

Abstract
Recent advances in our understanding of the structure and function of class B GPCRs provide multiple opportunities for targeted development of allosteric modulators. Given the pleiotrophic signaling patterns emanating from these receptors in response to a variety of natural agonist ligands, modulators have the potential to sculpt the responses to meet distinct needs of different groups of patients. In this review, we provide insights into how this family of GPCRs differs from the rest of this superfamily, how orthosteric agonists bind and activate these receptors, the potential for allosteric modulators to interact with various regions of these targets, and the allosteric influence of endogenous proteins on the pharmacology of these receptors that are important considerations when developing new therapies.
INTRODUCTION
G protein-coupled receptors (GPCRs) are the dominant target of currently approved drugs, with most of these directed to class A GPCRs. Class B1 GPCRs (secretin receptor family) are a small group of physiological, and pathologically very important potential drug targets (1). They encompass receptors for 15 peptide hormones, including adrenomedullin (AM), amylin (AMY), calcitonin (CT), calcitonin gene-related peptide (CGRP), corticotropin-releasing factor (CRF), glucagon (GCG), glucagon-like peptides 1 and 2 (GLP), glucose-dependent insulinotropic peptide (GIP), growth hormone-releasing hormone (GHRH), parathyroid hormone (PTH), and parathyroid hormone-related peptide (PTHrP), pituitary adenylate cyclase activating peptide (PACAP), secretin (SCT), and vasoactive intestinal polypeptide (VIP) (Table 1). All of the natural ligands for these receptors are moderate length peptides between 27 and 44 amino acids in length, with diffuse pharmacophoric domains, and with a propensity to form α-helical secondary structure. Members of this receptor family have been proposed as targets for the treatment of many clinically important problems, such as type 2 diabetes, obesity, bone disease, migraine, pain, psychiatric disorders, and cancer. Despite the importance of these targets, the only approved therapeutic agents directed to these receptors have been peptides representing variants of their natural agonists, and very recent approval of antibodies targeting receptor-containing complexes (2). The presence of allosteric binding sites within class B GPCRs may offer new opportunities for drug development.
Table 1.
Class B GPCRs and their natural ligands
| Natural peptide ligands | Receptors |
|---|---|
| Adrenomedullin | AM1 (CLR:RAMP2); AM2 (CLR:RAMP3) |
| Amylin | AMY1 (CTR:RAMP1); AMY2 (CTR:RAMP2); AMY3 (CTR:RAMP3) |
| Calcitonin | CTR |
| Calcitonin gene-related peptide | CGRPR (CLR:RAMP1) |
| Corticotropin-releasing factor (and urocortins 1,2,3) | CRF-1R; CRF-2R |
| Glucagon | GCGR |
| Glucagon-like peptide-1 (and exendin-4) | GLP-1R |
| Glucagon-like peptide-2 | GLP-2R |
| Glucose-dependent insulinotropic peptide | GIPR |
| Growth hormone-releasing hormone | GHRHR |
| Parathyroid hormone (and TIP39 for PTH2R) | PTH1R; PTH2R |
| Parathyroid hormone-related peptide | PTH1R |
| Pituitary adenylate cyclase activating peptide | PAC1R |
| Secretin | SCTR |
| Vasoactive intestinal polypeptide | VPAC1R; VPAC2R |
While there has been some success in the preclinical development of small molecule antagonists for class B1 GPCRs, it has been particularly difficult to develop small molecule agonists; such agonists are often the desired type of drug for these targets. With the relatively recent successes in structural characterization of intact receptors in this family (3; 4), it is finally becoming clear why the development of small molecule agonists targeting these receptors has been so challenging. Unlike the class A GPCRs that contain a small intrahelical pocket that is ideal for docking drug-like molecules, an analogous confined site for ligand docking is missing from class B GPCRs, in which the upper helical bundle is wide open in the activated state and devoid of distinct small molecule binding sites (seen in Fig. 1A and identified as orthosteric TM binding site in Fig 2B) (5; 6).
Figure 1. Structural characterization of class B GPCRs – two relatively stable domains with variable relative orientations.
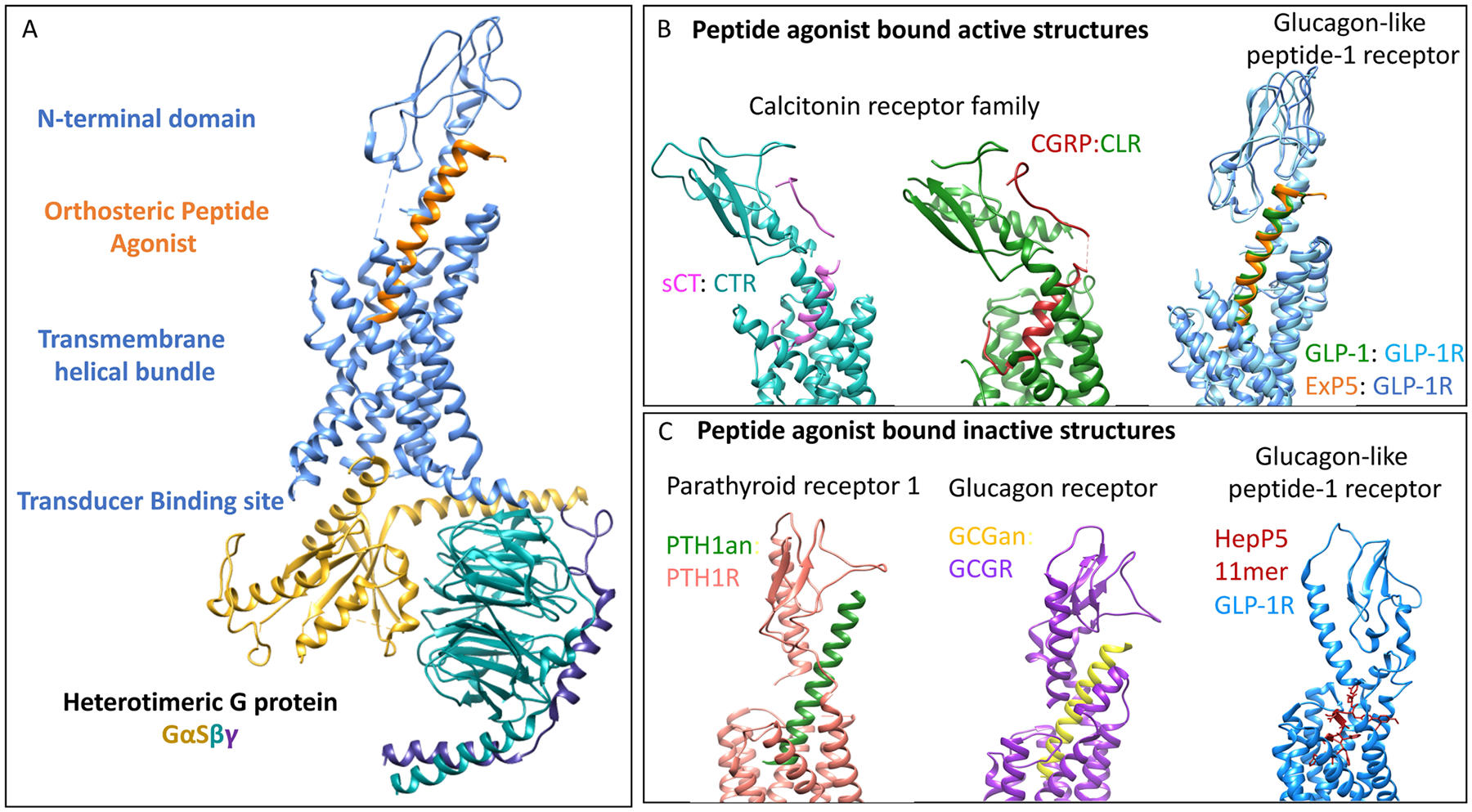
(A) Structural domains of class B GPCRs using the GLP-1R structure as an exemplar. The amino-terminal extracellular domain (ECD) of the class B GPCRs contains two central anti-parallel β-sheets (with the peptide-binding cleft in between) and an amino-terminal α-helix connected by a series of loops and stabilized by three disulfide bonds. The core transmembrane helical bundle domain is more open toward the extracellular side of the membrane than other families of GPCRs, and lacks distinct small molecule docking sites. The cytosolic side of the helical bundle is identified as the transducer binding site, shown here as binding the heterotrimeric G protein, GαSβγ. The orthosteric natural peptide ligand binding begins with the carboxyl terminus of the peptide occupying the ECD cleft, thereby directing the biologically active amino terminus of the peptide toward the junctional domain (J-domain); this comprises the two-site hypothesis with two distinct stages separated spatially and temporally. (B) Peptide agonist-bound active structures solved to date. Shown are structures of the CT family of receptors (CT receptor and CLR) binding CT or CGRP and peptides with GLP-1 agonist activity bound to the GLP-1 receptor. Note the differences in orientations of the receptor amino-terminal domain (ECD) relative to the core transmembrane helical bundle domain. (C) Peptide-bound inactive structures solved to date. Shown are structures of the PTH-1 receptor and glucagon receptor (GCGR) with peptide agonists bound, but stabilized in inactive receptor conformations, again highlighting the differences in orientation of the two key peptide-binding domains.
Figure 2. Class B GPCR ligand binding sites.
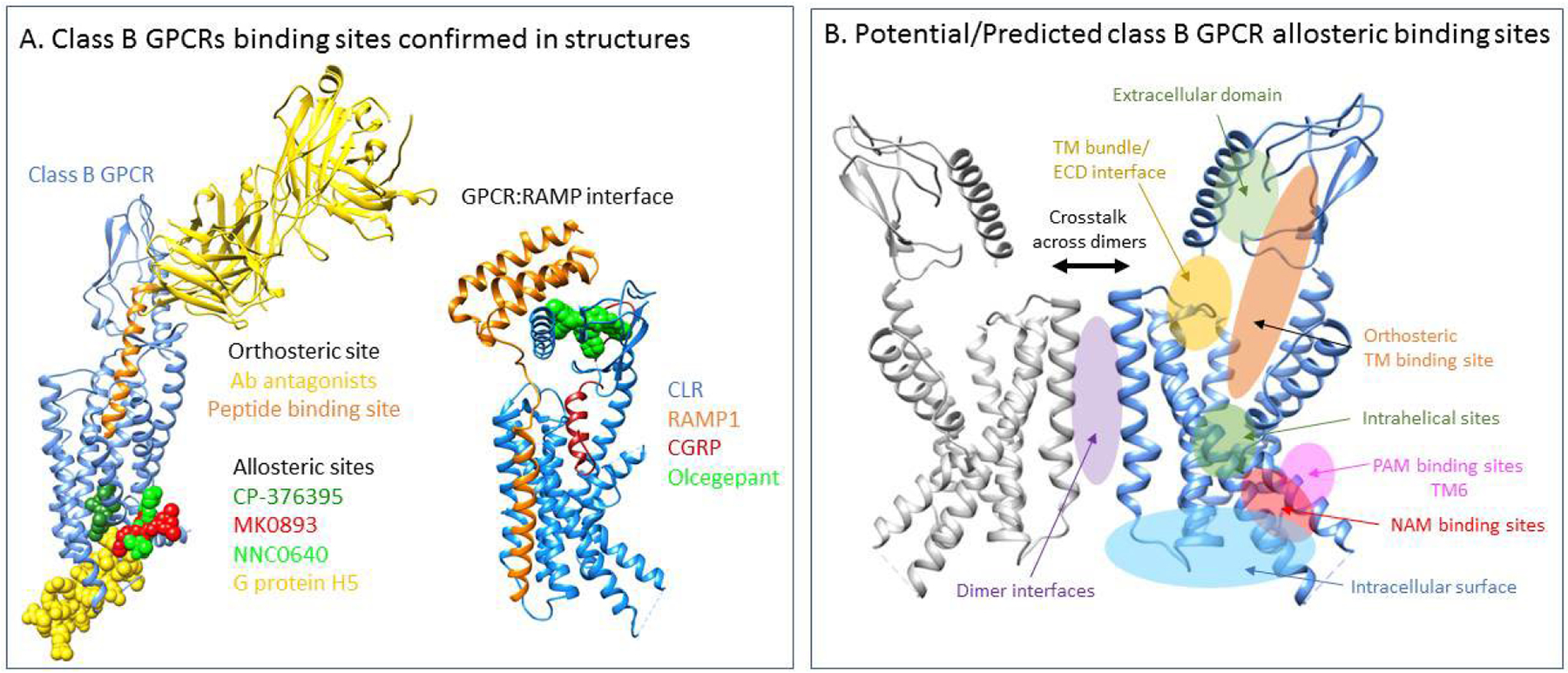
(A) Ligand-binding sites confirmed in existing structures. Shown are sites for the orthosteric peptide ligands, a well as an antibody partially directed to that site. Also shown are the location of allosteric ligand binding sites deep within the helical bundle and outside of this domain, as well as sites for G protein association, RAMP interaction, and sites of antagonists that recognize the RAMP:receptor ECD complex. (B) Broad theoretical classes of ligand-binding sites. Based on existing structures and what is currently understood about dynamics and functional domains of class B GPCRs, this illustrates potential sites of modulation by various types of ligands. In addition to what is shown, lipids could act anywhere around the intramembranous helical bundle and different RAMPs could interact differently than the prototype illustrated in panel A and could possess unique allosteric sites that could be targeted. Also, other endogenous molecules, such as ions, have potential sites of action too numerous to display.
Peptide and protein drugs often need to be administered parenterally, due to limited enteric absorption and degradation in a protease-rich environment, such as the gut. While there have been major advances in percutaneous delivery (7) and with automated injection devices for these types of molecules (8), facilitating their acceptance by patients, stable and bioavailable small molecules that can be administered orally continue to be preferred.
Allosteric modulators of receptors are ligands that bind anywhere other than the site of receptor interaction with the natural orthosteric ligands (9; 10). The power of such pharmacologic agents is finally being recognized and there are now examples of this type of drug being approved (11). The class B1 GPCRs seem to be ideal targets for allosteric modulation, having many potential sites of interaction and the opportunity to sculpt responses from a broad spectrum of activities. The functional impact of allosteric agents can range from agonism (allosteric agonist) to antagonism (allosteric inhibitor), including the possibility of having no intrinsic activity, while also possessing the ability to modulate the actions of orthosteric agonists of the receptor in a positive (PAM; positive allosteric modulator), negative (NAM; negative allosteric modulator), or neutral (NAL, neutral allosteric ligand) manner (12). Given the breadth of potential clinical applications, the potential for such agents to target this class of receptors is extraordinary.
Allosteric vs. orthosteric drugs
For many years, drugs targeting receptors were mainly thought of as switches, either turning on the post-receptor signaling events activated by the natural hormone or neurotransmitter with agonists or turning them off with antagonists. Natural ligands have provided excellent starting points for the development of these types of drugs to achieve receptor selectivity, taking advantage of the determinants of receptor binding that typically result in docking to the natural, orthosteric ligand-binding pocket. As noted above, allosteric agents interact with sites on the receptor that are spatially distinct from the orthosteric site of interaction with the natural agonist (12). Because both allosteric and orthosteric ligands can bind to the receptor simultaneously, the allosteric interaction has the potential to have a positive or negative modulatory effect on the action of the natural agonist or agonists (PAM and NAM, respectively), affecting binding affinity and/or efficacy (12). This occurs through conformational cooperativity between the two ligand-binding sites of the GPCR. When there are multiple natural agonists, such modulatory activity can also be different for each ligand. This is termed probe-dependence (12). In addition to this modulatory activity, allosteric agents can also possess a spectrum of intrinsic activity (PAM-agonist) (12). They can also have no intrinsic or modulatory activity (NAL) (12). To date, no physiologically-relevant endogenous allosteric modulators or class B GPCRs have been recognized.
GPCRs are a prime example of a natural allosteric protein since they allow signal transduction via communication between two spatially distinct, but conformationally-linked sites; the agonist binding site and intracellular transducer binding site. Activated class B GPCRs predominantly signal via the transducer heterotrimeric Gs protein, raising cAMP levels within the cell. However, they are pleiotropically coupled, capable of activating many diverse signaling pathways (13). This subjects them to biased agonism where different ligands acting at the same receptor have the potential to promote distinct signaling profiles. Biased agonism arises due to chemically diverse interactions formed by different ligands that govern differential allosteric propagation through the GPCR. This results in discrete subsets of conformational receptor ensembles available to interact with transducers, with each transducer predicted to couple to a distinct conformational state (14). While biased agonism is readily observed with orthosteric ligands that bind to an overlapping binding site within class B GPCRs, perhaps not surprisingly, allosteric ligands have the potential to display very distinct biased signaling profiles relative to orthosteric peptides (15). Due to the natural allosteric nature of GPCRs that promotes signal transduction via communication between agonist binding sites and intracellular transducer binding sites, the nature of the allosteric modulatory activity between two ligands can also differentially alter distinct signaling pathways, resulting in biased allosteric modulation (14). This broad spectrum of actions provides opportunities for allosteric agents to sculpt the biological responses emanating from a receptor.
Natural receptor agonists possess the potential advantages of often mediating physiologically relevant servomechanisms, being secreted at optimal times for activation of their targets and frequently having relatively short and finite durations of action limited by their clearance and/or degradation. A major advantage of allosteric modulators is their ability to modulate these physiologic events, maintaining action at the physiologically relevant time and duration in the case of pure modulators (12). This action of the physiological ligand, however, can be augmented or reduced by an allosteric modulator if therapeutically desirable (11). Allosteric agents with intrinsic efficacy can provide additional tone, and timing of effects different from the natural agonist, if this is beneficial.
Because allosteric modulators without intrinsic agonist activity are dependent on natural agonists for their action, the timing of action can be limited to physiologically-relevant settings and times. In addition, the impact of allosteric modulators is also typically saturable, with the effect reaching a plateau. These properties of allosteric modulators provide improvements in safety profiles when compared to pure agonists. For biological systems where it is desirable to limit the level or duration of activation, allosteric modulators are ideal.
Structural insights into class B GPCRs (Figure 1)
Like all groups of receptors in the GPCR superfamily, class B GPCRs possess seven hydrophobic segments that encode α-helical membrane-spanning segments. These seven helices form a bundle that is characteristic of the entire superfamily, but the class B GPCRs achieve this conformation without possessing the signature sequences present in the other families of GPCRs (16). As noted below, while this general topology is conserved, there are very important differences in the structure of this domain across the GPCR superfamily.
For most GPCRs, the natural ligands approach the membrane receptors from their ectodomain, outside the bilayer. There is extraordinary structural diversity for the natural ligands of these receptors, ranging from photons and odorants, to biogenic amines, peptides, and even large proteins (17). Evolution has accommodated this structural diversity by achieving specializations in ligand-binding domains, such as intrahelical pockets for small molecules, and extracellular loop and amino-terminal domains for larger peptides and proteins. The class B GPCRs bind the moderate length natural peptide ligands within a characteristic extracellular domain (ECD) that includes two central anti-parallel β-sheets and an amino-terminal α-helix connected by a series of loops and stabilized by three disulfide bonds. Peptide comprising this ECD was the first component of the class B GPCRs that was structurally characterized at high resolution, first solved using NMR in 2004 (18), and in subsequent NMR or crystallographic studes found to be highly conserved in at least eight other members of this family. The peptide binds between two β-sheets in a highly conserved fold within this amino-terminal ECD. Of note, however, the distal regions of the amino terminus, which are the functionally important regions for receptor activation, were not defined in any of the isolated domain structures.
In 2013, isolated helical bundle domains of two members of this family (the glucagon receptor and the CRF-1 receptor) were solved using x-ray crystallography. This was accomplished in the presence of inhibitor ligands and the use of modified receptors (via mutations or addition of fusion proteins) to stabilize the bundle to enable crystallization (5; 6). As noted above, these structures confirmed the predicted transmembrane heptahelical conformation. Of note, the cytosolic side of the helical bundle was remarkably similar to that of the many inactive state class A GPCRs previously structurally characterized (3). This is the region that couples to the heterotrimeric G proteins, a highly conserved function of most members of the GPCR superfamily. In contrast, the structure of the upper portion of the transmembrane spanning bundle, implicated in natural ligand binding and activity, was distinct from that of class A GPCRs. This region was more open toward the extracellular side of the membrane than other GPCR structures solved to date, providing a large solvent-filled cavity, and was devoid of distinct pockets where small molecules could be docked.
With methods documenting substantial success for solution of the crystal structure of intact class A GPCRs, and with evidence for conserved and stable conformations of the two key domains of class B GPCRs, the ECD and the core transmembrane helical bundle, it was informative that class B GPCR holostructures were so difficult to determine using x-ray crystallography. This likely reflects substantial mobility of these domains relative to each other. The two domains are tethered to each other through the continuous peptide backbone, but there are no interdomain disulfide bonds to provide additional fixation. When the natural peptides dock with these receptors, there are determinants for binding in both the ECD and the helical bundle, providing a second tethering influence, but it is not clear how limiting this feature might be on overall conformation.
A major advance came from the application of cryo-electron microscopy (cryo-EM) to solve the structures of intact holoreceptors in this family (3; 4; 19; 20) (Figure 1). This technique is compatible with the natural sequences of these receptors, particularly when in complexes with other proteins, such as G proteins. In contrast, most GPCR crystallography has required high affinity, thermostabilizing ligands, elimination of flexible regions by truncation, and stabilization of conformation by mutagenesis to provide stable structures for the crystallization (21). High resolution crystal structures of full-length class B GPCRs (in inactive conformations) bound to peptides have now also been solved (22–25), but these, too, include stabilizing manipulations that could affect the overall conformation of the receptor. Antibodies have also been used that bind to stabilized forms of receptors or that span ECD and core epitopes and have been useful tools to aid structure determination of class B receptors in inactive conformations.
The structures solved to date provide direct evidence for a diversity of orientations between ECD and helical bundle of class B GPCRs (Figure 1). The degree of flexibility observed is likely due to the nature of the interacting peptide. The amino terminus of the solved CT receptor structure is extremely mobile and with bound CT peptides possessing secondary structure in their amino-terminal domain, but being unstructured in the region that binds to the receptor ECD (3). In contrast, in all other full length peptide bound structures of members of this family, the peptides adopt a helical structure throughout the entire length of the peptide and this stabilizes a predominant conformation of the amino terminus (Figure 1B). We are just beginning to learn what roles these relative orientations might play. There are observations in the literaure that suggest the presence of non-covalent interactions between ECD and core helical bundle domains of class B GPCRs that may constrain an inactive conformation of the receptor, thus playing a role in the mode of receptor activation (26; 27). Antibodies directed to the ECD of the glucagon receptor have been observed to act as inverse agonists (26; 27), consistent with the presence of an interaction between these two domains in the basal state that may be stabilized by antibody binding. Mutation of three residues in ECL3 of the glucagon receptor disrupted the presumed interaction and eliminated the inverse agonist activity of this antibody, supporting the functional relevance of interdomain interaction, at least for this class B GPCR.
Comparisons between inactive and active receptor structures reveal important insights regarding how orthosteric peptides engage and activate their receptors via a conserved conformational rearrangement of the helical bundle (3; 4; 20; 28). Peptide engagement promotes conformational transitions that lead to an unwinding of transmembrane helix (TM) 6 around a central PXXG motif that promotes a large kink in the center of TM6, opening up the intracellular face of the receptor to allow G protein engagement. This is associated with conformational reorganization of ECL2 and movements within the upper portion of TM1, TM6 and TM7 that facilitate this peptide interaction and receptor activation. Key networks conserved across class B GPCRs facilitate these conformational transitions including a reorganization of a central polar network below the peptide binding site, reordering of a hydrophobic transmission switch, whereby interactions that maintain the inactive conformation are released by the unwinding of the TM6 PXXG motif and disruption of two polar networks within the intracellular portion of the receptor that are sufficient to open up the bundle so that it engages the G protein.
Orthosteric sites of class B GPCRs – two site hypothesis and dynamics (Figure 1)
Given that allosteric ligands bind to sites on the receptor that are topographically distinct from the orthosteric site, it is critical to understand the orthosteric site of binding and action of the natural peptide ligands for the class B GPCRs. Long before the structures of these receptors were understood, the presumed sites of interaction were mapped using peptide ligand substitution and modifications to the receptor protein in order to provide structure-activity relationships. It became clear that the carboxyl-terminal region of the ligands, particularly those tending to form an α-helical structure, interacts with the amino-terminal ECD, while the amino-terminal region of the ligands interacts with the extracellular-facing half of the helical bundle, a region termed the junctional-domain (J-domain) (29). All subsequent structural insights have been consistent with this dual mode of interaction (Figure 1). Kinetic studies have supported this occurring sequentially in two stages, with the peptide first interacting with the receptor ECD domain, followed by interaction with the core transmembrane helical bundle domain (30).
Peptide structure-activity studies revealed the native peptide interaction with the ECD contributes most of the binding affinity, and contributes little to biological activity (i.e., signal transduction) with truncated forms of peptides often behaving as antagonists (31). The interaction of the peptide amino terminus with the receptor core transmembrane helical bundle adds to the binding affinity and is responsible for essentially all of the biological activity. In this family, the affinity of natural peptide ligands of these receptors for the J-domain has been estimated as being as much as 100,000-fold weaker than its affinity for some intact receptors (29). As we have learned about the structure of the ECD domain of these receptors, it is clear that there is a cleft between two β-sheets in which amphipathic helical peptides can reside, and those physical features are the most critical to contribute to the initial docking of the natural ligands. This capture event yields a functionally-tethered peptide ligand to be directed toward the upper region of the helical bundle where even a low affinity binding event can yield docking and a biological response (32). This explains why peptides across this family with increasing degrees of amino-terminal truncation lose their biological activity, while isolated amino-terminal regions of these peptides rarely exhibit biological activity, due to their very low affinity interactions with the receptors. The shortest such peptides that have been engineered to retain potent biological activity have been nine or more residues in length (25).
Recent structural information is also beginning to shed light on how different peptide ligands binding to the same orthosteric site may achieve biased agonism (33–39). The largest conformational differences observed in class B GPCR active structures when compared with inactive receptor bundles occur within the J-domain with outward movements of TM1, TM6, and TM7. Biased agonism between peptide agonists has been extensively explored in the GLP-1R (15; 37; 40–42) and there are now two cryo-EM structures of this receptor; one bound to the endogenous ligand, GLP-1 (20), and the other to the G protein-biased agonist, exendin-P5 (4). Intrigingly, major differences were observed in the conformation of the extracellular portion of TM6 and extracellular loop (ECL) 3, with more subtle differences observed within the extracellular ends of TM1 and TM7, suggesting that these domains may be involved in modulating biased agonism for this receptor.
In addition to this direct structural information, there are a plethora of mutagenesis data that identify important determinants for peptide engagement and activation of class B GPCRs. It is quite interesting that mutagenesis studies have mapped important determinants in this region to several different portions of the helical bundle for different receptors in this family (36; 37), and even for different agonists targeting a single family member (37; 40; 43). These studies are consistent with observations from the cryo-EM structures, identifying ECL2 and ECL3 as important determinants for coupling of ligand interactions to activation of cAMP and pERK1/2 signaling and for biased agonism, however the exact pattern for these determinants differ across receptors within the class B subfamily.
Other fascinating obervations support the interpretation that different profiles of signaling events can be achieved by interactions with different regions of the extracellular portion of the helical bundle. A particularly interesting approach incorporated an azobenzene photoresponsive element into the center of liraglutide, a peptide analog of GLP-1 (44) to yield isomerization-induced biased signaling patterns, with the cis isomer favoring cAMP generation and the trans isomer favoring calcium events. Presumably, the amino terminus of both peptides was directed toward the region at the top of the helical bundle, with differences induced by the optical control. Much remains to be learned regarding how this region of the helical bundle controls receptor activation linked to distinct signaling events in class B GPCRs.
The two-stage docking process for peptide engagement with class B GPCRs, particularly with the first stage providing substantial binding energy, provides an operational quandary in the definition of allosteric ligands. It is quite possible to have a small molecule ligand that binds to the upper region of the helical bundle that can bind simulataneously with a natural peptide ligand, not competing with the binding interaction between the peptide and the ECD. Similarly, the open structure of the upper region of the helical bundles of the class B GPCRs also provides adequate space for small molecule ligands to dock in a site spatially distinct from where the natural ligands interact. Several of the indirect methods to define a ligand as acting in an allosteric manner, such as the completeness of competition binding and impact on ligand dissociation, may not define the mode of action of ligands binding in this region of the receptor. The best demonstration of the allosteric nature of small molecule ligands of class B GPCRs will be structural analysis of complexes of receptor with both ligands.
Opportunities for allosteric ligand sites of action (Figure 2)
ECD outside of peptide-binding cleft.
The most obvious conserved site for ligand binding to the ECD of class B GPCRs is within the peptide-binding groove (Figure 2). By definition, such a ligand would occupy a well defined orthosteric site. A high affinity interaction in that location would compete for natural peptide ligand binding and would likely represent a competitive antagonist. This is likely the mode of action of both antibodies and small molecule CGRP antagonists (45; 46). There are no current examples of a ligand targeted exclusively to the peptide-binding cleft within the ECD of a class B GPCR that possesses agonist activity. With the natural peptide occupying a specific interface within the ECD, and given the relatively large size of this domain (~100–150 amino acids), there are opportunities for ligands to bind other regions within the ECD. While the folding pattern of the ECD is highly conserved in class B GPCRs, the sequences are not, providing substantial opportunities for receptor specificity, such as might be achieved with monoclonal antibodies. As noted above, at least one such antibody has characteristics as an inverse agonist of the glucagon receptor (26; 27) and an antibody that behaves as an antagonist has also been described for the GLP-1 receptor (47). There are alsoapproved drugs of monoclonal antibodies directed toward the CGRP receptor, having an antigenic site that spans both the ectodomain of the ECD of CLR and the ECD of its associated receptor activity-modifying protein 1 (RAMP1) (erenumab) (2), as well as toward the CGRP ligand (fremanezumab and galcanezumab) (48). The CLR-RAMP1 ECD interface is also a good target for large, complex small molecules (olcegepant and telcagepant) (46). These reagents have been used as antagonists of CGRP signaling for the control of migraine.
ECD-helical bundle interface.
There are surfaces of both the base of the ECD and the top of the helical bundle domains that likely interact with each other during normal physiology. Targeting these surfaces would be expected to disrupt the interactions of these domains, although it is possible that a ligand binding to the top of the helical bundle could mimic the natural activation process. Such a ligand represents only a theoretical possibility, since no reagents like this have yet been described.
Inside the helical bundle.
Given the shape of the helical bundle region of the class B GPCRs, there are accessible sites of ligand binding high in the bundle that could be distinct from the site of natural orthosteric agonist interaction, as well as such sites deep in the bundle. The latter have already been demonstrated with the ligand, CP-376395 (a 2-aryloxy-4-alkylaminopyridine), which acts as an antagonist of the CRF1 receptor (5). This site was described as more than 15Å below most peptide-binding sites within GPCRs and 7Å deeper than the classical conserved small molecule-docking site in the helical bundle of class A GPCRs, only 4Å from the cytosolic surface (Figure 2). From what we know about the movement of the cytosolic side of class A GPCRs (and also observed with activated states of class B GPCRs), a tight interaction deep in the bundle could be expected to interfere with the movement of cytosolic face of the helical bundle that is critical for the activation process and fully explains the structural basis of CP-376395 as an antagonist.
The difficulties pharmaceutical companies have faced in developing high affinity drug-like small molecule agonists for this family suggest that achieving adequate affinity and potency for such molecules to act high within the helical bundle is challenging. A series of small molecule agonists of the class B CT receptor was developed, with their site of action proposed to be in the juxtamembranous hinge region between the helical bundle and the ECD (residues 150–153) (49). With the solution of the structure of this receptor (3), it is now clear that amino acids 150–153 reside within the helical bundle, high in TM1. The relationship of this site of action with that of the biologically active amino terminus of CT is not yet clear. A series of substituted azoanthracene compounds developed by Transtech Pharma, one of which is currently in clinical trials, are also proposed to bind high within the helical bundle of the GLP-1R (http://vtvtherapeutics.com/pipeline/ttp273) (50).
Outside of helical bundle.
There are multiple examples of small molecules directed toward the outside of the helical bundle of class B GPCRs. The group that is best understood includes the glucagons receptor antagonists, NNC0640, MK-0893, and PF-0637222 (22; 51; 52). These molecules straddle the outside of TM6 near the intracellular surface of the bundle (Figure 2), presumably interfering with its ability to move in an outward direction to allow the G protein H5 helix to be inserted into the helical bundle during activation. This chemotype of antagonists has been well defined for the glucagon receptor, and this site of action has been demonstrated in co-crystal structures (22; 51).
The outside of the helical bundle is also the dominant site of interactions of lipid, such as cholesterol, which increases thermal stability of many membrane proteins, acting through conserved cholesterol-association sequence motifs, such as the cholesterol recognition/interaction amino acid consensus pattern (CRAC) (53), the reversed CRAC motif (CARC) (54), and the cholesterol consensus motif (CCM) (55) with this lipid present in many crystal structures of membranes proteins. Unfortunately, the sites of cholesterol in crystal structures of many membrane proteins are not included in these motifs, and many times that these motifs are present, the crystal structures include no cholesterol (56). Using dynamic single-molecule force spectroscopy, cholesterol has been shown to increase the kinetic, energetic, and mechanical stability of many regions other than the structural core of the β2-adrenergic receptor (57). Cholesterol has been described to modify the function of several GPCRs (58), but this has been inconsistent, even when shown to bind to analogous regions of different receptors. To date, no clear demonstration of functionally-important cholesterol association has yet been described for a class B GPCR.
Intracellular surface.
Other small molecule agonists have been localized to the intracellular face of these receptors. Two such agents are PAM-agonists of the GLP-1 receptor: 6,7-dichloro-2-methylsulfonyl-3-tert-butylaminoquinoxaline (compound 2) (59) and 4-(3-(benzyloxyphenyl)-2-(ethylsulfinyl)-6-(trifluoromethyl)pyrimidine (BETP/compound B) (60), which form a covalent bond with an exposed cysteine residue (Cys347) at the junction between intracellular loop 3 and TM6 (61). The approach of azobenzene photoresponsive isomerization described above when applied to peptide agonists of the GLP-1 receptor, was also applied to BETP, an allosteric small molecule ligand (62). These constrained compounds exhibited differential impact on insulin secretion, demonstrating spatial specificity of action at this site. The profiles of signaling events stimulated by BETP and compound 2 were observed to be affected by the state of GLP-1 receptor dimerization (63), with disruption of these complexes by mutation of the TM4 interface eliminating the cAMP responses, while preserving the ERK phosphorylation events. At the same time the intrinsic agonist action of these compounds to stimulate cAMP was eliminated in the monomeric form of the GLP-1 receptor, their allosteric modulatory effect on peptides acting at the same protomer (cis action) was retained. This TM4 dimeric interface may also provide a structurally-specific complex as a potential target of allosteric modulators.
The intracellular face of these receptors is also known to interact with heterotrimeric G proteins and arrestins. This interface between the receptor and transducer molecules could also provide another interface to potentially target allosteric ligands (Figure 2).
Endogenous potential allosteric ligands
GPCRs are natural allosteric proteins interacting with ligands on the extracellular surface and transducers on the intracellular side of the cell. Like all GPCRs, class B GPCRs associate with heterotrimeric G proteins and can also couple to other effectors such as arrestins and kinases. These transducers are endogenous allosteric proteins that can alter the conformation of the helical bundle and in turn, the way that a GPCR responds to its ligands (64).
In addition to signaling partners, other endogenous allosteric regulators of receptor function have been increasingly recognized (65). These can include ions normally in the circulation as well as lipid components of the plasma membrane where they reside. Additionally, more specific interactions with distinct membrane or cytosolic proteins can also occur. A common GPCR interaction important for the function of class A GPCRs is with sodium ions that negatively modeulate receptor function through interaction with a highly conserved aspartate residue in TM2 (D2.50) and with a serine in TM3 (S3.39) (66; 67). A similar mechanism for sodium ion regulation has also been proposed for class B GPCRs (68), although this interaction has been suggested for residues in positions 3.43, 6.53, and 7.49. As noted above, cholesterol and other lipids withn the bilayer can also act as endogenous allosteric modulators.
Receptor activity-modifying proteins (RAMPs) are a family of three single transmembrane proteins that can modulate the pharmacology of numerous GPCRs. RAMP1 was first identified as required for physical interaction with the calcitonin-like receptor (CLR) to achieve responsiveness of the bimolecular complex to CGRP (69), whereas RAMPs 2 and 3, when coupled with CLR, yield functional adrenomedullin receptors. Extensions to these studies revealed these proteins could also interact with the CT receptor, switching its ligand specificity to become a high affinity amylin receptor and (with RAMP1 only) a second CGRP receptor (70). It quickly became clear that many class B GPCRs could interact with one or more of the three described RAMPs, with impact ranging from effects on receptor trafficking to the plasma membrane during biosynthesis, to affecting ligand specificity and biological responses, as well as a physical association without currently recognized functional significance. Receptor association with RAMPs has subsequently been extended to include a class A and class C GPCRs, and possibly even non-GPCR membrane receptors (71).
Structural characterization of the extracellular domains of CLR and RAMP1 demonstrated that this complex is stabilized by hydrophobic and electrostatic interactions between the amino-terminal helix of this receptor and helices 2 and 3 of the amino terminus of RAMP1 (46). Subsequently, the full length RAMP1, CLR complex bound to the peptide CGRP and the transducer Gs was solved using cryo-EM (19). This revealed minimal interactions of the RAMP with the ligand, confirming predictions that RAMPs act allosterically to switch ligand specificity. The RAMP transmembrane domain forms extensive interactions with TMs 3, 4 and 5 of the CLR and stabilizes ECL2, while the amino terminus of the RAMP stabilizes the orientation of the CLR extracellular domain. Interestingly, the amino-terminal domain of the related CT receptor is much more mobile than the RAMP1 complexed CLR (3; 19). This receptor binds CT peptides, however in the presence of RAMP1, it forms a high affinity CGRP receptor. These observations lead to speculation that for the CT family of receptors, RAMPs allosterically alter ligand selectivity via stabilization of the amino-terminal domain and ECL2, which are principal sites identified for peptide recognition and activation for this class of receptors.
The interface of RAMP1 with CLR within the ECD has proven to be a useful target for the development of both small molecules (olcegepant and telcagepant, with ubrogepant and rimegepant in advanced development) (Figure 2) and antibody reagents (erenumab) to inhibit the action of CGRP that have been helpful for the treatment of migraine. With new structural data now available, additional allosteric sites may be identified that could be targeted in the future.
Intrinsic membrane proteins contain hydrophobic cores responsible for their insertion and stability in the bilayer. This characteristic makes them amenable to aggregation, a property that is often concentration-dependent. There has been much debate about possible GPCR oligomerization (72), with some questioning whether this is a non-specific phenomenon and a possible artifact of over-expression. It is now clear that at least a subset of GPCRs can form functionally-significant dimers or higher-order oligomers with likely physiological roles. This seems to differ based on class of GPCR, with obligate, and sometimes covalently stabilized, dimers observed for class C GPCRs, and with more transient association events described for a subset of the class A GPCRs. Class B GPCRs seem to be intermediate between these two groups (73), with what appears to represent relatively stable dimers that are structurally specific along the lipid face of TM4 (74). These can have functional effects, such as to increase ligand affinity and potency, but this has not been present in all such complexes and the tendency to dimerize has been variable across this family. The secretin receptor has been the most stable of those studied to date. Of note, with this well defined interface, homodimers and heterodimers with other members of this family using the same TM4 interface have been described (75). Heterodimers have been shown to affect receptor internalization and trafficking, with agonist occupation of one protomer leading to the internalization of both receptors (76–78). Association events have also been described for class B GPCRs with class A GPCRs, although this appears to be less common and the examples of functional significance are quite small (79; 80).
Existing ligands – what is known about sites of action
Almost all of the drugs targeting class B GPCRs that have been approved to date are peptides, all representing derivatives of natural peptide ligands (or structurally-related peptides like exenatide) and, therefore, almost certainly orthosteric ligands (Table 2). The only exceptions are monoclonal antibodies targeting the CGRP receptor (erenumab). The peptides include agonists and antagonists, taking advantage of what is known (and described above) about structure-activity relationships. Marketed peptides include teriparatide, a PTH1 receptor agonist used for osteoporosis; pramlintide, an agonist of amylin 1–3 receptors used for types 1 and 2 diabetes mellitus; human and salmon CT, CT receptor agonists used for osteoporosis, hypercalcemia, and Paget’s disease; tesamorelin, an agonist of the GHRH receptor used for lipodystrophy; teduglutide, a GLP-2 receptor agonist used in short bowel syndrome; secretin, an agonist of the secretin receptor used in pancreaticobiliary and Zollinger Ellison syndrome diagnostics; and a series of GLP-1 receptor agonists used for type 1 diabetes mellitus, including exenatide, liraglutide, lixisenatide, albiglutide, dulaglutide, and semaglutide. Other peptide agents targeting these and other family members include agonists and antagonists targeting the glucagon receptor, dual glucagon/GLP-1 receptor agents (81), dual glucagon/GIP receptor agents (82), and even triple glucagon/GLP-1/GIP receptor agents (83; 84).
Table 2.
Marketed peptide ligands for class B GPCRs
| Receptors | Peptide ligands |
|---|---|
| Amylin receptors | pramlintide |
| CTR | salmon calcitonin, human calcitonin |
| GHRHR | tesamorelin |
| GLP-1R | Exenatide, liraglutide, lixisenatide, albiglutide, dulaglutide, semaglutide |
| GLP-2R | teduglutide |
| PTH1R | teriparatide |
| SCTR | secretin |
Substantial efforts have been exerted to identify small molecule ligands for these receptors, given the extensive potential clinical applications. Antagonists of the CRF1 receptor for depression, post traumatic stress disorder, and alcoholism (85); the glucagon receptor for type 2 diabetes mellitus (86; 87); and the CGRP receptor for migraine (88) have been identified, but have not reached the clinic. Agonists have been even more problematic to develop. Most of those reported to date have higher lipophilicity and molecular weight than optimal, likely resulting in inadequate bioavailability after oral administration (89). The GLP-1 receptor as a proven target of therapy for type 2 diabetes mellitus has been the major focus of these efforts, with no small molecule agents yet approved for clinical use.
We know much about the molecular basis of action of some antagonists of the CGRP, glucagon, and CRF1 receptors, due to the existence of high resolution structures. Above, we decribed the site of action of CP-376395, a 2-aryloxy-4-alkylaminopyridine inhibiting the CRF1 receptor by binding deep inside the helical bundle (5). Small molecule antagonists of the CGPR receptor (telcagepant and olcegepant) have been observed in crystal structures of the amino-terminal ectodomain of the CLR receptor and RAMP1 (46). This region is also quite important for peptide binding (90; 91), as well as for binding of the monoclonal antibody erenumab (45). These reagents are functional inhibitors, likely interfering with molecular motions important in activation of this receptor.
Different chemotypes of glucagon receptor antagonists have been developed and at least one site of action of some of these molecules has been described, although it is likely that at least one additional distinct site exists for other chemotypes (87). This group of ligands, including NNC0640 (92) and MK-089313 (93), is characterized by a pharmacophore with three components, one of which is a carboxylic acid or an isostere, projecting from a small core (e.g. urea or pyrazole). These compounds bind a site on the intracellular side of the receptor outside of the helical bundle at the lipid interface, spanning TM5, TM6, and TM7 (Figure 2A), and presumably preventing their activation-associated conformational rearrangement (22; 51).
Monoclonal antibodies targeting the ECD of the glucagon receptor and the GLP-1R that are functional antagonists have also been described (26; 27; 47). The antibody mAb23 (for the glucagon receptor) and mAb3F52 (for the GLP-1R) binds within the peptide cleft and competes for natural hormone binding and action, thus representing a competitive antagonist. Interestingly, mAb23 has inverse agonist activity (26). The glucagon receptor antibody mAb7 acts at two sites on the ECD outside of the peptide cleft, having a negative cooperative effect on glucagon binding and action, thus representing a NAM (27).
Small molecule ligands of the PTH-1 receptor, AH-3960 and SW106, that represent an agonist and partial agonist, respectively, have been shown to act on the receptor core, with its ECD deleted (94). The precise location of it action has not been established.
The best understood small molecule PAM-agonists of the GLP-1 receptor include quinoxalines, such as compound 2 (59), pyrimidines, such as BETP/compound B (60), and substituted cyclobutanes, such as Boc5 (1,3-bis[[4-tert-butoxy-carbonylamino)benzoyl]amino]-2,4-bis[3-methoxy-4-(thiophene-2-carbonyloxy)-phenyl]cyclobutane-1,3-dicarboxylic acid (95; 96). Transtech Pharma has communicated the development of compounds such as TT-15 and TTP273 only in patents and presentations (50). The first two compounds are strong nucleophiles and have been demonstrated to covalently modify a cysteine present in the cytosolic face of this receptor in intracellular loop 3 (Cys347) (61). Other electrophilic chemotypes also capable of derivatizing Cys347, also exhibited PAM activity (97). Unfortunately, reactive chemical moieties are quite uncommon in drugs, introducing problems for in vivo administration. The effect of Boc5 as an agonist of the GLP-1 receptor is inhibited by preincubation with the peptide antagonist, exendin(9–39) (95). This has been interpreted to suggest that its site of action is within the ECD, however, such an indirect observation could be explained in other ways, such as having the antagonist change the conformation or access to the helical bundle. The Transtech pharma compounds are reported as allosteric agonists, predicted to bind in the top of helical bundle, although the binding site has yet to be directly defined. Of note, TTP273 has entered into clinical trials as the first orally-available GLP-1R agonist and has been described as meeting its primary endpoint in a phase 2a clinical trial.
The probe dependency of PAMs is particularly well illustrated by some of these small molecules acting at the GLP-1 receptor (98). Compound 2 has been observed to have minimal impact on GLP-1(7–36)NH2 stimulation of cAMP, while increasing the cAMP response to oxyntomodulin 30-fold (43; 98). BETP also has a disproportionate positive impact on oxyntomodulin signaling (98). Unlike their minimal impact on signaling by full length GLP-1, both of these compounds have dramatic positive impact on the cAMP responses to the metabolite of GLP-1, GLP-1(9–36)NH2, increasing responses 400-fold (99; 100). Another interesting feature of this effect is that these compounds positively modulate the affinity of oxyntomodulin and GLP-1(9–36)NH2, however the functional manifestation of this modulation is largely limited to cAMP, with only weak enhancement of calcium mobilization and ERK1/2 phosphorylation (99).
Summary/Conclusions
The convergence between recognition of the remarkable potential of allosteric modulators and the increased insights into the structure of class B GPCRs, providing multiple sites of action for such drugs, promises an exciting future. The broad clinical relevance of this family of receptors, both to normal physiology and the pathophysiology of disease, provides opportunities to respond in a safe and selective manner to individualized needs. We expect to see many allosteric modulators of class B GPCRs introduced in the near future, as the druggable allosteric sites within these receptors are better recognized and as useful scaffolds to target them are identified.
Support:
This work was partially supported by the Mayo Clinic. D.W. is a Career Development Research Fellow of the National Health and Medical Research Council of Australia.
Abbreviations used:
- AM
adrenomedullin
- AMY
amylin
- CT
calcitonin
- CGRP
calcitonin gene-related peptide
- CARC
reversed CRAC motif
- CRAC
cholesterol recognition/interaction amino acid consensus pattern
- CCM
cholesterol consensus motif
- CLR
calcitonin-like receptor
- CRF
corticotropin-releasing factor
- CT
calcitonin
- ECD
extracellular domain
- GCG
glucagon
- GHRH
growth hormone-releasing hormone
- GIP
glucose-dependent insulinotropic peptide
- GLP
glucagon-like peptide
- GPCR
G protein-coupled receptor
- J-domain
junctional-domain
- NAL
neutral allosteric ligand
- NAM
negative allosteric modulator
- PACAP
pituitary adenylate cyclase activating peptide
- PAM
positive allosteric modulator
- PTH
parathyroid hormone
- PTHrP
parathryroid hormone-related peptide
- RAMP
receptor activity-modifying protein
- SCT
secretin
- TIP39
tuberoinfundibular peptide 39
- TM
transmembrane
- VIP
vasoactive intestinal polypeptide
References
- 1.Bortolato A, Dore AS, Hollenstein K, Tehan BG, Mason JS, Marshall FH. 2014. Structure of Class B GPCRs: new horizons for drug discovery. Br. J. Pharmacol 171:3132–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traynor K 2018. FDA approves licensing of erenumab-aooe to prevent migraine. Am. J. Health. Syst. Pharm 75:929–30 [DOI] [PubMed] [Google Scholar]
- 3.Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, et al. 2017. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546:118–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang YL, Khoshouei M, Glukhova A, Furness SGB, Zhao P, et al. 2018. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555:121–5 [DOI] [PubMed] [Google Scholar]
- 5.Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, et al. 2013. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499:438–43 [DOI] [PubMed] [Google Scholar]
- 6.Siu FY, He M, de Graaf C, Han GW, Yang D, et al. 2013. Structure of the human glucagon class B G-protein-coupled receptor. Nature 499:444–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dillon C, Hughes H, O’Reilly NJ, McLoughlin P. 2017. Formulation and characterisation of dissolving microneedles for the transdermal delivery of therapeutic peptides. Int. J. Pharm 526:125–36 [DOI] [PubMed] [Google Scholar]
- 8.Gudiksen N, Hofstatter T, Ronn BB, Sparre T. 2017. FlexTouch: An Insulin Pen-Injector with a Low Activation Force Across Different Insulin Formulations, Needle Technologies, and Temperature Conditions. Diabetes Technol. Ther 19:603–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wootten D, Miller LJ, Koole C, Christopoulos A, Sexton PM. 2017. Allostery and biased agonism at class B G protein-coupled receptors. Chem. Rev 117:111–38 [DOI] [PubMed] [Google Scholar]
- 10.Thal DM, Glukhova A, Sexton PM, Christopoulos A. 2018. Structural insights into G-proteincoupled receptor allostery. Nature 559:45–53 [DOI] [PubMed] [Google Scholar]
- 11.Iqbal J, Zaidi M, Schneider AE. 2003. Cinacalcet hydrochloride (Amgen). IDrugs 6:587–92 [PubMed] [Google Scholar]
- 12.Christopoulos A 2014. Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol 86:463–78 [DOI] [PubMed] [Google Scholar]
- 13.de Graaf C, Song G, Cao C, Zhao Q, Wang MW, et al. 2017. Extending the structural view of class B GPCRs. Trends Biochem. Sci 42:946–60 [DOI] [PubMed] [Google Scholar]
- 14.Kenakin TP. 2012. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br. J. Pharmacol 165:1659–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM. 2013. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc. Natl. Acad. Sci. U. S. A 110:5211–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cvicek V, Goddard WA 3rd, Abrol R. 2016. Structure-Based Sequence Alignment of the Transmembrane Domains of All Human GPCRs: Phylogenetic, Structural and Functional Implications. PLoS Comput. Biol 12:e1004805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vass M, Kooistra AJ, Yang D, Stevens RC, Wang MW, de Graaf C. 2018. Chemical Diversity in the G Protein-Coupled Receptor Superfamily. Trends Pharmacol. Sci 39:494–512 [DOI] [PubMed] [Google Scholar]
- 18.Grace CR, Perrin MH, DiGruccio MR, Miller CL, Rivier JE, et al. 2004. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl. Acad. Sci. U. S. A 101:12836–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang YL, Khoshouei M, Deganutti G, Glukhova A, Koole C, et al. 2018. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561:492–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Sun B, Feng D, Hu H, Chu M, et al. 2017. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546:248–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erlandson SC, McMahon C, Kruse AC. 2018. Structural basis for G protein-coupled receptor signaling. Annu. Rev. Biophys [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Qiao A, Yang D, Yang L, Dai A, et al. 2017. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 546:259–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Qiao A, Yang L, Van Eps N, Frederiksen KS, et al. 2018. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 553:106–10 [DOI] [PubMed] [Google Scholar]
- 24.Ehrenmann J, Schoppe J, Klenk C, Rappas M, Kummer L, et al. 2018. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat. Struct. Mol. Biol 25:1086–92 [DOI] [PubMed] [Google Scholar]
- 25.Jazayeri A, Rappas M, Brown AJH, Kean J, Errey JC, et al. 2017. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546:254–8 [DOI] [PubMed] [Google Scholar]
- 26.Koth CM, Murray JM, Mukund S, Madjidi A, Minn A, et al. 2012. Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U. S. A 109:14393–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukund S, Shang Y, Clarke HJ, Madjidi A, Corn JE, et al. 2013. Inhibitory mechanism of an allosteric antibody targeting the glucagon receptor. J. Biol. Chem 288:36168–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wootten D, Reynolds CA, Koole C, Smith KJ, Mobarec JC, et al. 2016. A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: Lessons from class B crystal structures. Mol. Pharmacol 89:335–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoare SR. 2005. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Disc. Today 10:417–27 [DOI] [PubMed] [Google Scholar]
- 30.Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP. 2005. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc. Natl. Acad. Sci. U. S. A 102:16084–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montrose-Rafizadeh C, Yang H, Rodgers BD, Beday A, Pritchette LA, Eng J. 1997. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J. Biol. Chem 272:21201–6 [DOI] [PubMed] [Google Scholar]
- 32.Hoare SR. 2007. Allosteric modulators of class B G-protein-coupled receptors. Curr. Neuropharmacol 5:168–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hager MV, Clydesdale L, Gellman SH, Sexton PM, Wootten D. 2017. Characterization of signal bias at the GLP-1 receptor induced by backbone modification of GLP-1. Biochem. Pharmacol 136:99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hager MV, Johnson LM, Wootten D, Sexton PM, Gellman SH. 2016. Beta-arrestin-biased agonists of the GLP-1 receptor from beta-amino acid residue incorporation into GLP-1 analogues. J. Am. Chem. Soc 138:14970–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furness SGB, Christopoulos A, Sexton PM, Wootten D. 2018. Differential engagement of polar networks in the glucagon-like peptide 1 receptor by endogenous variants of the glucagon-like peptide 1. Biochem. Pharmacol 156:223–40 [DOI] [PubMed] [Google Scholar]
- 36.Dal Maso E, Zhu Y, Pham V, Reynolds CA, Deganutti G, et al. 2018. Extracellular loops 2 and 3 of the calcitonin receptor selectively modify agonist binding and efficacy. Biochem. Pharmacol 150:214–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wootten D, Reynolds CA, Smith KJ, Mobarec JC, Koole C, et al. 2016. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell 165:1632–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koole C, Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM. 2012. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J. Biol. Chem 287:3642–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koole C, Wootten D, Simms J, Savage EE, Miller LJ, et al. 2012. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) differentially regulates orthosteric but not allosteric agonist binding and function. J. Biol. Chem 287:3659–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lei S, Clydesdale L, Dai A, Cai X, Feng Y, et al. 2018. Two distinct domains of the glucagon-like peptide-1 receptor control peptide-mediated biased agonism. J. Biol. Chem 293:9370–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wootten D, Reynolds CA, Koole C, Smith KJ, Mobarec JC, et al. 2016. A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: lessons from class B crystal structures. Mol. Pharmacol 89:335–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wootten D, Reynolds CA, Smith KJ, Mobarec JC, Furness SG, et al. 2016. Key interactions by conserved polar amino acids located at the transmembrane helical boundaries in Class B GPCRs modulate activation, effector specificity and biased signalling in the glucagon-like peptide-1 receptor. Biochem. Pharmacol 118:68–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koole C, Wootten D, Simms J, Valant C, Sridhar R, et al. 2010. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol. Pharmacol 78:456–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broichhagen J, Podewin T, Meyer-Berg H, von Ohlen Y, Johnston NR, et al. 2015. Optical control of insulin secretion using an incretin switch. Angew. Chem. Int. Ed. Engl 54:15565–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi L, Lehto SG, Zhu DX, Sun H, Zhang J, et al. 2016. Pharmacologic characterization of AMG 334, a potent and selective human monoclonal antibody against the calcitonin gene-related peptide receptor. J. Pharmacol. Exp. Ther 356:223–31 [DOI] [PubMed] [Google Scholar]
- 46.ter Haar E, Koth CM, Abdul-Manan N, Swenson L, Coll JT, et al. 2010. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure 18:1083–93 [DOI] [PubMed] [Google Scholar]
- 47.Hennen S, Kodra JT, Soroka V, Krogh BO, Wu X, et al. 2016. Structural insight into antibody-mediated antagonism of the Glucagon-like peptide-1 Receptor. Sci. Rep 6:26236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edvinsson L, Haanes KA, Warfvinge K, Krause DN. 2018. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat. Rev. Neurol 14:338–50 [DOI] [PubMed] [Google Scholar]
- 49.Dong M, Cox RF, Miller LJ. 2009. Juxtamembranous region of the amino terminus of the family B G protein-coupled calcitonin receptor plays a critical role in small-molecule agonist action. J. Biol. Chem 284:21839–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freeman J, Weaver S, Davis S, Rao M, Quada J, et al. 2016. TTP273: Oral, G protein pathway selective clinical-stage GLP-1 receptor (GLP-1R) agonist. Keystone Symposium, Keystone, CO February 22, 2016 [Google Scholar]
- 51.Jazayeri A, Dore AS, Lamb D, Krishnamurthy H, Southall SM, et al. 2016. Extra-helical binding site of a glucagon receptor antagonist. Nature 533:274–7 [DOI] [PubMed] [Google Scholar]
- 52.Song G, Yang D, Wang Y, de Graaf C, Zhou Q, et al. 2017. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546:312–315 [DOI] [PubMed] [Google Scholar]
- 53.Li H, Papadopoulos V. 1998. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 139:4991–7 [DOI] [PubMed] [Google Scholar]
- 54.Baier CJ, Fantini J, Barrantes FJ. 2011. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep 1:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, et al. 2008. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 16:897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Genheden S, Essex JW, Lee AG. 2017. G protein coupled receptor interactions with cholesterol deep in the membrane. Biochim. Biophys. Acta Biomembr 1859:268–81 [DOI] [PubMed] [Google Scholar]
- 57.Zocher M, Zhang C, Rasmussen SG, Kobilka BK, Muller DJ. 2012. Cholesterol increases kinetic, energetic, and mechanical stability of the human beta2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A 109:E3463–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desai AJ, Miller LJ. 2018. Changes in the plasma membrane in metabolic disease: impact of the membrane environment on G protein-coupled receptor structure and function. Br. J. Pharmacol 175:4009–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, et al. 2007. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc. Natl. Acad. Sci. U. S. A 104:937–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sloop KW, Willard FS, Brenner MB, Ficorilli J, Valasek K, et al. 2010. Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes 59:3099–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nolte WM, Fortin JP, Stevens BD, Aspnes GE, Griffith DA, et al. 2014. A potentiator of orthosteric ligand activity at GLP-1R acts via covalent modification. Nat. Chem. Biol 10:629–31 [DOI] [PubMed] [Google Scholar]
- 62.Broichhagen J, Johnston NR, von Ohlen Y, Meyer-Berg H, Jones BJ, et al. 2016. Allosteric Optical Control of a Class B G-Protein-Coupled Receptor. Angew. Chem. Int. Ed. Engl 55:5865–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harikumar KG, Wootten D, Pinon DI, Koole C, Ball AM, et al. 2012. Glucagon-like peptide-1 receptor dimerization differentially regulates agonist signaling but does not affect small molecule allostery. Proc. Natl. Acad. Sci. U. S. A 109:18607–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeVree BT, Mahoney JP, Velez-Ruiz GA, Rasmussen SG, Kuszak AJ, et al. 2016. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535:182–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. 2015. Endogenous allosteric modulators of G protein-coupled receptors. J. Pharmacol. Exp. Ther 353:246–60 [DOI] [PubMed] [Google Scholar]
- 66.Liu W, Chun E, Thompson AA, Chubukov P, Xu F, et al. 2012. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337:232–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.White KL, Eddy MT, Gao ZG, Han GW, Lian T, et al. 2018. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 26:259–69 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Selvam B, Shamsi Z, Shukla D. 2018. Universality of the Sodium Ion Binding Mechanism in Class A G-Protein-Coupled Receptors. Angew. Chem. Int. Ed. Engl 57:3048–53 [DOI] [PubMed] [Google Scholar]
- 69.Foord SM, Marshall FH. 1999. RAMPs: accessory proteins for seven transmembrane domain receptors. Trends Pharmacol. Sci 20:184–7 [DOI] [PubMed] [Google Scholar]
- 70.Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, et al. 2002. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol. Rev 54:233–46 [DOI] [PubMed] [Google Scholar]
- 71.Klein KR, Matson BC, Caron KM. 2016. The expanding repertoire of receptor activity modifying protein (RAMP) function. Crit. Rev. Biochem. Mol. Biol 51:65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sleno R, Hebert TE. 2019. Shaky ground - The nature of metastable GPCR signalling complexes. Neuropharmacology [DOI] [PubMed] [Google Scholar]
- 73.Harikumar K, Dong M, Miller L. 2010. Secretin receptor dimerization. A possible functionally-important paradigm for Family B G protein-coupled receptors. In: GPCR molecular pharmacology and drug targeting: Shifting paradigms and new directions. Gilchrist A (ed) John Wiley & Sons, Inc., 138–164. [Google Scholar]
- 74.Gao F, Harikumar KG, Dong M, Lam PC, Sexton PM, et al. 2009. Functional importance of a structurally distinct homodimeric complex of the family B G protein-coupled secretin receptor. Mol. Pharmacol 76:264–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harikumar KG, Morfis MM, Sexton PM, Miller LJ. 2008. Pattern of intra-family hetero-oligomerization involving the G-protein-coupled secretin receptor. J. Mol. Neurosci 36:279–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Roed SN, Nohr AC, Wismann P, Iversen H, Brauner-Osborne H, et al. 2015. Functional consequences of glucagon-like peptide-1 receptor cross-talk and trafficking. J. Biol. Chem 290:1233–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roed SN, Orgaard A, Jorgensen R, De Meyts P. 2012. Receptor oligomerization in family B1 of G-protein-coupled receptors: focus on BRET investigations and the link between GPCR oligomerization and binding cooperativity. Front Endocrinol 3:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Harikumar KG, Lau S, Sexton PM, Wootten D, Miller LJ. 2017. Coexpressed class B G protein-coupled secretin and GLP-1 receptors self- and cross-associate: Impact on pancreatic islets. Endocrinology 158:1685–700 [DOI] [PubMed] [Google Scholar]
- 79.Lee LT, Ng SY, Chu JY, Sekar R, Harikumar KG, et al. 2014. Transmembrane peptides as unique tools to demonstrate the in vivo action of a cross-class GPCR heterocomplex. FASEB J. 28:2632–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harikumar KG, Augustine ML, Lee LT, Chow BK, Miller LJ. 2016. Structure and function of cross-class complexes of G protein-coupled secretin and angiotensin 1a receptors. J. Biol. Chem 291:17332–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, et al. 2013. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl. Med 5:209ra151. [DOI] [PubMed] [Google Scholar]
- 82.Frias JP, Bastyr EJ 3rd, Vignati L, Tschop MH, Schmitt C, et al. 2017. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090–2746, in patients with type 2 diabetes. Cell Metab. 26:343–52 e2 [DOI] [PubMed] [Google Scholar]
- 83.Finan B, Yang B, Ottaway N, Smiley DL, Ma T, et al. 2015. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med 21:27–36 [DOI] [PubMed] [Google Scholar]
- 84.Knerr PJ, Finan B, Gelfanov V, Perez-Tilve D, Tschop MH, DiMarchi RD. 2018. Optimization of peptide-based polyagonists for treatment of diabetes and obesity. Bioorg. Med. Chem 26:2873–81 [DOI] [PubMed] [Google Scholar]
- 85.Fahmy H, Kuppast B, Ismail MT. 2017. Structure and Function of Small Non-Peptide CRF Antagonists and their Potential Clinical Use. Curr. Mol. Pharmacol 10:270–81 [DOI] [PubMed] [Google Scholar]
- 86.Scheen AJ, Paquot N, Lefebvre PJ. 2017. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin. Investig. Drugs 26:1373–89 [DOI] [PubMed] [Google Scholar]
- 87.Sammons MF, Lee EC. 2015. Recent progress in the development of small-molecule glucagon receptor antagonists. Bioorg. Med. Chem. Lett 25:4057–64 [DOI] [PubMed] [Google Scholar]
- 88.Maasumi K, Michael RL, Rapoport AM. 2018. CGRP and Migraine: The Role of Blocking Calcitonin Gene-Related Peptide Ligand and Receptor in the Management of Migraine. Drugs 78:913–28 [DOI] [PubMed] [Google Scholar]
- 89.Willard FS, Bueno AB, Sloop KW. 2012. Small molecule drug discovery at the glucagon-like peptide-1 receptor. Exper. Diab. Res 2012:709893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Booe JM, Walker CS, Barwell J, Kuteyi G, Simms J, et al. 2015. Structural basis for receptor activity-modifying protein-dependent selective peptide recognition by a G protein-coupled receptor. Mol. Cell 58:1040–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gingell JJ, Simms J, Barwell J, Poyner DR, Watkins HA, et al. 2016. An allosteric role for receptor activity-modifying proteins in defining GPCR pharmacology. Cell Discov. 2:16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lau J, Behrens C, Sidelmann UG, Knudsen LB, Lundt B, et al. 2007. New beta-alanine derivatives are orally available glucagon receptor antagonists. J. Med. Chem 50:113–28 [DOI] [PubMed] [Google Scholar]
- 93.Xiong Y, Guo J, Candelore MR, Liang R, Miller C, et al. 2012. Discovery of a novel glucagon receptor antagonist N-[(4-{(1S)-1-[3-(3, 5-dichlorophenyl)-5-(6-methoxynaphthalen-2-yl)-1H-pyrazol-1-yl]ethyl}phenyl)carbo nyl]-beta-alanine (MK-0893) for the treatment of type II diabetes. J. Med. Chem 55:6137–48 [DOI] [PubMed] [Google Scholar]
- 94.Carter PH, Dean T, Bhayana B, Khatri A, Rajur R, Gardella TJ. 2015. Actions of the small molecule ligands SW106 and AH-3960 on the type-1 parathyroid hormone receptor. Mol. Endocrinol 29:307–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen D, Liao J, Li N, Zhou C, Liu Q, et al. 2007. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc. Natl. Acad. Sci. U. S. A 104:943–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Q, Li N, Yuan Y, Lu H, Wu X, et al. 2012. Cyclobutane derivatives as novel nonpeptidic small molecule agonists of glucagon-like peptide-1 receptor. J. Med. Chem 55:250–67 [DOI] [PubMed] [Google Scholar]
- 97.Bueno AB, Showalter AD, Wainscott DB, Stutsman C, Marin A, et al. 2016. Positive allosteric modulation of the glucagon-like peptide-1 receptor by diverse electrophiles. J. Biol. Chem 291:10700–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wootten D, Savage EE, Willard FS, Bueno AB, Sloop KW, et al. 2013. Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol. Pharmacol 83:822–34 [DOI] [PubMed] [Google Scholar]
- 99.Li N, Lu J, Willars GB. 2012. Allosteric modulation of the activity of the glucagon-like peptide-1 (GLP-1) metabolite GLP-1 9–36 amide at the GLP-1 receptor. PloS one 7:e47936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wootten D, Savage EE, Valant C, May LT, Sloop KW, et al. 2012. Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol. Pharmacol 82:281–90 [DOI] [PubMed] [Google Scholar]