

Abstract
Background
The Brazilian endemic clone Pseudomonas aeruginosa ST277 carries important antibiotic resistance determinants, highlighting the gene coding for SPM-1 carbapenemase. However, the resistance and persistence of this clone is apparently restricted to the Brazilian territory. To understand the differences between Brazilian strains from those isolated in other countries, we performed a phylogenetic analysis of 47 P. aeruginosa ST277 genomes as well as analyzed the virulence and resistance gene profiles. Furthermore, we evaluated the distribution of genomic islands and assessed in detail the characteristics of the CRISPR-Cas immunity system in these isolates.
Results
The Brazilian genomes presented a typical set of resistance and virulence determinants, genomic islands and a high frequency of the CRISPR-Cas system type I-C. Even though the ST277 genomes are closely related, the phylogenetic analysis showed that the Brazilian strains share a great number of exclusively SNPs when compared to other ST277 genomes. We also observed a standard CRISPR spacers content for P. aeruginosa ST277, confirming a strong link between sequence type and spacer acquisition. Most CRISPR spacer targets were phage sequences.
Conclusions
Based on our findings, P. aeruginosa ST277 strains circulating in Brazil characteristically acquired In163 and PAGI-25, which can distinguish them from strains that do not accumulate resistance mechanisms and can be found on the Asian, European and North American continents. The distinctive genetic elements accumulated in Brazilian samples can contribute to the resistance, pathogenicity and transmission success that characterize the ST277 in this country.
Keywords: Pseudomonas aeruginosa, ST277, Comparative genomics, CRISPR
Background
Pseudomonas aeruginosa is an important pathogen that shows a strong potential for development of multidrug resistance and is frequently implicated in healthcare-associated infections. Since the first report in 2002, SPM-1 metallo-β-lactamase is the main carbapenemase associated with P. aeruginosa in Brazil [1, 2]. To date, the blaSPM-1 is restricted to P. aeruginosa and there are only two confirmed cases outside of Brazil, both of which received medical treatment while in this country [3, 4]. Although SPM-1-producing P. aeruginosa has been mainly isolated from nosocomial settings, reports of this multidrug-resistant bacterium in urban rivers and microbiota of migratory birds in Brazil alert to the dispersion of this important resistance mechanism [5, 6]. Usually, the blaSPM-1 gene is inserted in the mobile genetic element ICETn43716061 [7] and, in turn, this ICE is located in P. aeruginosa’s Genomic Island 15 (PAGI-15) [8].
Most SPM-1-producing P. aeruginosa strains descend from a common ancestor, a clone belonging to ST277 [2]. This clone has been characterized as a resistance-enriched ST [9], and the expression of SPM-1 generates resistance to all β-lactams, except for aztreonam [8]. Besides SPM-1, other genetic determinants have been associated with ST277: i) the class 1 integron In163 carrying three resistance genes (aacA4, blaOXA-56 and aadA7); ii) rmtD gene that confers high-level resistance to most aminoglycosides; and iii) the type I-C of Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) and associated proteins [9–11].
The CRISPR family of repetitive DNA sequences, together with a group of CRISPR-associated (cas) genes, encodes a unique defense mechanism that acts against invading genetic elements such as viruses and plasmids. CRISPR loci consist of an array of short and partially palindromic, repetitive sequences interspaced by equally short ‘spacer’ sequences from viral or plasmid origin [12–14]. The Class 1 system is widespread among Archaea and Bacteria, and includes type I, type III, as well as the rare type IV. The cas8c gene is the signature gene for subtype I-C which includes other six cas genes [15]. P. aeruginosa has emerged as a major CRISPR-Cas model system, with types I-F and I-E being the CRISPR-Cas system most commonly found in this species [9].
Here, we examine the phylogenetic distribution and conservation of genetic determinants among ST277 P. aeruginosa genomes available at NCBI. We aim to provide in depth evidence about the genetic determinants that have contributed for its widespread resistance and persistence in Brazil as opposed to other countries.
Results
We compared the genome sequence of 47 strains to understand the genomic diversity of P. aeruginosa ST277. According to NCBI’s BioSample records, strains from a 21-year period were included in this study (1997–2018). The vast majority was obtained from Brazil (35/47), and overall, they represent human-derived isolates (32/47). The other countries represented are United States (6), China (2), United Kingdom (1), Mexico (1), Thailand (1), and Belgium (1). Based on phylogenetic analysis and SNP differences, we can divide the strains into four important groups. One group with strains from China and Mexico (sharing 4054 exclusively SNPs); another one with strains from the United States, Thailand, and Belgium (sharing 299 exclusively SNPs); a main clade that includes all Brazilian strains plus four strains from the US and one from UK (sharing 1025 exclusively SNPs); and finally a branch containing the Chinese strain (PA298) that share 95 exclusively SNPs with the main clade. Overall, the genomes’ phylogenetic relationships do not seem to be associated to the year of isolation (Fig. 1).
Fig. 1.

Whole-genome SNP-based parsimony tree of 47 ST277 P. aeruginosa isolates and the reference genome PAO1 generated by kSNP3.0. The branch lengths are expressed in terms of changes per number of SNPs. The tree was visualized using Dendroscope. Labels in the internal nodes (red) are the number of SNPs that are exclusively shared by the descendants of that node. The panel shows the presence (black) and absence (white) of the genetic determinants surveyed. The purple bars represent an additional mutation in the gyrA gene (D87N). BR: Brazil, CH: China, MX: Mexico, USA: United Stated of America, UK: United Kingdom, BEL: Belgium, THA: Thailand, Hum: Human, Env: Environmental, −: not available
We searched in silico for resistance genes in all ST277 strains. These data demonstrate that 83% (39/47) of the genomes are positive for In163, 62% (29/47) for blaSPM-1 (carbapenem resistance), 57% (27/47) for rmtD (aminoglycoside resistance), and 53% (25/47) for crpP (ciprofloxacin resistance) and aac (6′)-IIc (aminoglycoside resistance). All ST277 strains carry the genes blaOXA-50 (beta-lactam resistance), blaPDC-5 (beta-lactam resistance, Pseudomonas-derived cephalosporinases), catB7 (chloramphenicol resistance), bcr1 (bicyclomycin resistance), fosA (fosfomycin resistance) and aph (3′)-IIb (aminoglycoside resistance). The prevalence of blaOXA-50, blaPDC-5, catB7, bcr1, fosA and aph (3′)-IIb, among 2576 P. aeruginosa whole-genome shotgun assemblies available at NCBI, are higher than 97% [16]. Therefore, these genes are related to the P. aeruginosa species, and not exclusively to the ST277 (Fig. 1, Additional file 1).
Non-synonymous mutations were found in the following resistance related genes: mexT, oprD, mexZ, nalC, pmrA, gyrA and phoQ. All strains presented an 8 nucleotide deletion on mexT (240–247), besides 39 strains (83%) with 2 nucleotide deletion on oprD (379–380) and 19 bp deletion on mexZ (437–456), which caused a frameshift. All ST277 strains have the same amino acid alterations in OprD (T103S, K115T), NalC (S209R, G71E), and PmrA (L71R). Forty-two strains have the T83I mutation on GyrA in addition to a D87N mutation on the same protein observed for 7 strains (1.5%), and one strain has a mutation on PhoQ (V260G) (Fig. 1, Additional file 1).
Mutations in the virulence genes were analyzed using the genes from P. aeruginosa PAO1 as a reference. Seventeen strains (36%) have different point mutations or one nucleotide deletion in the lasR gene, 5 strains (11%) have the same non-synonymous mutation on rhlR (K222T), and a fragment with 11 nucleotides was absent in algB (382–393) for 3 strains (6%). All ST277 strains carry the virulence genes exoS, exoT and exoY (Fig. 1, Additional file 1).
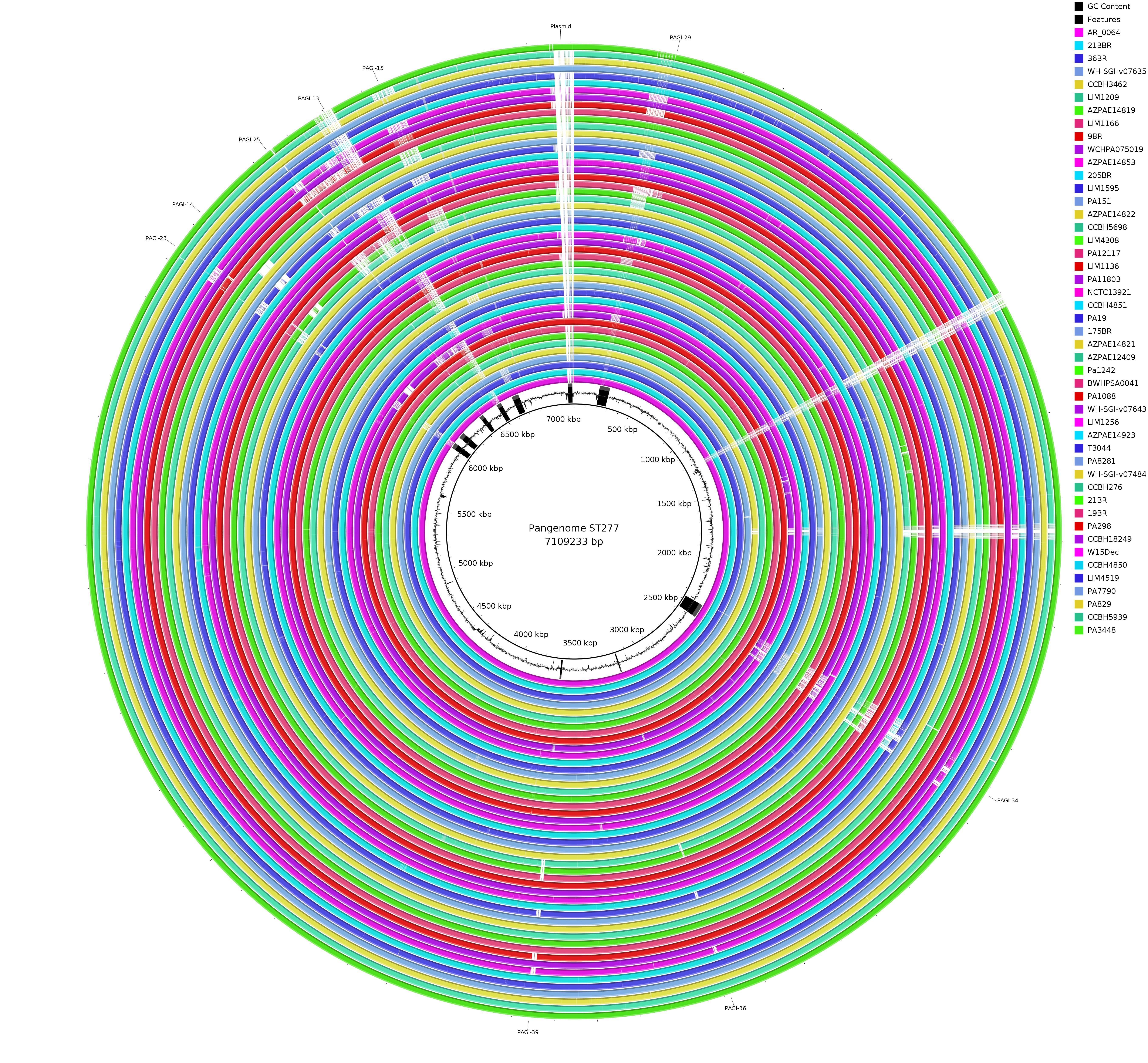
Genomic comparisons using the pangenome as a reference visually showed that some regions are not shared by all ST277 genomes (Additional file 2). We verified that these regions were PAGIs, already described in previous works [8, 17], or a 49 Kb plasmid, present in 4 strains (36BR, 9BR, PA7790 and PA3448). Samples from countries other than Brazil markedly show the absence of islands such as PAGI-14, PAGI-15, PAGI-25, PAGI-29, PAGI-34, PAGI-36 and PAGI-39 (Fig. 1). Some of these PAGI harbor typical ST277 genetic determinants. For all strains positive for the blaSPM-1 gene, it is inserted inside PAGI-15. For those strains that carry the class 1 integron In163 and/or rmtD gene, they are contained in PAGI-25.
In silico analysis identified intact type I-C CRISPR-Cas systems in 33 genomes from ST277 (70%) (Fig. 1, Additional file 1). Two P. aeruginosa isolates (AZPAE14819 and AZPAE14822) have the complete Cas operon, but the CRISPR associated was not detected. No other type of CRISPR-Cas system was found among the ST277 strains. When present, the type I-C CRISPR-Cas system was uniformly localized on PAGI-34 in the ST277 genomes analyzed here.
We inspected the whole spacers content present in type I-C CRISPR-Cas system identified in P. aeruginosa genomes. Among the ST277 strains with intact type I-C CRISPR-Cas systems, 91% (30/33) have the same 39 spacers content, considered by us the standard spacers content of this ST (Additional file 3). This data suggests a strong link between clonality (ST) and spacer content. Strain 213BR has a CRISPR array of 22 and another of 11 spacers, separated by 200 bp from each other and near the Cas operon. These 33 spacers are included in the ST277 standard spacers content. We detected 3 CRISPR arrays in the strain PA19, with 12, 13 and 6 spacers, all of them included in the ST277 standard spacers content. The first CRISPR array (12 spacers) is associated with the Cas operon. Strain PA298 has a set of 36 spacers, associated with the Cas operon and included in the ST277 standard spacers content. We noticed that 64% (25/39) of the spacers are present in all genomes from this ST. We need to consider that possible sequencing and assembly errors may have influenced the spacers identification on 213BR and PA19 partially assembled genomes, so the percentage can be higher than 64%. The ST277 spacers at both ends are conserved between the strains, and the deleted/unidentified spacers are positioned in CRISPR central region (Fig. 2).
Fig. 2.

Spacer content among ST277 P. aeruginosa carrying type I-C CRISPR-Cas system. Light gray spacers are those not shared between all strains
Analyzing BLAT results between the ST277 standard spacers content and the NCBI plasmid and phage databases, targets were assigned for 8% (3/39) and 38% (15/39) of the unique spacers, respectively. In some cases, a unique spacer presented homology with more than one phage and plasmid. We observed that the spacers with the greatest number of matches are distributed throughout the CRISPR array (Fig. 3, Additional file 4).
Fig. 3.

Number of ST277 spacers’ matches against public sequences of plasmids and phages. N: Number of matches
Among the available plasmid and phage in Genbank, 6 plasmids and 40 phages were targeted by some spacers (Additional file 4). Two target plasmids harbor an integron encoding resistance to aminoglycosides, beta-lactam, and sulfonamides (pPB354_1 and pPB353_1). Among the phages, 15 perform a lysogenic cycle, 2 a lytic cycle, 2 lytic-lysogenic switch, and for 21 we could not find this information using the Genbank accession numbers.
We searched for anti-CRISPR genes (acr) on phage and plasmids targeted by CRISPR spacers. Proteins were identified as candidate anti-CRISPRs in 3 plasmids (3/6) and 20 phages (20/40) (Additional file 5, 6 and 7, respectively). The 2 plasmids harboring resistance genes described above also code for putative Acr proteins. Between the 23 anti-CRISPRs candidate genes found in this study, 67% were identified as known groups of acr genes, all of them characterized as anti-type I-F (Additional file 4).
Discussion
P. aeruginosa ST277 is a clone spread throughout the Brazilian territory and related to high mortality rates [2, 10, 18]. A greater similarity within Brazilian strains was found when compared to ST277 genomes isolated from other countries, showing that strains from Brazil share a high number of exclusively SNPs.
It has been reported that In163, rmtD and blaSPM-1 are frequently associated to ST277 [2, 10, 17, 18]. Together, these mechanisms cause resistance to the main antimicrobials used against P. aeruginosa in clinical practice. In163 carries 3 gene cassettes that confer resistance to aminoglycosides and cephalosporins [10], and in this work, it was absent only in strains from countries other than Brazil, proving to be a relevant genetic acquisition of ST277 circulating in this territory. The rRNA methylase RmtD promotes a high-level resistance to all clinically available aminoglycosides and was observed in more than half of the ST277 strains studied [11]. SPM-1 producing P. aeruginosa is almost exclusively reported in Brazil, mostly belonging to ST277 [2]. Analyzing genomes available from Genbank, we identified that blaSPM-1-negative ST277 strains are mainly from countries other than Brazil (61%). Only one non-Brazilian sample was positive for blaSPM-1 (WH-SGI-V-07484, from USA), and more sample information is needed to discuss this observation.
According to our findings, the arsenal of acquired antibiotic resistant genes characteristic of ST277 seems relatively restricted (3 β-lactamase families and 4 aminoglycoside resistant genes) and strains from this clone were mostly isolated in Brazil (74%). This scenario differs from other P. aeruginosa high-risk clones such as ST235, which shows a worldwide distribution (more than 20 countries) and possesses a great diversity of resistant genes including 9 β-lactamase families and 22 aminoglycoside transferases genes [19].
We analyzed mutational events in some genes that can contribute to enhance the bacterial resistance phenotype. Among these genetic alterations, those already described as related to increased resistance are: activator deletion of mexT that leads to low sensitivity to chlorophenylcholine and fluoroquinolones [20]; mexZ nucleotide deletion associated to aminoglycoside, cefepime, tetracyclines and macrolides resistance [8, 21]; mutation in PhoQ linked to colistin-resistant phenotype [22]; GyrA alterations creating quinolone-resistant strains [23]; and oprD frameshift related to imipenem resistance [8, 24]. On the other hand, the literature does not associate the PmrA and NalC amino acid changes observed here to resistant strains [25, 26]. In addition, the 20 oprD mutations identified for all strains may be a feature of ST277, as specific amino acid substitutions in OprD have been potentially associated to MLST profiles [24].
Changes in genes encoding virulence-related proteins have shown the possibility of increased virulence such as, for example, by the efflux of molecules from the quorum sense signaling system through MexEF-OprN (caused by mexT deletion in all strains analyzed) [20]. Some strains presented alteration on lasR and rhlR that can affect virulence and a wide range of metabolic functions by quorum sensing (QS) regulation [27]. However, other factors found may decrease the virulence of strains, such as the deletion on algB in a few strains, that can decrease strain virulence as AlgB is related to the mucoid phenotype [28] and exoS positivity for all strains, which is correlated with a less virulent profile than exoU strains [29].
The presence of the type I-C CRISPR-Cas system was first reported in P. aeruginosa just recently, mainly associated to STs 277 and 235. This system is located at PAGI-34, a pKLC102-like ICE, the first putative mobile genetic element with a CRISPR-Cas system in a Pseudomonas species [9]. Our results confirmed the high prevalence of this subtype on ST277 (70%).
Being aware of the evolutionary and immunological information provided by CRISPR spacers [14], we analyzed the whole spacers content present in type I-C CRISPR-Cas subtype identified in P. aeruginosa genomes. We observed a standard spacers content for ST277, and this strong link between sequence types and spacer content in P. aeruginosa clinical isolates was already reported previously [9]. This set varied a little for only 3 strains. We believe this may be associated with problems in the sequencing and/or assembly steps of the genomes deposited at NCBI. The exception is PA298, an isolate from China of which the lack of 3 spacers may be related to geographical distance.
We also analyzed the phages and plasmids that were the possible spacers’ targets. The rates of spacers with assigned targets were close to those found by Belkum and coworkers, who used the same identification strategy [9]. Most spacers with matches originated from phage sequences (38%), which are in accordance with the origin of the CRISPR-Cas system, primarily engaged in antivirus defense [30].
A trend related to the presence of a lower fraction of spacers with matches going from the beginning to the end of CRISPR arrays has been described [30]. We found an irregular distribution of the spacers with the greatest number of matches throughout the CRISPR array. As we are studying a specific Cas subtype in a particular P. aeruginosa clone this may be influencing the uncommon distribution found here.
Strains with intact CRISPR-Cas systems can be phenotypically CRISPR-Cas incompetent if they encode a cognate anti-CRISPR protein (Acr) that deactivates the corresponding CRISPR-Cas system [9]. The anti-CRISPR-associated protein 1 (Aca1) is a highly conserved sequence located downstream of the acr gene, the latter with variable sequences classified in different families [31, 32].
We did not find homologues to the unique anti-type I-C gene described until now (acrIC1 from Moraxella bovoculi) [33]. However, the dual specificity of some anti-CRISPR proteins has already been described. AcrF6Pae can inactivate both type I-F and I-E CRISPR-Cas systems [31], while AcrVA3.1 may inhibit the type I-C as well as type V-A system [33]. So, all putative Acr proteins identified in this work, even those with anti-type I-F proven activity, should be tested for activity in strains of P. aeruginosa possessing the type I-C system.
Conclusion
The published literature on ST277 P. aeruginosa points to a great similarity between these strains, but these studies are always restricted to the Brazilian strains, mostly SPM-1 producers [2, 8, 34]. The hypothesis that ST277 carries an “intrinsic resistome” has been speculated [34]. However, our findings suggest that the proposed “chromosomal pack of resistance genes” is a characteristic of the ST277 strains harboring In163 and PAGI-25, circulating mainly in Brazil and accumulating other resistance determinants. More information is required about the USA and UK registered strains that have the same profile to establish a possible correlation between them and the Brazilian strains.
It was still impossible to confirm if this ST appeared as a high-risk clone in this country or if it had already arrived in Brazil with important mechanisms of resistance. The strain collection explored here is of limited size and suffers from a bias in geographical distribution, with a high representation of Brazilian strains. However, this is the most comprehensive genomic analysis of ST277 possible considering the genomes available at the time.
Methods
Selection of ST277 P. aeruginosa isolates
Seven clinical P. aeruginosa ST277 isolates deposited in the Culture Collection of Hospital-Acquired Bacteria (CCBH) located at the Laboratório de Pesquisa em Infecção Hospitalar Hospital/Fiocruz (LAPIH-IOC) (WDCM947; CGEN022/2010) were sequenced on an Illumina MiSeq instrument (Illumina Inc., San Diego, CA, United States) at the Genomic Lab located in the Departamento de Bioquimica, UERJ (Rio de Janeiro, Brazil). The reads were de novo assembled using A5 assembly pipeline [35]. Four of these samples are part of a previous study (CCBH276, CCBH3462, CCBH4851 and CCBH5939) [17]. Posteriorly, CCBH4851 was fully assembled (to be published) (CP021380.1).
Until September 2019, there were 4910 P. aeruginosa assembled genomes deposited in NCBI. To identify the genomes belonging to ST277, we picked up the exact matches resulting from BLAST (blastn) [36] searches with the seven alleles for ST277 (retrieved from the MLST databases, https://pubmlst.org/paeruginosa/). By this analysis, 47 genomes were assigned to ST277 (0.95%) and included in this study.
ST277 phylogeny and Pangenome analysis
For not-completely closed P. aeruginosa ST277 genomes, the contigs were reordered using the “reorder contigs” option in Mauve, under the default parameters [37]. The strain P. aeruginosa CCBH4851 (CP021380.1), deposited in CCBH-IOC, was used as the reference genome [38].
Phylogenetic analysis of the 47 ST277 genomes and the P. aeruginosa PAO1 (NC_002516.2) was performed using kSNP3.0 software [39]. The program Kchooser, which is part of the kSNP package, was used to identify the optimal kmer length (21). Dendroscope was used for visualizing and rooting the parsimony tree by using P. aeruginosa PAO1 [40].
The GView Server was used to obtain the pangenome of the P. aeruginosa PAO1 and the 47 ST277 isolates with a minimum identity of 90% and an e < 10–5 [41]. The strain CCBH4851 was used as seed genome. Then, genome comparisons were performed with the BLAST Ring Image Generator (BRIG, 0.95) using the pangenome as a reference [42]. The ST277 PAGIs described in previous works were labeled in this comparison using the coordinates obtained by BLAST (blastn) [36] against the pangenome [8, 17].
Screening of resistance and virulence genes in P. aeruginosa ST277 genomes
The prediction of the resistome was made using the Comprehensive Antibiotic Resistance database (CARD) [16]. In163 was verified by BLASTn [36] searches using GenBank accession AY660529.1. Besides that, BLAST (blastn) alignment results using PAO1 genes as query were analyzed to identify mutations in some genes related to resistance (not included in CARD analyzes) and virulence. Genes were obtained from Pseudomonas genome database (http://www.pseudomonas.com) [oprD (PA0958), mexT (PA2492), mexZ (PA2020) lasR (PA1430), rhlR (PA3477), and algB (PA5483)]. Moreover, BLAST (blastn) was applied to check the presence of the virulence genes exoS (AY029246.1), exoU (KX641457.1), exoT (PA0044, PAO1), and exoY (PA2191, PAO1).
CRISPR-Cas and anti-CRISPR annotation in P. aeruginosa ST277 genomes
From reordered contigs or complete genomes, CRISPR arrays and their associated proteins (Cas) were predicted using CRISPRCasFinder [43]. The .json file output for each genome was downloaded and used to extract the CRISPR spacers applying in-house Perl scripts. These scripts considered the orientation of the direct repeat and CRISPR to determine the correct orientation of the corresponding spacer sequences. All the CRISPR arrays with more than two spacers had their spacers clustered together using CD-HIT [44] at a 90% identity threshold to obtain a non-redundant spacer set.
Putative anti-CRISPR (acr) genes were identified based on BLAST (blastn) [36] matches to nucleotide sequences of anti-CRISPR-associated protein 1 (Aca1, accession YP_007392343). The proteins encoded by genes immediately upstream of putative Aca1 homologue were selected [31]. Eighteen curated acr genes were retrieved from previous studies and aligned to the putative acr identified here using MUSCLE software [45] [anti-I-F (n = 12), anti-I-E (n = 4), anti-I-C (n = 1) and anti-VA (n = 1)] [31, 33, 46, 47]..
Target identification for spacer from type I-C CRISPR-Cas system located at P. aeruginosa ST277 genomes
When using the non-redundant spacer set as BLAT query (version: 36, parameters: blat -tileSize = 8 -minScore = 28 [reference] [query] [results]) [48], following Belkum et al. 2015 [9], we identified the spacer matches against two categories of sequence databases: 1) phage genomes from NCBI (n = 4634 phage files, source: ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/viral/) (downloaded on April 2018), and 2) Refseq plasmids from NCBI (n = 13,428 sequences, source: ftp://ftp.ncbi.nlm.nih.gov/refseq/release/plasmid/ -downloaded on April 2018-).
Supplementary information
Additional file 1. General information about ST277 P. aeruginosa NCBI genomes.
{kind=link}
Additional file 2. P. aeruginosa ST277 pangenome.
Additional file 3. Spacers from ST277 P. aeruginosa CRISPR-Cas system type I-C.
Additional file 4. BLAT output from P. aeruginosa ST277 spacers against plasmids and phages.
Additional file 5. Acr putative sequences.
Additional file 6. Acr nucleotide sequences from plasmids.
Additional file 7. Acr nucleotide sequences from phages.
Acknowledgments
MCS recognizes CNPq for supporting her with a scholarship. We thank CNPq, CAPES and FAPERJ financial support.
Abbreviations
- CCBH
Culture Collection of Hospital-Acquired Bacteria
- CRISPR
Clustered Regularly Interspaced Short Palindromic Repeat
- ICE
Integrative and Conjugative Element
- MLST
Multilocus Sequence Typing
- PAGI
Pseudomonas aeruginosa genomic island
- SNP
Single Nucleotide Polymorphism
- ST
Sequence type
Authors’ contributions
APDC conceived the research; MCS and RMA participated in method design and data handling; MCS performed the data analyses; APDC, MCS and ICOS participated in the selection of sequenced strains; APDC, MCS, CMRS wrote parts and edited the complete manuscript. All authors have read and approved the manuscript.
Funding
This manuscript has not received specific funding.
Availability of data and materials
The genomes are available at GenBank - NCBI, and accession numbers are in Fig. 1. Most other relevant data are contained in Additional files. Other datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable. We have no human or animal data involved.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-020-6650-9.
References
- 1.Murphy TA, Simm AM, Toleman MA, Jones RN, Walsh TR. Biochemical characterization of the acquired metallo-β-lactamase SPM-1 from Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2003;47:582–587. doi: 10.1128/AAC.47.2.582-587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silva FM, Carmo MS, Silbert S, Gales AC. SPM-1-producing Pseudomonas aeruginosa : analysis of the ancestor relationship using multilocus sequence typing, pulsed-field gel electrophoresis, and automated Ribotyping. Microb Drug Resist. 2011;17:215–220. doi: 10.1089/mdr.2010.0140. [DOI] [PubMed] [Google Scholar]
- 3.El Salabi A, Toleman MA, Weeks J, Bruderer T, Frei R, Walsh TR. First report of the metallo-β-lactamase SPM-1 in Europe. Antimicrob Agents Chemother. 2010;54:582. doi: 10.1128/AAC.00719-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hopkins KL, Meunier D, Findlay J, Mustafa N, Parsons H, Pike R, et al. SPM-1 metallo-β-lactamase-producing Pseudomonas aeruginosa ST277 in the UK. JMM Corresp. 2016;65:696–697. doi: 10.1099/jmm.0.000269. [DOI] [PubMed] [Google Scholar]
- 5.Turano H, Gomes F, Medeiros M, Oliveira S, Fontes LC, Sato MIZ, et al. Presence of high-risk clones of OXA-23-producing Acinetobacter baumannii (ST79) and SPM-1-producing Pseudomonas aeruginosa (ST277) in environmental water samples in Brazil. Diagn Microbiol Infect Dis. 2016;86:80–82. doi: 10.1016/j.diagmicrobio.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Martins WMBS, Narciso AC, Cayô R, Santos SV, Fehlberg LCC, Ramos PL, et al. SPM-1-producing Pseudomonas aeruginosa ST277 clone recovered from microbiota of migratory birds. Diagn Microbiol Infect Dis. 2018;90:221–227. doi: 10.1016/j.diagmicrobio.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Fonseca EL, Marin MA, Encinas F, Vicente ACP. Full characterization of the integrative and conjugative element carrying the metallo-β-lactamase blaSPM-1 and bicyclomycin bcr1 resistance genes found in the pandemic Pseudomonas aeruginosa clone SP/ST277. J Antimicrob Chemother. 2015;70:2547–2550. doi: 10.1093/jac/dkv152. [DOI] [PubMed] [Google Scholar]
- 8.Nascimento APB, Ortiz MF, Martins WMBS, Morais GL, Fehlberg LCC, Almeida LGP, et al. Intraclonal genome stability of the metallo-β-lactamase SPM-1-producing Pseudomonas aeruginosa ST277, an endemic clone disseminated in Brazilian hospitals. Front Microbiol. 2016;7:1946. doi: 10.3389/fmicb.2016.01946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Belkum A, Soriaga LB, Lafave MC, Akella S, Veyrieras J, Barbu EM, et al. Phylogenetic distribution of CRISPR-Cas Systems in Antibiotic- Resistant Pseudomonas aeruginosa. MBio. 2015;6:1–13. doi: 10.3391/mbi.2015.6.1.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvalho APD, Albano RM, Oliveira DN, Cidade DAP, Teixeira LM, Marques EA. 2006. Characterization of an epidemic Carbapenem-resistant Pseudomonas aeruginosa producing SPM-1 Metallo-β-lactamase in a hospital located in Rio de Janeiro, Brazil. Microb Drug Resist. 2006;12:103–108. doi: 10.1089/mdr.2006.12.103. [DOI] [PubMed] [Google Scholar]
- 11.Doi Y, Adams-Haduch JM, Paterson DL. Genetic environment of 16S rRNA methylase gene rmtD. Antimicrob Agents Chemother. 2008;52:2270–2272. doi: 10.1128/AAC.00037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jansen R, van Embden JDA, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 13.Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. doi: 10.1038/nature15386. [DOI] [PubMed] [Google Scholar]
- 14.McGinn J, Marraffini LA. Molecular mechanisms of CRISPR–Cas spacer acquisition. Nat Rev Microbiol. 2019;17:7–12. doi: 10.1038/s41579-018-0071-7. [DOI] [PubMed] [Google Scholar]
- 15.Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. doi: 10.1016/j.mib.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silveira MC, Albano RM, Asensi MD, Carvalho-Assef APDA. Description of genomic islands associated to the multidrug-resistant Pseudomonas aeruginosa clone ST277. Infect Genet Evol. 2016;42:60–65. doi: 10.1016/j.meegid.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 18.Chaves L, Tomich LM, Salomão M, Leite GC, Ramos J, Martins RR, et al. High mortality of bloodstream infection outbreak caused by carbapenem-resistant P. aeruginosa producing SPM-1 in a bone marrow transplant unit. J Med Microbiol. 2017;66:1722–1729. doi: 10.1099/jmm.0.000631. [DOI] [PubMed] [Google Scholar]
- 19.Treepong P, Kos VN, Guyeux C, Blanc DS, Bertrand X, Valot B, et al. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin Microbiol Infect. 2018;24:289–294. doi: 10.1016/j.cmi.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Maseda H, Uwate M, Nakae T. Transcriptional regulation of the mexEF-oprN multidrug efflux pump operon by MexT and an unidentified repressor in nfxC-type mutant of Pseudomonas aeruginosa. FEMS Microbiol Lett. 2010;311:36–43. doi: 10.1111/j.1574-6968.2010.02063.x. [DOI] [PubMed] [Google Scholar]
- 21.Llanes C, Llanes C, Hocquet D, Vogne C, Benali-baitich D, Neuwirth C, et al. Clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob Agents Chemother. 2004;48:1797–1802. doi: 10.1128/AAC.48.5.1797-1802.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyle B, Fernandez L, Laroche J, Kukavica-Ibrulj I, Mendes CMF, Hancock RW, et al. Complete genome sequences of three Pseudomonas aeruginosa isolates with phenotypes of polymyxin B adaptation and inducible resistance. J Bacteriol. 2012;194:529–530. doi: 10.1128/JB.06246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yonezawa M, Takahata M, Matsubara N, Watanabe Y, Narita H. DNA Gyrase gyrA mutations in quinolone-resistant clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1995;39:1970–1972. doi: 10.1128/AAC.39.9.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CH, Kang HY, Kim BR, Jeon H, Lee YC, Lee SH, et al. Mutational inactivation of OprD in carbapenem-resistant Pseudomonas aeruginosa isolates from Korean hospitals. J Microbiol. 2016;54:44–49. doi: 10.1007/s12275-016-5562-5. [DOI] [PubMed] [Google Scholar]
- 25.Lee JY, Lim MH, Heo ST, Ko KS. Repeated isolation of Pseudomonas aeruginosa isolates resistant to both polymyxins and carbapenems from 1 patient. Diagn Microbiol Infect Dis. 2012;72:267–271. doi: 10.1016/j.diagmicrobio.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Horna G, López M, Guerra H, Saénz Y, Ruiz J. Interplay between MexAB-OprM and MexEF-OprN in clinical isolates of Pseudomonas aeruginosa. Sci Rep. 2018;8:1–11. doi: 10.1038/s41598-018-34694-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luján AM, Moyano AJ, Segura I, Argaraña CE, Smania AM. Quorum-sensing-deficient (lasR) mutants emerge at high frequency from a Pseudomonas aeruginosa mutS strain. Microbiology. 2007;153:225–237. doi: 10.1099/mic.0.29021-0. [DOI] [PubMed] [Google Scholar]
- 28.Goldberg JB, Ohman DE. Construction and characterization of Pseudomonas aeruginosa algB mutants: role of algB in high-level production of alginate. J Bacteriol. 1987;169:1593–1602. doi: 10.1128/JB.169.4.1593-1602.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aditi, Shariff M, Chhabra SK, Rahman MU. Similar virulence properties of infection and colonization associated Pseudomonas aeruginosa. J Med Microbiol. 2017;66:1489–1498. doi: 10.1099/jmm.0.000569. [DOI] [PubMed] [Google Scholar]
- 30.Shmakov SA, Sitnik V, Makarova KS, Wolf YI, Severinov KV, Koonin EV. The CRISPR spacer space is dominated by sequences from species-specific Mobilomes. MBio. 2017;8:1–18. doi: 10.1128/mBio.01397-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pawluk A, Davidson AR, RHJ S, Taylor C, BNJ W, et al. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol. 2016;1:16085. doi: 10.1038/nmicrobiol.2016.85. [DOI] [PubMed] [Google Scholar]
- 32.Sontheimer EJ, Davidson AR. Inhibition of CRISPR-Cas systems by mobile genetic elements. Curr Opin Microbiol. 2017;37:120–127. doi: 10.1016/j.mib.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marino, et al. Discovery of widespread Type I and Type V CRISPR-Cas inhibitors. Science. 2018;362:240–242. doi: 10.1126/science.aau5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galetti R, Andrade LN, Varani AM, Darinia ALC. SPM-1-producing Pseudomonas aeruginosa ST277 carries chromosomal pack of acquired resistance genes: an example of high-risk clone associated to “intrinsic resistome”. J Glob Antimicrob Resist. 2019;16:183–186. doi: 10.1016/j.jece.2018.06.051. [DOI] [PubMed] [Google Scholar]
- 35.Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Ilumina MiSeq data. Bioinformatics. 2015;31:587–589. doi: 10.1093/bioinformatics/btu661. [DOI] [PubMed] [Google Scholar]
- 36.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silveira M, Albano R, Asensi M, Assef APC. The draft genome sequence of multidrug-resistant Pseudomonas aeruginosa strain CCBH4851, a nosocomial isolate belonging to clone SP (ST277) that is prevalent in Brazil. Mem Inst Oswaldo Cruz. 2014;109:1086–1087. doi: 10.1590/0074-0276140336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gardner SN, Slezak T, Hall BG. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics. 2015;31:2877–2878. doi: 10.1093/bioinformatics/btv271. [DOI] [PubMed] [Google Scholar]
- 40.Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61:1061–1067. doi: 10.1093/sysbio/sys062. [DOI] [PubMed] [Google Scholar]
- 41.Petkau A, Stuart-Edwards M, Stothard P, van Domselaar G. Interactive microbial genome visualization with GView. Bioinformatics. 2010;26:3125–3126. doi: 10.1093/bioinformatics/btq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–W251. doi: 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL, Davidson AR. A new Group of Phage Anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio. 2014;5:1–7. doi: 10.1128/mBio.00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. General information about ST277 P. aeruginosa NCBI genomes.
Additional file 2. P. aeruginosa ST277 pangenome.
Additional file 3. Spacers from ST277 P. aeruginosa CRISPR-Cas system type I-C.
Additional file 4. BLAT output from P. aeruginosa ST277 spacers against plasmids and phages.
Additional file 5. Acr putative sequences.
Additional file 6. Acr nucleotide sequences from plasmids.
Additional file 7. Acr nucleotide sequences from phages.
Data Availability Statement
The genomes are available at GenBank - NCBI, and accession numbers are in Fig. 1. Most other relevant data are contained in Additional files. Other datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.