

Abstract
The pathogenesis of colorectal cancer (CRC) involves different mechanisms, such as genomic and microsatellite instabilities. Recently, a contribution of the base excision repair (BER) pathway in CRC pathology has been emerged. In this context, the involvement of APE1 in the BER pathway and in the transcriptional regulation of genes implicated in tumor progression strongly correlates with chemoresistance in CRC and in more aggressive cancers. In addition, the APE1 interactome is emerging as an important player in tumor progression, as demonstrated by its interaction with Nucleophosmin (NPM1). For these reasons, APE1 is becoming a promising target in cancer therapy and a powerful prognostic and predictive factor in several cancer types. Thus, specific APE1 inhibitors have been developed targeting: i) the endonuclease activity; ii) the redox function and iii) the APE1-NPM1 interaction. Furthermore, mutated p53 is a common feature of advanced CRC. The relationship between APE1 inhibition and p53 is still completely unknown. Here, we demonstrated that the inhibition of the endonuclease activity of APE1 triggers p53-mediated effects on cell metabolism in HCT-116 colon cancer cell line. In particular, the inhibition of the endonuclease activity, but not of the redox function or of the interaction with NPM1, promotes p53 activation in parallel to sensitization of p53-expressing HCT-116 cell line to genotoxic treatment. Moreover, the endonuclease inhibitor affects mitochondrial activity in a p53-dependent manner. Finally, we demonstrated that 3D organoids derived from CRC patients are susceptible to APE1-endonuclease inhibition in a p53-status correlated manner, recapitulating data obtained with HCT-116 isogenic cell lines. These findings suggest the importance of further studies aimed at testing the possibility to target the endonuclease activity of APE1 in CRC.
Keywords: colorectal cancer, BER, APE1, APE1-inhibitors, p53, organoids
Graphical Abstract
1. Introduction
Colorectal cancer (CRC) is considered the third most common cancer and the fourth most common cause of cancer-related death worldwide [1]. CRC is a multistep process involving a series of histo-morphological and genetic changes, that accumulate over time in the epithelial layer of the intestinal tract. Interestingly, genetic and epigenetic modifications lead to the activation of oncogenes and/or the inactivation of tumor suppressor genes, as formulated by Volgestein and Fearon [2,3]. Both genetic and environmental factors are essential in the etiology of the disease. About 70% of CRC patients suffer from a sporadic form, whereas 10-30% present a familial predisposition and only 5-7% exhibit an inherited trait [4]. However, the most common, often overlapping, mechanisms involved in the pathogenesis of CRC are represented by the chromosome and microsatellite instabilities, the CpG island methylator phenotype and the deletion of the long arm of chromosome 18 [1,5]. In the landscape of chromosome instability, the most common mutations occur in specific tumor suppressor genes (e.g. APC, PTEN, SMAD4, TGFBR2, TP53) or oncogenes (e.g. BRAF, KRAS, PIK3CA). Recently, the involvement of DNA repair genes has been demonstrated to be associated with the pathogenesis of CRC [6-8]. In particular, both endogenous (e.g. metabolic activity of the cells) and exogenous factors (e.g. food intake) are involved in DNA damage, which requires the activation of the DNA repair mechanisms. In particular, the base excision repair (BER) pathway is involved in repairing DNA chemical modifications, such as deamination, oxidation, and alkylation [9]. Interestingly, BER has been found altered in CRC, as demonstrated by the presence of single nucleotide polymorphisms (SNPs) in several BER genes including DNA glycosylases, apurinic/apyrimidinic endonuclease 1 (APE1) and DNA Polymerase β (Pol β) [10]. In BER, APE1 cleaves the DNA phosphodiester backbone on the 5′ side of an abasic apurinic/apyrimidinic (AP) site, previously produced by the action of damage-specific glycosylases, generating a nick in the DNA and leaving 3′ hydroxyl and 5′ dRP free termini, which are processed and replaced by a correct nucleotide by Pol β [11]. Recent molecular snapshots of the endonuclease-reaction clearly defined the APE1 catalytic mechanism of action [12]. The catalytic site of the enzymes involves a Mg2+ ion coordinated by Asp70, Glu96 and a water molecule in contact with non-bridging oxygen of the phosphate. Additionally, the nucleophilic water is in position for in-line attack of the phosphorus atom and is coordinated by Asn212 and Asp210. APE1 is also endowed of an exonuclease activity through the removal of 3’ end groups of a mismatched or DNA damaged bases to generate substrates that are processed by the downstream repair enzymes [13]. Recently, we demonstrated that APE1 plays an important role in the recognition and processing of ribose monophosphate AP sites and oxidized ribonucleotides embedded in DNA through a classical AP endonuclease activity and a nucleotide incision repair (NIR) activity, respectively [14].
As widely described, APE1 is implicated in cancer gene expression regulation due to its role as a redox co-activator of several transcription factors, such as Egr-1, NF-κB, p53, STAT3, HIF-1α, CREB, AP-1, and Pax-5/8 [15]. APE1 is considered as a unique nuclear redox-signaling factor bearing seven Cys residues. Three of the Cys residues, C65, C93, and C99, are essential for its redox activity, that involves a redox cycle through the potential formation of intermolecular disulfide bonds with the protein target [16-20]. While C65 acts as the nucleophilic cysteine, C93 and C99 likely play roles in resolving disulfide bonds that are formed in APE1 upon oxidation. Structural studies demonstrated that APE1 exists in both native and partially unfolded conformations, controlled by Thioredoxin (TRX) [16], in which the partially unfolded state of APE1 represents the redox active intermediate of the enzyme.
Recently, it has been demonstrated that APE1 is not only involved in DNA repair mechanisms and transcriptional regulation, but also in miRNA metabolism [21]. Furthermore, the interaction between APE1 and Nucleophosmin (NPM1) is essential for the subcellular localization of APE1 modulating its endonuclease activity [22]. Interestingly, this interaction was found altered in ovarian cancer promoting tumor aggressiveness and resistance. Finally, it has been demonstrated that in the majority of cases, there is a positive correlation between upregulated expression of APE1 and the development of several tumors, such as: colon [23,24], breast [25], hepatic [26], prostate [27], pancreatic [28], ovarian [29], lung cancers [30], leukemias [31] and many others [32]. Furthermore, APE1 overexpression is associated with the onset of chemoresistance phenomena [22]. For these reasons, APE1 is considered a promising prognostic and predictive biomarker [33] and several strategies have been developed to inhibit its functions in cancer cells [34], leading to the development of ongoing clinical trials.
In recent years, a great effort has been put in the development of specific inhibitors targeting the different functions of APE1, i.e. Compound #3, APX2009, Spiclomazine, Fiduxosin and SB206553. Compound #3 blocks the endonuclease activity of APE1 acting as a competitive inhibitor by binding the active site of the enzyme and consequently leading to a decrease of APE1-DNA complex formation in a dose-dependent manner [35]. The inhibition promotes, in turn, an increase of the unrepaired AP sites accumulation in genomic DNA, that, in combination with an alkylating agent treatment, such as methyl methanesulfonate (MMS), induces high levels of cellular death [36,37]. APX2009, an APE1 specific redox inhibitor, is the second generation molecules of the drug APX3330, previously named E3330 [38]. APX3330 inhibits the activator function of APE1 on different transcription factors such as NF-κB, AP-1, HIF-1 and STAT3 [18,39,40]. In particular, APX3330 increases disulfide bonds involving C65 and/or C93 residues in APE1, impairing its redox activity [41]. APX3330 recently completed cancer phase I clinical trials with demonstrated safety, response and APE1 target engagement [42,43]. Finally, Spiclomazine, Fiduxosin, and SB206553 molecules inhibit the interaction of APE1 with NPM1, a protein involved in rRNA biogenesis, by directly binding to the N-terminal domain of APE1. We demonstrated that these inhibitors alter the localization and the endonuclease activity of APE1, but not the rRNA maturation [22] and induce apoptosis in different tumor cell lines [22,44]. To our knowledge, only one study investigated the ability of the APE1-redox inhibitor (APX3330) to affect colon cancer stem cells (CCSCs) growth in vitro and to enhance the effect of the chemotherapeutic agent 5-Fluorouracil (5-FU) in CCSCs xenograft mice [23]. Thus, the importance of exploring the effect of different APE1 inhibitors in CRC models is apparent. Here, we used the well-known HCT-116 colon cancer cell model, to explore the relevance of p53 upon APE1 inhibition, and extended our findings using a 3D organoid cultures model derived from CRC affected patients.
Due to the intricate mechanisms that characterize the CRC etiology, research has focused on personalized precision medicine of CRC. The generation of patient-derived 3D tumor organoids will greatly enhance our understanding of the disease complexity and the heterogeneity in order to develop patient-specific therapies [45]. Organoids have a special property to mirror the key-features of the original patient’s tissue [46], representing an ideal tool to develop patient-specific therapies by performing drug screenings.
Similarly to APE1, the well-known tumor suppressor gene TP53 has been found altered in most tumors [47]. The wild-type p53 protein is a transcription factor involving in cell cycle arrest, senescence and apoptosis, besides being a key player in the DNA Damage Response (DDR) to single-strand breaks (SSBs) and double-strand break (DSBs) accumulation. Among all the mutated genes promoting CRC, p53 has an important role [48]. Indeed, loss of p53 function stimulates the development of the late stage of CRC and is associated with poor prognosis [49]. p53 plays a role not only as a modulator of the cell cycle to guarantee genome stability, but it is also directly involved in the activation of proteins that are associated with DNA repair processes [50]. In particular, it has been demonstrated that p53 prevents genomic instability through a BER gene expression regulation [51]. Importantly, p53 regulates DNA glycosylases (OGG1 and MUTYH) [52,53], APE1 [54,55], Pol β [56] expression and acts as a transcriptional repressor of DNA polymerase δ [57]. However, it is unknown whether p53 is part of DDR starting from AP sites accumulation as a consequence of APE1 inactivation or inhibition. The present study was aimed at addressing this issue. Furthermore, data on whether APE1 inhibitors may affect cell viability of colon cancer cells through p53-induced cell response are the focus of the work presented here.
2. Materials and methods
2.1. Cell cultures.
HCT-116 p53+/+ and HCT-116 p53−/− (ATCC®) cells were grown in Dulbecco’s modified Eagle’s medium (EuroClone, Milan, Italy) supplemented with 10% fetal bovine serum (EuroClone). CH12F3+/+/Δ and CH12F3Δ/Δ/Δ cells were grown in RPMI 1640 (EuroClone) supplemented with 10% fetal bovine serum (EuroClone), 1X non Essential Amino Acids (EuroClone), 1 mM Sodium Pyruvate (EuroClone), 25 mM HEPES (EuroClone) and 50 μM β-mercaptoethanol (Promega, Madison, WI, USA). OCI-AML2 cells were grown in α-MEM (Invitrogen, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum (EuroClone). HCC70 cells were grown in RPMI 1640 (EuroClone) supplemented with 10% fetal bovine serum (EuroClone). All culturing media were also supplemented with 2 mM GlutaMAX (EuroClone), 100 U/ml penicillin and 10 μg/ml streptomycin (EuroClone).
2.2. Human tissues.
Colonic tissues were obtained upon surgical resection from the University Hospital Santa Maria della Misericordia of Udine. All patients were diagnosed with colorectal cancer. This study was approved by the ethical committee of University Hospital Santa Maria della Misericordia of Udine (CEUR-2017-PR-048-UNIUD) and all samples were obtained prior to informed consent.
2.3. Organoid culture.
The generation of patient-derived tumor organoids was performed as described by [58] with some modifications. Tumor intestinal tissue was washed with ice-cold PBS supplemented with 100 U/ml penicillin and 10 μg/ml streptomycin (EuroClone) several times. Tumor tissue was homogenized with scissors and then digested with 0.26 U/ml Liberase (Sigma-Aldrich, St. Louis, MO, USA) in basal medium, composed of Advanced DMEM/F12 (Life Technologies, Carlsbad, CA, USA) supplemented with 2 mM Glutamax (Life Technologies), 10 mM HEPES (Life Technologies), 100 U/ml penicillin and 10 μg/ml streptomycin (EuroClone) complemented with 100 μg/ml Primocin (InvivoGen, San Diego, CA, USA) and 10 μM Y-27632 (Abcam, Cambridge, UK) for 1 h at 37 °C shaking at 250 rpm. The resulting fraction was passed through a 100-mm cell strainer. The filtered solution was centrifuged at 1,200 rpm for 5 min at 4°C. The cell pellet was resuspended with Red Blood Cell (RBC) Lysis Buffer (BioLegend, San Diego, CA, USA) and incubated for 10 min at room temperature (RT) in the dark. RBC buffer was neutralized adding basal medium complemented with 10% fetal bovine serum (BioWest, Nuaillé, France). The resulting solution was centrifuged at 1,200 rpm for 5 min at 4°C and the cell pellet was resuspended with 3 ml of basal medium. Cells were counted with a Burker chamber and 200,000 cells/10 μl were mixed with 20 μl of Matrigel and 30 μl drop was plated in a single well of 24-well. After polymerization of Matrigel (Corning®, Corning, NY, USA) for 10-15 min at 37°C, 500 μl of culture medium was added. Tumor organoids were cultured in basal medium containing 1X B27 (Life Technologies), 1.15 mM N-acetylcysteine (Sigma-Aldrich), 10 mM Nicotinamide (Sigma-Aldrich), 10 nM Gastrin I (Tocris Bioscience, Bristol, UK), 10 nM Prostaglandin E2 (Sigma-Aldrich), 500 nM A83-01 (R&D System, Minneapolis, MN, USA), 50 ng/ml mEGF (PeproTech, London, UK), 3 μM SB202190 (Sigma-Aldrich), 10% Noggin Conditioned Medium, 20% R-Spondin1 Conditioned Medium, 10 μM Y-27632 (Abcam) and 100 μg/ml Primocin (InvivoGen). The medium was refreshed every two/three days. Tumour organoids were passaged 1:4 every one/two weeks.
2.4. Treatments.
Compound #3 was kindly provided by the National Center for Advancing Translational Sciences. APX2009 was kindly provided by Professor Mark R. Kelley (Indiana University, School of Medicine). Fiduxosin, SB206553, Spiclomazine were purchased as previously described [22]. All compounds were solved in dimethyl sulfoxide (DMSO). IC50 values were calculated using Combenefit 2.021 Software.
2.5. RNA interference.
One day before transfection, cells were seeded in 60-mm plates at a density of 600,000 cells per plate. Cells were then transiently transfected with 100 pmol siRNA APE1 5ʹ-UACUCCAGUCGUACCAGACCU-3ʹ or the scramble control siRNA 5ʹ-CCAUGAGGUCAUGGUCUGdTdT-3ʹ (Dharmacon, Lafayette, CO, USA) using DharmaFECT reagent (Dharmacon). After 72 h upon transfection, cells were collected and whole cell extracts were prepared.
2.6. Preparation of the cell extracts and protein quantification.
For the preparation of whole cell lysates, 200,000 HCT-116 cells were plated on 6-well plates and, the following day, cells were treated with Compound #3 or APX2009. After 48 h cells were collected by trypsinization and centrifuged at 250 x g for 5 min. The supernatant was removed, and the pellet was washed once with PBS and then centrifuged again as described before. Cell pellet was resuspended in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% w/v Triton X-100 supplemented with 1 mM protease inhibitor cocktail (Sigma-Aldrich), 1 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM NaF and 1 mM Na3VO4 for 30 min at 4 °C. After centrifugation at 13,000 rpm for 20 min at 4 °C, the supernatant was collected as a whole cell lysate. The protein concentration was determined by using Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA, USA).
2.7. Antibodies and Western blotting analysis.
For Western blotting analyses, whole cell lysates were prepared and 30 μg of proteins were resolved on 12% SDS–PAGE, transferred onto nitrocellulose membranes (Schleicher & Schuell Bioscience, Keene, NH, USA) and incubated with antibodies for p53 1:1000 (#sc-126, Santa Cruz Biotechnology, Dallas, TX, USA), APE1 1:1000 (#NB-100116, Thermo Fisher Scientific, Waltham, MA, USA), p21 1:1000 (#2947, Cell Signaling Technology, Danvers, MA, USA) and actin 1:2000 (#A2066, Sigma-Aldrich). The corresponding secondary antibodies labelled with IR-Dye (goat anti-rabbit IgG IRDye 680 and goat anti-mouse IgG IRDye 800, LI-COR Biosciences, Lincoln, NE, USA) were used (1:10,000). Blots were developed by using the NIR Fluorescence technology (LI-COR Biosciences). Images were acquired and quantified by using an Odyssey CLx Infrared Imaging System (LI-COR Biosciences).
2.8. Metabolic activity.
Metabolic activity of HCT-116 cell lines was measured through the MTS CellTiter 96 AQueous One Solution Cell Proliferation Assay and through RealTime-Glo™ MT Cell Viability Assay (Promega) according to the manufacturer’s instructions. In details, 5,000 cells were plated on transparent or black 96-well plates, as needed, and the day after, cells were treated with the indicated drugs for 48 h. Metabolic activity of CH12F3 cell lines was measured with CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega) according to the manufacturer’s instructions. In details, 10,000 cells were plated on black 96-well plates and the day after cells were treated with increasing amounts of Compound #3 or APX2009 at the indicated concentrations for 48 h. Metabolic activity of patient-derived tumor organoids was measured through the RealTime-Glo™ MT Cell Viability Assay (Promega) according to the manufacturer’s instructions. In this case, 3,000 cells derived from tumor organoids were plated together with Matrigel on black 96-well plates and incubated for three days for allowing the generation of the organoids. Tumor organoids were treated with increasing amounts of Compound #3 for 48 h. For metabolic assay, Y-27632 was removed from the culture medium.
2.9. AP sites measurements.
For the AP sites measurements, 200,000 HCT-116 cells were plated on 6-well plates and, the following day, cells were treated with Compound #3. After 48 h, genomic DNA was extracted from the collected cells using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), and concentration and purity were determined by NanoDrop Microvolume Spectrophotometer (Thermo Fisher Scientific). Samples of 100 μg/ml of genomic DNA were analyzed to quantify abasic sites in DNA using the DNA Damage Quantification Kit -AP Site Counting- (Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer’s instructions.
2.10. Comet assay analysis.
The comet assay was performed as previously described [59]. Specifically, 25,000 cells were plated on 24-well plates and the day after cells were treated with 0.5 μM Compound #3. After 48 h, cells were collected and mixed with low melting point agarose at 37 °C, and the mixture was applied to a agarose pre-coated glass coverslip to form a thin layer. Cold lysis buffer was added to lyse cells at 4 °C. After 1 h, the glass coverslip was moved into alkaline electrophoresis buffer for 30 min to unwind DNA. Electrophoresis was carried out at 25 V and 300 mA for 30 min. The glass coverslip was washed with neutralizing buffer for three times and stained by Sybr Gold 1X (Thermo Fisher Scientific) for 30 min in dark. Comets were recorded by laser scanning confocal microscope (LEICA TCS SP2, Leica Microsystems, Wetzlar, Germany). The tail moment was analyzed for 100 cells at random by OpenComet software.
2.11. Quantitative real-time reverse transcriptase-PCR.
For RNA extraction, 200,000 HCT-116 cells were plated on 6-well plates and, the following day, were treated with Compound #3 or APX2009. After 48 h, total RNA was extracted from the collected cells using the NUCLEOSPIN RNA II (MACHEREY-NAGEL GmbH & Co., Duren, Germany). The quality of the RNA samples was tested on an agarose gel. Then, single-strand cDNA was obtained from 1 μg of purified RNA using SensiFAST cDNA Synthesis Kit (Bioline Meridian Bioscience, Cincinnati, OH, USA). qRT-PCR was performed using SensiFAST SYBR No-ROX Kit (Bioline Meridian Bioscience) and a CFX96 Real-Time System (Bio-Rad). Each sample analysis was performed in triplicate. Samples without template and without reverse transcriptase were used as negative control,. The cycling parameters contemplate a first denaturation at 95 °C for 10 s and then 40-cycles of annealing/extension at 60 °C for 30 s. In order to verify the specificity of the amplification, a melting-curve analysis was performed, immediately after the amplification protocol. Relative gene expression was calculated with the 2-ΔΔCt method. The sequences of the primers used are the following: APEX1 For 5’-CCTGGACTCTCTCATCAATACTGG-3’, APEX1 Rev 5’- AGTCAAATTCAGCCACAATCACC-3’, BIRC5 For 5ʹ- ACCGCATCTCTACATTCAAG-3ʹ, BIRC5 Rev 5ʹ-CAAGTCTGGCTCGTTCTC-3ʹ, DNA polδ For 5’-GCTCCGCTCCTACACGCTCAA-3’, DNA polδ Rev 5’-GTCTGGTCGTTCCCATTCTGC-3’ and Actin For 5ʹ-CGCCGCCAGCTCACCATG-3ʹ, Actin Rev 5ʹ-CACGATGGAGGGGAAGACGG-3ʹ.
2.12. Caspases activity assay.
The activities of caspases 3/7 were examined via a fluorescence-based assay using Apo-ONE® Homogeneous Caspase-3/7 Assay (Promega) according to the manufacturer’s instructions. In details, 5,000 cells were plated on white 96-well plates and the day after cells were treated with increasing amounts of Compound #3 or APX2009 for 48 h.
2.13. Cell Mito Stress test.
Analyses of the mitochondrial function of HCT-116 cells were performed by using XFe Extracellular Flux Analyzer (Seahorse, Agilent Technologies, Santa Clara, CA, USA) as previously described [60]. In details, 5,000 cells were seeded and cultured in XF Cell Culture Microplates (Agilent Technologies). Before the measurements, the culture medium was removed from each well and replaced by 500 μl of Seahorse XF Base Medium (Agilent Technologies), pre-warmed at 37 °C and supplemented with 10 mM glucose (Sigma-Aldrich), 1 mM pyruvate (EuroClone), 2 mM glutamine (Sigma-Aldrich), at pH 7.4. Cells were incubated in a CO2-free incubator at 37 °C for 1 h and OCR (oxygen consumption rate) was detected under basal conditions. The following compounds were prepared at appropriate concentrations for each injection port to reach the final concentration of 0.5 μM oligomycin A, 0.5 μM FCCP, 1 μM rotenone and 1 μM antimycin A (Sigma-Aldrich). Volumes of respectively 56, 62, 69 and 73 μl of compounds were added to each injection port. Then, after a 3-min pause, 3 response measurements were taken between each addition. OCR values were normalized to the protein content (μg) of each well obtained by the Bradford method.
2.14. Immunofluorescence.
Patient-derived tumor organoids were grown on a slide and fixed in 4% paraformaldehyde for 20 min at 37°C, then washed with PBS 1X, treated with 0.1 M glycine in PBS 1X for 10 min and permeabilized with 0.5% Triton X-100 in PBS 1X for 5 min. After washing with PBS 1X and blocking for 1 h with 1% BSA, 10% FBS, 0.5% Triton X-100 in Washing Buffer (10 mM Tris HCl pH7.4, 150 mM NaCl and 0.01% Tween 20) organoids were incubated with primary antibodies diluted in blocking solution o/n at 4°C. After several washes in PBS 1X, organoids were incubated with labeled secondary antibodies for 2 h at RT. F-actin was stained with 0.165 μM Alexa Fluor™ 594 Phalloidin (Thermo Fisher Scientific) for 20 min at RT. For nuclear staining, organoids were incubated with 14.3 mM DAPI (Thermo Fisher Scientific) for 5 min at RT. Organoids were washed and mounted with Mowiol mounting medium. The following antibodies were used: anti-Ki67 1:200 (#AB92742, Abcam), anti-OLFM4 1:200 (#14369S, Cell Signaling Technology), anti-E-Cadherin 1:50 (#610404, BD Biosciences, San Jose, CA, USA) and anti-Lysozyme 1:200 (#A009902-2, Agilent Technologies). For detection, Alexa Fluor-488 (#711-546-152 and #715-546-150 Jackson ImmunoResearch, Ely, UK) antibodies were used. Fluorescent images were collected using a laser scanning confocal microscope (LEICA TCS SP8, Leica Microsystems). Brightfield images were collected using a microscope (LEICA MC170 HD, Leica Microsystems).
2.15. Library preparation and next-generation sequencing.
Genomic DNA from tumor organoid was extracted using the QIAamp DNA Mini Kit (Qiagen) and quantified by using the Quant-iT™ PicoGreen™ dsDNA Assay Kit (LifeTechnologies). Briefly, barcoded libraries were generated from 50 ng of DNA per sample (N=3) using the Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific) and two premixed pools of 952 primer pairs (Thermo Fisher Scientific), according to manufacturer’s instructions. Clonal amplification of libraries was performed by emulsion PCR on an Ion Chef Instrument. The prepared libraries were then sequenced on an Ion S5 GeneStudio Sequencer using a Ion 530 Chip and the Ion 510/520/530 kit–Chef (all Thermo Fisher Scientific).
2.16. Data analysis and Variant prioritization.
We analyzed data using Variant Caller v5.10 (Thermo Fisher Scientific). Variant caller format files were annotated with Ion Reporter 5.10 (Thermo Fisher Scientific) and wANNOVAR. Somatic variants were called when a position was covered at least 100X. We set the clinical sensitivity of point mutations and indels at 5%. Variant prioritization for TP53 was based on population frequency, quality values and functional consequences. Synonymous variants were excluded. Variants were filtered based on their frequency ExAC datasets (http://exac.broadinstitute.org) and on clinical associations (ClinVar database) (https://www.ncbi.nlm.nih.gov/clinvar).
2.17. Bioinformatics analysis.
Analysis of APEX1 gene expression in colon cancer was performed through TCGA data retrieval from cbioportal and CMS class information were accessed from the original Consensus Molecular Stratification of colon cancer. Differential gene expression analysis was conducted by Mann Whitney test. APE1 interactors analysis in colon cancer was performed using the differential gene expression results from TCGA and normal datasets (GTEX data) for the genes encoding the interacting partners of APE1 for colon cancer (COAD). Data was obtained via the GDC data portal hub (https://portal.gdc.cancer.gov/, last accessed July 2018). The RUVSeq package inside the R/Bioconductor environment was used to eliminate the batch effect coming from the combination of two data sources [61]. In order to better estimate the differentially expressed genes between the tumor and the normal corresponding datasets, we obtained “in-silico empirical” negative controls, i.e., the least significantly DE genes based on a first-pass DE analysis performed prior to RUVg normalization [61]. Differentially expressed genes (multiple correction adjustment using the Benjamini-Hochberg method, p<0.05; absolute log fold change difference≥1) were used to perform enrichment and survival analyses. Enrichment analysis of the DE genes was performed using the DAVID functional annotation tool based on GO biological process and cellular component terms [62]. For the survival analysis, Kaplan-Meier plots were drawn using the RTCGA Bioconductor package [63], which uses maximally selected rank statistics (maxstat) to determine the optimal cutpoint for continuous variables. Division of the samples was done within the 30-70% percentile range of gene expression by the optimal cutpoint value. The Benjamini-Hochberg method was used for p-value correction of Kaplan-Meier plots.
2.18. Statistical analyses.
All reported values are represented as the mean ± SD or mean ± SEM of at least three biological replicates. Statistical analyses were performed using the Student’s t-test. P<0.05 was considered as statistically significant.
3. Results
3.1. APE1 is overexpressed in colorectal cancer and the inhibition of its endonuclease activity triggers p53-mediated effects on cell metabolism
APE1 overexpression is correlated with tumor progression of many cancers, but little information is available on colorectal cancer (CRC) [23,24,64]. In order to further determine the involvement of APE1 in CRC, we took advantage of the information available from TCGA data sets of CRC. By performing differential gene expression (DEG) analysis on the TCGA COAD RNA-seq V2 data, we compared the expression profile of the four consensus molecular subtypes (CMS) of CRC versus normal control samples [PMC4636487]. The DEG was computed by the non-parametric Mann Whitney test. A significant upregulation of the APEX1 gene in the four CMS classes was identified compared to the normal one (Fig. 1A) (p-value=1.7e−15, 4e−11, 7.2e−14, 3.5e−11, for the CMS class 1-4, respectively), however there was no significant difference between the CMS classes analysed. These data demonstrate that APE1 is generally overexpressed in CRC, supporting the hypothesis that it could represent a novel target for the treatment of this cancer type.
Fig. 1. APE1 is overexpressed in colorectal cancer and the inhibition of its endonuclease activity triggers p53-mediated effects on cell metabolism.

(A) Gene expression level of APEX1 in the CRC samples of the four CMS classes and normal controls (CMS1 MSI Immune n=71; CMS2 Canonical n=130; CMS3 Metabolic n=58; CMS4 Mesenchymal n=98; control n=42). For left to right, the violin plots illustrate the log(FPKM+1) normalized gene expression level in the CMS 1-4 class CRC and normal control samples, respectively. (B) Schematic representation of different functions of APE1. The specific inhibitors of the different functions of APE1 are also indicated. (C) Western blot analysis of p53 and APE1 protein levels in HCT116 p53+/+ and p53−/− cell lines. Actin was used as a loading control. MTS assay on HCT-116 p53+/+ and p53−/− cell lines. Cells were treated with increasing amounts of APE1 inhibitors (Compound #3, APX2009, Fiduxosin, SB206553 and Spiclomazine) at the indicated concentrations for 48 h. Untreated cells were treated with DMSO. In graph, the percentage of metabolic activity relative to untreated cells, arbitrary set to 100%, is represented. Values are mean±SD (n≥3). (D) RealTime Glo assay on HCT-116 p53+/+ and p53−/− cell lines. Cells were treated with increasing amounts of Compound #3 or APX2009 for 48 h. Untreated cells were treated with DMSO. In graph, the percentage of metabolic activity relative to untreated cells, arbitrary set to 100%, is represented. Values are mean±SD (n=3). (E) Western blot analysis of APE1 in CH12F3+/+/Δ and CH12F3Δ/Δ/Δ cell lines. Actin was used as a loading control. CellTiter assay on CH12F3+/+/Δ and CH12F3Δ/Δ/Δ cell lines. Cells were treated with increasing amounts of Compound #3 or APX2009 at the indicated concentrations for 48 h. The percentages of viable cells relative to untreated cells, arbitrary set to 100%, are represented in graph. Values are mean±SD (n=3). Data were evaluated statistically by two-tails Student t-test. Resulting p-value is indicated (* p<0.05, ** p<0.01, *** p<0.005).
We analyzed the effects of APE1 inhibition on colon cancer cell lines using a number of APE1 specific small molecule inhibitors: Compound #3, which blocks APE1-endonuclease activity thus leading to genome instability [35]; APX2009, which inhibits APE1-redox function, leading to cell-cycle arrest and apoptosis in different cancer cell models [38], and Fiduxosin, SB206553 and Spiclomazine, as inhibitors of the well-known interaction between APE1 and NPM1, leading to an alteration of the APE1-endonuclease activity and to apoptosis in different tumor cell lines [22,44]. A schematic representation of the APE1-inhibitors used in this study is shown in Fig. 1B.
To determine the p53 contribution in APE1 inhibitors induced effects, we took advantage of the well-known HCT-116 p53+/+ and HCT-116 p53−/− isogenic colon cancer cell lines, which are wild-type and knock-out for p53 gene expression, respectively [65] (Fig. 1C, top left panel). Experimentally, HCT-116 cells were treated with increasing concentrations of different APE1 inhibitors for 48 hours. The metabolic activity of viable cells was measured by using the MTS assay, a colorimetric assay which measures the reduction of MTS tetrazolium compound into formazan in mitochondrial metabolically active cells. The treatment with the APE1-endonuclease inhibitor Compound #3 demonstrated that HCT-116 p53+/+ cell line was significantly more sensitive, at the doses of 0.25 μM and 0.5 μM, than HCT-116 p53−/− (Fig. 1C, top in the middle panel and Table 1 for IC50 values). In order to understand if the inhibition of APE1-redox activity could have some effects on HCT-116 cell lines, we treated cells with the APX2009 compound. We did not find a significant difference between HCT-116 p53+/+ and HCT-116 p53−/− cell metabolism upon APX2009 treatment, even if a mild effect can be observed (Fig. 1C, top right panel and Table 1 for IC50 values). Similarly, no significant differences, between the two cell lines, were found when inhibitors of APE1 and NPM1 interaction were used, with Fiduxosin being the most toxic for both cell lines (Fig. 1C, bottom panel and Table 1 for IC50 values). These findings were in agreement with the presence of no significant difference in the expression levels of both APE1 and NPM1, as measured at the mRNA and protein levels (Fig. S1A, B). Moreover, to further confirm the differential effect of Compound #3 and APX2009 on cell metabolism of HCT-116 p53+/+ and HCT-116 p53−/− cell lines, we performed a luminescence-based metabolic assay (RealTime-Glo). Compared to MTS assay, previously used, RealTime-Glo is a more sensitive assay based on the measurement of the reducing potential of mitochondrial metabolic active cells, considering the luminescent signal produced from the reduction of the substrate used by the luciferase, and thus mitochondrial metabolism. RealTime-Glo assay demonstrated that HCT-116 p53+/+ cells presented an impairment of metabolic activity in comparison to HCT-116 p53−/− upon Compound #3 treatment, especially at the dose of 0.5 μM, confirming the data previously obtained through MTS assay (Figure 1D, right panel and Table 1 for IC50 values). Interestingly, RealTime-Glo assay revealed that HCT-116 p53−/− cells underwent a major loss of metabolic activity compared to HCT-116 p53+/+ cells upon APX2009 administration in a dose-dependent manner, particularly at the doses of 2.5 μM and 5 μM (Fig. 1D, right panel and Table 1 for IC50 values). These data demonstrated that the small difference in metabolic activity that we observed with MTS assay (Fig 1C, top right panel) was much more apparent when the more sensitive RealTime-Glo assay was used. in place of the MTS assay. In order to exclude that the toxic effect could be associated with an alteration of the doubling time of the HCT-116 p53+/+ and p53−/− cell lines, we calculated it (HCT-116 p53+/+ 38±2.2 h, HCT-116 p53−/− 35±2.4 h) and no difference was observed. This excludes an alteration of the proliferation index as the cause of the effects on cell viability between the two cell lines.
Table 1: List of IC50 values obtained from each model used.
The analysis was carried out using the Combenefit 2.021 software allowing to calculate the IC50 values for each drug tested on the models analyzed. Cell models (cell lines and patient-derived 3D tumor organoids), types of assays, drugs and time points are reported. n.d. indicates that the goodness of fit value is too low or a standard Hill equation does not correctly account for the specific agent used. In some cases, the expected interpolated IC50 value has been reported.
| Model used | Drug | Time (hours) | IC50 (∝M) | Assay |
|---|---|---|---|---|
| CH12F3+/+/Δ | Compound #3 | 48 | 1.44 | CellTiter |
| CH12F3Δ/Δ/Δ | 1.9 | |||
| HCT-116 p53+/+ | 0.58 | MTS | ||
| HCT-116 p53−/− | 0.69 | |||
| HCT-116 p53+/+ | 1.04 | RealTime-Glo | ||
| HCT-116 p53−/− | 1.50 | |||
| CH12F3+/+/Δ | APX2009 | 6.61 | CellTiter | |
| CH12F3Δ/Δ/Δ | 9.64 | |||
| HCT-116 p53+/+ | 11.7 | MTS | ||
| HCT-116 p53−/− | 11.6 | |||
| HCT-116 p53+/+ | 10.0 | RealTime-Glo | ||
| HCT-116 p53−/− | 6.39 | |||
| HCT-116 p53+/+ | Fiduxosin | 16.1 | MTS | |
| HCT-116 p53−/− | 15.6 | |||
| HCT-116 p53+/+ | SB206553 | n.d. (>100) | ||
| HCT-116 p53−/− | n.d. (>100) | |||
| HCT-116 p53+/+ | Spiclomazine | 30.4 | ||
| HCT-116 p53−/− | 31.2 | |||
| HCT-116 p53+/+ | MMS | 8 | 721 | |
| HCT-116 p53−/− | 938 | |||
| HCT-116 p53+/+ | Compound #3 | 40 + 8 | 0.373 | |
| HCT-116 p53−/− | 0.58 | |||
| tumor organoid P12 | Compound #3 | 48 | 1.36 | RealTime-Glo |
| tumor organoid P14 | 2.8 | |||
| tumor organoid P16 | 0.71 |
In order to further validate the specificity of Compound #3 and APX2009 as APE1 inhibitors in the range of doses used in the present study, we took advantages of CH12F3+/+/Δ and CH12F3Δ/Δ/Δ cell lines [66], a recently developed murine cell model containing two and zero copies of APEX1 alleles, respectively (Fig. 1E, left panel). Both CH12F3 cell lines were treated with increasing concentrations of Compound #3 or APX2009 for 48 hours and then, the cellular metabolism of viable cells was measured by using a luminescence-based assay (CellTiter-Glo), which evaluates the amount of total ATP produced by cells. As shown in Fig. 1E, APE1-endonuclease and -redox inhibitors specifically affect CH12F3+/+/Δ but not CH12F3Δ/Δ/Δ cell line, as expected, confirming the high specificity of the inhibitors, under the experimental conditions used (Table 1 for IC50 values). In the case of the effects of Fiduxosin, SB206553 and Spiclomazine, their specificity was previously demonstrated by us [22,44].
In conclusion, data obtained indicate that the sensitivity of HCT-116 cells to Compound #3 and APX2009 treatment was dependent on their p53 status. Moreover, the inhibition of APE1-NPM1 interaction impairs cell metabolism independently of p53 status. Interestingly, while the toxic effect of Compound #3 was dependent on the expression of p53, a more toxic effect of APX2009 was apparent in p53 knock-out cells.
3.2. Inhibition of APE1-endonuclease activity promotes p53 activation
We then evaluated whether APE1 inhibition could promote p53 activation. We focused only on Compound #3 and APX2009, since the APE1-NPM1 inhibitors did not display any significant difference in terms of biological effects exerted on HCT-116 cell lines. p53 activation is part of DDR triggered by single-strand breaks (SSBs) and double-strand breaks (DSBs) formation. We determined whether the amount of AP sites and SSBs could be affected by the treatment with Compound #3 in HCT-116 cell lines. AP sites measurements (Fig 2A) and comet assay analyses (Fig. 2B and C) clearly demonstrated that Compound #3 treatment promoted a significant increase of AP sites and SSBs formation in both HCT-116 p53+/+ and HCT-116 p53−/− cell lines independently of the p53 status at both the doses tested (0.5 μM and 3 μM). The non-significant difference in the levels of AP sites and SSBs between both HCT-116 cell lines is in agreement with the non-differential expression of APE1 between the two cell lines used (Fig. S1).
Fig. 2. Inhibition of APE1-endonuclease activity promotes p53 activation.

HCT-116 p53+/+ and p53−/− cell lines were treated with Compound #3 (#3) at the indicated concentrations for 48h. Untreated cells were treated with DMSO (A-F, L). (A) The amount of abasic (AP) sites was measurement. Values are mean±SD (n=3). (B) Representative fluorescence confocal microscope images of in vitro comet assay. Scale bars, 25 μm. (C) Tail moment was analysed for 100 cells at random by OpenComet software. Untreated cells was used as reference and set to 1. Values are mean±SD. (D) Western blot analysis of p53, p21, APE1 and Nucleophosmin (NPM1) protein levels. Actin was used as a loading control (n=3). (E) Densitometric analysis of p53, p21, APE1 and NPM1 protein expression. Levels were normalized to actin. Fold change values relative to untreated control cells, arbitrary set to 100%, are shown. Values are mean±SD (n=3). (F) Expression level of DNA Polimerase δ (DNA Pol δ) was determined by qRT-PCR analysis. DNA Pol δ levels were normalized to β-actin. Fold change values relative to untreated control cells, arbitrary set to 1, are shown. Values are mean±SD (n=3). (G) HCT116 p53+/+ cell lines silenced for APE1 expression. Western blot analysis of p53 and APE1 protein levels. Actin was used as a loading control. Fold change values relative to untreated control cells, arbitrary set to 1, are shown. (H and I) Western blot analysis of p53 protein levels upon Compound #3 (#3) at the indicated concentrations of AML2 cell lines treated with for 48 h (H) and HCC70 cell lines treated for 24 h (I). Actin was used as a loading control. Fold change values relative to untreated control cells, arbitrary set to 1, are shown (n=3). (L) Apo-ONE assay was used to quantify relative levels of apoptosis. The activities of caspases 3/7 were examined using a fluorescence-based assay. Untreated control cells were used as reference and set to 100%. Values are mean±SD (n=2). Data were evaluated statistically by two-tails Student t-test. Resulting p-value is indicated (* p<0.05, ** p<0.01).
In order to get a better understanding of the relationship existing between p53, its gene target genes and the inhibition of APE1 enzymatic activity in HCT-116 cell lines, we analyzed the protein and mRNA expression levels of p53 and those of some p53-target genes upon Compound #3 treatment. In details, HCT-116 p53+/+ and HCT-116 p53−/− cells were treated with increasing concentrations of Compound #3 (0.25 μM and 0.5 μM) and cells were collected and tested 48 hours later. These doses, with limited effects on cell viability, allowed to avoide epiphenomena due to general toxic effects and allowed comparison to data obtained with those obtained with the redox-inhibitor (see below). Upon the administration of Compound #3, we analyzed the protein levels of APE1, p53, and p21, one of the major downstream targets of p53 [47], through Western blotting. We observed a significant increase of p53 and p21 protein levels upon Compound #3 administration in HCT-116 p53+/+ cells only, indicating a p53 activation possibly due as a consequence of cell DDR induction (Fig. 2D). Moreover, the inhibition of APE1-endonuclease activity did not considerably alter neither APE1 nor NPM1 protein levels in both cell lines, exerting only slight effects. Densitometric analysis of p53, p21, APE1, and NPM1 protein levels, normalized to actin levels, is shown in Fig. 2E. Moreover, to further confirm the unaltered levels of APE1 gene expression upon Compound #3 treatment, we analyzed its mRNA expression. No statistically significant changes in APE1 transcript were observed (Fig. S1C), confirming that the higher sensitivity of HCT-116 p53+/+ cell line to Compound #3 is not due to an impairment of APE1 expression. Furthermore, to test a possible impact of the activation of the p53 pathway on another known p53-target gene, negatively regulated by p53 and involved in BER [11,57,67-69], we analyzed the mRNA expression level of DNA Polymerase d (DNA Pold). As it is shown in Fig. 2F, the level of transcripts of DNA Pold decreased upon Compound #3 treatment in HCT-116 p53+/+ cells only, in line with p53 induction. These data support the hypothesis that APE1-endonuclease inhibition causes the functional activation of p53 protein acting both as a transcriptional activator and repressor of different target genes.
In order to further support that APE1 inhibition may result in p53 activation, a knockdown approach, through specific siRNA targeting APE1, was used. As shown in Fig 2G, knock down of APE1 promoted an increased expression of p53 in HCT-116 p53+/+ cell line, demonstrating a mutual inverse relationship between APE1 and p53 (Fig. 2G). Finally, to generalize the stimulatory effect by Compound #3 on p53 expression, we treated two additional tumor cell lines, i.e. AML2 as a model of acute myeloid leukemia cells [70] and HCC70 cell line, a triple negative breast cancer cell line [71]. These cell lines harbor a wild-type form [70] and a missense mutation (p.R248Q) [71] of the TP53 gene, respectively. TP53 p.R248Q is a gain-of-function mutation that causes an aberrant overexpression of the p53 protein. AML2 and HCC70 cell lines showed an increased expression of p53 upon Compound #3 treatment, demonstrating that the activation of p53 upon inhibition of APE1-endonuclease activity could be a generalized phenomenon across different cancer cell lines (Fig. 2H and I) and not only colon specific. Altogether, these data confirm that APE1-endonuclease inhibitor treatment of different cancer cell lines promotes p53 expression and its functional induction.
DNA damage activating the p53-p21 pathway may lead to cell apoptosis through caspase activation [47]. Therefore, we performed a caspase activity assay to measure apoptosis induction by Compound #3 treatment (Fig 2L). After 48 hours of treatment with increasing concentrations of Compound #3, Apo-ONE assay was used to quantify relative levels of apoptosis. The treatment with APE1 inhibitor induced the activation of the caspase pathway in both colon cancer cell lines (Fig. 2L) occurring only at the highest dose (3 μM) of treatment. However, no differences were apparent between the two cell lines, confirming that Caspase activation was independent of p53-activation and was present only at highest dose of treatment with Compound #3. Data not shown also demonstrated that Compound #3-treatment did not exert any significant differential effect on cell cycle of the two isogenic HCT-116 cell lines.
These experiments, coupled with the metabolic assays, indicate that the inhibition of the APE1-endonuclease activity promoted a p53-induced response involving mitochondria, which may explain the higher susceptibility of p53+/+ cell metabolism to Compound #3 treatment in the absence of significant Caspase-3 activation and of differential effects on cell cycle between the two HCT-116 isogenic cell lines.
3.3. Inhibition of APE1-redox activity does not affect p53 expression
We analyzed whether inhibition of APE1-redox activity could affect p53 expression. We checked the efficacy of APX2009 treatment on HCT-116 colon cancer cell lines, through qRT-PCR on the baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5), also known as Survivin, that was already demonstrated to be a target of APE1-redox function [40]. Treatment with increasing concentrations of APX2009 (Fig. 3A) promoted a decrease in the expression levels of BIRC5 transcript in both HCT-116 cell lines (Fig. 3A), in line with previously demonstrated data on different cell lines [40].
Fig. 3. Inhibition of APE1-redox activity does not affect p53 expression.
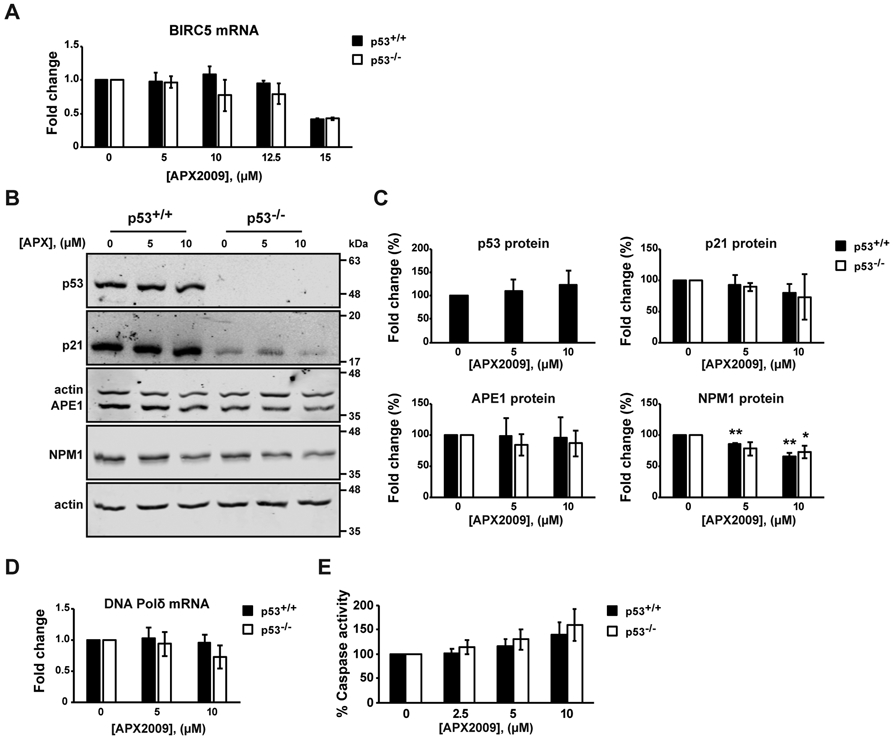
HCT-116 p53+/+ and HCT-116 p53−/− cells were treated with APX2009 (APX) at the indicated concentrations for 48h. Untreated cells were treated with DMSO. (A) Expression level of BIRC5 was determined by qRT-PCR analysis. BIRC5 levels were normalized to β-actin. Fold change expression values relative to untreated cells, arbitrary set to 1, are shown. Values are mean±SD (n=3). (B) Western blot analysis of p53, p21, APE1 and Nucleophosmin (NPM1) protein levels. Actin was used as a loading control (n=3). (C) Densitometric analysis of p53, p21, APE1 and NPM1 protein expression. Levels were normalized to actin. Fold change values relative to untreated control cells, arbitrary set to 100%, are shown. Values are mean±SD (n=3). (D) Expression level of DNA Polimerase δ (DNA Pol δ) was determined by qRT-PCR analysis. DNA Pol δ levels were normalized to β-actin. Fold change expression values relative to untreated control cells, arbitrary set to 1, are shown. Values are mean±SD (n=3). (E) Apo-ONE assay was used to quantify relative levels of apoptosis. The activities of caspases 3/7 were examined using a fluorescence-based assay. Untreated control cells were used as reference and set to 100%. Values are mean±SD (n=3). Data were evaluated statistically by two-tails Student t-test. Resulting p-value is indicated (* p<0.05, ** p<0.01).
To assess whether APE1-redox inhibitor stimulates the p53-pathway, we performed the same set of experiments, as with Compound #3, but with APX2009. We used APX2009 at concentrations of 5 μM and 10 μM, that promoted a reduction in cell viability of less than 40% similar to the conditions used with Compound #3 treatment. As shown in Fig. 3B and C, no differences in p53 and in the expression of the p21 target gene were observed upon the administration of the redox inhibitor. Moreover, DNA Pold transcript did not significantly change following APX2009 administration, in agreement with the lack of any stimulatory effects by the APE1-redox inhibition on p53 levels (Fig. 3D). Moreover, the caspase activity assay demonstrated a dose-dependent activation of the caspase pathway indistinctly in both colon cancer cell lines (Fig. 3E).
Taken together, these experiments demonstrate that the inhibition of the APE1-redox activity does not activate a p53-mediated cell response and this is in agreement with data on cell metabolism.
3.4. Inhibition of APE1-endonuclease activity impairs mitochondrial activity in a p53-dependent manner
Data obtained so far, through MTS and RealTime-Glo analyses, point to a major role of the p53-dependent effect of Compound #3 on cell viability due to metabolic effects associated with mitochondrial toxicity. In order to better characterize this aspect, we directly analyzed mitochondrial activity using the Cell Mito Stress Test [72] upon Compound #3 treatment. Specifically, HCT-116 p53+/+ and HCT-116 p53−/− cell lines were treated with 0.5 μM of Compound #3 for 48 hours and were analysed for mitochondrial respiration. The parameters of basal oxygen consumption, respiration coupled to ATP production, spare respiratory capacity and maximal respiration were obtained through oxygen consumption rate (OCR) profile. As represented in Fig. 4A, OCR profile indicates that Compound #3 impaired the mitochondrial activity of HCT-116 p53+/+ cells, while the treatment did not significantly alter the mitochondrial function of HCT-116 p53−/− cells. In particular, the OCR values were comparable between the two colon cancer cell lines under basal conditions. Interestingly, Compound #3 treatment affected the majority of the respiratory parameters in a p53-dependent manner (Fig. 4B). Among them, the basal and ATP-coupled respiration was significantly lower in HCT-116 p53+/+ than the isogenic p53−/− counterpart upon treatment. Moreover, the spare respiratory capacity and maximal respiration were impaired in HCT-116 p53+/+ treated cells in comparison to the untreated condition. On the other hand, we did not observe any difference in mitochondrial function in p53-null colon cancer cell line upon treatment. The OCR profiles of HCT-116 p53+/+ and p53−/− cell lines with Compound #3 at different concentrations (0.25 μM and 1 μM) are shown in Fig. S2, confirming the obtained results, particularly at the dose of 1 μM.
Fig. 4. Inhibition of APE1-endonuclease activity impairs mitochondrial activity in a p53-dependent manner.
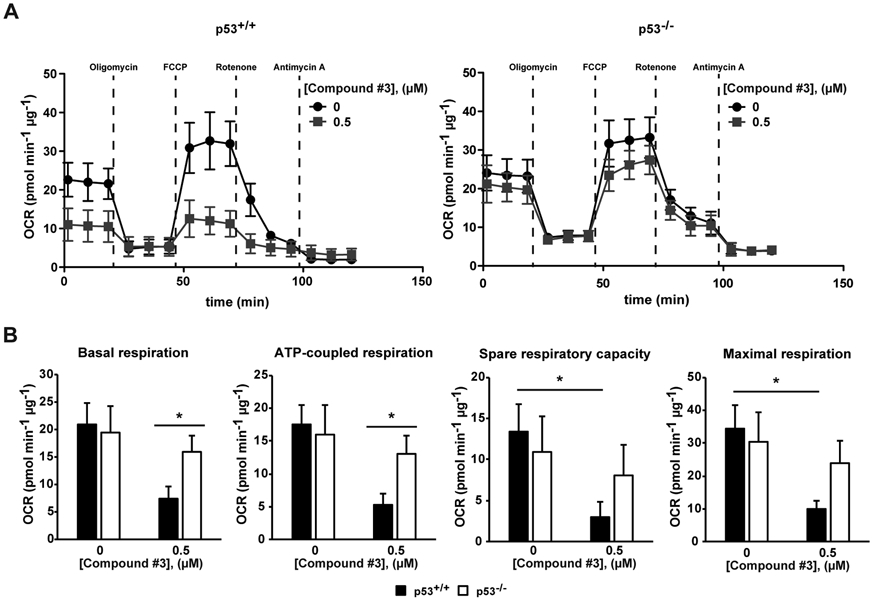
(A and B) HCT-116 p53+/+ and p53−/− cells were seeded in a Seahorse XF-24 analyzer and treated with 0.5 μM of Compund#3 for 48 h. Untreated cells were treated with DMSO. (A) Real-time oxygen consumption rate (OCR) was determined during sequential treatments with oligomycin (ATP-synthase inhibitor), FCCP (uncoupler of oxidative phosphorylation), rotenone (complex I inhibitor) and antimycin-A (complex III inhibitor). (B) The rates of basal respiration, ATP-coupled respiration, spare respiratory capacity and maximal respiration were quantified by normalization of OCR level to total protein content. Data represent means±SEM (n=3). Data were evaluated statistically by two-tails Student t-test. Resulting p-value is indicated (* p<0.05, *** p<0.005).
Overall, the data obtained suggest that the expression of p53 is responsible for the observed effects on cell metabolism, upon inhibition of APE1-endonuclease activity, through an involvement of mitochondrial respiratory function.
3.5. Inhibition of APE1-endonuclease activity sensitizes p53 expressing cell lines to MMS treatment
Based on the results obtained so far, we analyzed the effect of p53 on cell metabolism in combination with a genotoxic insult, which is specifically repaired through the enzymatic activity of APE1 and BER [73]. We used methyl methanesulfonate (MMS), an alkylating agent generating damages specifically repaired through BER [73]. Experimentally, HCT-116 cells were treated with increasing concentrations of Compound #3 (0.25 μM and 0.5 μM) for 40 hours and in combination with MMS (range from 200 μM to 600 μM) for additional 8 hours (Fig. 5). After a total of 48 hours of treatment, cell metabolism was measured using the MTS assay (Table 1 for IC50 values). The graphs in Fig. 5A-C clearly shows that the two isogenic cell lines responded similarly sensitive to MMS. Interestingly, when Compound #3 was used in combination with MMS, the sensitivity to the drug treatment increased (Fig. 5A-C); however, the p53-null colon cancer cells were significantly less sensitive to combination treatment than the isogenic counterpart (Table 1 for IC50 values), in particular at the dose of 600 μM of MMS, in agreement with the stimulatory effect of Compound #3 on p53 expression demonstrated above.
Fig. 5. Inhibition of APE1-endonuclease activity sensitises p53 expressing cell lines to MMS treatment.
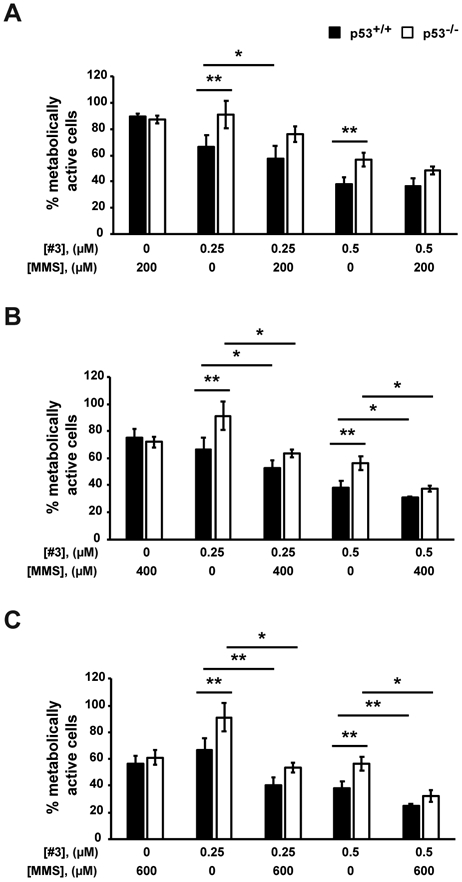
MTS assay on HCT-116 p53+/+ and HCT-116 p53−/− cells. Cells were treated with Compound #3 (#3) at the indicated concentrations for 40 h and in combination with 200 μM (A), 400 μM (B) or 600 μM (C) methyl methanesulfonate (MMS) for additional 8 h. Untreated cells were treated with DMSO. In graph, the percentage of metabolic activity relative to untreated cells, arbitrary set to 100%, is represented. Values are mean±SD (n=3). Data were evaluated statistically by two-tails Student t-test. Resulting p-value is indicated (* p<0.05, ** p<0.01).
These results suggest that p53-status is essential for cellular resistance to genotoxic stress upon the inhibition of APE1-endonuclease activity.
3.6. Inhibition of APE1-endonuclease activity exerts toxic effects on patient-derived tumor organoids metabolism and is associated with the p53-mutational status
In order to further validate the possible opportunity of targeting the APE1-endonuclease activity in CRC and further supporting our data previously obtained with HCT-116 isogenic cancer cell lines, we performed targeted experiments by using 3D tumor organoids derived from three different patients affected by colon cancer (P12, P14 and P16). The molecular and morphological characterization, obtained through immunofluorescence, is shown in Fig. 6A. Patient-derived tumor organoids were positive for the most common intestinal markers, such as Ki67 (proliferating cells), OLFM4 (intestinal stem cells), E-Cadherin (intestinal epithelial cells) and Lysozyme (Paneth cells). Representative images of the molecular morphology of the patient-derived tumor organoids are shown in Fig. 6B. To check the p53-functional status of these model organoids, genomic DNA was isolated from tumor organoids and used as a template for next generation sequencing analysis, using the Ion Torrent S5 GeneStudio NGS platform. After variant calling filtration and annotation, we detected a c.586C>T stop-gain mutation (p.R196X) in P14 and a c.524G>A missense mutation (p.R175H) in P16, the latter being a structural aminoacidic substitution causing a TP53 gain-of-function and wild-type alleles in P12 (Fig. 6C). All samples did not bear any pathogenic alteration of APEX1 gene sequence. The western blotting analysis demonstrated that p53 was expressed in P12 and P16 but not in P14, in agreement with sequencing data (Fig. 6D). No major differences in APE1 protein expression level were assessed among tumor organoids (Fig. 6E), with only a mild increase in P14 and P16 with respect to P12. We treated the different tumor organoids with increasing concentrations of Compound #3 (range from 0.5 to 5 μM) and cell metabolism was measured by using RealTime-Glo assay upon 48 hours of treatment. Interestingly, the treatment with APE1-endonuclease inhibitor impaired the cell metabolism of all tumor organoids in a dose-dependent manner but to different extents (Fig. 6F and Table 1 for IC50 values). The reduction of cell metabolism was observed within a range of 40-65% at the concentration of 5 μM. Interestingly, we observed a different cytotoxic sensitivity of the three tumor organoids to Compound #3 that was dependent on their p53 status. In particular, tumor organoids expressing wild-type p53 (P12) and the R175H gain-of-function mutant (P16) were more sensitive to APE1 inhibition than the P14 null-mutant, in agreement with the data obtained with the colon cancer cell lines.
Fig. 6. Inhibition of APE1-endonuclease activity exerts toxic effects on patient-derived tumor organoids metabolism and is associated with the p53 mutational status.

(A) Fluorescence confocal microscope imaging of patient-derived organoids. Patient-derived organoids were stained with Ki67 (proliferating cells), OLFM4 (intestinal stem cell), E-Cadherin (intestinal epithelial cells) or Lysozyme (Paneth cells). F-actin was stained with Phalloidin. Nuclei were visualized with DAPI. Scale bars, 50 μm (Ki67 and Lysozyme) and 25 μm (OLFM4 and E-Cadherin). (B) Brightfield representative images of colorectal cancer organoids derived from different patients (P12, P14 and P16). Scale bars, 200 μm. (C) TP53 mutational status in colorectal cancer organoids was assessed by NGS. Patient P12, represented by the green box, harboured a wild-type p53, patient P14, represented by the blue box, beared a stop-gain mutation while patient P16, represented by the orange box, harboured a missense one. (D) Western blot analysis of p53 protein level in CRC patient-derived organoids. The graph shows the densitometric analysis of p53 protein expression in patient-derived organoids. Fold change values relative to P12, arbitrary set to 1, are shown. Values are mean of 3 measurements±SD. (E) Western blot analysis of APE1 protein level in CRC patient-derived organoids. The graph shows the densitometric analysis of APE1 protein expression in patient-derived organoids. Fold change values relative to P12, arbitrary set to 1, are shown. Values are mean of 3 measurements±SD. (F) RealTime-Glo assay on patient-derived tumor organoids. Patient-derived tumor organoids were treated with increasing amounts of Compound #3 (range from 0.5 μM to 5 μM) for 48 h. In graph, the percentage of metabolic activity relative to untreated organoids, arbitrary set to 100%, is represented (n=1). Values are mean of 5 measurements±SD.
Even though the limited number of sample patients, these results indicate that the inhibition of APE1-endonuclease activity significantly hampers the patients-derived tumor organoids metabolism and is associated with the p53 mutational status, in agreement with data obtained with HCT-116 cell lines.
4. Discussion
Human cells are constantly subjected to potentially damaging events to the stability of nucleic acid, with tens of thousands of DNA lesions occurring per day [74]. The intestinal tract is exposed to multiple insults coming from metabolic activity of the microenvironment [75] and from the diet [7], that can lead to mutations or deletions in the DNA of epithelial cells. In the intestinal tract, the involvement of the direct reversal of DNA damage by O6-methylguanine-DNA methyltransferase (MGMT), nucleotide excision repair (NER) and base excision repair (BER) have been described [7]. Alterations of these repair mechanisms are clearly associated with tumor development but also represent an emerging ‘Achilles’ heel’ for the development of new anticancer strategies [76]. Indeed, a dysregulation of BER has been observed in different tumors, such as breast, liver, melanoma, bladder and colorectal cancer (CRC) [6,77-81]. In the case of CRC, an increased expression of BER enzymes is associated with a poor prognosis and contributes to chemoresistance [6]. In particular, higher levels of N-methylpurine-DNA glycosylase (MPG), 8-oxoguanine-DNA glycosylase (OGG1), APE1, PARP1, DNA polymerase β (Polβ) and XRCC1 are associated with adverse outcomes in patients with sporadic CRC [6,81]. Interestingly, the tumor suppressor p53, frequently mutated in the late stages of CRC [48], is involved in the regulation of DNA glycosylases (OGG1 and MUTYH) [52,53], APE1 [54,55], Pol β [56] and Pol δ [57] expression. Generally, upon genotoxic lesions, p53 regulates the expression of DNA repair genes [68], as in the case of APE1 that is negatively regulated by p53 through modulation of Sp1 stability [57]. Therefore, impaired p53 results in a loss of transcriptional regulation thus leading to BER imbalance and genome instability. Moreover, the DNA repair capacity of BER has been taken into account as a prognostic factor to 5-Fluorouracil (5-FU) treatment in CRC-affected patients, as demonstrated by a recent publication showing that a good therapy response is correlated with an observed higher activity of BER in non-malignant adjacent mucosa and a lower BER activity in tumor tissue [82]. To date, the therapeutic effect of the only APE1-redox inhibitor has been highlighted in different cancer cell lines. For example, APX3330 inhibits cell growth in tumor endothelium/endothelial progenitor cells [83], leukemia [31], pancreatic [84,85], breast [86], hepatocellular [87], prostate cancer cells [88] and colon [23]. It should be noted that APE1-redox inhibitor APX3330 recently completed a cancer Phase I clinical trial (https://clinicaltrials.gov/ct2/show/NCT03375086) [42,43]. Thus, discovering new drugs able to inhibit the activity of APE1 in a CRC context could be a promising field in the precision medicine. However, no studies targeting the APE1-endonuclease activity and its effect on p53 activation have been published. Here, we demonstrated the anticancer effects of different APE1 inhibitors in colon cancer cells and their relationship with p53 activation. By using different technical approaches, we observed a p53-dependent metabolic impairment, impacting on mitochondrial respiratory mechanisms, of HCT-116 cell lines upon treatment with APE1-endonuclease inhibitor, proving the effectiveness of the treatments also in a CRC context. It is noteworthy to mention that the APX2009 redox inhibitor resulted more toxic to p53 knock-out cells than wild-type counterpart. This observation will need additional studies to understand the molecular basis for this effect.
In our case, the increased p53 level, following the DNA damage (AP site accumulation and SSBs generation) caused by APE1-endonuclease inhibition, was not associated with any changes in APE1 mRNA and protein levels, in contrast to what was previously demonstrated in other papers, in which p53 was demonstrated to negatively regulate APE1 gene expression [55]. Moreover, we did not observe any p53 stimulation caused by APX2009 treatment, therefore suggesting the possibility to target different APE1 activities by acting through different response mechanisms either involving the DNA-repair or the redox-functions. We also observed that the inhibition of APE1-endonuclease activity promotes p53 activation not only in colon cancer cells, but also in breast and leukemia cancer cell lines, suggesting that the APE1 inactivation and the consequent accumulation of DNA damage converge toward p53 activation. Thus, the use of APE1-endonuclease inhibitors could be particularly relevant considering the p53-functional status of the specific tumor.
Another relevant finding regards the effect of APE1-endonuclease inhibitor on CRC patient-derived tumor organoids. Our study demonstrates that Compound #3 impairs the viability of CRC patient-derived organoids in a dose-dependent manner and in a p53-dependent mutational status, which is in agreement with our findings obtained from cancer cell lines.
To our knowledge, the observation that the APE1-endonuclease inhibitor effects are possibly related to the p53 role on mitochondrial metabolism is extremely important. Besides cell cycle arrest, apoptosis and senescence, p53 plays an important function also in mitochondrial respiration [89]. As a regulator, p53 induces the expression of cytochrome c oxidase 2 (SCO2), ferredoxin reductase (FDXR) and glutamine 2 (GLS2), thus modulating oxidative phosphorylation and tricarboxylic acid (TCA) cycle [89,90]. Moreover, the involvement of BER in mitochondrial homeostasis has been extensively demonstrated. Indeed, p53 regulates mitochondrial BER in removing oxidized bases of the mtDNA generated by reactive oxygen species (ROS) metabolism [91]. Notably, Compound #3 sensitizes p53-expressing colon cancer cells to genotoxic treatment, such as MMS. Due to the relevance of APE1 inhibition on mitochondrial functionality in a p53-dependent manner, we performed additional bioinformatics analyses that clearly pointed to a major role of mitochondrial toxicity correlated with APE1 functional dysregulation in tumors. It is also possible that the effect of Compound #3 could be also exerted through inhibition of mitochondrial APE1 [92-94], whose role is still debated. An important confirmation of the mitochondrial role of APE1 linking to p53 activation is also supported by our recent findings (Ayyildiz D. et al., submitted). In fact, when characterizing the APE1 protein-protein interactome (PPI) in different cancers cells, we found that the differential expression status of APE1 PPI was clearly associated with bad prognosis signatures in cancers (Ayyildiz D. et al., submitted). TCGA analyses demonstrated that in CRC, 79% (n=287) of APE1 PPI resulted in upregulation, while 21% (n=77) of the genes altered in expression were downregulated (Fig. 7A). Based on the relationship existing between these differentially expressed interactors, by focusing on their subcellular localizations through functional enrichment analysis based on GO CC terms, we observed that 24% (n=70) of the upregulated and 12% (n=9) of the downregulated interactors were mitochondria-associated proteins (Fig. 7A). The subcellular localizations and enrichment results of these differentially-regulated physical-interactors of APE1 are shown in Fig. 7B. The enrichment analysis highlighted pathways that can improve our understanding of the importance of APE1 for mitochondrial activity. Among them, mitochondrial RNA processing, fatty-acid oxidation, respiratory complex III and apoptosis represented the most important ones (Fig. 7B). All these processes are essential for cancer metabolism [95,96], as in the case of the mitochondrial respiratory complex III, that plays a role in CRC progression [97]. Interestingly, some of these interactors were found to be significantly regulated in our previously published microarray data [98], such as AK2, CKB, CLIC4, DNAJA1, DUT, HSPA1A, IDH1, KYNU, MTCH2, SHMT2, SIRT1 and YWHAH (marked in red circle in Fig. 7B). SIRT1, which regulates BER modulating the acetylation status of APE1 [73], has been found upregulated in various cancer cell lines, including HCT-116 cell line [99]. The survival signature of the 79 interactors were also confirmed by Kaplan-Meier analysis in TCGA colon cancer dataset (COAD) and BCAP31, CDKN2A, DUT, NIF3L1 and SHMT2 were associated to poor prognosis (marked with red star in Fig. 7B). Together, this evidences support our findings, namely the mitochondrial impairment related to p53 upon Compound #3 treatment. The experimental data we provide, together with these bioinformatics analyses, support the hypothesis to target mitochondrial function in cancer cells through APE1-endonuclease inhibitors. More tailored studies are needed along these lines.
Fig. 7. Differentially expressed interactors of APE1 in colorectal cancer.

(A) Differentially expressed genes (DEGs) coding APE1 interactors in colon cancer (COAD) are in the middle. Cellular locations of upregulated interactors in upper panel and downregulated interactors in lower panel. The colour codes are as follows: green colour for up-regulation, orange colour for down-regulation, blue colour for mitochondrial location and grey colour for other cellular locations. (B) Representation of DEGs on mitochondrial scheme. Grey shaded boxes indicate mitochondrial locations while boxes without any shade indicate enriched pathways. Green upside arrows indicate up-regulation while red downside arrows indicate down-regulation. Genes that were also regulated in previously published microarray data were shown in red circles. Poor prognostic interactors were marked with circled skull symbol.
In conclusion, our data highlights novel relevant aspects regarding developing new strategies for targeting BER, and especially APE1 functions, in CRC. Therefore, the opportunity to use APE1-endonuclease inhibitors, such as Compound #3, in cancer therapy should not be underestimated. Finally, greater treatment efficacy and reduced drug resistance onset mechanisms have been demonstrated when different anti-cancer drugs are combined together [100], thus the possibility of studying the effects of the different combination of APE1 inhibitors with well-known chemotherapeutic agents may open new perspectives in cancer biology research.
Supplementary Material
Fig. S1. APE1 mRNA and protein levels remain stable in basal condition and upon APE1 endonuclease inhibition in colon cancer cell lines. (A) Expression level of APE1 were determined by qRT-PCR analysis. APE1 levels were normalized to β-actin. Fold change expression values relative to HCT-116 p53+/+ cells, arbitrary set to 1, are shown. Values are mean±SD (n=3). (B) Densitometric analysis of basal APE1 expression. Levels were normalized to actin. HCT-116 p53+/+ cells were used as reference and set to 100%. Values are mean±SD (n=6). (C) HCT116 p53+/+ and HCT116 p53−/− cells were treated with Compound #3 (#3) at the indicated concentration for 48 h. Expression level of APE1 were determined by qRT-PCR analysis. APE1 levels were normalized to β-actin. Fold change expression values relative to untreated control cells, arbitrary set to 1, are shown. Values are mean±SD (n=3).The p-value was calculated using Student’s two-tailed t-test. Resulting p-value is indicated (NS, not significant).
Fig. S2. Inhibition of APE1-endonuclease activity impairs mitochondrial activity in a p53-dependent manner. HCT-116 p53+/+ and HCT-116 p53−/− cells were seeded in a Seahorse XF-24 analyzer and treated with 0.25 μM (A) and 1 μM (B) of Compound #3 at the indicated concentrations for 48 h. Untreated cells were treated with DMSO. Real-time oxygen consumption rate (OCR) was determined during sequential treatments with oligomycin (ATP-synthase inhibitor), FCCP (uncoupler of oxidative phosphorylation), rotenone (complex I inhibitor) and antimycin-A (complex III inhibitor). Values are mean of 5 measurements±SD.
Acknowledgments
We thank all the members of the GT lab for thought-provoking discussions. We acknowledge David Maloney and Anton Simeonov (National Center for Advancing Translational Sciences) for providing Compound #3. We thank Prof. Calvin Kuo (Stanford University) for providing Rspo1 producer cells and Prof. Jeroen den Hertog (Hubrecht Institute) for providing Noggin producer cells.
Funding
This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (IG19862) and from the European Union, European Regional Development Fund and Interreg V-A Italia-Austria 2014-2020 ITAT1096-P (Program PRECANMED ITAT1009 CUP G22F16000890006) to G.T.. M.R.K. was supported by NIH RO1 CA205166, NIH R01 CA231267, NIH R01 CA167291, NIH R01 CA167291-06S1 and NIH R01HL140961.
Footnotes
Conflict of interest
Mark R. Kelley is Chief Scientific Officer of Apexian Pharmaceuticals, the biotech company that has licensed APX2009 used in these studies. Apexian Pharmaceuticals had neither control nor oversight of the studies, interpretation, or presentation of the data in this manuscript. They did not have to approve the manuscript in any way prior to its submission. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- [1].Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, van de Velde CJH, Watanabe T, Colorectal cancer., Nat. Rev. Dis. Prim 1 (2015) 15065. doi: 10.1038/nrdp.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Smits AMM, Bos JL, Smits AM, Bos JL, Genetic Alterations during Colorectal-Tumor Development, N. Engl. J. Med 319 (1988) 525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- [3].Smith G, Carey FA, Beattie J, Wilkie MJV, Lightfoot TJ, Coxhead J, Garner RC, Steele RJC, Wolf CR, Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer, Proc. Natl. Acad. Sci 99 (2002) 9433–9438. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].De Rosa M, Pace U, Rega D, Costabile V, Duraturo F, Izzo P, Delrio P, Genetics, diagnosis and management of colorectal cancer (Review)., Oncol. Rep 34 (2015) 1087–96. doi: 10.3892/or.2015.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tariq K, Ghias K, Colorectal cancer carcinogenesis: a review of mechanisms., Cancer Biol. Med 13 (2016) 120–35. doi: 10.28092/j.issn.2095-3941.2015.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Leguisamo NM, Gloria HC, Kalil AN, Martins TV, Azambuja DB, Meira LB, Saffi J, Leguisamo NM, Gloria HC, Kalil AN, Martins TV, Azambuja DB, Meira LB, Saffi J, Base excision repair imbalance in colorectal cancer has prognostic value and modulates response to chemotherapy, Oncotarget. 8 (2017) 54199–54214. doi: 10.18632/oncotarget.14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fahrer J, Kaina B, Impact of DNA repair on the dose-response of colorectal cancer formation induced by dietary carcinogens., Food Chem. Toxicol 106 (2017) 583–594. doi: 10.1016/j.fct.2016.09.029. [DOI] [PubMed] [Google Scholar]
- [8].Alhadheq AM, Purusottapatnam Shaik J, Alamri A, Aljebreen AM, Alharbi O, Almadi MA, Alhadeq F, Azzam NA, Semlali A, Alanazi M, Bazzi MD, Reddy Parine N, The Effect of Poly(ADP-ribose) Polymerase-1 Gene 3’Untranslated Region Polymorphism in Colorectal Cancer Risk among Saudi Cohort., Dis. Markers 2016 (2016) 8289293. doi: 10.1155/2016/8289293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Krokan HE, Bjoras M, Base Excision Repair, Cold Spring Harb. Perspect. Biol 5 (2013) a012583–a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ray D, Kidane D, Gut Microbiota Imbalance and Base Excision Repair Dynamics in Colon Cancer., J. Cancer 7 (2016) 1421–30. doi: 10.7150/jca.15480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Antoniali G, Malfatti MC, Tell G, Unveiling the non-repair face of the Base Excision Repair pathway in RNA processing: A missing link between DNA repair and gene expression?, DNA Repair (Amst). 56 (2017) 65–74. doi: 10.1016/j.dnarep.2017.06.008. [DOI] [PubMed] [Google Scholar]
- [12].Freudenthal BD, Beard WA, Perera L, Shock DD, Kim T, Schlick T, Wilson SH, Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide, Nature. 517 (2015) 635–639. doi: 10.1038/nature13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Whitaker AM, Flynn TS, Freudenthal BD, Molecular snapshots of APE1 proofreading mismatches and removing DNA damage, Nat. Commun 9 (2018) 399. doi: 10.1038/s41467-017-02175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Malfatti MC, Balachander S, Antoniali G, Koh KD, Saint-Pierre C, Gasparutto D, Chon H, Crouch RJ, Storici F, Tell G, Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2, Nucleic Acids Res. 45 (2017) 11193–11212. doi: 10.1093/nar/gkx723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tell G, Quadrifoglio F, Tiribelli C, Kelley MR, The many functions of APE1/Ref-1: not only a DNA repair enzyme., Antioxid. Redox Signal 11 (2009) 601–20. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Luo M, Zhang J, He H, Su D, Chen Q, Gross ML, Kelley MR, Georgiadis MM, Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease., Biochemistry. 51 (2012) 695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang J, Luo M, Marasco D, Logsdon D, LaFavers KA, Chen Q, Reed A, Kelley MR, Gross ML, Georgiadis MM, Inhibition of apurinic/apyrimidinic endonuclease I’s redox activity revisited., Biochemistry. 52 (2013) 2955–66. doi: 10.1021/bi400179m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, Borch RF, Qiao X, Georgiadis MM, Kelley MR, Kelley MR, Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1., Antioxid. Redox Signal 10 (2008) 1853–67. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Su D, Delaplane S, Luo M, Rempel DL, Vu B, Kelley MR, Gross ML, Georgiadis MM, Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme., Biochemistry. 50 (2011) 82–92. doi: 10.1021/bi101248s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR, Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease., Mutat. Res 643 (2008) 54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Antoniali G, Serra F, Lirussi L, Tanaka M, D’Ambrosio C, Zhang S, Radovic S, Dalla E, Ciani Y, Scaloni A, Li M, Piazza S, Tell G, Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism, Nat. Commun 8 (2017) 797. doi: 10.1038/s41467-017-00842-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Poletto M, Malfatti MC, Dorjsuren D, Scognamiglio PL, Marasco D, Vascotto C, Jadhav A, Maloney DJ, Wilson DM, Simeonov A, Tell G, Tell G, Inhibitors of the apurinic/apyrimidinic endonuclease 1 (APE1)/nucleophosmin (NPM1) interaction that display anti-tumor properties., Mol. Carcinog 55 (2016) 688–704. doi: 10.1002/mc.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lou D, Zhu L, Ding H, Dai H-Y, Zou G-M, Aberrant expression of redox protein Ape1 in colon cancer stem cells., Oncol. Lett 7 (2014) 1078–1082. doi: 10.3892/ol.2014.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Noike T, Miwa S, Soeda J, Kobayashi A, Miyagawa S, Increased expression of thioredoxin-1, vascular endothelial growth factor, and redox factor-1 is associated with poor prognosis in patients with liver metastasis from colorectal cancer, Hum. Pathol 39 (2008) 201–208. doi: 10.1016/j.humpath.2007.04.024. [DOI] [PubMed] [Google Scholar]
- [25].Coskun E, Jaruga P, Reddy PT, Dizdaroglu M, Extreme Expression of DNA Repair Protein Apurinic/Apyrimidinic Endonuclease 1 (APE1) in Human Breast Cancer As Measured by Liquid Chromatography and Isotope Dilution Tandem Mass Spectrometry, Biochemistry. 54 (2015) 5787–5790. doi: 10.1021/acs.biochem.5b00928. [DOI] [PubMed] [Google Scholar]
- [26].Di Maso V, Mediavilla MG, Vascotto C, Lupo F, Baccarani U, Avellini C, Tell G, Tiribelli C, Crocè LS, Transcriptional Up-Regulation of APE1/Ref-1 in Hepatic Tumor: Role in Hepatocytes Resistance to Oxidative Stress and Apoptosis., PLoS One. 10 (2015) e0143289. doi: 10.1371/journal.pone.0143289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kelley MR, Cheng L, Foster R, Tritt R, Jiang J, Broshears J, Koch M, Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer., Clin. Cancer Res 7 (2001) 824–30. [PubMed] [Google Scholar]
- [28].Jiang Y, Zhou S, Sandusky GE, Kelley MR, Fishel ML, Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression., Cancer Invest. 28 (2010) 885–95. doi: 10.3109/07357907.2010.512816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Moore DH, Michael H, Tritt R, Parsons SH, Kelley MR, Alterations in the expression of the DNA repair/redox enzyme APE/ref-1 in epithelial ovarian cancers., Clin. Cancer Res 6 (2000) 602–9. [PubMed] [Google Scholar]
- [30].Yoo DG, Song YJ, Cho EJ, Lee SK, Park JB, Yu JH, Lim SP, Kim JM, Jeon BH, Alteration of APE1/ref-1 expression in non-small cell lung cancer: The implications of impaired extracellular superoxide dismutase and catalase antioxidant systems, Lung Cancer. 60 (2008) 277–284. doi: 10.1016/j.lungcan.2007.10.015. [DOI] [PubMed] [Google Scholar]
- [31].Ding J, Fishel ML, Reed AM, McAdams E, Czader MB, Cardoso AA, Kelley MR, Ref-1/APE1 as a Transcriptional Regulator and Novel Therapeutic Target in Pediatric T-cell Leukemia, Mol. Cancer Ther 16 (2017) 1401–1411. doi: 10.1158/1535-7163.MCT-17-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shah F, Logsdon D, Messmann RA, Fehrenbacher JC, Fishel ML, Kelley MR, Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: from bench to clinic., NPJ Precis. Oncol 1 (2017). doi: 10.1038/s41698-017-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang S, He L, Dai N, Guan W, Shan J, Yang X, Zhong Z, Qing Y, Jin F, Chen C, Yang Y, Wang H, Baugh L, Tell G, Wilson DM, Li M, Wang D, Wang D, Serum APE1 as a predictive marker for platinum-based chemotherapy of non-small cell lung cancer patients., Oncotarget. 7 (2016) 77482–77494. doi: 10.18632/oncotarget.13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fishel ML, Kelley MR, The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target, Mol. Aspects Med 28 (2007) 375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- [35].Rai G, Vyjayanti VN, Dorjsuren D, Simeonov A, Jadhav A, Wilson DM, Maloney DJ, Maloney DJ, Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors., J. Med. Chem 55 (2012) 3101–12. doi: 10.1021/jm201537d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wyatt MD, Pittman DL, Methylating Agents and DNA Repair Responses: Methylated Bases and Sources of Strand Breaks, Chem. Res. Toxicol 19 (2006) 1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman ASH, Helleday T, Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks., Nucleic Acids Res. 33 (2005) 3799–811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, Fishel ML, Vasko MR, Identification and Characterization of New Chemical Entities Targeting Apurinic/Apyrimidinic Endonuclease 1 for the Prevention of Chemotherapy-Induced Peripheral Neuropathy., J. Pharmacol. Exp. Ther 359 (2016) 300–309. doi: 10.1124/jpet.116.235283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fishel ML, Jiang Y, Rajeshkumar NV, Scandura G, Sinn AL, He Y, Shen C, Jones DR, Pollok KE, Ivan M, Maitra A, Kelley MR, Impact of APE1/Ref-1 Redox Inhibition on Pancreatic Tumor Growth, Mol. Cancer Ther 10 (2011) 1698–1708. doi: 10.1158/1535-7163.MCT-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y, Maitra A, Kelley MR, Fishel ML, APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival., PLoS One. 7 (2012) e47462. doi: 10.1371/journal.pone.0047462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Laev SS, Salakhutdinov NF, Lavrik OI, Inhibitors of nuclease and redox activity of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/Ref-1)., Bioorg. Med. Chem 25 (2017) 2531–2544. doi: 10.1016/j.bmc.2017.01.028. [DOI] [PubMed] [Google Scholar]
- [42].M.R.K. Shahda afi, Lakhani Nehal J., O’Neil Bert, Rasco Drew W., Wan Jun, Mosley Amber L, Liu Hao, A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors. ∣ 2019 ASCO Annual Meeting Abstracts, (2019). http://abstracts.asco.org/239/AbstView_239_257841.html (accessed May 30, 2019).
- [43].R.A.M. Chu incy, Anderson Amanda K. L., Landers Mark Andrew, Wang Yipeng, Kelley Mark R., CTC enumeration and characterization as a pharmacodynamic marker in the phase I clinical study of APX3330, an APE1/Ref-1 inhibitor, in patients with advanced solid tumors. ∣ 2019 ASCO Annual Meeting Abstracts, (2019). http://abstracts.asco.org/239/AbstView_239_257889.html (accessed May 30, 2019).
- [44].Zhao W, Li D, Liu Z, Zheng X, Wang J, Wang E, Spiclomazine induces apoptosis associated with the suppression of cell viability, migration and invasion in pancreatic carcinoma cells., PLoS One. 8 (2013) e66362. doi: 10.1371/journal.pone.0066362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cristobal A, van den Toorn HWP, van de Wetering M, Clevers H, Heck AJR, Mohammed S, Personalized Proteome Profiles of Healthy and Tumor Human Colon Organoids Reveal Both Individual Diversity and Basic Features of Colorectal Cancer., Cell Rep. 18 (2017) 263–274. doi: 10.1016/j.celrep.2016.12.016. [DOI] [PubMed] [Google Scholar]
- [46].Ohta Y, Sato T, Intestinal tumor in a dish., Front. Med 1 (2014) 14. doi: 10.3389/fmed.2014.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kastenhuber ER, Lowe SW, Putting p53 in Context., Cell. 170 (2017) 1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li X-L, Zhou J, Chen Z-R, Chng W-J, P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation., World J. Gastroenterol 21 (2015) 84–93. doi: 10.3748/wjg.v21.i1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Iacopetta B, Russo A, Bazan V, Dardanoni G, Gebbia N, Soussi T, Kerr D, Elsaleh H, Soong R, Kandioler D, Janschek E, Kappel S, Lung M, Leung C-SS, Ko JM, Yuen S, Ho J, Leung SY, Crapez E, Duffour J, Ychou M, Leahy DT, O’Donoghue DP, Agnese V, Cascio S, Di Fede G, Chieco-Bianchi L, Bertorelle R, Belluco C, Giaretti W, Castagnola P, Ricevuto E, Ficorella C, Bosari S, Arizzi CD, Miyaki M, Onda M, Kampman E, Diergaarde B, Royds J, Lothe RA, Diep CB, Meling GI, Ostrowski J, Trzeciak L, Guzińska-Ustymowicz K, Zalewski B, Capellá GM, Moreno V, Peinado MA, Lönnroth C, Lundholm K, Sun XF, Jansson A, Bouzourene H, Hsieh L-L, Tang R, Smith DR, Allen-Mersh TG, Khan ZAJ, Shorthouse AJ, Silverman ML, Kato S, Ishioka C, TP53-CRC Collaborative Group, Functional categories of TP53 mutation in colorectal cancer: results of an International Collaborative Study, Ann. Oncol 17 (2006) 842–847. doi: 10.1093/annonc/mdl035. [DOI] [PubMed] [Google Scholar]
- [50].Harris SL, Levine AJ, The p53 pathway: positive and negative feedback loops, Oncogene. 24 (2005) 2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- [51].Williams AB, Schumacher B, p53 in the DNA-Damage-Repair Process., Cold Spring Harb. Perspect. Med 6 (2016). doi: 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chatterjee A, Mambo E, Osada M, Upadhyay S, Sidransky D, The effect of p53 -RNAi and p53 knockout on human 8-oxoguanine DNA glycosylase (hOgg1) activity, FASEB J. 20 (2006) 112–114. doi: 10.1096/fj.04-3423fje. [DOI] [PubMed] [Google Scholar]
- [53].Oka S, Leon J, Tsuchimoto D, Sakumi K, Nakabeppu Y, MUTYH, an adenine DNA glycosylase, mediates p53 tumor suppression via PARP-dependent cell death, Oncogenesis. 3 (2014) e121–e121. doi: 10.1038/oncsis.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cun Y, DAI N, LI M, XIONG C, ZHANG Q, SUI J, QIAN C, WANG D, APE1/Ref-1 enhances DNA binding activity of mutant p53 in a redox-dependent manner, Oncol. Rep 31 (2014) 901–909. doi: 10.3892/or.2013.2892. [DOI] [PubMed] [Google Scholar]
- [55].Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S, Bhakat KK, Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage, Nucleic Acids Res. 36 (2008) 1555–1566. doi: 10.1093/nar/gkm1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhou J, Ahn J, Wilson SH, Prives C, A role for p53 in base excision repair, EMBO J. 20 (2001) 914–923. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Antoniali G, Marcuzzi F, Casarano E, Tell G, Cadmium treatment suppresses DNA polymerase δ catalytic subunit gene expression by acting on the p53 and Sp1 regulatory axis, DNA Repair (Amst). 35 (2015) 90–105. doi: 10.1016/j.dnarep.2015.08.007. [DOI] [PubMed] [Google Scholar]
- [58].Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, Ohta Y, Toshimitsu K, Nakazato Y, Kawasaki K, Uraoka T, Watanabe T, Kanai T, Sato T, A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis., Cell Stem Cell. 18 (2016) 827–38. doi: 10.1016/j.stem.2016.04.003. [DOI] [PubMed] [Google Scholar]
- [59].Meng Q, Shi S, Liang C, Liang D, Xu W, Ji S, Zhang B, Ni Q, Xu J, Yu X, Diagnostic and prognostic value of carcinoembryonic antigen in pancreatic cancer: a systematic review and meta-analysis., Onco. Targets. Ther 10 (2017) 4591–4598. doi: 10.2147/OTT.S145708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Comelli M, Pretis I, Buso A, Mavelli I, Mitochondrial energy metabolism and signalling in human glioblastoma cell lines with different PTEN gene status, J. Bioenerg. Biomembr 50 (2018) 33–52. doi: 10.1007/s10863-017-9737-5. [DOI] [PubMed] [Google Scholar]
- [61].Risso D, Ngai J, Speed TP, Dudoit S, Normalization of RNA-seq data using factor analysis of control genes or samples, Nat. Biotechnol 32 (2014) 896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat. Protoc 4 (2009) 44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- [63].Marcin Kosinski PB, Bioconductor - RTCGA, (n.d.). doi: 10.18129/B9.bioc.RTCGA. [DOI] [Google Scholar]
- [64].Kakolyris S, Kaklamanis L, Engels K, Turley H, Hickson ID, Gatter KC, Harris AL, Human apurinic endonuclease 1 expression in a colorectal adenoma-carcinoma sequence., Cancer Res. 57 (1997) 1794–7. [PubMed] [Google Scholar]
- [65].Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B, Requirement for p53 and p21 to sustain G2 arrest after DNA damage., Science. 282 (1998) 1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- [66].Masani S, Han L, Yu K, Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination., Mol. Cell. Biol 33 (2013) 1468–73. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Poletto M, Legrand AJ, Fletcher SC, Dianov GL, p53 coordinates base excision repair to prevent genomic instability, Nucleic Acids Res. 44 (2016) 3165–3175. doi: 10.1093/nar/gkw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Christmann M, Kaina B, Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation, Nucleic Acids Res. 41 (2013) 8403–8420. doi: 10.1093/nar/gkt635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li B, Lee MY, Transcriptional regulation of the human DNA polymerase delta catalytic subunit gene POLD1 by p53 tumor suppressor and Sp1., J. Biol. Chem 276 (2001) 29729–39. doi: 10.1074/jbc.M101167200. [DOI] [PubMed] [Google Scholar]
- [70].Sutcliffe T, Fu L, Abraham J, Vaziri H, Benchimol S, A functional wild-type p53 gene is expressed in human acute myeloid leukemia cell lines., Blood. 92 (1998) 2977–9. [PubMed] [Google Scholar]
- [71].Shtraizent N, Matsui H, Polotskaia A, Bargonetti J, Hot Spot Mutation in TP53 (R248Q) Causes Oncogenic Gain-of-Function Phenotypes in a Breast Cancer Cell Line Derived from an African American patient., Int. J. Environ. Res. Public Health 13 (2015) ijerph13010022. doi: 10.3390/ijerph13010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Plitzko B, Loesgen S, Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism, BIO-PROTOCOL. 8 (2018). doi: 10.21769/BioProtoc.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yamamori T, DeRicco J, Naqvi A, Hoffman TA, Mattagajasingh I, Kasuno K, Jung S-B, Kim C-S, Irani K, SIRT1 deacetylates APE1 and regulates cellular base excision repair, Nucleic Acids Res. 38 (2010) 832–845. doi: 10.1093/nar/gkp1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lindahl T, Barnes DEE, Repair of endogenous DNA damage., Cold Spring Harb. Symp. Quant. Biol 65 (2000) 127–33. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- [75].Turgeon M-O, Perry NJS, Poulogiannis G, DNA Damage, Repair, and Cancer Metabolism., Front. Oncol 8 (2018) 15. doi: 10.3389/fonc.2018.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Meek DW, Tumour suppression by p53: a role for the DNA damage response?, Nat. Rev. Cancer 9 (2009) 714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- [77].Ali R, Rakha EA, Madhusudan S, Bryant HE, DNA damage repair in breast cancer and its therapeutic implications, Pathology. 49 (2017) 156–165. doi: 10.1016/j.pathol.2016.11.002. [DOI] [PubMed] [Google Scholar]
- [78].Mattar M-AM, Zekri A-RN, Hussein N, Morsy H, Esmat G, Amin MA, Polymorphisms of base-excision repair genes and the hepatocarcinogenesis, Gene. 675 (2018) 62–68. doi: 10.1016/j.gene.2018.06.056. [DOI] [PubMed] [Google Scholar]
- [79].Abbotts R, Jewell R, Nsengimana J, Maloney DJ, Simeonov A, Seedhouse C, Elliott F, Laye J, Walker C, Jadhav A, Grabowska A, Ball G, Patel PM, Newton-Bishop J, Wilson DM, Madhusudan S, Targeting human apurinic/apyrimidinic endonuclease 1 (APE1) in phosphatase and tensin homolog (PTEN) deficient melanoma cells for personalized therapy., Oncotarget. 5 (2014) 3273–86. doi: 10.18632/oncotarget.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kumar M, Shukla VK, Misra PK, Raman MJ, Dysregulated Expression and Subcellular Localization of Base Excision Repair (BER) Pathway Enzymes in Gallbladder Cancer., Int. J. Mol. Cell. Med 7 (2018) 119–132. doi: 10.22088/IJMCM.BUMS.7.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Azambuja DB, Leguisamo NM, Gloria HC, Kalil AN, Rhoden E, Saffi J, Prognostic impact of changes in base excision repair machinery in sporadic colorectal cancer, Pathol. - Res. Pract 214 (2018) 64–71. doi: 10.1016/j.prp.2017.11.012. [DOI] [PubMed] [Google Scholar]
- [82].Vodenkova S, Jiraskova K, Urbanova M, Kroupa M, Slyskova J, Schneiderova M, Levy M, Buchler T, Liska V, Vodickova L, Vymetalkova V, Collins A, Opattova A, Vodicka P, Base excision repair capacity as a determinant of prognosis and therapy response in colon cancer patients, DNA Repair (Amst). 72 (2018) 77–85. doi: 10.1016/j.dnarep.2018.09.006. [DOI] [PubMed] [Google Scholar]
- [83].Zou G-M, Karikari C, Kabe Y, Handa H, Anders RA, Maitra A, The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: Therapeutic implications in tumor angiogenesis, J. Cell. Physiol 219 (2009) 209–218. doi: 10.1002/jcp.21666. [DOI] [PubMed] [Google Scholar]
- [84].Zou G-M, Maitra A, Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration, Mol. Cancer Ther 7 (2008) 2012–2021. doi: 10.1158/1535-7163.MCT-08-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Logsdon DP, Shah F, Carta F, Supuran CT, Kamocka M, Jacobsen MH, Sandusky GE, Kelley MR, Fishel ML, Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival, Sci. Rep 8 (2018) 13759. doi: 10.1038/s41598-018-32034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Guerreiro PS, Corvacho E, Costa JG, Saraiva N, Fernandes AS, Castro M, Miranda JP, Oliveira NG, The APE1 redox inhibitor E3330 reduces collective cell migration of human breast cancer cells and decreases chemoinvasion and colony formation when combined with docetaxel, Chem. Biol. Drug Des 90 (2017) 561–571. doi: 10.1111/cbdd.12979. [DOI] [PubMed] [Google Scholar]
- [87].Saitou Y, Shiraki K, Yamanaka T, Miyashita K, Inoue T, Yamanaka Y, Yamaguchi Y, Enokimura N, Yamamoto N, Itou K, Sugimoto K, Nakano T, Augmentation of tumor necrosis factor family-induced apoptosis by E3330 in human hepatocellular carcinoma cell lines via inhibition of NF kappa B., World J. Gastroenterol 11 (2005) 6258–61. doi: 10.3748/wjg.v11.i40.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].McIlwain DW, Fishel ML, Boos A, Kelley MR, Jerde TJ, APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells, Oncotarget. 9 (2018) 10962–10977. doi: 10.18632/oncotarget.23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Fields J, Hanisch JJ, Choi JW, Hwang PM, Is There an Answer? How Does p53 Regulate Mitochondrial Respiration?, (n.d.). doi: 10.1080/15216540601185021. [DOI] [PubMed] [Google Scholar]
- [90].Hashimoto N, Nagano H, Tanaka T, The role of tumor suppressor p53 in metabolism and energy regulation, and its implication in cancer and lifestyle-related diseases, Endocr. J 66 (2019) 485–496. doi: 10.1507/endocrj.EJ18-0565. [DOI] [PubMed] [Google Scholar]
- [91].Park J-H, Zhuang J, Li J, Hwang PM, p53 as guardian of the mitochondrial genome, FEBS Lett. 590 (2016) 924. doi: 10.1002/1873-3468.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tell G, Crivellato E, Pines A, Paron I, Pucillo C, Manzini G, Bandiera A, Kelley MR, Di Loreto C, Damante G, Mitochondrial localization of APE/Ref-1 in thyroid cells., Mutat. Res 485 (2001) 143–52. doi: 10.1016/s0921-8777(00)00068-9. [DOI] [PubMed] [Google Scholar]
- [93].Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S, Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells, Nucleic Acids Res. 34 (2006) 2067. doi: 10.1093/NAR/GKL177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Joo HK, Lee YR, Park MS, Choi S, Park K, Lee SK, Kim C-S, Park JB, Jeon BH, Mitochondrial APE1/Ref-1 suppressed protein kinase C-induced mitochondrial dysfunction in mouse endothelial cells, Mitochondrion. 17 (2014) 42–49. doi: 10.1016/J.MITO.2014.05.006. [DOI] [PubMed] [Google Scholar]
- [95].Wallace DC, Mitochondria and cancer, Nat. Rev. Cancer 12 (2012) 685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Carracedo A, Cantley LC, Pandolfi PP, Cancer metabolism: fatty acid oxidation in the limelight, Nat. Rev. Cancer 13 (2013) 227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kim H-C, Chang J, Lee HS, Kwon HJ, Mitochondrial UQCRB as a new molecular prognostic biomarker of human colorectal cancer, Exp. Mol. Med 49 (2017) e391–e391. doi: 10.1038/emm.2017.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Vascotto C, Cesaratto L, Zeef LAH, Deganuto M, D’Ambrosio C, Scaloni A, Romanello M, Damante G, Taglialatela G, Delneri D, Kelley MR, Mitra S, Quadrifoglio F, Tell G, Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells., Proteomics. 9 (2009) 1058–74. doi: 10.1002/pmic.200800638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Stünkel W, Peh BK, Tan YC, Nayagam VM, Wang X, Salto-Tellez M, Ni B, Entzeroth M, Wood J, Function of the SIRT1 protein deacetylase in cancer, Biotechnol. J 2 (2007) 1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- [100].Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H, Combination therapy in combating cancer., Oncotarget. 8 (2017) 38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. APE1 mRNA and protein levels remain stable in basal condition and upon APE1 endonuclease inhibition in colon cancer cell lines. (A) Expression level of APE1 were determined by qRT-PCR analysis. APE1 levels were normalized to β-actin. Fold change expression values relative to HCT-116 p53+/+ cells, arbitrary set to 1, are shown. Values are mean±SD (n=3). (B) Densitometric analysis of basal APE1 expression. Levels were normalized to actin. HCT-116 p53+/+ cells were used as reference and set to 100%. Values are mean±SD (n=6). (C) HCT116 p53+/+ and HCT116 p53−/− cells were treated with Compound #3 (#3) at the indicated concentration for 48 h. Expression level of APE1 were determined by qRT-PCR analysis. APE1 levels were normalized to β-actin. Fold change expression values relative to untreated control cells, arbitrary set to 1, are shown. Values are mean±SD (n=3).The p-value was calculated using Student’s two-tailed t-test. Resulting p-value is indicated (NS, not significant).
Fig. S2. Inhibition of APE1-endonuclease activity impairs mitochondrial activity in a p53-dependent manner. HCT-116 p53+/+ and HCT-116 p53−/− cells were seeded in a Seahorse XF-24 analyzer and treated with 0.25 μM (A) and 1 μM (B) of Compound #3 at the indicated concentrations for 48 h. Untreated cells were treated with DMSO. Real-time oxygen consumption rate (OCR) was determined during sequential treatments with oligomycin (ATP-synthase inhibitor), FCCP (uncoupler of oxidative phosphorylation), rotenone (complex I inhibitor) and antimycin-A (complex III inhibitor). Values are mean of 5 measurements±SD.