three regimens in rhesus macaques, and evaluated the predictive value of the rhesus macaque as a model for HCV vaccine immunogenicity by comparing the immune response observed in rhesus macaques after DNA-MVA to those observed in chimpanzees after the exact same DNA-MVA vaccine regimen.20
Results
All regimens induced lymphoproliferative responses to NS3
Three regimens were tested in three groups of four animals (Figure 1). Group I received the DNA vaccine twice followed by two immunizations with HuAd5, group II received the DNA vaccine twice followed by two immunizations with MVA and group III received the rSFV vaccine twice followed by two immunizations with MVA. HCV-specific lymphoproliferative responses were tested using recombinant proteins: Core and NS3 from genotoype 1a and E1 and E2 from genotype 1b. Responses were induced after the two DNA or SFV injections (Figure 2, individual stimulation index (SI) cumulated for all four antigens below 32), and were directed mainly to C (Figure 2, top pie charts and Supplementary Figure 1), but no significant differences were observed between DNA (both DD groups) and SFV-immunized (SS) animals.
Figure 1.

Immunization schedule. Three groups of four macaques per group were immunized at the four time points indicated by arrows (at weeks 0, 6, 14 and 20), with the vaccine immunogens as indicated, further referred to as DNA+HuAd5 (DDAA), DNA+MVA (DDMM) and SFV+MVA (SSMM). The composition of vectors and their transgenes used for each immunization (DNA, HuAd5, MVA or SFV) is indicated.
Figure 2.

Lymphoproliferation responses induced against all four HCV proteins (graph) and the proportional contribution of each antigen (pie charts) after priming and boosting. The cumulated SI to all four HCV antigens of each animal is shown, each diamond representing one animal, 2 weeks after the two DNA or SFV injections, and 2 weeks after HuAd5 or MVA injections. Individual values mentioned in the text are indicated. The geometric mean+s.d. of each group at both time points is also represented as horizontal bars. The contribution of each antigen-specific lymphoproliferation is represented on top of each vaccine regimen, each pie chart representing the geometric mean lymphoproliferation of each group to each antigen as indicated in the legend. Lymphoproliferation was performed using recombinant proteins.
Lymphoproliferative responses were clearly boosted by the HuAd5 or MVA immunizations to similar high levels (Figure 2, cumulative SI up to 168). Analysis of the contribution of each individual HCV vaccine antigen showed that after Ad or MVA boosting, the NS3-specific lymphoproliferation dominated, with individual SI ranging from 8 to 55 (Supplementary Figure 1). The combination SSMM (SFV+MVA) induced significantly higher proliferation to E2 (average SI of 28) as compared with the other vaccines (average SI of 6 in both DNA-primed groups, P=0.04, Supplementary Figure 1).
All regimens induced a Th1-biased T-cell response
We enumerated the C and NS3 (genotype 1a), E1, E2 (genotype 1b)-induced secretion of IFN-γ, IL-2 and IL-4, the respective representative Th1 and Th2 cytokines (Figure 3). Although the CD4+ cells were not separated from the peripheral blood mononuclear cells (PBMCs), the enzyme-linked immunospot (ELISPOT) assay was performed with recombinant proteins as stimulus; therefore, the responses detected are presumed to represent mainly CD4+ T cells.21 After immunization with either DNA or SFV, while lymphoproliferation was detected, only marginal cytokine production was induced: only 2 animals out of 12 mounting a detectable IFN-γ response, and very low levels of IL-2 and IL-4 responses were detected (<50 and <34 spot forming units (SFU)/million cells respectively against all antigens, data not shown). No difference was observed between groups (not shown).
Figure 3.
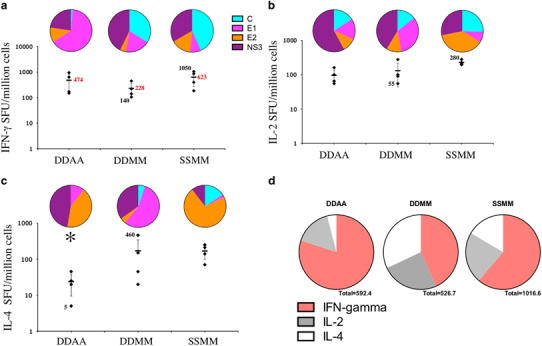
Enumeration of IFN-γ (a), IL-2 (b) and IL-4 (c) producing cells by ELISPOT against all four HCV proteins (graphs) and the proportional contribution of each antigen (pie charts) after HuAd5 or MVA-booster injections. (a–c) The cumulated HCV-specific Spot Forming Units/million cells per animal are shown after two HuAd5 or MVA injections as indicated, each diamond representing one animal. The mean+s.d. of each group is also represented as horizontal bars. Some individual values (black) and average group values (red) are indicated for ease of comparison. * indicates statistical significant difference by ANOVA. The contribution of each antigen-specific cytokine production is represented on top of each regimen, each pie chart representing the mean of IFN-γ (a), IL-2 (b) and IL-4 (c) producing cell number of the group to the corresponding antigen as indicated in the colored legend. (d) Pie charts of the geometric mean cytokine-producing cells showing the contribution of each cytokine response to all four antigens for each regimen. The ELISPOT assays were performed using recombinant proteins.
After the booster immunizations with HuAd5 or MVA, all three regimens induced strong HCV-specific IFN-γ production in all animals, with numbers of specific IFN-γ producing cells ranging from 140 to 1050 SFU/million cells (Figure 3a). There was no statistical difference between the three groups (Figure 3a). All four antigens contributed to the IFN-γ responses (Figure 3a, pie charts and Supplementary Figure 2), and no statistically significant difference was observed between the groups, even for the apparent higher C-specific response observed after SFV+MVA injections (P=0.08). The apparent higher E1 response in the group DDAA is also not statistically significant, due to the high variability of the E1-specific responses in this group (P=0.19, Supplementary Figure 2). In contrast, and similarly to the lymphoproliferative response after the two MVA injections, SSMM induced a significantly higher IFN-γ response to E2 (with an average of 95 SFU/million cells) as compared with the other vaccine strategies (with averages of 54 and 10 SFU/million cells in the DDAA and in the DDMM groups, respectively, P=0.006, Supplementary Figure 2).
The number of HCV-specific IL-2 secreting cells detected after HuAd5 or MVA injections was lower than for IFN-γ, ranging from 55 to 280 SFU/million cells, and was equivalent between groups (Figure 3b). All four antigens contributed to the HCV-specific IL-2 response in all three groups (Figure 3b, pie charts and Supplementary Figure 3), however again the SFV+MVA regimen (SSMM) induced a significantly higher response to E2 (P=0.0005, Supplementary Figure 3).
After HuAd5 or MVA boosts, all animals elicited IL-4 responses, overall lower than IFN-γ responses (up to 460 SFU/million cells, Figure 3c). The DNA+HuAd5 regime induced lower IL-4 responses than the two other regimen involving MVA (P=0.04), and no response to C. All vaccine antigens contributed to the IL-4 response (except for C in the DDAA group), and again the SFV+MVA vaccinated animals elicited significantly higher responses to E2 (P=0.01, Supplementary Figure 4). In addition, DNA-MVA induced significantly higher E1 responses than the other regimens (P=0.048, Supplementary Figure 4).
Taken together, these results demonstrate that the three prime-boost vaccine regimens were able to induce C, E1, E2 and NS3-specific T-cell responses after the HuAd5 or MVA injections, that included higher IFN-γ production than IL-4 (Figure 3d). Notably, animals receiving two HuAd5 boosts induced lower IL-4 response as compared with the other groups boosted with MVA, suggestive of a stronger Th1 bias (Figure 3d). The group immunized with the SFV+MVA regimen elicited higher E2-specific lymphoproliferation and cytokine responses as compared with the two other vaccine combinations.
Induction of IFN-γ and IL-2 responses to E1 and NS3 peptide pools
The IFN-γ ELISPOT was also performed with overlapping peptides covering E1 and NS3 from the genotype 1b J strain used in the vaccine vectors. Peptide pools (pp) containing 24 peptides for E1 (aa 193–377) and 39 peptides for NS3 (NS3pp1 covering aa 1028–1346 and NS3pp2 covering aa 1340–1659) were used. Only three animals mounted a detectable E1 or NS3 peptide-specific IFN-γ response after DNA injections, while none responded after the SFV injections (data not shown). The low NS3pp-specific responses in DDAA and DDMM had waned by week 12, 6 weeks post last DNA injection (Figure 4c). In contrast, HuAd5 booster injections induced E1 peptide-specific responses in all 4 animals (20–690 SFU/million cells, Figure 4a), while MVA induced lower E1 peptide-specific responses, in 2/4 animals in the DNA+MVA group and in 3/4 animals in the SFV+MVA group (Figure 4a). Both HuAd5 and MVA boosts induced high NS3-specific responses in all animals, ranging from 265 to 1525 SFU/million cells for the HuAd5-induced responses, 340 to 1240 in the DNA+MVA-immunized animals and 215 to 2180 after the SFV+MVA regimen (Figure 4b). The NS3-specific responses were remarkably similar between groups in total levels (Figure 4b), in kinetic (Figure 4c) and in the contribution of the N-and C-terminal ends of the protein (Figure 4d).
Figure 4.

Individual E1 (a) and NS3 (b–d) peptide pool-specific IFN-γ production by PBMCs. IFN-γ production to E1 or NS3 peptide pools as tested by ELISPOT is represented for each animal from each group post HuAd5 or MVA immunization (a and b, respectively). Results are expressed as mean number of spots of triplicate assay per one million cells minus the mean number of spots obtained with the medium cultured cells (also in triplicate assays)+2s.d. (c) The time course of NS3-specific response is shown for the three groups as indicated in the legend, as the geometric mean numbers of cytokine-producing cells per group. Gray arrows indicate the timing of DNA or SFV injections, and black arrows indicate the timing of HuAd5 or MVA injections. (d) Individual responses to NS3pp1 covering aa 1028–1346 and NS3pp2 covering aa 1340–1659 after HuAd5 or MVA immunizations are shown, along with the geometric mean of the group.
The DDAA vaccine induced superior E2-specific antibody response
The capacity of the three different prime/boost combinations to induce HCV envelope-specific B-cell responses was analyzed by ELISA using recombinant E1 and E2 proteins from genotype 1b and neutralization assays. Virtually, no antibody responses to E1 were detected at any time point (Figure 5a). In contrast, E2-specific antibody responses were detected in all three groups (Figure 5b). The DNA or SFV injections were poor inducers of antibody responses. After two HuAd5 injections, strong E2-specific responses were elicited in all four animals previously primed with DNA (Figures 5b and c), while little or no antibody responses were generated in the eight animals that received MVA-booster injections (Figure 5c). None of the sera had neutralizing activity in a E1-E2 pseudoparticle assay (data not shown), using pseudoparticles with 100% homology in the hyper variable region 1 (HVR1 containing the neutralization epitope, Supplementary Figure 5).
Figure 5.

Serum antibody responses to E1 and E2 recombinant proteins. The geometric mean titers+s.d. of the E1- (a) and E2 (b) -specific antibody responses in each of the three groups is represented over time. Black arrows indicate the time points of immunizations (DNA or SFV at weeks 0 and 6, HuAd5 or MVA at weeks 14 and 20). (c) Individual responses to E2 at week 22, each dot representing one animal and the horizontal lines representing the geometric mean of the group. Differences of statistical significance between groups are indicated by *. ELISA was performed with recombinant proteins.
Discussion
In this study, the immunogenicity of three HCV prime-boost vaccine regimens was evaluated in non-human primates. DNA was compared with SFV immunization, followed by immunizations with either adenovirus or MVA in rhesus macaques. All vaccine regimens induced Th1 and IFN-gamma T-cell responses. Only the DNA+HuAd5 regime induced antibody responses to the envelope protein E2, while SFV+MVA induced stronger E2-specific T-cell responses.
The comparison of DNA and SFV was not conclusive. While lymphoproliferative responses were induced in all animals, only a limited subset of animals elicited detectable antigen-specific cytokine responses after DNA or SFV priming. The DNA immunization may not have provided an optimal priming, as use of specific formulations or delivery devices such as electroporation would have induced superior priming.15, 22 Notably, higher E2-specific lymphoproliferation and E2-specific cytokine production was induced after MVA boosting in the group immunized with SFV+MVA as compared with DNA+MVA, indicating that SFV priming was responsible for this effect, even though this did not lead to an antibody response to E2 in this group. This could be due to differences in the constructs rather than the delivery vector: for technical reasons, the E2 gene was inserted in a E1-E2 expressing SFV particle as opposed to a combined C-E1-E2 as in the DNA, adenovirus and MVA vectors. Therefore, differences may be due to different levels of antigen expression from each vector, or whether the antigens are presented on the cell surfaces. Combined expression of three antigens from the same vector may have compromised independent expression, or processing or presentation within antigen presenting cells.
Other questions addressed by this study included: (1) was HuAd5 ‘boosting’ superior to MVA after DNA priming? and (2) were rhesus macaque immune responses predictive of the protective HCV responses observed in chimpanzees challenged with HCV? Regarding the boosting potential of HuAd5, T-cell responses were relatively similar between the DNA+HuAd5 and DNA+MVA groups. Trends were observed indicating higher proliferation with MVA-boosted and higher IFN-γ responses in the HuAd5-boosted groups, but these were not statistically significant. The absence of significant differences may be due to the low number of animals per group, a limitation that is a direct consequence of working with non-human primates. In addition, the respective immunogenicity of adenovirus and MVA vaccines may be partially antigen dependent, as the effect of each antigen immunomodulatory property may also influence the cytokine microenvironment induced by the vector. For example, HuAd5 and MVA have been compared in the context of other antigens: HuAd5 was found superior to poxviruses for induction of IFN-γ T-cell responses to malaria antigens,23 while MVA encoding the MTB 85A antigen induces stronger IFN-γ producing T cells after a BCG prime.24
All three vaccine strategies induced Th1-biased immune responses, with IFN-γ responses largely exceeding the IL-4 responses. A striking difference observed, however, was the lower IL-4 response in the HuAd5-immunized animals, revealing a more Th1 bias. However, while MVA has been shown to induce antibody response to different antigens such as CMV,25 SARS,26 Flu27 and blood-stage malaria,28 in this study only the HuAd5 vector induced a strong but transient antibody response to one of the two envelope proteins. This is in agreement with other recent HCV vaccine developments demonstrating the capacity of adenovirus boosts to support envelope-specific antibodies,29 but confirm that caution should be taken when trying to use results from different immunogens to influence the design of vaccines for other diseases. However, the antibody response elicited by HuAd5 immunization in our study was not neutralizing and was short-lived, maybe inducing antibodies masking the neutralization epitopes as recently described.5, 30 The absence of functional activity or persistence of antibodies using viral vectors was also previously observed.31, 32, 33
This study offers the unique opportunity to compare the immune response elicited by the DNA+MVA regimen between rhesus macaques and chimpanzees. The exact same constructs and regimen were used to immunize four chimpanzees.20 In chimpanzees, immunization induced robust multi-specific immune responses in all four animals, which, following HCV1b exposure, was associated with a drastic reduction in the peak viremia and RNA levels in liver during the acute phase of infection. The vaccine regimen induced IFN-γ and IL-2 responses as detected by ELISPOT, with a similar pattern to the one observed in rhesus macaques (strong IFN- γ responses and lower IL-2 responses, especially to NS3). Contrary to rhesus macaques, however, the chimpanzees mounted more potent IL-4 responses with numbers of antigen-specific IL-4 producing cells comparable to the number of IFN-γ producing cells in three out of the four animals.20 The most striking difference between the two primate species was that DNA+MVA regime induced a robust antibody response in all chimpanzees against both E1 and E2. It is possible that the stronger IL-4 response observed in chimpanzees supported the more potent B-cell response, but the reason for the difference is unknown, and the IL-4 response was not absent in rhesus macaques, just lower than the IFN-γ response. This underlines the difficulty to predict the immunogenicity of a specific vaccine regime based on only one primate species, and to unravel the relative contribution of cellular and humoral immunity to prevent acute or chronic disease. With the availability of highly efficient antiviral drug treatments able to cure a large proportion of HCV infections, the relevance and cost of HCV vaccine development is questioned. However, the treatments are extremely expensive, particularly for developing countries, and thus a vaccine for preventing or treating HCV infection still has an indication in high prevalence countries.34, 35 While the strong restrictions on research involving chimpanzees will restrict efficacy studies, expensive phase II clinical trials will become the only possibility for evaluating the efficacy of vaccine candidates. Therefore, studies identifying approaches that are or not optimal, and investigating the predictive value of non-human primates are highly relevant in this context.
Materials and methods
Animals
Twelve outbred, purpose bred naive male rhesus macaques (Macaca mulatta), 3 to 6 years old, imported from China, seronegative for adenovirus preponderant genus (including serotype 5) and poxviruses (IgG anti-vaccinia virus IgG), were selected following a comprehensive health check, and were randomly assigned into three groups. The study and all experimental procedures were approved by the local animal ethical and use committee and were performed in accordance to the Dutch and international standards for the use of animals in science. Serum and PBMCs were isolated from blood samples collected at regular time points under sedation using aseptic techniques (Becton Dickinson, Breda, The Netherlands, Vacutainer systems), and were not blinded. Body weight, temperature, hematology and biochemistry values were monitored at routine intervals.
Generation of vaccine constructs
The gene inserts encoding the proteins C, E1, E2 and NS3 were based on genotype 1b, J strain of HCV (GenBank: D90208.1)36 for all the vectors used for immunization and described below. All vectors encoding NS3 (DNA, SFV and HuAd5) were described previously.37 The plasmid DNA-C-E1-E2, obtained by inserting the C-E1-E2 (aa 1–746) gene sequences into pgWiz (Gene Therapy System Inc., San Diego, CA, USA), was described previously.20 The SFV-C and SFV-E1-E2 particles were prepared by inserting the corresponding fragments isolated by PCR into an SFV expression vector, and packaging of the recombinant RNA into SFV particles.38 Production of HCV proteins by the rSFV particles was confirmed by immunofluorescence and by in vitro transfection of BHK cells, metabolic labeling (pulse-chase) and immunoprecipitation followed by SDS-PAGE (data not shown). SFV-NS3, SFV-core and SFV-E1-E2 were mixed for immunization. The C-E1-E2 expressing human type 5 replication-defective adenovirus (HuAd5-C-E1-E2) was described earlier.39 Human adenovirus serotype 5 was chosen as a prototypic adenovirus for this study. Both the recombinant MVA-C-E1-E2 and MVA-NS3 vectors were constructed by transient host range selection using the HCV1b structural (amino acids, aa 1–830) and non-structural 3 (aa 1028–1658) genes respectively, as previously described.20 All vectors used for immunization and their transgenes are described in Figure 1.
Peptides and recombinant proteins used for in vitro assays
The C polypeptide (a.a. 1–120) and NS3 helicase (NS3h, aa 1193–1458), derived from HCV genotype 1a,40 were expressed in E. coli and purified on Ni-NTA column as described previously.39 The envelope proteins E1 and E2, deleted from their transmembrane domain, were derived from the HCV1b sequence cloned into pT-alpha vector, and were described previously.20 Fifteen-mer peptides, with overlaps of seven amino acids covering the C, E1, E2 and NS3 sequences (genotype 1b, J strain36), were purchased from Clonestar Biotech (Brno, Czech Republic).
Immunizations
The immunizations with DNA or rSFV consisted of two injections at weeks 0 and 6, and HuAd5 and MVA were administered at weeks 14 and 20 (Figure 1). For each DNA immunization, 2 mg DNA-C-E1-E2 and 2 mg DNA-NS3 dissolved in saline buffer were equally divided and administered both intramuscularly and intradermally. For each SFV immunization, 5 × 109 p.f.u. of each construct dissolved in saline were injected subcutaneously (SC). Animals from group I were boosted subcutaneously twice with 5 × 1010 p.f.u. of each HuAd5 construct. Animals from groups II and III were boosted twice with 5 × 108 p.f.u. MVA of each construct, again administered both intramuscularly and intradermally. The constructs were not mixed, the C, E1 and E2 constructs were injected on the left side and the NS3 on the right. Therefore, the vaccine vectors have been administered via different routes: the route for each vector was selected to induce the strongest immune response. Because routes may impact on magnitude and quality of resulting immune responses, the aim of this study was not to compare the vector’s potency by a single route, but to identify the optimal vaccine regimen, with each vector injected by its optimal route.
Analysis of the humoral immune responses
Quantification of anti-adenovirus antibodies was performed in frozen serum by an independent hospital laboratory (Erasmus MC—Virology, Rotterdam, The Netherlands), with a quantitative enzyme immunoassay (SERION ELISA classic Adenovirus IgG/IgA), detecting antibodies to eight preponderant genus-specific epitopes.
Anti-HCV antibody responses in frozen sera were measured using ELISA with HCV C (0.5 mg ml−1), E1 (4 μg ml−1), E2 (1 μg ml−1) or NS3-helicase proteins (0.5 mg ml−1). ELISA was performed as described previously.20 For each experiment, the cutoff was determined as the mean value plus three times the standard deviations obtained with serum of three random naive serum samples.
The capacity of the frozen sera to neutralize HCV was analyzed using HCV pseudoparticles with E1-E2 glycoproteins of strain CG1b in infection assays on Huh-7 target cells as previously described.41 Control neutralizations were performed using pseudoparticles generated with glycoproteins derived from the feline endogenous retrovirus RD114 (RD114pp).
Analysis of the cellular immune responses by lymphoproliferation and ELISPOT
Lymphoproliferation was measured by 3H-thymidine incorporation as previously described, using fresh PBMCs.42 The results were expressed as SI, calculated as c.p.m. with antigen divided by c.p.m. with medium alone. Lymphoproliferation was considered positive when the SI exceeded 2. Quantification of specific cytokine secreting cells was performed by IFN-γ, IL-2 and IL-4 ELISPOT assays using fresh PBMCs according to the manufacturer’s instructions (U-Cytech, Utrecht, The Netherlands), using concanavaline A (ConA), or recombinant proteins or peptides (5 μg ml−1), or medium alone. The results were expressed as mean spot forming units of triplicate assay per one million PBMCs minus the mean number of spots obtained with the cells cultured in medium alone (also in triplicate assays). Results superior to 5 SFC/million PBMCs were considered as positive response.
Statistical analysis
To compare the immunogenicity of the different vaccine combinations, ANOVA, with P-values calculated by exact methods and two-tailed, was used. Difference was considered as statistically significant when P⩽0.05. Prism 6 for Mac OS X was used.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by European Union contract QLK2-CT-1999-00356, by the Biomedical Primate Research Centre, The Netherlands, and by the Swedish Research Council. We are grateful to Alexander van den Berg for technical assistance with the ICS, to our colleagues from Animal Science Department for technical assistance and expert care of the macaques, to the participants of the European HCVacc Cluster who provided help and support, and to Thomas Darton (Oxford Vaccine Group, UK) for input and advice on the manuscript. CSR is an Oxford Martin fellow and a Jenner Institute Investigator.
Competing interests
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Gene Therapy website
References
- 1.Choo QL, Kuo G, Ralston R, Weiner A, Chien D, Van Nest G. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc Natl Acad Sci USA. 1994;91:1294–1298. doi: 10.1073/pnas.91.4.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forns X, Payette PJ, Ma X, Satterfield W, Eder G, Mushahwar IK. Vaccination of chimpanzees with plasmid DNA encoding the hepatitis C virus (HCV) envelope E2 protein modified the infection after challenge with homologous monoclonal HCV. Hepatology. 2000;32:618–625. doi: 10.1053/jhep.2000.9877. [DOI] [PubMed] [Google Scholar]
- 3.Rollier C, Depla E, Drexhage JA, Verschoor EJ, Verstrepen BE, Fatmi Control of heterologous hepatitis C virus infection in chimpanzees is associated with the quality of vaccine-induced peripheral T-helper immune response. J Virol. 2004;78:187–196. doi: 10.1128/JVI.78.1.187-196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoll-Keller F, Barth H, Fafi-Kremer S, Zeisel MB, Baumert TF. Development of hepatitis C virus vaccines: challenges and progress. Expert Rev Vaccines. 2009;8:333–345. doi: 10.1586/14760584.8.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keck ZY, Girard-Blanc C, Wang W, Lau P, Zuiani A, Rey FA. Antibody response to the hypervariable region-1 interferes with broadly neutralizing antibodies to hepatitis C virus. J Virol. 2016;90:3112–3122. doi: 10.1128/JVI.02458-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verstrepen BE, Boonstra A, Koopman G. Immune mechanisms of vaccine induced protection against chronic hepatitis C virus infection in chimpanzees. World J Hepatol. 2015;7:53–69. doi: 10.4254/wjh.v7.i1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahari H, Feinstone SM, Major ME. Meta-analysis of hepatitis C virus vaccine efficacy in chimpanzees indicates an importance for structural proteins. Gastroenterology. 2010;139:965–974. doi: 10.1053/j.gastro.2010.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fauvelle C, Lepiller Q, Felmlee DJ, Fofana I, Habersetzer F, Stoll-Keller F. Hepatitis C virus vaccines—progress and perspectives. Microb Pathog. 2013;58:66–72. doi: 10.1016/j.micpath.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Swadling L, Klenerman P, Barnes E. Ever closer to a prophylactic vaccine for HCV. Expert Opin Biol Ther. 2013;13:1109–1124. doi: 10.1517/14712598.2013.791277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med. 2014;6:261ra153. doi: 10.1126/scitranslmed.3009185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AV. Viral vectors as vaccine platforms: deployment in sight. Curr Opin Immunol. 2011;23:377–382. doi: 10.1016/j.coi.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature. 2013;501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukh J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology. 2012;142:1279–1287 e3. doi: 10.1053/j.gastro.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Desombere I, Fafi-Kremer S, Van Houtte F, Pessaux P, Farhoudi A, Heydmann L. Monoclonal anti-envelope antibody AP33 protects humanized mice against a patient-derived hepatitis C virus challenge. Hepatology. 2015;63:1120–1134. doi: 10.1002/hep.28428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latimer B, Toporovski R, Yan J, Pankhong P, Morrow MP, Khan AS. Strong HCV NS3/4a, NS4b, NS5a, NS5b-specific cellular immune responses induced in Rhesus macaques by a novel HCV genotype 1a/1b consensus DNA vaccine. Hum Vaccin Immunother. 2014;10:2357–2365. doi: 10.4161/hv.29590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen B, Deng Y, Chen H, Guan J, Chuai X, Ruan L. The novel replication-defective vaccinia virus (Tiantan strain)-based hepatitis C virus vaccine induces robust immunity in macaques. Mol Ther. 2013;21:1787–1795. doi: 10.1038/mt.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lang Kuhs KA, Ginsberg AA, Yan J, Wiseman RW, Khan AS, Sardesai NY. Hepatitis C virus NS3/NS4A DNA vaccine induces multiepitope T cell responses in rhesus macaques mimicking human immune responses [corrected] Mol Ther. 2012;20:669–678. doi: 10.1038/mt.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Arriaza J, Perdiguero B, Heeney J, Seaman M, Montefiori DC, Labranche C. Head-to-head comparison of poxvirus NYVAC and ALVAC vectors expressing identical HIV-1 clade C immunogens in prime-boost combination with Env protein in nonhuman primates. J Virol. 2015;89:8525–8539. doi: 10.1128/JVI.01265-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Himoudi N, Abraham JD, Fournillier A, Lone YC, Joubert A, Op De Beeck Comparative vaccine studies in HLA-A2.1-transgenic mice reveal a clustered organization of epitopes presented in hepatitis C virus natural infection. J Virol. 2002;76:12735–12746. doi: 10.1128/JVI.76.24.12735-12746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rollier CS, Paranhos-Baccala G, Verschoor EJ, Verstrepen BE, Drexhage JA, Fagrouch Z. Vaccine-induced early control of hepatitis C virus infection in chimpanzees fails to impact on hepatic PD-1 and chronicity. Hepatology. 2007;45:602–613. doi: 10.1002/hep.21573. [DOI] [PubMed] [Google Scholar]
- 21.Gerlach JT, Diepolder HM, Jung MC, Gruener NH, Schraut WW, Zachoval R. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 1999;117:933–941. doi: 10.1016/S0016-5085(99)70353-7. [DOI] [PubMed] [Google Scholar]
- 22.Wise MC, Hutnick NA, Pollara J, Myles DJ, Williams C, Yan J. An enhanced synthetic multiclade DNA prime induces improved cross-clade-reactive functional antibodies when combined with an adjuvanted protein boost in nonhuman primates. J Virol. 2015;89:9154–9166. doi: 10.1128/JVI.00652-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reyes-Sandoval A, Wyllie DH, Bauza K, Milicic A, Forbes EK, Rollier CS. CD8+ T effector memory cells protect against liver-stage malaria. J Immunol. 2011;187:1347–1357. doi: 10.4049/jimmunol.1100302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goonetilleke NP, McShane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J Immunol. 2003;171:1602–1609. doi: 10.4049/jimmunol.171.3.1602. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, La Rosa C, Maas R, Ly H, Brewer J, Mekhoubad S. Recombinant modified vaccinia virus Ankara expressing a soluble form of glycoprotein B causes durable immunity and neutralizing antibodies against multiple strains of human cytomegalovirus. J Virol. 2004;78:3965–3976. doi: 10.1128/JVI.78.8.3965-3976.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Z, Zhang L, Qin C, Ba L, Yi CE, Zhang F. Recombinant modified vaccinia virus Ankara expressing the spike glycoprotein of severe acute respiratory syndrome coronavirus induces protective neutralizing antibodies primarily targeting the receptor binding region. J Virol. 2005;79:2678–2688. doi: 10.1128/JVI.79.5.2678-2688.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreijtz JH, Suezer Y, de Mutsert G, van Amerongen G, Schwantes A, van den Brand JM. MVA-based H5N1 vaccine affords cross-clade protection in mice against influenza A/H5N1 viruses at low doses and after single immunization. PLoS One. 2009;4:e7790. doi: 10.1371/journal.pone.0007790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Draper SJ, Cottingham MG, Gilbert SC. Utilizing poxviral vectored vaccines for antibody induction-progress and prospects. Vaccine. 2013;31:4223–4230. doi: 10.1016/j.vaccine.2013.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar A, Das S, Mullick R, Lahiri P, Tatineni R, Goswami D. Immune responses against hepatitis C virus genotype 3a virus-like particles in mice: a novel VLP prime-adenovirus boost strategy. Vaccine. 2015;34:1115–1125. doi: 10.1016/j.vaccine.2015.11.061. [DOI] [PubMed] [Google Scholar]
- 30.Kachko A, Frey SE, Sirota L, Ray R, Wells F, Zubkova I. Antibodies to an interfering epitope in hepatitis C virus E2 can mask vaccine-induced neutralizing activity. Hepatology. 2015;62:1670–1682. doi: 10.1002/hep.28108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koblin BA, Casapia M, Morgan C, Qin L, Wang ZM, Defawe OD. Safety and immunogenicity of an HIV adenoviral vector boost after DNA plasmid vaccine prime by route of administration: a randomized clinical trial. PLoS One. 2011;6:e24517. doi: 10.1371/journal.pone.0024517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keefer MC, Gilmour J, Hayes P, Gill D, Kopycinski J, Cheeseman H. A phase I double blind, placebo-controlled, randomized study of a multigenic HIV-1 adenovirus subtype 35 vector vaccine in healthy uninfected adults. PLoS One. 2012;7:e41936. doi: 10.1371/journal.pone.0041936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Cassan SC, Draper SJ. Recent advances in antibody-inducing poxviral and adenoviral vectored vaccine delivery platforms for difficult disease targets. Expert Rev Vaccines. 2013;12:365–378. doi: 10.1586/erv.13.11. [DOI] [PubMed] [Google Scholar]
- 34.Scott N, McBryde E, Vickerman P, Martin NK, Stone J, Drummer H. The role of a hepatitis C virus vaccine: modelling the benefits alongside direct-acting antiviral treatments. BMC Med. 2015;13:198. doi: 10.1186/s12916-015-0440-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker CM, Grakoui A. Hepatitis C virus: why do we need a vaccine to prevent a curable persistent infection? Curr Opin Immunol. 2015;35:137–143. doi: 10.1016/j.coi.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci USA. 1990;87:9524–9528. doi: 10.1073/pnas.87.24.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rollier C, Verschoor EJ, Paranhos-Baccala G, Drexhage JA, Verstrepen BE, Berland JL. Modulation of vaccine-induced immune responses to hepatitis C virus in rhesus macaques by altering priming before adenovirus boosting. J Infect Dis. 2005;192:920–929. doi: 10.1086/432517. [DOI] [PubMed] [Google Scholar]
- 38.Smerdou C, Liljestrom P. Two-helper RNA system for production of recombinant Semliki forest virus particles. J Virol. 1999;73:1092–1098. doi: 10.1128/jvi.73.2.1092-1098.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arribillaga L, de Cerio AL, Sarobe P, Casares N, Gorraiz M, Vales Vaccination with an adenoviral vector encoding hepatitis C virus (HCV) NS3 protein protects against infection with HCV-recombinant vaccinia virus. Vaccine. 2002;21:202–210. doi: 10.1016/S0264-410X(02)00456-5. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Vitvitski L, Tong S, Trepo C. PCR detection of HCV RNA among French non-A, non-B hepatitis patients. Arch Virol Suppl. 1992;4:234–237. doi: 10.1007/978-3-7091-5633-9_51. [DOI] [PubMed] [Google Scholar]
- 41.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mooij P, Nieuwenhuis IG, Knoop CJ, Doms RW, Bogers WM, Ten Haaft PJ. Qualitative T-helper responses to multiple viral antigens correlate with vaccine-induced immunity to simian/human immunodeficiency virus infection. J Virol. 2004;78:3333–3342. doi: 10.1128/JVI.78.7.3333-3342.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.