

Abstract
1-Substituted and 1,1-disubstituted tetrahydro-β-carbo-lines undergo sodium periodate oxidative ring expansion in the presence of formaldehyde and other aldehydes to form 5,6-dihydro-7H-1,4-methanobenzo[e][1,4]diazonine-2,7(3H)-diones in 30–81% yield. In most cases, the reaction to form this new 6/8/5-tricyclic ring system proceeds with high diastereoselectivity. These benzannulated medium-ring keto imidazolidin-4-ones expand the menu of tetrahydro-β-carboline oxidation products.
Grapical Abstract

Although medium-ring compounds (8 to 11 atoms) feature in a variety of biologically active natural products, these compounds are relatively rare in compound libraries.1 In addition, medium-sized rings are typically difficult to synthesize and have unfavorable transannular strain that is absent in macrocycles and small rings.1–3 However, a variety of ring-expansion approaches show promise in medium-ring synthesis.1,3,4 In particular, we were interested in Witkop5 oxidative ring expansion of cycloalkylindoles to form the corresponding medium-ring keto lactams.
As a first step, we treated tetrahydro-β-carboline 1a6 with 4 equiv of sodium periodate in aq. MeOH (2:1 MeOH/H2O) following the method of Gatta and Misiti.7 Rather than obtain the anticipated 1,4-diazonine-2,7-dione 2a, we determined that tricyclic imidazolidin-4-one 3a had formed by proton NMR data characteristic of a diastereotopic aminal-type methylene: the presence of two doublets at 4.22 and 4.92 ppm with an 11.4 Hz coupling constant (Scheme 1). We surmised that in situ oxidation of MeOH had generated formaldehyde, which then condensed with the secondary amine of 2a to form an iminium ion (vide infra) followed by cyclization8 to form 3a in 21% yield.9 Indeed, one of the most widely used methods to synthesize imidazolidin-4-ones is by reaction of α-amino amides with formaldehyde (and other aldehydes and ketones).10–13 Under the same reaction conditions, adding 4 equiv of 37% aq. formaldehyde increased the yield of 3a to 65%. Additional formaldehyde did not improve the yield. We also found that the reaction was complete within 2 h. Reducing the molar equivalents of sodium periodate from 4 to 3 or 2 reduced the yield to 42% and 25%, respectively. Substituting MeOH with CH3CN decreased the yield to 22% suggesting that a protic solvent is optimal, possibly by accelerating the formation of or stabilizing a polar reaction intermediate (vide infra). Finally, adding 2 equiv of AcOH lowered the yield to 52%.
Scheme 1.

Conversion of Tetrahydro-β-carboline 1a to Tricyclic Imidazolidin-4-one 3a via Intramolcular Ring Closure
To confirm that the reaction had proceeded by initial formation of a keto lactam (1,4-diazonin-2,7-dione), we converted achiral tetrahydro-β-carboline 1b14 to the corresponding 1,4-diazonin-2,7-dione 2b via ozonolysis of its Boc derivative 4 (Scheme 2). Exposure of keto lactam 2b to aq. formaldehyde afforded imidazolidin-4-one 3b in 82% yield. Similarly, exposure of 1b to the optimized sodium periodate reaction conditions furnished 3b in one step in 50% yield.
Scheme 2.

Synthesis of Tricyclic Imidazolidin-4-one 3b from Tetrahydro-β-carboline 1b and 1,4-Diazonin-2,7-dione 2b
As we had observed for 3a, 3b was formed as a single syn diastereomer. An X-ray crystallographic structure of 3b (Scheme 2) revealed that the newly formed aminal methylene carbon of the imidazolidin-4-one was on the same face (syn) as the ketone; the distance between the aminal methylene and ketone was 3.04 Å. We did not observe formation of the anti isomer of 3b.
To assess potential participation of the ketone functional group in this ring-forming reaction, we synthesized 5, an oxime ether derivative of 2b. Exposure of 5 to aq. formaldehyde furnished 6 in high yield (Scheme 3). This result indicates that imidazolidin-4-one (3) formation is independent of the newly formed ketone in the intermediate 1,4-diazonin-2,7-dione periodate oxidation products (2) of the tetrahydro-β-carbo-lines (1).
Scheme 3.

Conversion of 2b to Tricyclic Imidazolidin-4-one Oxime Ether 6 via 7-Methoxyimino-1,4-diazonin-2-one 5
In order to assess potential mechanisms underlying this diastereospecific reaction, we modeled the potential energy surface (PES) for the conversion of 2b to 3b using density functional theory. The ambident amide nucleophile can react at either the O or N atom. Amide anions generally react at nitrogen, whereas in neutral to slightly acidic conditions both O and N attack is observed; in this case, the reaction medium in the presence of periodate is slightly acidic (pH 5).15,16 Models of the hemiaminal conformers derived from 2b showed the close proximity of the amide hydrogen to the hydroxy of the hemiaminal for the syn vs anti conformer. We explored the possibility of an intramolecular proton transfer between the hemiaminal hydroxy group and amide hydrogen to form a zwitterion followed by ring closure to the aminal (Figure S11, Supporting Information). A viable PES was observed; however, syn and anti proton transfers displayed similar kinetics and lead to a high-energy zwitterion intermediate.
Amide tautomerization to an iminol is another route to a nucleophilic amide nitrogen. When both enol and iminol tautomers are possible, the latter is preferred involving an endergonic equilibrium of approximately 14 kcal/mol.17 Starting with the iminol iminium ion C derived from 2b, we modeled ring closure to aminal D for both syn and anti conformers (Figure 1). While the starting anti conformer of C is lower in energy than its syn conformer, ring closure to D is both kinetically and thermodynamically favored for the syn vs anti conformer. The difference in energy between syn and anti transition states is comprised mainly of conformational strain.
Figure 1.

Modeled PES (M062x/aug-cc-pvtz/M062x/cc-pvdz) for the iminol mechanism in the conversion of 2b to 3b.
Using the optimized reaction conditions, we next investigated the scope of the imidazolidin-4-one-forming reaction with respect to the tetrahydro-β-carboline structure (Scheme 4). The known tetrahydro-β-carboline starting materials 1a–1o6,7,14,18–23 were synthesized by Pictet–Spengler reactions following standard protocols. It is evident that the reaction is relatively insensitive to the substitution pattern at the 1-position of the tetrahydro-β-carboline starting materials 1. The reaction failed only for tetrahydro-β-carbolines substituted with a trifluoromethyl group at the 1-position. The unsubstituted 1j formed imidazolidin-4-one 3j in 57% yield. Similarly, a variety of 1-aryl tetrahydro-β-carbolines (1f–1i) formed the corresponding imidazolidin-4-ones 3f–3i in moderate to good yields. Like 3a, 3f–3i were formed as single diastereomers with pseudoequatorial aryl substituents. Similarly, 3d with its 1-cyclohexyl substituent was formed as a single diastereomer. X-ray crystallographic data (Figure S2, Supporting Information) show that cycloalkyl (3d) and aryl (3f, 3i) substituents are on the same face as the aminal methylene carbon of the imidazolidin-4-one. Insight into the diastereoselectivity observed at C3 for products 3a, 3f, 3i, and 3d was explored by modeling the syn ring closure of 3f. The starting iminol, transition state, and aminal product for the exo isomer was preferred to that of the endo isomer (Figure S12, Supporting Information). We propose that a pseudoequatorial preference for the larger arene substituent is the source of the observed diastereoselectivity. Only entries 3c and 3e that lack a significant steric difference of C3 substitutents exhibited diminished diastereoselectivity; these were formed as 2:1 mixtures of diasteromers. For 3c, it was possible to isolate both of the stereoisomers; for 3e, we were only able to isolate the major isomer. For the racemic tetrahydro-β-carboline precursors of these two imidazolidin-4-ones, we note that they contain one (1c) or two (1e) unbranched methylene carbons at the 1-position. The reaction also works well for 1,1-spiroalkyl (3k, 3n) and 1,1-spiroheteroalkyl (3l, 3m) tetrahydro-β-carbolines.
Scheme 4.

Tetrahydro-β-carboline Substrate Scope
As evidenced by the 52% and 65% yield of 3o and 3p from the tryptophan-derived indoles 1o and 1p, ester functional groups on the indole substrate are well-tolerated in this reaction (Scheme 5) and afford the imidazolidin-4-one products as single diastereomers. Next, using tetrahydro-β-carboline 1b as substrate, we determined if we could replace formaldehyde with other aldehydes or ketones. Starting material 1b reacted with acetaldehyde, hexanal, and 3-phenylpropanal to form 3q–3s in 30%, 45%, and 77% yield, respectively (Scheme 6). However, the reaction failed when formaldehyde was replaced with acetone, 1,3-difluoroacetone, or trifluoroacetone. The observed diastereoselectivity for 3q–3s may be due to unfavorable 1,3-diaxial-like interactions between alkyl substituents on carbons 1 and 3 of the “envelope-like” imidazolidin-4-one heterocycle that would arise in the unobserved epimers.
Scheme 5.
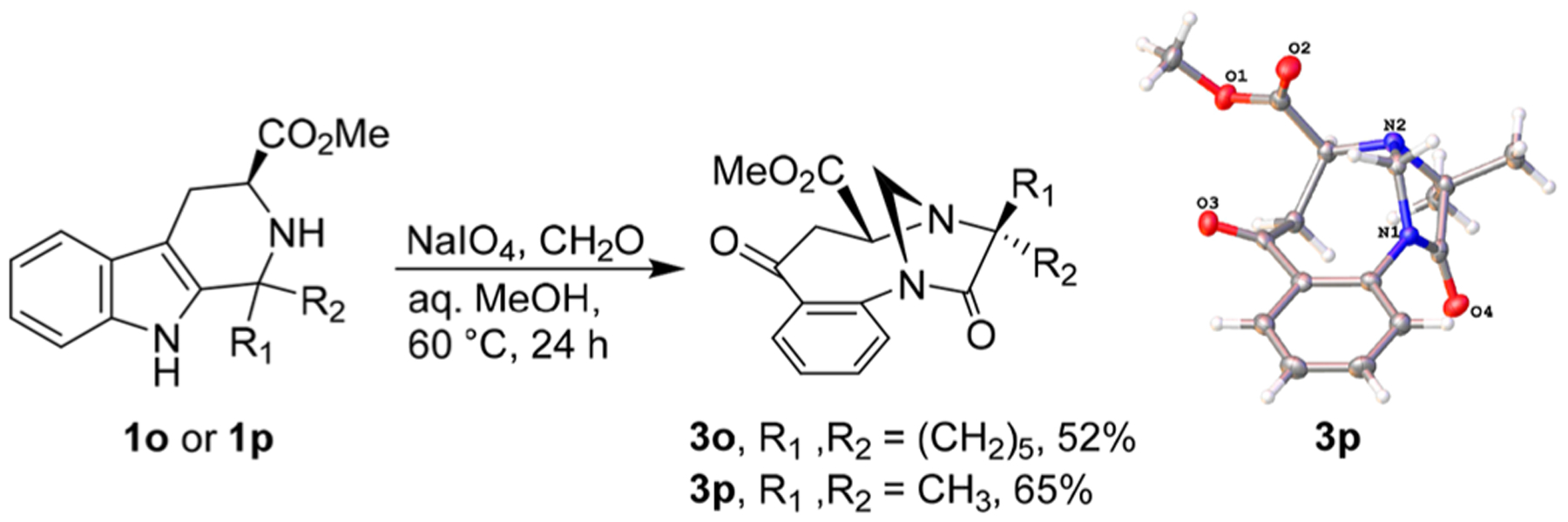
Further Reaction Scope; Thermal Ellipsoid Plot of 3p
Scheme 6.

Aldehyde Scope; Thermal Ellipsoid Plot of 3q
We next began to explore what further chemistry might be possible with these new tricyclic imidazolidin-4-ones (Scheme 7). Imidazolidin-4-one 3a was stable to aq. Na2CO3 but not 1 M aq. NaOH at rt for 24 h; only unidentified products were observed for the latter. Imidazolidin-4-one 3m was converted to oxime 7 in 78% yield. As determined by HMBC data and ROESY crosspeaks between the oxime OH and hydrogens on the neighboring methylene carbons, 7 was obtained as the E isomer. However, all attempts to coax 7 to undergo a Beckmann rearrangement failed.24 The ketone functional group of 3a underwent diastereospecific reduction with NaBH4 to the corresponding benzylic alcohol 8 in 70% yield. We attribute this face-selective reduction to steric hindrance by the aminal methylene carbon of the imidazolidin-4-one preventing approach of borohydride from the other face. Subsequent attempts to deoxygenate 8 with triethylsilanetrifluoroacetic acid failed as did attempts to convert the ketone functional group of 3a to the corresponding methylene with H2/Pd–C in various carboxylic acid solvents; only starting material was isolated. We were also not able to convert the ketone to the geminal difluoride with deoxyfluor. Finally, Schmidt reactions of 3a and other imidazolidin-4-ones failed.
Scheme 7.

Reactions of Imidazolidin-4-ones 3a and 3m; Key 13C–1H HMBC (Black Arrow), 15N–1H HMBC (Blue Arrows), and ROESY (Red Arrows) Correlations of 7; Thermal Ellipsoid Plot of 8
In summary, we discovered that tetrahydro-β-carbolines undergo oxidative ring expansion in the presence of aldehydes to form 5,6-dihydro-7H-1,4-methanobenzo[e][1,4]diazonine-2,7(3H)-diones, a new 6/8/5-tricyclic ring system.25 In most cases, this conformation-directed cyclization proceeds with high diastereoselectivity. These benzannulated medium-ring keto imidazolidin-4-ones26 expand the menu of tetrahydro-β-carboline oxidation products beyond oxindoles and quinolones.5 We anticipate that these compounds will be a useful new chemotype for organic and medicinal chemistry.27,28
EXPERIMENTAL SECTION
Melting points are uncorrected. 1D 1H and 13C NMR spectra were recorded on 400, 500, and 600 MHz spectrometers using CDCl3 or DMSO-d6 as solvents. All chemical shifts are reported in parts per million (ppm) and are relative to internal (CH3)4Si (0 ppm) for 1H and CDCl3 (77.2 ppm) or DMSO-d6 (39.5 ppm) for 13C{1H} NMR. EI GC-MS data were obtained using a quadrupole mass spectrometer with 30 m DB-XLB type columns and a He flow rate of 1 mL/min. Silica gel (sg) particle size 32–63 μm was used for all flash column chromatography. Reported reaction temperatures are those of the oil bath.
General Procedure for Synthesis of Imidazolidin-4-ones 3a–3s.
Tetrahydro-β-carboline 1 (3 mmol), NaIO4 (12 mmol), MeOH (20 mL), and H2O (10 mL) were stirred at rt for 10 min. Then 37% aq. formaldehyde (1.1 mL, 13.3 mmol) was added, and the solution was heated to 60 °C. Once the starting material tetrahydro-β-carboline 1 disappeared via TLC or GC/MS (typically within 2 h), the solution was cooled to rt and water (60 mL) was added. The resultant precipitate was filtered and dried in vacuo. If no solid precipitated, the aq. solution was extracted with DCM (3 × 40 mL) and the combined organic layers were washed with brine and dried with Na2SO4 with subsequent solvent removal. The crude material was purified by silica gel (sg) flash chromatography unless stated otherwise.
3,3-Dimethyl-3,4,5,6-tetrahydro-1H-benzo[e][1,4]-diazonine-2,7-dione (2b). Step 1.
In a procedure adapted from Sigaut et al.,29 a solution of 4 (861 mg, 3.71 mmol) in DCM (40 mL) and CH3OH (10 mL) was exposed to ozone for 20 min at −78 °C. Then (CH3)2S (3 mL) was added followed by water (50 mL). The aq. phase was extracted with DCM (2 × 30 mL). The combined organic layers were dried with brine (50 mL) and MgSO4, and the solvent was removed to give a crude residue which was dissolved in DCM (10 mL) and TFA (10 mL). The solution was stirred at rt for 12 h. The solvent was evaporated in vacuo and the residue was purified by sg chromatography (EA/hexane/TEA 74:25:1) to give 2b (266 mg, 40%) as a white solid. Mp 159–161 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.79 (s, 1H), 7.58–7.50 (m, 1H), 7.38–7.30 (m, 2H), 7.25 (d, J = 7.8 Hz, 1H), 3.21 (s, 1H), 2.81 (s, 1H), 2.57 (s, 3H), 1.23 (d, J = 138.4 Hz, 6H). 13C{1H} NMR (125 MHz, DMSO-d6) δ 204.1, 176.9, 139.6, 137.5, 131.3, 127.5, 127.0, 126.3, 58.0, 46.0, 41.1, 26.4, 22.1. HRMS (ESI-TOF) m/z: [M]+ calcd for C13H16N2O2 232.1212; found 232.1223.
3-(4-Chlorophenyl)-5,6-dihydro-7H-1,4-methanobenzo[e]-[1,4]diazonine-2,7(3H)-dione (3a). Step 1.
The crude precipitate was dissolved in CH3CN (3 mL), and MsOH (1 equiv) was added. After sonication for 10 min, the solution was cooled and filtered. The filtered solid was washed with ether and dried to give the MsOH salt of 3a. Mp 215–216 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.64 (td, J = 7.6, 1.7 Hz, 1H), 7.59 (d, J = 8.3 Hz, 2H), 7.55 (dd, J = 7.7, 1.6 Hz, 1H), 7.51 (d, J = 8.3 Hz, 2H), 7.47 (t, J = 7.5 Hz, 1H), 7.28 (d, J = 7.7 Hz, 1H), 4.91–4.78 (m, 2H), 4.26 (d, J = 11.8 Hz, 1H), 3.71 (t, J = 13.2 Hz, 1H), 3.46 (dd, J = 14.3, 5.3 Hz, 1H), 3.35 (td, J = 12.7, 2.3 Hz, 1H), 2.41 (s, 4H), 2.37 (dd, J = 13.2, 5.0 Hz, 1H). 13C{1H} NMR (125 MHz, DMSO-d6) δ 204.4, 175.2, 138.0, 135.8, 133.2, 133.1, 132.6, 129.1, 128.8, 128.7, 128.4, 128.2, 73.1, 68.1, 56.5, 39.1. Step 2. The MsOH salt of 3a was then sonicated (20 min) in sat. Na2CO3(5 mL), filtered, and washed with water (100 mL) to give 3a (637 mg, 65%) as a white solid. Mp 227–228 °C. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.2 Hz, 2H), 7.63–7.53 (m, 2H), 7.45–7.36 (m, 3H), 7.25 (d, J = 9.7 Hz, 1H), 4.92 (d, J = 11.4 Hz, 1H), 4.46 (s, 1H), 4.22 (d, J = 11.3 Hz, 1H), 3.94 (t, J = 13.6 Hz, 1H), 3.59 (td, J = 12.8, 2.2 Hz, 1H), 3.36 (dd, J = 14.7, 5.4 Hz, 1H), 2.42 (dd, J = 12.8, 4.3 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ 205.2, 175.8, 138.1, 136.1, 134.3, 133.6, 131.8, 129.7, 129.1, 128.7, 128.6, 128.1, 73.5, 69.7, 57.7, 39.6. HRMS (ESI-TOF) m/z: [M]+ calcd for C18H15ClN2O2 326.0822; found 326.0818.
3,3-Dimethyl-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]-diazonine-2,7(3H)-dione (3b).
The reaction product was purified by sg chromatography (EA/hexane 1:1) to give 3b (366 mg, 50%) as a white solid. Mp 141–142 °C. 1H NMR (500 MHz, CDCl3) δ 7.61–7.54 (m, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.29 (d, J = 8.1 Hz, 1H), 5.13 (d, J = 11.5 Hz, 1H), 4.14 (d, J = 11.4 Hz, 1H), 3.63–3.52 (m, 1H), 3.43 (dd, J = 15.1, 5.8 Hz, 1H), 3.30 (td, J = 12.4, 1.9 Hz, 1H), 2.28 (dd, J = 12.2, 5.3 Hz, 1H), 1.47 (s, 3H), 1.44 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ 205.7, 179.3, 138.4, 135.7, 133.4, 129.3, 128.4, 128.0, 72.2, 63.3, 51.7, 39.4, 22.1, 19.9. HRMS (ESI-TOF) m/z: [M]+ calcd for C14H16N2O2 244.1212; found 244.1218. Synthesis of 3b from 2b. 2b (266 mg, 1.15 mmol) was dissolved in a stirred solution of CH3OH (2 mL) and water (1 mL) at rt. Then, CH2O (0.35 mL, 37% aq. solution) was added and the reaction was warmed to 60 °C for 1 h. The solution was cooled, and water (10 mL) was added. The precipitate was filtered and dried to give 3b (229 mg, 82%) as a white solid.
3-Pentyl-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]-diazonine-2,7(3H)-dione (3c).
The crude product 3c (455 mg, 53%) was isolated an orange gel composed of two diastereomers (2:1 via GC/MS and 1H NMR) which were separable and purified by sg chromatography (EA/hexane 3:7) to give 3c (455 mg, 53%). Less polar major diastereomer (300 mg). Mp 181–183 °C. 1H NMR (600 MHz, CDCl3) δ 7.62–7.53 (m, 2H), 7.38 (td, J = 7.6, 1.1 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 5.06 (dd, J = 11.4, 1.3 Hz, 1H), 4.16 (d, J = 11.4 Hz, 1H), 3.78 (t, J = 13.1 Hz, 1H), 3.44 (td, J = 12.6, 2.2 Hz, 1H), 3.23 (dd, J = 9.8, 5.3 Hz, 1H), 3.14 (dd, J = 14.5, 5.1 Hz, 1H), 2.31 (ddd, J = 12.8, 5.7, 1.8 Hz, 1H), 1.88–1.71 (m, 2H), 1.68–1.57 (m, 1H), 1.57–1.45 (m, 1H), 1.43–1.29 (m, 4H), 0.98–0.86 (m, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.2, 178.0, 138.3, 135.9, 133.4, 129.5, 128.5, 128.2, 73.3, 68.9, 57.9, 38.9, 31.5, 28.0, 26.8, 22.5, 14.1. HRMS (ESI-TOF) m/z: [M]+ calcd for C17H22N2O2 286.1681; found 286.1681. More polar minor diastereomer (155 mg). 1H NMR (500 MHz, CDCl3) δ 7.62–7.54 (m, 2H), 7.40 (t, J = 7.7 Hz, 1H), 7.30 (d, J = 7.8 Hz, 1H), 5.05 (d, J = 11.1 Hz, 1H), 4.30 (d, J = 11.0 Hz, 1H), 3.84 (s, 1H), 3.60 (t, J = 13.8 Hz, 1H), 3.34 (dd, J = 15.1, 5.7 Hz, 1H), 3.24 (t, J = 12.8 Hz, 1H), 2.36 (dd, J = 13.1, 5.7 Hz, 1H), 2.13–1.98 (m, 1H), 1.72–1.51 (m, 3H), 1.48–1.27 (m, 4H), 0.92 (t, J = 6.6 Hz, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 204.3, 175.5, 137.9, 135.4, 133.5, 129.4, 128.8, 128.4, 73.0, 63.8, 48.3, 38.6, 31.5, 26.3, 25.3, 22.4, 14.0. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H22N2O2Na+ 309.1579; found 309.1584.
3-Cyclohexyl-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]-diazonine-2,7(3H)-dione (3d).
The crude product was crystallized from ether/DCM/hexane 1:1:8 (10 mL) to give 3d (671 mg, 75%) as a white crystalline solid. Mp 154–156 °C. 1H NMR (600 MHz, CDCl3) δ 7.60–7.54 (m, 2H), 7.37 (td, J = 7.6, 1.3 Hz, 1H), 7.27 (d, J = 7.0 Hz, 1H), 5.01 (d, J = 11.3 Hz, 1H), 4.12 (d, J = 11.4 Hz, 1H), 3.77 (td, J = 12.6, 1.3 Hz, 1H), 3.45 (td, J = 12.6, 2.2 Hz, 1H), 3.09 (dd, J = 14.6, 5.2 Hz, 1H), 2.95 (d, J = 8.6 Hz, 1H), 2.28 (ddd, J = 12.7, 5.6, 1.8 Hz, 1H), 2.05 (d, J = 12.7 Hz, 1H), 2.00–1.91 (m, 1H), 1.89–1.75 (m, 3H), 1.70 (d, J = 12.0 Hz, 1H), 1.36–1.18 (m, 4H), 1.14 (qd, J = 12.1, 3.4 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.5, 177.5, 138.6, 135.9, 133.3, 129.5, 128.7, 128.0, 74.2, 73.7, 58.6, 39.0, 37.5, 31.4, 29.8, 26.2, 26.0, 25.9. HRMS (ESI-TOF) m/z [M]+ calcd for C18H22N2O2 298.1681; found 298.1686.
3,4,5′,6′-Tetrahydro-1H,2′H,7′H-spiro[naphthalene-2,3′-[1,4]methanobenzo[e][1,4]diazonine]-2′,7′-dione (3e).
The reaction product was obtained as a mixture of two diastereomers (2:1 via GC/MS and 1H NMR) which were purified by sg chromatography (hexane to EA) to give one pure isomer and a mixture of the two isomers as white solids (598 mg, 60% combined yield). Less polar single isomer 3e (97 mg). Mp 215–217 °C. 1H NMR (600 MHz, CDCl3) δ 7.62–7.56 (m, 2H), 7.39 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.20–7.10 (m, 4H), 5.17 (d, J = 11.6 Hz, 1H), 4.16 (d, J = 11.5 Hz, 1H), 3.55–3.41 (m, 2H), 3.37 (d, J = 12.3 Hz, 1H), 3.23–3.10 (m, 2H), 2.97–2.84 (m, 2H), 2.41–2.26 (m, 2H), 2.04 (dd, J = 12.4, 5.9 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.7, 178.6, 138.4, 135.7, 135.4, 133.4, 132.5, 129.4, 128.9, 128.8, 128.5, 128.1, 126.5, 126.1, 72.1, 65.1, 51.5, 39.9, 32.5, 26.4, 26.0. HRMS (ESI-TOF) m/z: [M]+ calcd for C21H20N2O2332.1525; found 332.1533. Mixture of two isomers (2:1). Mp 201–202 °C. 1H NMR (600 MHz, CDCl3) δ 7.63–7.54 (m, 2H), 7.41–7.34 (m, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.17–7.09 (m, 4H), 5.20 (d, J = 11.6 Hz, 0.62 H), 5.15 (d, J = 11.5 Hz, 0.36 H), 4.17 (d, J = 11.6 Hz, 0.62 H), 4.14 (d, J = 11.5 Hz, 0.37 H), 3.62–3.35 (m, 3H), 3.30–2.87 (m, 4H), 2.46–2.28 (m, 2H), 2.16–1.99 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.6, 205.4, 178.5, 178.5, 138.4, 138.2, 135.7, 135.6, 135.3, 134.4, 133.4, 133.3, 132.54, 132.45, 129.4, 129.3, 129.3, 128.8, 128.7, 128.7, 128.5, 128.2, 128.0, 126.5, 126.4, 126.2, 126.0, 72.8, 72.0, 65.0, 64.6, 51.5, 51.1, 39.9, 39.7, 33.0, 32.4, 26.4, 26.0, 25.3, 24.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C21H20N2O2Na+355.1422; found 355.1436.
3-(4-Nitrophenyl)-5,6-dihydro-7H-1,4-methanobenzo[e]-[1,4]diazonine-2,7(3H)-dione (3f).
MsOH (1 equiv) was added to the crude precipitate dissolved in CH3CN (3 mL). After sonication for 10 min, the solution was cooled and filtered. The filtered solid was washed with ether and dried. The MsOH salt was partially dissolved in acetone (2 mL), and sat. Na2CO3(10 mL) was added followed by sonication for 10 min and cooling. The resulting solid was filtered and washed with water (100 mL) to give 3f (1.012 g, 72%) as a tan solid. Mp 227–229 °C. 1H NMR (600 MHz, CDCl3) δ 8.28 (d, J = 8.3 Hz, 2H), 7.93 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 7.7 Hz, 1H), 7.59 (t, J = 7.7 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.25 (t, J = 7.0 Hz, 1H), 4.92 (d, J = 11.5 Hz, 1H), 4.56 (s, 1H), 4.27 (d, J = 11.5 Hz, 1H), 3.98 (t, J = 13.7 Hz, 1H), 3.59 (t, J = 12.8 Hz, 1H), 3.39 (dd, J = 15.0, 5.4 Hz, 1H), 2.46 (dd, J = 13.0, 5.4 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 204.7, 174.7, 147.9, 140.4, 137.7, 135.9, 133.5, 129.6, 128.6, 128.4, 127.4, 124.0, 73.5, 69.5, 57.6, 39.4. HRMS (ESI-TOF) m/z: [M]+ calcd for C18H15N3O4337.1063; found 337.1074.
3-(Pyridin-3-yl)-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]-diazonine-2,7(3H)-dione (3g).
The product was purified by sg chromatography (hexane to EA) to give 3g (370 mg, 42%) as pale yellow microcrystals. Mp 198–200 °C. 1H NMR (600 MHz, CDCl3) δ 8.94 (s, 1H), 8.63 (d, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.62 (dd, J = 7.5, 1.8 Hz, 1H), 7.59 (td, J = 7.6, 1.7 Hz, 1H), 7.42 (tt, J = 7.5, 1.4 Hz, 1H), 7.36 (dd, J = 7.8, 4.9 Hz, 1H), 7.29–7.26 (m, 1H), 4.94 (dd, J = 11.4, 1.7 Hz, 1H), 4.53 (s, 1H), 4.26 (d, J = 11.5 Hz, 1H), 3.96 (td, J = 12.9, 1.6 Hz, 1H), 3.60 (td, J = 13.2, 1.9 Hz, 1H), 3.39 (dd, J = 14.2, 4.8 Hz, 1H), 2.45 (ddd, J = 12.9, 5.5, 1.9 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ 205.0, 175.3, 149.8, 148.3, 138.0, 136.1, 134.5, 133.6, 129.7, 129.2, 128.7, 128.6, 123.7, 73.5, 68.5, 57.8, 39.5. HRMS (ESI-TOF) m/z: [M]+ calcd for C17H15N3O2 293.1164; found 293.1151.
3-(Thiophen-2-yl)-5,6-dihydro-7H-1,4-methanobenzo[e]-[1,4]diazonine-2,7(3H)-dione (3h).
The product was purified by sg chromatography (EA/hexane 1:1) to give an orange gel which was crystallized from ether/hexane 1:9 (10 mL) to give 3h (465 mg, 52%) as a yellow solid. Mp 140–141 °C. 1H NMR (500 MHz, CDCl3) δ 7.65–7.53 (m, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.31 (dd, J = 17.8, 6.4 Hz, 2H), 7.26 (d, J = 3.7 Hz, 1H), 7.07 (dd, J = 5.2, 3.5 Hz, 1H), 5.11 (d, J = 11.3 Hz, 1H), 4.64 (s, 1H), 4.23 (d, J = 11.3 Hz, 1H), 3.89 (t, J = 13.2 Hz, 1H), 3.54 (td, J = 12.7, 2.2 Hz, 1H), 3.40 (dd, J = 14.8, 5.4 Hz, 1H), 2.41 (ddd, J = 12.9, 5.6, 1.9 Hz, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ 204.8, 174.5, 137.9, 136.9, 135.9, 133.5, 129.5, 128.5, 128.4, 127.5, 125.8, 125.0, 73.8, 67.6, 57.3, 39.3. HRMS (ESI-TOF) m/z: [M]+ calcd for C16H14N2O2S 298.0776; found 298.0769.
3-Methyl-3-(4-nitrophenyl)-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]diazonine-2,7(3H)-dione (3i).
The product was purified by sg chromatography (EA/hexane 3:7) followed by crystallization from ether/hexane 1:9 (10 mL) to give 3i (738 mg, 70%) as a white solid. Mp 212–213 °C. 1H NMR (500 MHz, CDCl3) δ 8.26 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.64–7.55 (m, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 7.5 Hz, 1H), 4.65 (d, J = 11.5 Hz, 1H), 4.13 (d, J = 11.6 Hz, 1H), 3.86–3.75 (m, 1H), 3.61 (dd, J = 15.4, 5.6 Hz, 1H), 3.45 (t, J = 12.5 Hz, 1H), 2.42 (dd, J = 12.8, 5.7 Hz, 1H), 1.66 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.3, 176.4, 147.8, 147.6, 137.8, 135.7, 133.5, 129.4, 128.5, 128.4, 127.1, 1234.0, 72.1, 69.6, 51.0, 39.7, 22.6. HRMS (ESI-TOF) m/z: calcd for C19H17N3O4 351.1219; found 351.1216.
5,6-Dihydro-7H-1,4-methanobenzo[e][1,4]diazonine-2,7(3H)-dione (3j).
The product was purified by sg chromatography (hexane to EA) to give 3j (469 mg, 57%) as a white solid. Mp 153–155 °C. 1H NMR (600 MHz, CDCl3) δ 7.64–7.54 (m, 2H), 7.39 (td, J = 7.6, 1.2 Hz, 1H), 7.30 (dd, J = 8.2, 1.2 Hz, 1H), 5.00 (dd, J = 11.1, 1.4 Hz, 1H), 4.26 (dd, J = 11.1, 2.0 Hz, 1H), 3.87 (d, J = 16.7 Hz, 1H), 3.72 (ddd, J = 14.5, 12.6, 1.8 Hz, 1H), 3.47 (td, J = 12.7, 2.2 Hz, 1H), 3.30 (dd, J = 16.7, 2.1 Hz, 1H), 3.15 (ddt, J = 14.6, 5.5, 1.7 Hz, 1H), 2.31 (ddd, J = 12.8, 5.6, 1.8 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.1, 175.7, 138.2, 135.8, 133.4, 129.5, 128.7, 128.3, 75.6, 57.9, 56.8, 38.5. HRMS (ESI-TOF) m/z: [M]+ calcd for C12H12N2O2 216.0899; found 216.0900.
5′,6′-Dihydro-2′H,7′H-spiro[cyclohexane-1,3′-[1,4]-methanobenzo[e][1,4]diazonine]-2′,7′-dione (3k).
The reaction product was purified by sg chromatography (EA/hexane 1:1) to give 3k (469 mg, 55%) as a white solid. Mp 110–112 °C. 1H NMR (400 MHz, CDCl3) δ 7.60–7.53 (m, 2H), 7.36 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 6.0 Hz, 1H), 5.09 (dd, J = 11.5, 1.5 Hz, 1H), 4.12 (d, J = 11.4 Hz, 1H), 3.63–3.50 (m, 1H), 3.39 (dd, J = 15.0, 6.0 Hz, 1H), 3.27 (td, J = 12.4, 1.9 Hz, 1H), 2.29 (dd, J = 12.5, 5.7 Hz, 1H), 2.05–1.92 (m, 2H), 1.90–1.63 (m, 5H), 1.61–1.38 (m, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ 206.0, 179.2, 138.5, 135.7, 133.3, 129.3, 128.6, 127.9, 72.3, 65.6, 50.8, 39.7, 29.1, 28.0, 25.6, 22.1, 21.9. HRMS (ESI-TOF) m/z: [M]+ calcd for C17H20N2O2 284.1525; found 284.1538.
5′,6′-Dihydro-2′H,7′H-spiro[oxetane-3,3′-[1,4]methanobenzo[e][1,4]diazonine]-2′,7′-dione (3l).
The reaction product was purified by sg chromatography (hexane to EA) to give 3l (310 mg, 40%) a pale yellow solid. Mp 155–157 °C. 1H NMR (400 MHz, CDCl3) δ 7.66–7.54 (m, 2H), 7.41 (td, J = 7.6, 1.2 Hz, 1H), 7.32 (d, J = 7.9 Hz, 1H), 5.11 (d, J = 7.1 Hz, 1H), 4.93–4.85 (m, 3H), 4.82 (dd, J = 11.3, 1.5 Hz, 1H), 4.17 (d, J = 11.3 Hz, 1H), 3.64 (ddd, J = 14.8, 12.8, 1.8 Hz, 1H), 3.39 (ddt, J = 15.2, 5.8, 1.8 Hz, 1H), 3.01 (td, J = 12.8, 2.2 Hz, 1H), 2.23 (ddd, J = 12.9, 5.8, 1.8 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3) δ 204.7, 175.1, 138.1, 135.7, 133.7, 129.7, 128.7, 128.7, 78.0, 72.3, 71.5, 67.0, 50.5, 39.0. HRMS (ESI-TOF) m/z: [M]+ calcd for C14H14N2O3 258.1004; found 258.1017.
Ethyl 2′,7′-Dioxo-6′,7′-dihydro-2′H,5′H-spiro[piperidine-4,3′-[1,4]methanobenzo[e][1,4]diazonine]-1-carboxylate (3m).
The reaction product was digested with ether/DCM/hexane 1:1:8 (10 mL), and 3m (826 mg, 77%) precipitated as a pale yellow solid that was filtered and dried. Mp 147–149 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.64 (td, J = 7.6, 1.6 Hz, 1H), 7.52 (dd, J = 7.7, 1.6 Hz, 1H), 7.45 (td, J = 7.6, 1.1 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H), 5.09 (d, J = 11.8 Hz, 1H), 4.13 (d, J = 11.8 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.94 (dt, J = 13.8, 4.3 Hz, 1H), 3.77 (dt, J = 13.6, 4.2 Hz, 1H), 3.47–3.33 (m, 2H), 3.26 (s, 1H), 3.16–2.95 (m, 2H), 2.26 (dd, J = 12.6, 5.3 Hz, 1H), 2.00 (d, 1H), 1.92–1.77 (m, 2H), 1.57 (ddd, J = 14.1, 10.8, 4.0 Hz, 1H), 1.20 (t, J = 7.0 Hz, 3H). 13C{1H} NMR (150 MHz, DMSO-d6) δ 204.6, 177.2, 154.5, 138.1, 135.3, 133.0, 128.9, 128.5, 127.9, 71.8, 63.0, 60.7, 50.2, 39.1, 28.0, 26.8, 14.5. HRMS (ESI-TOF) m/z: [M]+ calcd for C19H23N3O4 357.1689; found 357.1690.
5,5′,6,6′,8,9-Hexahydro-2′H,7′H-spiro[benzo[7]annulene-7,3′-[1,4]methanobenzo[e][1,4]diazonine]-2′,7′-dione (3n).
The reaction product was filtered and dried to give 3n (842 mg, 81%) as a white solid. Mp 185 °C dec 1H NMR (600 MHz, CDCl3) δ 7.61–7.53 (m, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 7.9 Hz, 1H), 7.14 (s, 4H), 5.17 (d, J = 11.6 Hz, 1H), 4.19 (d, J = 11.7 Hz, 1H), 3.69 (t, J = 13.4 Hz, 1H), 3.49 (dd, J = 15.1, 5.9 Hz, 1H), 3.43–3.28 (m, 2H), 3.06 (t, J = 12.7 Hz, 1H), 2.83 (s, 1H), 2.70 (dd, J = 14.9, 7.7 Hz, 1H), 2.42–2.29 (m, 2H), 2.19–2.03 (m, 2H), 1.70 (t, J = 13.1 Hz, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 205.8, 178.6, 142.4, 141.6, 138.3, 135.7, 133.3, 129.2, 128.8, 128.7, 128.5, 128.0, 126.4, 126.4, 72.1, 68.9, 51.3, 40.1, 30.2, 30.1, 30.0, 29.2. HRMS (ESI-TOF) m/z: [M]+ calcd for C22H22N2O2 346.1681; found 346.1691.
Methyl (1′R,4′R,5′S)-2′,7′-Dioxo-6′,7′-dihydro-2′H,5′H-spiro[cyclohexane-1,3′-[1,4]methanobenzo[e][1,4]diazonine]-5′-carboxylate (3o).
The reaction product was purified by sg chromatography (EA/hexane 1:1) to give 3o (534 mg, 52%) as a colorless glass. Mp 88–89 °C. 1H NMR (500 MHz, CDCl3) δ 7.60–7.51 (m, 2H), 7.34 (t, J = 7.6 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 5.13 (d, J = 12.4 Hz, 1H), 4.63 (d, J = 12.5 Hz, 1H), 4.10 (s, 1H), 3.85 (s, 3H), 3.38 (dd, J = 12.8, 3.4 Hz, 1H), 2.80 (dd, J = 12.7, 4.9 Hz, 1H), 2.14–1.99 (m, 2H), 1.92–1.84 (m, 1H), 1.83–1.63 (m, 5H), 1.52–1.34 (m, 2H). 13C{1H} NMR (125 MHz, CDCl3) δ 202.0, 178.4, 171.0, 138.4, 135.2, 133.5, 129.3, 128.3, 127.8, 69.8, 66.2, 58.4, 52.3, 40.7, 29.3, 28.2, 25.5, 22.0, 21.8. HRMS (ESI-TOF) m/z: [M]+ calcd for C19H22N2O4 342.1580; found 342.1578.
Methyl (1S,4R,5R)-3,3-Dimethyl-2,7-dioxo-2,3,6,7-tetrahydro-5H-1,4-methanobenzo[e][1,4]diazonine-5-carboxylate (3p).
The reaction product was purified by sg chromatography (EA/hexane 1:1) to give 3p (590 mg, 65%) as an orange solid. Mp 153–155 °C. 1H NMR (600 MHz, CDCl3) δ 7.58 (td, J = 7.7, 1.6 Hz, 1H), 7.54 (dd, J = 7.7, 1.6 Hz, 1H), 7.36 (td, J = 7.6, 1.1 Hz, 1H), 7.30 (dd, J = 7.8, 1.1 Hz, 1H), 5.18 (dd, J = 12.5, 1.3 Hz, 1H), 4.64 (d, J = 12.5 Hz, 1H), 4.14 (t, J = 4.0 Hz, 1H), 3.85 (s, 3H), 3.40 (dd, J = 12.8, 3.5 Hz, 1H), 2.80 (dd, J = 12.8, 4.9 Hz, 1H), 1.54 (s, 3H), 1.44 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 201.8, 178.6, 170.9, 138.2, 135.1, 133.6, 129.4, 128.2, 127.9, 69.8, 64.0, 59.7, 52.4, 40.3, 22.2, 20.0. HRMS (ESI-TOF) m/z: [M]+ calcd for C16H18N2O4302.1267; found 302.1277.
(12S)-3,3,12-Trimethyl-5,6-dihydro-7H-1,4-methanobenzo-[e][1,4]diazonine-2,7(3H)-dione (3q).
Using the general procedure, EtOH (20 mL) and acetaldehyde (586 mg, 13.3 mmol) were substituted for CH3OH and CH2O, respectively, giving the crude product which was purified by sg chromatography (hexane to hexane/EA/TEA 25:74:1) to give 3q (232 mg, 30%) as a pale yellow solid. Mp 136–137 °C. 1H NMR (500 MHz, CDCl3) δ 7.75 (dd, J = 7.7, 1.6 Hz, 1H), 7.60 (td, J = 7.6, 1.6 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 5.36 (q, J = 7.0 Hz, 1H), 3.49 (dd, J = 15.8, 11.7 Hz, 1H), 3.34–3.18 (m, 2H), 2.34 (dd, J = 12.7, 6.6 Hz, 1H), 1.57 (s, 3H), 1.48 (s, 3H), 1.22 (d, J = 7.0 Hz, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ 204.9, 180.6, 137.8, 135.7, 133.8, 130.3, 129.5, 128.6, 71.2, 64.1, 42.9, 39.8, 22.0, 20.8, 14.3. HRMS (ESI-TOF) m/z: [M]+ calcd for C15H18N2O2 258.1368; found 258.1381.
(12S)-3,3-Dimethyl-12-pentyl-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]diazonine-2,7(3H)-dione (3r).
Using the general procedure, isopropanol (20 mL) and hexanal (1.332 g, 13.3 mmol) were substituted for CH3OH and CH2O, respectively, giving a crude reaction product that was purified by sg chromatography (hexane to hexane/EA/TEA 25:74:1) to give 3r (424 mg, 45%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.74 (dd, J = 7.8, 1.6 Hz, 1H), 7.59 (td, J = 7.6, 1.6 Hz, 1H), 7.41 (td, J = 7.6, 1.2 Hz, 1H), 7.23 (dd, J = 7.8, 1.2 Hz, 1H), 5.12 (dd, J = 8.4, 5.7 Hz, 1H), 3.41 (dd, J = 15.3, 11.6 Hz, 1H), 3.29–3.18 (m, 2H), 2.33 (ddd, J = 12.5, 6.5, 1.2 Hz, 1H), 1.65–1.57 (m, 1H), 1.56 (s, 3H), 1.47 (s, 3H), 1.38–1.23 (m, 2H), 1.19–1.08 (m, 4H), 1.08–0.98 (m, 1H), 0.80–0.74 (m, 3H). 13C{1H} NMR (125 MHz, CDCl3) δ 204.6, 180.6, 137.5, 136.0, 133.8, 130.3, 129.5, 128.5, 75.6, 63.9, 42.7, 39.7, 31.3, 28.0, 25.2, 22.2, 21.9, 20.7, 13.8. HRMS (ESI-TOF) m/z: [M]+ calcd for C19H26N2O2 314.1994; found 314.1997.
(12S)-3,3-Dimethyl-12-phenethyl-5,6-dihydro-7H-1,4-methanobenzo[e][1,4]diazonine-2,7(3H)-dione (3s).
Using the general procedure, isopropanol (20 mL) and 3-phenypropanal (1.785 g, 13.3 mmol) were substituted for CH3OH and CH2O, respectively, giving a crude reaction product that was purified by sg chromatography (hexane to hexane/EA/TEA 25:74:1) to give 3s (765 mg, 77%) as a yellow oil. 1H NMR (600 MHz, CDCl3) δ 7.76 (dd, J = 7.8, 1.6 Hz, 1H), 7.60 (td, J = 7.6, 1.6 Hz, 1H), 7.43 (td, J = 7.6, 1.2 Hz, 1H), 7.26–7.19 (m, 3H), 7.19–7.14 (m, 1H), 6.97 (d, J = 7.1 Hz, 2H), 5.11 (dd, J = 8.5, 6.4 Hz, 1H), 3.47 (dd, J = 15.7, 11.4 Hz, 1H), 3.34–3.22 (m, 2H), 2.57 (ddd, J = 14.3, 9.1, 5.7 Hz, 1H), 2.44–2.30 (m, 2H), 2.01–1.86 (m, 1H), 1.69–1.60 (m, 1H), 1.51 (s, 3H), 1.48 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 204.6, 180.6, 139.8, 137.5, 135.9, 134.0, 130.4, 129.6, 128.6, 128.5, 128.4, 126.4, 74.4, 64.0, 42.8, 39.7, 31.5, 29.9, 21.9, 20.7. HRMS (ESI-TOF) m/z: [M]+ calcd for C22H24N2O2348.1838; found 348.1847.
tert-Butyl 1,1-Dimethyl-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indole-2-carboxylate (4).
(Boc)2O (1.733 g, 7.94 mmol) dissolved in DCM (15 mL) was added dropwise to a stirred solution of 1b (1.4 g, 6.98 mmol), TEA (1.449 g, 14.32 mmol) in DCM (20 mL) at rt under N2. The solution was stirred for 12 h, and the ammonium salts were filtered. The organic layer was then washed with sat. K2CO3(30 mL) and water (40 mL). The organic layer was dried with brine (40 mL) and MgSO4 to give 4 as a white solid (861 mg, 41%). Mp 220–221 °C. 1H NMR (600 MHz, CDCl3) δ 7.72 (s, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.16 (td, J = 6.9, 0.7 Hz, 1H), 7.11 (t, J = 7.6 Hz, 1H), 3.80 (t, J = 5.5 Hz, 2H), 2.76 (t, J = 5.4 Hz, 2H), 1.78 (s, 6H), 1.53 (s, 9H). 13C NMR{1H} (150 MHz, CDCl3) δ 155.7, 134.0, 135.9, 126.5, 121.8, 119.6, 118.2, 110.7, 108.6, 80.0, 56.0, 42.4, 28.6, 27.4, 21.6. HRMS (ESI-TOF) m/z: [M]+ calcd for C18H24N2O2 300.1838; found 300.1840.
(E)-7-(Methoxyimino)-3,3-dimethyl-1,3,4,5,6,7-hexahydro-2H-benzo[e][1,4]diazonin-2-one (5).
To a solution of 2b (906 mg, 3.9 mmol) in EtOH (10 mL) was added pyridine (3.093 g, 39.1 mmol), and the mixture was stirred for 10 min. Methoxyamine hydrochloride (654 mg, 7.8 mmol) was added, and the mixture was stirred for 24 h. Sat. NaHCO3 (30 mL) was added, and the mixture was extracted with DCM (3 × 30 mL), washed with brine (100 mL), and dried with Na2SO4 to give 5 (515 mg, 51%) as a white foam. As determined via GC-MS and 1H NMR, 5 was obtained as an 85:15 mixture of E/Z isomers and was used directly for the synthesis of 6. For the major E isomer of 5: 1H NMR (500 MHz, DMSO-d6) δ 9.64 (d, 1H), 7.43 (td, J = 1.5, 7.6 Hz, 1H), 7.32 (dd, J = 1.5, 7.6 Hz, 1H), 7.26 (td, J = 1.0, 7.5 Hz, 1H), 7.22 (d, J = 7.6 Hz, 1H), 3.89 (s, 3H), 3.76 (s, 1H), 2.97 (s, 1H), 2.71 (s, 1H), 2.36 (t, J = 4.9 Hz, 2H), 1.23 (s, 6H). 13C{1H} NMR (125 MHz, DMSO-d6) δ 178.5, 157.7, 137.2, 134.7, 129.4, 128.9, 128.1, 126.1, 61.5, 57.5, 40.4, 32.3, 27.2, 21.1. HRMS (ESI-TOF) m/z: [M]+ calcd for C14H19N3O2 261.1477; found 261.1466.
(E)-7-(Methoxyimino)-3,3-dimethyl-6,7-dihydro-5H-1,4-methanobenzo[e][1,4]diazonin-2(3H)-one (6).
Diazonine oxime ether 5 (515 mg, 1.97 mmol) was dissolved in a stirred solution of CH3OH (2 mL) and water (1 mL) at rt. Then, CH2O (2.0 mL, 37% aq. solution) was added and the reaction was warmed to 60 °C for 24 h. Water (20 mL) was added, and the mixture was extracted with DCM (3 × 25 mL), washed with brine (50 mL), and dried with Na2SO4 to give 6 as a white solid (484 mg, 90%). As determined via GC-MS and 1H NMR, 6 was obtained as a 4:1 mixture of E/Z isomers. Mp 158–161 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.61 (dd, J = 1.1, 7.8 Hz, 0.22H), 7.48 (t, J = 7.0 Hz, 1.64H), 7.44 (dd, J = 1.3, 7.7 Hz, 0.17H), 7.33–7.38 (m, 0.88H), 7.31 (d, J = 7.6 Hz, 0.15H), 7.21 (d, J = 7.9 Hz, 1H), 4.96 (dd, J = 7.7, 11.0 Hz, 1H), 4.27 (dd, J = 11.3, 13.9 Hz, 1H), 3.96 (s, 2.43H), 3.85 (s, 0.60H), 3.34–3.46 (m, 0.42H), 3.15–3.31 (m, 1.49H), 2.86 (dd, J = 4.6, 13.5 Hz, 0.83H), 2.41 (dd, J = 2.9, 15.0 Hz, 0.22H), 2.11 (t, J = 12.9 Hz, 0.84H), 1.97 (t, J = 12.0 Hz, 0.84H), 1.32 (s, 2.24H), 1.31 (s, 0.73H), 1.24 (s, 2.24H), 1.22 (s, 0.71H). 13C{1H} NMR (major E isomer, 125 MHz, DMSO-d6) δ 179.7, 157.2, 136.2, 131.8, 130.1, 129.2, 128.8, 127.4, 68.4, 62.2, 62.0, 48.9, 24.6, 21.9, 19.9. 13C{1H} NMR (minor Z isomer, 125 MHz, DMSO-d6) δ 179.2, 154.8, 135.4, 131.2, 130.0, 128.4, 127.9, 126.2, 67.7, 61.9, 61.7, 53.9, 29.2, 21.7, 20.0. HRMS (ESI-TOF) m/z: [M]+ calcd for C15H19N3O2 273.1477; found 273.1484.
Ethyl (E)-7′-(Hydroxyimino)-2′-oxo-6′,7′-dihydro-2′H,5′H-spiro[piperidine-4,3′-[1,4]methanobenzo[e][1,4]diazonine]-1-carboxylate (7).
To 3m (358 mg, 1 mmol) was added NH2OH (5 mL), and the reaction was heated to 80 °C for 24 h. Water was then added (20 mL), and the resulting solid was filtered and dried in vacuo to give 7 (291 mg, 78%) as a white solid. Mp 204–206 °C. 1H NMR (500 MHz, DMSO-d6) δ 11.67 (s, 1H), 7.49 (dd, J = 7.7, 1.6 Hz, 1H), 7.45 (td, J = 7.6, 1.6 Hz, 1H), 7.35 (td, J = 7.6, 1.3 Hz, 1H), 7.19 (dd, J = 7.7, 1.3 Hz, 1H), 4.95 (d, J = 11.3 Hz, 1H), 4.27 (d, J = 11.2 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 3.98–3.85 (m, 1H), 3.80–3.68 (m, 1H), 3.48–3.37 (m, 1H), 3.23 (dd, J = 15.2, 5.4 Hz, 2H), 3.08 (s, 1H), 2.96 (dd, J = 13.6, 4.7 Hz, 1H), 1.97 (d, J = 13.6 Hz, 1H), 1.88 (t, J = 12.5 Hz, 1H), 1.85–1.70 (m, 2H), 1.55 (ddd, J = 14.1, 10.8, 3.9 Hz, 1H), 1.19 (t, J = 7.1 Hz, 3H). 13C{1H} NMR (125 MHz, DMSO-d6) δ 178.3, 155.5, 154.5, 135.7, 132.7, 129.5, 128.9, 128.6, 127.4, 68.3, 62.6, 60.6, 47.9, 28.2, 24.0, 14.5. HRMS (ESI-TOF) m/z: [M]+ calcd for C19H24N4O4 372.1798; found 372.1796.
3-(4-Chlorophenyl)-7-hydroxy-6,7-dihydro-5H-1,4-methanobenzo[e][1,4]diazonin-2(3H)-one (8).
3a (121 mg, 0.37 mmol), NaBH4 (35 mg, 0.93 mmol), and CH3OH (10 mL) were stirred at rt for 24 h. Sat. Na2CO3 (15 mL) was added, and the precipitate was filtered and dried to afford 8 (73 mg, 60%) as a white solid. Mp 230–231 °C. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.2 Hz, 2H), 7.43–7.32 (m, 5H), 7.23–7.18 (m, 1H), 5.08 (d, J = 9.1 Hz, 1H), 4.98 (d, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.30 (s, 1H), 4.05 (ddd, J = 14.6, 12.6, 1.8 Hz, 1H), 3.05 (dd, J = 15.0, 4.9 Hz, 1H), 2.43–2.33 (m, 1H), 2.20–2.09 (m, 1H), 2.05 (s, 1H). 13C{1H} NMR (125 MHz, CDCl3) δ 177.8, 139.4, 135.6, 133.8, 132.9, 132.7, 129.9, 129.7, 128.8, 128.7, 128.1, 71.4, 70.1, 69.8, 51.7, 27.2. HRMS (ESI-TOF) m/z: [M]+ calcd for C18H17ClN2O2 328.0979; found 328.0969.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the U.S. National Institutes of Health (AI116723-01) for financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.9b03402.
NMR spectra for 1a–1o and all new compounds; computational data for the production of Figures 1, S11, and S12 (PDF)
X-ray crystallographic data (CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.9b03402
The authors declare no competing financial interest.
Contributor Information
Douglas E. Stack, University of Nebraska at Omaha, Omaha, Nebraska.
Jonathan L. Vennerstrom, University of Nebraska Medical Center, Omaha, Nebraska.
REFERENCES
- (1).Bauer R; Wenderski TA; Tan DS Biomimetic diversity-oriented synthesis of benzannulated medium rings via ring expansion. Nat. Chem. Biol 2013, 9, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Roxburgh CJ Syntheses of medium sized rings by ring expansion reactions. Tetrahedron 1993, 49, 10749–10784. [Google Scholar]
- (3).Donald JR; Unsworth WP Ring-expansion reactions in the synthesis of macrocycles and medium-sized rings. Chem. - Eur. J 2017, 23, 8780–8799. [DOI] [PubMed] [Google Scholar]
- (4).Stach H; Hesse M Synthesis of macrocyclic compounds by ring enlargement. Tetrahedron 1988, 44, 1573–1590. [Google Scholar]
- (5).(a) Witkop B; James B; Patrick JB; Rosenblum MJ Ring effects in autoxidation. A new type of Camps reaction. J. Am. Chem. Soc 1951, 73 (6), 2641–2647. [Google Scholar]; (b) Mentel M; Breinbauer R The Witkop-Winterfeldt oxidation of indoles. Curr. Org. Chem 2007, 11, 159–176. [Google Scholar]
- (6).Wang L-N; Shen S-L; Qu J Simple and efficient synthesis of tetrahydro-β-carbolines via the Pictet-Spengler reaction in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP). RSC Adv. 2014, 4, 30733–30741. [Google Scholar]
- (7).Gatta F; Misiti D Sodium periodate oxidation of tetrahydro-β-carboline derivatives. J. Heterocycl. Chem 1989, 26, 537–539. [Google Scholar]
- (8).Johnson CD Stereoelectronic effects in the formation of 5-and6-membered rings: The role of Baldwin’s rules. Acc. Chem. Res 1993, 26, 476–482. [Google Scholar]
- (9). The corresponding carboline and other unidentified reaction products formed by overoxidation reactions were observed by GCMS.
- (10).Blackmore TR; Thompson PE Imidazolidin-4-ones, their syntheses and applications. Heterocycles 2011, 83, 1953–1975. [Google Scholar]
- (11).Chen F-L; Sung K An exception of Eschweiler-Clarkemethylation: Cyclocondensation of A-aminoamides with formaldehyde and formic acid. J. Heterocycl. Chem 2004, 41, 697–700. [Google Scholar]
- (12).Wei L; Gan X; Zhong J; Alliston KR; Groutas WC Noncovalent inhibitors of human leukocyte elastase based on the 4-imidazolidinone scaffold. Bioorg. Med. Chem 2003, 11, 5149–5153. [DOI] [PubMed] [Google Scholar]
- (13).Ho B; Crider AM; Stables JP Synthesis and structure-activity relationships of potential anticonvulsants based on 2-piperidinecarboxylic acid and related pharmacophores. Eur. J. Med. Chem 2001, 36, 265–286. [DOI] [PubMed] [Google Scholar]
- (14).Horiguchi Y; Nakamura M; Kida A; Kodama H; Saitoh T; Sano T A facile synthesis of 1,1-disubstituted 1,2,3,4-tetrahydro-β-carbolines via trifluoroacetic acid catalyzed Pictet-Spengler reaction using titanium(IV) isopropoxide as an imination reagent. Heterocycles 2003, 59, 691–705. [Google Scholar]
- (15).Challis BC; Challis JA Reactions of the carboxamide group In The Chemistry of Amides; Zabicky J, Ed.; Interscience: London, 1970; pp 731–858. [Google Scholar]
- (16).Stirling CJM Intramolecular reactions of amides. Part II. Cyclisation of amides of ω-bromo-carboxylicacids. J. Chem. Soc 1960, 0, 255–262. [Google Scholar]
- (17).Sklenák S; Apeloig Y; Rappoport Z Calculated pKEnol values for enols of carboxylic acid derivativesHC = C(OH)X (X = OH, NH2, NMe2, OMe, OCHO, F, Cl, Br). J. Am. Chem. Soc 1998, 120, 10359–10364. [Google Scholar]
- (18).Domany G; Gizur T; Barta-Szalai G; Schon I; Szantay C Jr; Hegedus B Synthesis of new guanidine derivatives from 2′,3′,4′,9′-tetrahydrospiro[piperidine-4,1′-[1H]pyrido[3,4-b]indole]. J. Heterocycl. Chem 1994, 31, 1657–1665. [Google Scholar]
- (19).Johnson KW; Nelson DLG; Phebus LA 5-HT-2 antagonists, and preparation thereof, for treating or ameliorating the symptoms of common cold or allergic rhinitis. U.S. Patent US5869497, 9 February 1999. [Google Scholar]
- (20).Eagon S; Anderson MO Microwave-assisted synthesis of tetrahydro-β-carbolines and β-carbolines. Eur. J. Org. Chem 2014, 2014, 1653–1665. [Google Scholar]
- (21).Beasley BO; Alli-Balogun A; Clarkson GJ; Shipman M Pictet-Spengler reactions of oxetan-3-ones and related heterocycles. Tetrahedron Lett. 2014, 55, 541–543. [Google Scholar]
- (22).Spindler A; Stefan K; Wiese M Synthesis and investigation of tetrahydro-β-carboline derivatives as inhibitors of the breast cancer resistance protein (ABCG2). J. Med. Chem 2016, 59, 6121–6135. [DOI] [PubMed] [Google Scholar]
- (23).Murai K; Kobayashi T; Miyoshi M; Fujioka H Oxidative rearrangement of secondary amines using hypervalent iodine(III) reagent. Org. Lett 2018, 20, 2333–2337. [DOI] [PubMed] [Google Scholar]
- (24). A Beckmann rearrangement with the oxime derivative of 3a also failed.
- (25). In a SciFinder Scholar substructure search of this imidazolidin-4-one 6/8/5-tricyclic ring system, only one other ring system was identified: a hexahydropyrroloimidazoindole 6/5/5/5-tetracyclic ring system described in:Gonzalez-Vera JA; Garcia-Lopez MT; Herranz R Unprecedented stereospecific synthesis of a novel tetracyclic ring system, a hybrid of tetrahydropyrrolo[2,3-b]indole and tetrahydroimidazo[1,2-a]indole, via a domino reaction upon a tryptophan-derived amino nitrile. Org. Lett 2004, 6, 2641–2644.
- (26). Crystallographic data (excluding structure factors) for 2b (CCDC 1946931), 3b (CCDC 1946932), 3d (CCDC 1946933), 3f (CCDC 1946934), 3i (CCDC 1946935), 3p (CCDC 1946930), 3q (CCDC 1946936), and 8 (CCDC 1946944) have been deposited with the Cambridge Crystallographic Data Centre. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033 or deposit@ccdc.cam.ac.uk).
- (27).Drabina P; Horakova E; Ruzickova Z; Sedlak M Henry reaction catalyzed by new series of imidazolidine-4-one Cu-complexes. Tetrahedron: Asymmetry 2015, 26, 141–147. [Google Scholar]
- (28).Zhao X; Liang S; Fan X; Yang T; Yu W Iron-catalyzed intramolecular C-H amination of α-azidyl amides. Org. Lett 2019, 21, 1559–1563. [DOI] [PubMed] [Google Scholar]
- (29).Sigaut F; Didierdefresse B; Levy J Indole as a tool in synthesis. Algorithmicconstruction of a family of compounds with all ring sizes ranging from 10 to 16. Tetrahedron 2000, 56, 9641–9646. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.