

Abstract
The molecular mechanisms mediating herpes simplex virus type 1 (HSV-1) gene silencing during latent infection are not clear. Five copies of early growth response gene 1 (Egr-1) binding elements were identified in the intron of HSV-1 ICP22 (infected cell protein No. 22) gene, leading to the hypothesis that Egr-1 binds to the viral genome and regulates the viral gene expression. Transient co-transfection assays indicated that Egr-1 negatively regulated the transcription of both full-length and intron-removed ICP22 promoters. The same assays also revealed that Egr-1 repressed ICP4 (infected cell protein No. 4) promoter activity in a dose-dependent manner but showed less inhibition when the intron was removed. Histone deacetylation was not involved in this regulation since histone deacetylase inhibitor trichostatin A did not exhibit any effect on Egr-1-mediated repression. Chromatin immunoprecipitation assays showed that Egr-1 reduced the binding of Sp1 to the promoters and that the co-repressor Nab2 (NGFI-A/EGR1-binding protein) was recruited to the proximity of ICP4 in the presence of Egr-1. These results suggested that the multifunctional transcription factor Egr-1 can repress HSV-1 immediate-early gene expression through the recruitment of co-repressor Nab2 and reduction of Sp1 occupancy, and thus may play a critical role in HSV-1 gene silencing during latency.
Keywords: HSV-1, latency, Egr-1, Sp1, Nab2, transcription regulation
Introduction
Herpes simplex virus type 1 (HSV-1) infection is a very prevalent disease affecting 90% of the adult population in the United States 1. The infection commonly appears around the eyes, esophagus, trachea, and brain. Upon infecting the host through epithelium, the virus enters nerve cells in the lower layers of skin tissue. Toward the end of the lytic infection, virions are transported by axons from the skin to ganglia, where the virus persists in a latent episomal form until it is re-activated 2. During latency, the viral genome is associated with nucleosomes, acute gene expression is repressed, and no viral peptide is detected 3. The molecular mechanisms mediating viral latency and silencing are not understood.
Examination of the HSV-1 genome for possible cis-elements that could contribute to gene silencing revealed the presence of five copies of the Egr-1 binding element CGCCC(A/C)CGC 4 in the HSV-1 ICP22 (infected cell protein No. 22) intron (Box 1). Egr-1 (also named NGFI-a, Zif268, or Krox24) belongs to the family of early growth response (EGR) transcription factors 4, 5, 6, 7. It was first identified from the differentiated PC-12 cells stimulated by the nerve growth factor (NGF) 6. Egr-1 has been shown to control a variety of divergent cellular responses ranging from survival to apoptosis, growth to growth arrest, differentiation to transformation, etc. 8. Egr-1 and other related EGR proteins such as Krox20, NGFI-c, and Egr-3 are now characterized as zinc-finger DNA-binding proteins induced by a wide variety of extracellular stimuli 5. Previous reports suggested that the expression of EGR proteins is correlated with viral infections such as gammaherpesvirus Epstein-Barr virus 9, 10, murine coronavirus 11, and HSV-1 12.
We hypothesized that Egr-1 can regulate the transcription of ICP22 and ICP4 (infected cell protein No. 4). To test the hypothesis, nucleosomal-associated episomal plasmids were constructed, containing full-length ICP4 or ICP22 (pICP4 or pICP22) promoters as well as intron-deleted promoters (pICP22-ID or pICP4-ID) (Figure 1). Our co-transfection experiments showed that Egr-1 repressed the activity of both ICP22 and ICP4 promoters. However, the results from intron-deleted mutant promoters indicated that the Egr-1-mediated inhibition was reduced in ICP4-ID, but not in ICP22-ID. Histone deacetylase (HDAC) inhibitor trichostatin A (TSA) failed to reverse Egr-1 mediated inhibition, suggesting that histone deacetylation is not involved in the regulation. Finally, our chromatin immunoprecipitation (ChIP) assays revealed that the co-repressor Nab2 (NGFI-A/EGR1-binding protein) was recruited to the proximity of ICP4 promoter, and that the occupancy of Sp-1 was reduced in the presence of Egr-1. These results raise the possibility that Egr-1 plays roles in regulating HSV-1 gene expression and controls viral latency and reactivation.
Figure 1.
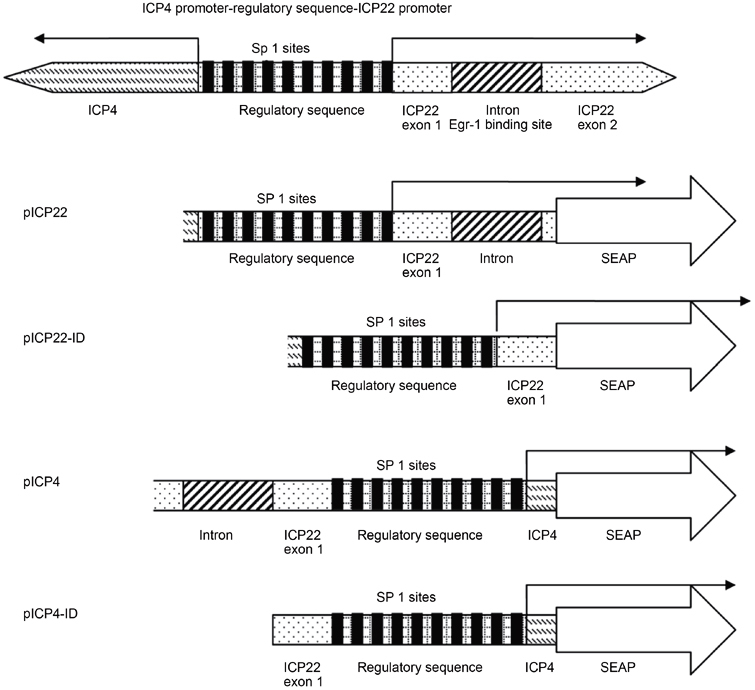
Schematic representation of promoter regions in plasmids. All plasmids are constructed based on the pREP-SEAP vector, and promoters were introduced into the multiple cloning sites. ICP4 and ICP22 promoters share the same regulatory sequence but direct transcription in opposite orientations. The Sp1 binding sites in the regulatory sequence are shown in black boxes.
Box 1: Comparison of Egr-1 consensus sequence to HSV-1 Egr-1 binding elements in ICP22 intron.

Materials and Methods
Construction of plasmids and PCR amplification
The episomal vector pREP-SEAP is based on the vector pREP-7 (Invitrogen, Carlsbad, CA, USA) with the removal of RSV promoter and the introduction of reporter gene SEAP. In short, the RSV promoter was removed from pREP-7 by digestion with XbaI and PvuII. A 0.3-kb XbaI-PvuII fragment from pcDNA-3 (Invitrogen) containing the bovine growth hormone polyadenylation signal was ligated to the XbaI-PvuII vector fragment of pREP-7 to block the expression from cryptic upstream promoters. This plasmid was designated pREP-bghpA. The gene encoding a secreted form of alkaline phosphatase (SEAP) was excised from pSEAP2-Basic (BD Biosciences, San Jose, CA, USA) by digesting the plasmid with NotI and SalI and ligating the 2.0 kb fragment containing the gene into NotI- and SalI-digested pREP-bghpA to form pREP-SEAP. To construct HSV-1 promoter plasmids, DNA fragments containing the ICP4 and ICP22 promoter/leader region were amplified by PCR and cloned in pCR2.1-TOPO (Invitrogen). The plasmid pAA3, containing the ICP4 and ICP22 genes, was used as a template. PCR was performed using primer 8834 (5′-GGT ACC CCA GAA AAC GTC CCG GAG-3′) and primer ICP4B5 (5′-GGT ACC CGA GCG TCT GAC GGT CTG TCT C-3′) at 55 °C. The 1208 bp PCR fragment, containing the regulatory sequence of ICP22 and ICP4 promoters with opposite orientation, was inserted into the KpnI site of the pREP-SEAP. As a result, there are two types of reporter plasmids. The resulting plasmid with SEAP driven by ICP22 was named pICP22. The pICP4 contains SEAP, which is driven by the ICP4 promoter (Figure 1). To construct the intron-deleted mutant promoter, primer 9104 (5′-GAC GTT CCC CCG TGC AAA AAA GGC GG-3′) and primer ICP4 B5 were used. The resulting plasmid with SEAP driven by ICP22 promoter without intron was named as pICP22-ID. The pICP4-ID has SEAP reporter driven by the intron-less ICP4 promoter. The map of plasmids is summarized in Figure 1. The Egr-1 expression vector pJDM948 was a generous gift provided by Dr Jeffery Milbrandt at Washington University in St Louis, MO, USA. The plasmid pH4-2, based on pUC19, contains the Hind III restriction fragment (non-prototype structure) covering the entire long and short internal repeats. This plasmid contains the genes of ICP4, ICP0, and LAT (latency-associated transcripts). Plasmid pGL3-basic (Promega, Madison, WI, USA) was used as a control for transfection.
Cell culture and transfection
The HEK293 cell line was purchased from the American Type Culture Collection (ATCC) and maintained in DMEM medium supplemented with 10% fetal bovine serum. It was used because of its high transfection efficiency and relationship of phenotype to neurons 13. For transfection, cultures of cells were prepared for transfection by plating 5×105 cells in 60 mm culture dishes. After overnight incubation, cells were transfected with the plasmid DNA complexed with Superfect Transfection reagent (Qiagen, Valencia, CA, USA), according to the procedures recommended by the manufacturer. PC-12 cell line was a gift from Dr Ying-hsiu Su (Drexel University, Doylestown, PA, USA). Cells were maintained using the culture condition provided by ATCC. TSA was purchased from Sigma (Cat#: T8552, St Louis, MO, USA).
Western blot analysis
HEK293 cells (2×106) were transfected with 1 μg of pJDM948. Protein extract was subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The blots were blocked using PBS with 5% (wt/vol) non-fat dry milk and washed in PBS. Goat anti-Egr1 polyclonal antibody (Chemicon, Temecula, CA, USA) was used at a dilution of 1:1 000. The membranes were washed as before and visualized using enhanced-chemiluminescence reagents (Pierce), according to the manufacturer's protocol. Anti-α-tubulin mouse antibody (Calbiochem, Cat#: CP06, San Diego, CA, USA) was added at a dilution of 1:10 000 as a control.
Reverse transcriptase PCR (RT-PCR)
For RT-PCR, total RNA from cultured cells was isolated by Trizol reagent (Invitrogen). RT-PCR was performed using Superscript One-Step RT-PCR (Invitrogen) with 0.5 μg of total RNA and two primer sets per reaction tube: one set for the actin as a control and the other for the experimental sample. Their sequences are as follows: Actin: 5′-ATT CCT ATG TGG GCG ACG AG-3′ and 5′-TGG ATA GCA ACG TAC ATG GC-3′;
LAT: 5′-CGG CGA CAT CCT CCC CCT AAG C-3′ and 5′-GAC AGA CGA ACG AAA CGT TCC G-3′; ICP4: 5′-CGA CAC GGA TCC ACG ACC C-3′ and 5′-GAT CCC CCT CCC GCG CTT CGT CCG-3′; ICP0: 5′-TTC GGT CTC CGC CTG AGA GT-3′ and 5′-GAC CCT CCA GCC GCA TAC GA-3′. The RT-PCR reaction was carried out at 45 °C for 20 min followed by 25 cycles of 94 °C for 30 s, 55 °C for 30 s, and 68 °C for 30 s. The RT-PCR products were analyzed by 2% agarose gel electrophoresis. The Kodak Gel-Logic 100 system was utilized for quantification.
Reporter SEAP assays
The promoter activity was analyzed by measuring SEAP reporter gene activity using GreatEscape kit according to the manufacturer's protocol (BD Biosciences). The chemiluminescent signal was measured by a 20/20n Luminometer (Turner Biosystems, Sunnyvale, CA, USA) after a 10-min incubation. Each construct was tested by using a minimum of three replicates and the data were collected and normalized as SEAP units relative to the controls.
ChIP assays
Chromatin was isolated using a modified protocol as described previously 14. Briefly, the cell monolayers were treated with 1% formaldehyde solution for 10 min at room temperature. Cells were then scraped into 15 ml tubes and subjected to the nuclei isolation protocol as described elsewhere 15. The lysed samples were centrifuged for 10 min at 13 000 r.p.m. with a refrigerated Eppendorf microfuge at 4 °C, and the supernatant was diluted 10-fold with dilution buffer (25 ml; 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, and 167 mM NaCl) containing protease inhibitor as described above. Immunoprecipitation was then performed with a ChIP assay kit essentially as described by the manufacturer (Chemicon) with antibodies against Egr-1 (Chemicon), Nab2 (Cat#: ab11987, Abcam Ltd, Cambridge, UK), or Sp1 (Chemicon). To analyze immunoprecipitated DNA, PCR amplification was performed with primers (5′-TGG GGT GGG CGG GTC TTT C-3′ and 5′-ACG AAC GAC GGG AGC GGC TG-3′). For each reaction, a 50-μl, 25-cycle PCR was carried out in the presence of 10 pmol of the primers. Each cycle consisted of 1 min at 94 °C, 40 s at 50 °C, and 1 min at 72 °C. Each experiment was repeated at least twice. The products were analyzed by 2% agarose gel electrophoresis.
Results
Identification of Egr-1 binding elements within the ICP22 intron
Examination of transcription factor binding sites on HSV-1 immediate-early gene (IE) promoters revealed that putative Egr-1 binding elements (EBEs) are repeated five times within the intron from 132 400 to 132 508 according to the HSV-1 complete sequence (accession number X14112; Box 1). The HSV-1 ICP22 and ICP4 shared the regulatory sequences, and these EBEs are located about 300 bp downstream of ICP22 transcription initiation site and 970 bp upstream of ICP4 transcription initiation site. The promoters of ICP4 and ICP22 and their intron-deleted versions were introduced into the episomal reporter vector pREP-SEAP (Figure 1).
Over-expression of Egr-1 in HEK293 cells
To analyze the endogenous expression and over-expression of Egr-1 in HEK293 cells, we performed Western blotting using the anti-Egr-1 antibody. The data revealed that cells transfected with the control plasmid showed no signal, and cells transfected with 0.5 μg of pJDM948 exhibited significant expression of Egr-1 (Figure 2). Immunoblotting using anti-α-tubulin antibody detected similar signal intensity, demonstrating that equal amounts of cellular extracts were applied in the experiments. These results indicated that endogenous Egr-1 level is very low and Egr-1 was over-expressed upon pJDM948 transfection in HEK293 cells.
Figure 2.
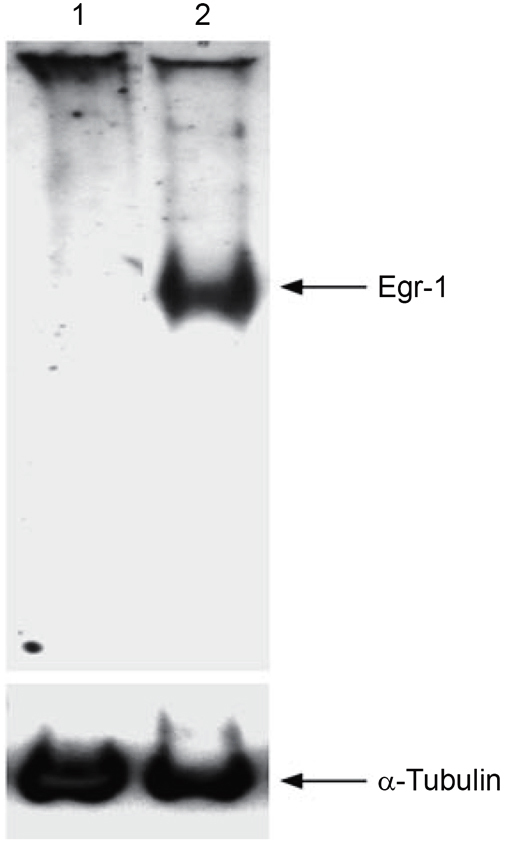
Over-expression of Egr-1 in HEK293 cells. HEK293 cells were transfected with the Egr-1 expression vector pJDM948. Lane 1: Cells transfected with the control plasmid. Lane 2: Cells transfected with pJDM948. Immunoblot was performed using anti-Egr-1 antibody and anti-α-tubulin antibody as a control.
Egr-1 repressed ICP4 expression but had no regulatory effect on the gene expression of LAT and ICP0
To test the regulatory effect of Egr-1 on HSV-1 genes, co-transfection of plasmid pH4-2 (see Materials and Methods) with or without pJDM948 was performed in HEK293 cells. The RT-PCR data showed that Egr-1 had only a mild effect (∼24% reduction) on the LAT promoter activity (Figure 3). The ICP0 promoter activity was not affected by Egr-1 (Figure 3). In contrast, the activity of ICP4 promoter was significantly repressed by Egr-1 (65% reduction; Figure 3). These results suggested that Egr-1 displayed regulatory effect on the ICP4 gene expression but not on LAT and ICP0 expression.
Figure 3.
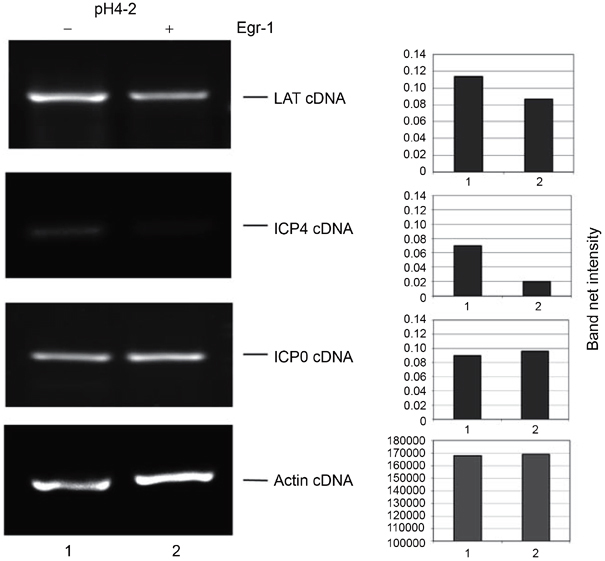
Regulatory effect of Egr-1 on HSV-1 promoters. HEK293 cells were co-transfected with pH4-2 and pJDM948. Lane 1: Cells tarnsfected with pH4-2 and the control plasmid (molar ratio, 1:1). Lane 2: Cells transfected with pH 4-2 and pJDM948 (molar ratio, 1:1). The cDNA of LAT, ICP4, ICP0, and actin were labeled. The quantitative analysis of RT-PCR signals was performed by Kodak Gel Logic 100 system.
Egr-1 repressed the promoter activity of ICP22 and ICP4
To confirm the regulatory effect of Egr-1 on ICP4, transient co-transfection was performed using pJDM948 and reporter plasmids pICP4 or pICP4-ID in HEK293 cells. The SEAP assays showed that Egr-1 preferentially repressed the full-length ICP4 promoter compared to the intron-deleted version. At molar ratios of 1:0.5 and 1:1.5 (reporter plasmid: pJDM948), 51.2% and 82.5% of the full-length promoter activities were repressed, respectively (Figure 4A, bars 1-3). However, under similar conditions Egr-1-mediated inhibitions of pICP4-ID were only at 11.4% and 36.8% (Figure 4A, bars 4-6). Similar experiments were performed to test the regulatory effect of Egr-1 on the neighboring gene ICP22. The SEAP reporter assays indicated that Egr-1 repressed the activity of full-length ICP22 promoter in a dose-dependent manner (Figure 4B). A similar reduction of activity was observed for the ICP22 promoter without intron (ICP22-ID). These results indicated that Egr-1 is able to inhibit the promoter activity of ICP4 and ICP22, and the intron is more important for the inhibition of ICP4 promoter by Egr-1.
Figure 4.
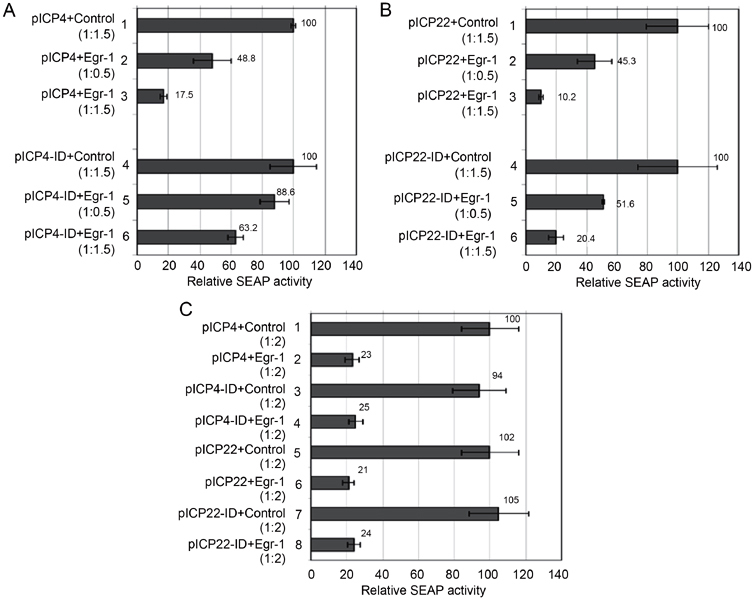
Regulation of ICP22 and ICP4 promoter activity by Egr-1 in HEK293 cells and neuronal PC-12 cells. (A) Co-transfection of pICP4 or pICP4-ID with different amounts of Egr-1 expression vector was performed followed by SEAP assays to analyze the regulatory effect of Egr-1 on promoters. 1, pICP4 and control plasmid (1:1.5); 2, pICP4 and pJDM948 (1:0.5); 3, pICP4 and pJDM948 (1:1.5); 4, pICP4-ID and control plasmid (1:1.5); 5, pICP4-ID and pJDM948 (1:0.5); 6, pICP4-ID and pJDM948 (1:1.5). Note that the SEAP activities of control in lanes 1 and 4 were arbitrarily set to 100 and the SEAP activities of other lanes were normalized against the controls. The error bars represent SDs and the data were calculated and graphed using Microsoft Excel. (B) Co-transfection of pICP22 or pICP22-ID with different amounts of pJDM948 plasmids was performed followed by SEAP assays. 1, pICP22 and control plasmid (1:1.5); 2, pICP22 and pJDM948 (1:0.5); 3, pICP22 and pJDM948 (1:1.5); 4, pICP22-ID and control plasmid (1:1.5); 5, pICP22-ID and pJDM948 (1:0.5); 6, pICP22-ID and pJDM948 (1:1.5). The data were analyzed by the same strategy as described in (A). (C) Co-transfection of all promoters with the Egr-1 expression vector pJDM948 at the molar ratio of 1:1 was performed followed by SEAP assay to analyze the regulatory effect of Egr-1 in neuronal cells PC-12. The arrangement was labeled as shown in the figure. Note that the SEAP activity in lane 1 was arbitrarily set to 100 and the SEAP activity of other lanes was normalized against that of lane 1. The error bars represent SDs and the data were calculated and graphed using Microsoft Excel.
Additional studies on neuronal PC-12 cells showed that Egr-1 induced similar inhibitory effect on all promoters (Figure 4C). These data supported our previous observation that Egr-1 mediated the regulation in neuronal cells as well.
Histone deacetylation is not involved in the regulation by Egr-1
Histone acetylation is suggested to play regulatory roles in HSV-1 transcription during lytic and latent infection 16, 17, 18. To test whether Egr-1 mediated repression might involve the recruitment of HDAC to the promoters, co-transfections of reporter plasmids and pJDM948 in the presence of HDAC inhibitor TSA (100 nM) were performed since TSA at this concentration was proved to be functional in this system (data not shown). The data showed that TSA did not have much effect on Egr-1 mediated repression of ICP22 and ICP22-ID (Figure 5A) or ICP4 and ICP4-ID promoters (Figure 5B). These results indicated that histone deacetylation was not a factor in the Egr-1 mediated regulation.
Figure 5.
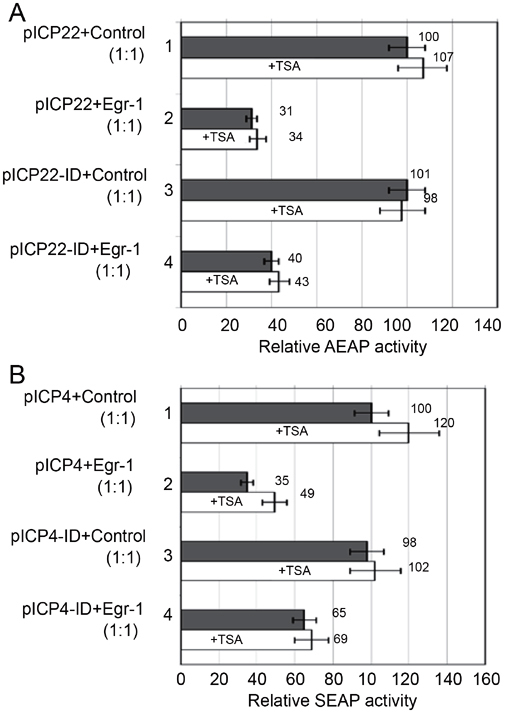
Regulation of ICP4 and ICP22 promoter activity by Egr-1 in the presence of TSA. (A) HEK293 cells were co-transfected with pICP22 or pICP22-ID and the control plasmid or pJDM948 in the presence (white bar) or absence (gray bar) of 100 nM TSA. 1, pICP22 co-transfected with control plasmid; 2, pICP22 co-transfected with pJDM948; 3, pICP22-ID co-transfected with control; 4, pICP22-ID co-transfected with pJDM948. The SEAP activity in lane 1 without TSA was arbitrarily set to 100 and the SEAP activity of others was normalized against this sample. The error bars represent SDs and the data were calculated and graphed using Microsoft Excel. (B) The effects of TSA on Egr-1 mediated regulation of ICP4 and ICP4-ID promoters were accessed using the strategy in (a) except that pICP4 and pICP4-ID were used instead.
ChIP revealed that Egr-1 diminished Sp1 occupancy and recruited co-repressor Nab2 to the proximity of its regulatory sequence
To test whether Egr-1 physically interacted with the HSV-1 promoters, ChIP assays were performed on HEK293 cells transfected with pICP4 and pJDM948 using the anti-Egr-1 antibody. Strong signals were detected from the samples when pICP4 or pICP4-ID was co-transfected with pJDM948 into the cells (Figure 6). These results indicated the in vivo interaction of Egr-1 with the ICP4 promoter. Additional ChIP studies using the antibody against Nab2 (a known co-repressor of Egr-1) showed that the recruitment of Nab2 to ICP4 promoters was increased in the presence of Egr-1 (Figure 6). These results suggested the participation of co-repressor Nab2 in Egr-1 mediated regulation of the ICP4 promoter.
Figure 6.
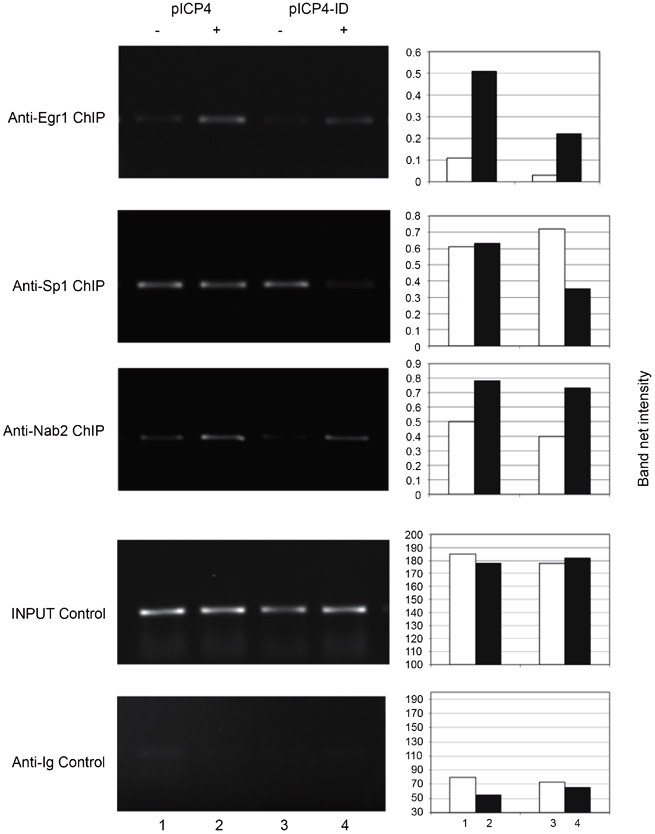
Co-repressor Nab2 was recruited to the ICP4 promoter in the presence of Egr-1 and Egr-1 significantly reduced the occupancy of Sp1 to the promoter when the intron was removed. HEK293 cells were co-transfected with the pICP4 (or pICP4-ID) and pJDM948. Lane 1, pICP4 and control; lane 2, pICP4 and pJDM948; lane 3, pICP4-ID and control plasmid, lane 4, pICP4-ID and pJDM948. The antibodies used for immunoprecipitation were labeled. Input control indicated the PCR products from samples prior to immunoprecipitation. Anti-Ig control confirmed the specificity of antibodies used in the experiments. The quantitative analysis was performed by Kodak Gel Logic 100 system. White bar: no Egr-1; gray bar: plus Egr-1.
Egr-1 binding elements share high similarity with Sp1 sites. To address the possibility that Egr-1 competes and reduces the binding of Sp1 to its binding sites, ChIP assays using anti-Sp1 antibody in the presence of transfected Egr-1 were performed. The data demonstrated that in the presence of Egr-1, the binding of Sp1 was reduced to 50% in pICP4-ID but not significantly altered in the full-length ICP4 promoter, suggesting the competitive binding of Egr-1 and Sp1 to the Sp1 binding elements in the absence of the intron (Figure 6). Taken together, these results support a hypothesis that Egr-1-mediated repression of HSV-1 IE promoters is due to the recruitment of co-repressor Nab2 and reduced occupancy of Sp-1.
Discussion
Existing literatures have suggested links between EGR proteins and the herpesviruses. In EBV, Egr-1 can be induced by EBV lytic transactivator Zta, suggesting a role of Egr-1 in the EBV reactivation 10. Tatarowicz et al. 12 indicated that the EGR proteins are rapidly induced by stimuli that also induce HSV-1 to reactivate from latency. The authors also showed that Egr-2, but not Egr-1 and -3, inhibited the transcription of HSV-1 LAT, which represents the only major transcript during latency. These studies suggested that EGR proteins are involved during the reactivation from latency. However, the effect of Egr-1 on HSV-1 IE genes was not shown. The present study indicates that Egr-1 negatively regulates ICP4 and ICP22 transcription, and thus may have a role in the establishment of latency. It is likely that Egr-1, but not other EGR proteins, exert negative effect on these promoters upon infection to cause viral gene silencing. It is also possible that the use of our model system, which involves circular plasmids associated with chromatin, may have contributed to uncovering this novel regulatory mechanism.
Members of the co-repressor Nab family are involved in Egr-1-mediated regulation 19, 20, 21, 22, 23. A recent study demonstrated that Nab2 interacts with the nucleosome remodeling/deacetylase complex and inhibits transcription 7. These results prompted us to hypothesize that Nab2 may participate in the Egr-1-mediated repression of HSV-1 IE genes. Results shown here suggest that Nab2 was recruited to the IE promoters in the presence of Egr-1. Although the intron is located closer to the transcription initiation site of ICP22 promoter than the ICP4 promoter, Egr-1 induced a greater repressive effect on ICP4 transcription. It is likely that repression of the more distant ICP4 promoter by Egr-1 relies on the chromatin remodeling complex associated with Nab2. More experiments to test this hypothesis are under way.
Egr-1 was suggested to function by competing with Sp1 owing to the great similarity of their binding sites 24. There are 10 Sp1 elements in the regulatory region between ICP4 and ICP22 25. ChIP assays demonstrated that the binding of Sp1 to full-length ICP4 promoter was not altered by the presence of Egr-1. However, studies with pICP4-ID in which the intron was removed from the promoter showed that Egr-1 greatly reduced the Sp1 occupancy, indicating that Egr-1 displaced the binding of Sp1. It is possible that Egr-1 changed its binding from EBEs to the Sp1 sites when the intron was removed. Previous study indicated that binding of Egr-1 to the LAT promoter inhibited the binding of TATA-binding proteins (TBP), and thus represses transcription 12. TBP has been shown to interact with Sp1 26 suggesting the possibility that Egr-1 reduced the participation of TBP at the ICP4 promoter via Sp1 displacement. Overall, these results indicated that Egr-1 can modulate the binding of Sp1 to HSV-1 promoter and the repression of ICP4 and ICP22 expression may be in part owing to the dislocation of Sp1 and TBP.
These results suggest a two-step mechanism for Egr-1 mediated regulation (Figure 7). First, the binding of Egr-1 to the Sp-1 sites reduced the binding of Sp-1 to its genuine elements and caused a reduction in transcription, presumably by diminishing the action of TBP and the assembly of polymerase complexes. Second, the binding of Egr-1 recruited co-repressor Nab2 to the proximity of promoters and caused gene silencing, possibly via chromatin remodeling. This novel pathway may contribute to HSV-1 gene silencing and have implications during the establishment of viral latency.
Figure 7.
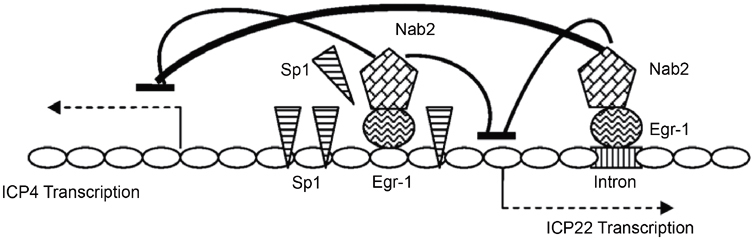
Proposed mechanism of Egr-1 action. Egr-1 has two binding locations, the intron or the Sp1 sites. Egr-1 recruits the co-repressor Nab2, exerts a negative effect on gene expression, and participates in gene silencing possibly via chromatin modeling. The competition of Egr-1 binding to the Sp1 sites may reduce the Sp1 occupancy to the promoter and also contributes to the inhibition of transcription.
Acknowledgements
We thank Dr Jeffery Milbrandt (Washington University, St Louis, MO, USA) for the Egr-1 expression vector pJDM948. We thank Dr James Hill, Dr Thomas Holland, and Dr Yun-Bo Shi for helpful suggestions. We appreciate Dr Paul Sylvester for thorough reading and discussions. Supports from University of Louisiana, Monroe and NCRR/NIH are acknowledged. This publication was made possible by NIH Grant P20RR16456. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Glossary
- (Egr-1)
early growth response gene 1
- (HSV-1)
herpes simplex virus type 1
- (IE gene(s))
immediate-early gene(s)
- (ICP22)
infected cell protein No. 22
- (ICP4)
infected cell protein No. 4
- (HDACs)
histone deacetylase complexes
- (Nab2)
NGFI-A/EGR1-binding protein
- (ChIP)
chromatin immunoprecipitation
- (SEAP)
secreted alkaline phosphatase
Accession codes
Accessions
GenBank/EMBL/DDBJ
References
- 1.Peng W, Henderson G, Inman M. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal ganglia of acutely infected mice. J Virol. 2005;79:6162–6171. doi: 10.1128/JVI.79.10.6162-6171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roizman B, Knipe DM . In: Howley PM, Knipe DM, eds. Herpes simplex viruses and their replication from fundamental fields virology. 4th Edition. Lippincott Williams & Wilkins, 2001: 2399–2460.
- 3.Amelio AL, McAnany PK, Bloom DC. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol. 2006;80:2358–2368. doi: 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swirnoff AH, Milbrandt J. DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol Cell Biol. 1995;15:2275–2287. doi: 10.1128/MCB.15.4.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russo MW, Sevetson BR, Milbrandt J. Identification of NAB1, a repressor of NGFI-A- and Krox20-mediated transcription. Proc Natl Acad Sci USA. 1995;92:6873–6877. doi: 10.1073/pnas.92.15.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milbrandt J. A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science. 1987;238:797–799. doi: 10.1126/science.3672127. [DOI] [PubMed] [Google Scholar]
- 7.Srinivasan R, Mager GM, Ward RM, Mayer J, Svaren J. NAB2 represses transcription by interacting with the CHD4 subunit of the nucleosome remodeling and deacetylase (NuRD) complex. J Biol Chem. 2006;281:15129–15137. doi: 10.1074/jbc.M600775200. [DOI] [PubMed] [Google Scholar]
- 8.Adamson E, de Belle I, Mittal S. Egr1 signaling in prostate cancer. Cancer Biol Ther. 2003;2:617–622. doi: 10.4161/cbt.2.6.671. [DOI] [PubMed] [Google Scholar]
- 9.Calogero A, Cuomo L, D'Onofrio M. Expression of Egr-1 correlates with the transformed phenotype and the type of viral latency in EBV genome positive lymphoid cell lines. Oncogene. 1996;13:2105–2112. [PubMed] [Google Scholar]
- 10.Chang Y, Lee HH, Chen YT. Induction of the early growth response 1 gene by Epstein-Barr virus lytic transactivator Zta. J Virol. 2006;80:7748–7755. doi: 10.1128/JVI.02608-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai Y, Liu Y, Zhang X. Induction of transcription factor Egr-1 gene expression in astrocytoma cells by Murine coronavirus infection. Virology. 2006;355:152–163. doi: 10.1016/j.virol.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatarowicz WA, Martin CE, Pekosz AS. Repression of the HSV-1 latency-associated transcript (LAT) promoter by the early growth response (EGR) proteins: involvement of a binding site immediately downstream of the TATA box. J Neurovirol. 1997;3:212–224. doi: 10.3109/13550289709018296. [DOI] [PubMed] [Google Scholar]
- 13.Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 14.Hsia SC, Shi YB. Chromatin disruption and histone acetylation in regulation of the human immunodeficiency virus type 1 long terminal repeat by thyroid hormone receptor. Mol Cell Biol. 2002;22:4043–4052. doi: 10.1128/MCB.22.12.4043-4052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsia SC, Wang H, Shi YB. Involvement of chromatin and histone acetylation in the regulation of HIV-LTR by thyroid hormone receptor. Cell Res. 2001;11:8–16. doi: 10.1038/sj.cr.7290061. [DOI] [PubMed] [Google Scholar]
- 16.Herrera FJ, Triezenberg SJ. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J Virol. 2004;78:9689–9696. doi: 10.1128/JVI.78.18.9689-9696.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kent JR, Zeng PY, Atanasiu D, Gardner J, Fraser NW, Berger SL. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J Virol. 2004;78:10178–10186. doi: 10.1128/JVI.78.18.10178-10186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muggeridge MI, Fraser NW. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J Virol. 1986;59:764–767. doi: 10.1128/jvi.59.3.764-767.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucerna M, Mechtcheriakova D, Kadl A. NAB2, a corepressor of EGR-1, inhibits vascular endothelial growth factor-mediated gene induction and angiogenic responses of endothelial cells. J Biol Chem. 2003;278:11433–11440. doi: 10.1074/jbc.M204937200. [DOI] [PubMed] [Google Scholar]
- 20.Kumbrink J, Gerlinger M, Johnson JP. Egr-1 induces the expression of its corepressor nab2 by activation of the nab2 promoter thereby establishing a negative feedback loop. J Biol Chem. 2005;280:42785–42793. doi: 10.1074/jbc.M511079200. [DOI] [PubMed] [Google Scholar]
- 21.Houston P, Campbell CJ, Svaren J, Milbrandt J, Braddock M. The transcriptional corepressor NAB2 blocks Egr-1-mediated growth factor activation and angiogenesis. Biochem Biophys Res Commun. 2001;283:480–486. doi: 10.1006/bbrc.2001.4810. [DOI] [PubMed] [Google Scholar]
- 22.Svaren J, Sevetson BR, Apel ED, Zimonjic DB, Popescu NC, Milbrandt J. NAB2, a corepressor of NGFI-A (Egr-1) and Krox20, is induced by proliferative and differentiative stimuli. Mol Cell Biol. 1996;16:3545–3553. doi: 10.1128/MCB.16.7.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swirnoff AH, Apel ED, Svaren J. Nab1, a corepressor of NGFI-A (Egr-1), contains an active transcriptional repression domain. Mol Cell Biol. 1998;18:512–524. doi: 10.1128/MCB.18.1.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Sarraj A, Day RM, Thiel G. Specificity of transcriptional regulation by the zinc finger transcription factors Sp1, Sp3, and Egr-1. J Cell Biochem. 2005;94:153–167. doi: 10.1002/jcb.20305. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen-Huynh AT, Schaffer PA. Cellular transcription factors enhance herpes simplex virus type 1 oriS-dependent DNA replication. J Virol. 1998;72:3635–3645. doi: 10.1128/jvi.72.5.3635-3645.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emili A, Greenblatt J, Ingles CJ. Species-specific interaction of the glutamine-rich activation domains of Sp1 with the TATA box-binding protein. Mol Cell Biol. 1994;14:1582–1593. doi: 10.1128/MCB.14.3.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.