

Abstract
Background:
ER (endoplasmic reticulum) stress leads to decreased complex I activity in cardiac mitochondria. The aim of the current study is to explore the potential mechanisms by which ER stress leads to the complex I defect. ER stress contributes to intracellular calcium overload and oxidative stress that are two key factors to induce mitochondrial dysfunction. Since oxidative stress is often accompanied by intracellular calcium overload during ER stress in vivo, the role of oxidative stress and calcium overload in mitochondrial dysfunction was studied using in vitro models. ER stress results in intracellular calcium overload that favors activation of calcium-dependent calpains. The contribution of mitochondrial calpain activation in ER stress-mediated complex I damage was studied.
Methods:
Thapsigargin (THAP) was used to induce acute ER stress in H9c2 cells and C57BL/6 mice. Exogenous calcium (25 μM) and H2O2 (100 μM) were used to induce modest calcium overload and oxidative stress in isolated mitochondria. Calpain small subunit 1 (CAPNS1) is essential to maintain calpain 1 and calpain 2 (CPN1/2) activities. Deletion of CAPNS1 eliminates the activities of CPN1/2. Wild type and cardiac-specific CAPNS1 deletion mice were used to explore the role of CPN1/2 activation in calcium-induced mitochondrial damage.
Results:
In isolated mitochondria, exogenous calcium but not H2O2 treatment led to decreased oxidative phosphorylation, supporting that calcium overload contributes a key role in the mitochondrial damage. THAP treatment of H9c2 cells decreased respiration selectively with complex I substrates. THAP treatment activated cytosolic and mitochondrial CPN1/2 in C57BL/6 mice and led to degradation of complex I subunits including NDUFS7. Calcium treatment decreased NDUFS7 content in wild type but not in CAPNS1 knockout mice.
Conclusion:
ER stress-mediated activation of mitochondria-localized CPN1/2 contributes to complex I damage by cleaving component subunits
Keywords: electron transport chain, calpain, CPNS1 knockout mice
Graphical Abstract
Schematic figure for oxidative stress and calcium crosstalk
Mitochondria and endoplasmic reticulum (ER) are closely connected through mitochondrial associated membranes (MAM) that contribute a key role in rapid calcium communication between mitochondria and ER. Calcium overload can lead to the degradation of metabolic enzymes by activating mitochondrial calpain 1 (mCPN1). Calcium increases ROS generation by impairing respiratory enzymes in the electron transport chain (ETC) including complex I and III. ROS generation can feedback to increase mitochondrial calcium overload by inhibiting the SERCA calcium ATPase pump that is responsible for restoration of intracellular calcium homeostasis by transferring calcium into the ER for storage. Protein targets of mCPN1 in the mitochondrial matrix and intermembrane space are shown.
1. INTRODUCTION
Mitochondrial dysfunction occurs in many pathological conditions including aging and heart failure [1]. An impaired mitochondrial electron transport chain (ETC) contributes to cardiac injury by decreasing ATP production, increasing the generation of reactive oxygen species (ROS), and sensitizing to mitochondrial permeability transition pore (MPTP) opening [1, 2]. Oxidative stress and mitochondrial calcium overload are key factors to induce the mitochondrial damage [3, 4]. There is a strong interaction between oxidative stress and mitochondrial calcium overload. An increased oxidative stress leads to mitochondrial calcium overload in cardiomyocytes [5]. Calcium overload, in turn, can also increase the production of ROS [6]. Therefore, it is difficult to differentiate the role of oxidant stress and calcium overload in mitochondrial damage in vivo. In contrast, it is feasible to separate the role of oxidative stress from calcium overload in mitochondrial damage in vitro [7]. We first used exogenous calcium with or without H2O2 to clarify the role of calcium or oxidative stress in mitochondrial damage in isolated cardiac mitochondria.
The endoplasmic reticulum (ER) is an intracellular organelle that functions in protein folding, lipid synthesis, and calcium homeostasis [8]. The ER is closely connected with mitochondria through mitochondria associated membranes (MAM) [9], leading to a strong functional interaction between the ER and mitochondria [9]. For example, the induction of the acute ER stress (ER dysfunction) impairs mitochondrial function in mouse and rat hearts [10–12], whereas inhibition of mitochondrial respiration using antimycin A [13] or rotenone [14] also increases the ER stress. ER is a key site of intracellular calcium storage, and ER stress leads to intracellular calcium overload by disrupting calcium homeostasis [15]. ER stress-mediated calcium overload contributes to mitochondrial damage in myocardial infarction-induced heart failure [16] and in hearts following ischemia-reperfusion [17]. In addition to calcium overload, the ER stress also increases ROS generation directly through NADPH oxidase 4 [8], and secondarily through impairment of mitochondrial electron transport [10, 12, 18]. An increased mitochondrial antioxidant capacity leads to decreased calcium overload in myocardial infarction-induced heart failure [16]. These results indicate that there is a positive feedback loop between ER stress and mitochondrial dysfunction.
The acute induction of ER stress using thapsigargin (THAP) leads to decreased oxidative phosphorylation in cardiac mitochondria oxidizing complex I substrates [10]. THAP-induced ER stress leads to decreased complex I activity in mouse heart mitochondria [10]. However, the mechanisms of the ER stress-mediated complex I damage are unclear. Complex I is a key site of the regulation of oxidative phosphorylation [19] and a component of the respirasome [20]. Complex I defects contribute to cell injury in many pathological conditions including aging [21], ischemia-reperfusion [2, 22–24], and heart failure [25]. Thus, understanding the mechanism of the complex I defect is a critical step to develop therapeutic strategies to protect complex I and decrease cardiac dysfunction and injury in pathological conditions.
Calpains are calcium-dependent cysteine-proteases. Calpain 1 (CPN1) and calpain 2 (CPN2) are two ubiquitous calpain isoforms [26]. Both CPN1 and CPN2 exist in cytosol and mitochondria [27–29]. Activation of cytosolic CPN1/2 increases cell injury in traumatic brain injury [30] and cardiac ischemia-reperfusion injury [28, 31, 32]. Mitochondria-localized CPN1 (mCPN1) and CPN2 (mCPN2) are involved in mitochondrial damage during ischemia and reperfusion [33–36], diabetic cardiomyopathy [37], doxorubicin-induced cardiotoxicity [38, 39], and heart failure [40]. Activation of the mCPN1 during ischemia-reperfusion decreases complex I activity by cleaving its subunit-NDUFS7 [41], whereas activation of the mCPN2 leads to degradation of ND6 subunit in complex I [29]. In addition, ischemia-reperfusion also leads to decreased complex I NDUFB1 content in cardiac mitochondria [42]. ER stress leads to intracellular calcium overload [8]. Therefore, we asked if ER stress leads to complex I dysfunction by cleaving its subunits through activation of mCPN1/2 in cardiac mitochondria. CPN1 and CPN2 contain one large subunit (78 KD) and one small regulatory subunit (CAPNS1) [43]. Genetic deletion of CAPNS1 eliminates the activities of CPN1 and CPN2 [43, 44]. Thus, CAPNS1 knockout mice are used to study the role of mCPN1/2 activation in mitochondrial damage.
The aim of the current study is to elucidate the mechanism of complex I damage during ER stress. We first asked if ER stress decreased complex I activity by cleaving complex I subunits. Next, we studied the effect of calcium overload or oxidative stress in mitochondrial damage. Thirdly, we tested the role of mCPN1 activation in calcium-induced complex I damage.
2. METHODS AND MATERIALS
All chemicals were reagent grade, purchased from Sigma Chemical (St. Louis, MO) and Fisher Scientific (Waltham, MA). The experimental procedures of animal studies conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of Virginia Commonwealth University (VCU) and the McGuire Department of Veterans Affairs Medical Center.
2.1. Induction of the ER stress in C57BL/6 mice
C57BL/6 mice (male, 2–3 months) were purchased from Jackson Laboratories (Bar Harbor, ME). THAP (3 mg/kg) was used to induce ER stress through one-time i.p. injection. THAP was first dissolved in DMSO and diluted with saline for injection [12]. DMSO and saline solution were used as vehicle (DMSO) treatment. After 48 hours vehicle or THAP treatment, mice were anesthetized with pentobarbital sodium (100 mg/kg, i.p.) and the heart was harvested for mitochondrial isolation [27].
2.2. CAPNS deletion mice
Cardiac specific calpain small subunit 1 (CAPNS1) deletion mice (CAPNS1P/P) are generated by breeding CAPNS1PZ/PZ mice in a C57BL/6 background crossed with mice bearing cardiac specific Cre [WT·CRE+/−, B6.FVB-Tg(Myh6-cre)2182Mds/J] (purchased from Jackson Laboratory (Bar Harbor, ME). The CAPNS1PZ/PZ mice were a generous gift from Dr. Peter Greer of Queen’s University Cancer Research Institute, Kingston, Ontario, Canada [43]. The CAPNS1PZ/PZ mice were used as control mice and CAPNS1P/P mice were used as deletion mice [43]. The CAPNS1 mice were genotyped with PCR primer: P1 (GTC AGG CTA GAT GCC ATG TTC C), P2 (CGA CTA TCC GAG CGC TGC C), and P3 (GTT CAC TTG GAT CTG TCC GGT GCC). The primer used for cre detection was: P1 (ATA TCT CAC GTA CTG ACG GTG GG) and P2 (CTG TTT CAC TAT CCA GGT TAG GG).
2.3. Isolation of cytosol and mitochondria from a single mouse heart
Heart mitochondria were isolated as previously described [45]. Mice were anesthetized with pentobarbital sodium (100 mg/kg, i.p.). The heart was quickly excised and blotted dry, weighed, and minced in cold buffer A (composition in mM: 100 KCl, 50 MOPS [3-(N-morpholino) propanesulfonic acid], 1 EGTA, 5 MgSO4, and 1 mM ATP). The minced heart tissue was homogenized using a polytron tissue homogenizer at 10,000 rpm for 2.5 seconds in the presence of trypsin (5 mg/g tissue). The trypsin was used to increase mitochondrial protein yield and remove potential cytosolic contamination. The homogenate was incubated for 15 min at 4°C, and then diluted with the same volume of buffer B [buffer A + 0.2% bovine serum albumin (BSA)]. The mixture was centrifuged at 500×g for 10 min. The supernatant was further centrifuged at 3000×g to pellet mitochondria. The mitochondrial pellet was washed with KME buffer (100 mM KCl, 50 mM MOPS, 0.5 mM EGTA), and centrifuged at 3000×g to yield the final mitochondrial pellet. Mitochondria were re-suspended in KME for functional study [45].
A small piece of myocardium was fixed for electron microcopy analysis of mitochondrial morphology alteration. Myocardial samples were immersed into 3% buffered glutaraldehyde. The myocardium tissue was processed into resin and cut for transmission electron microscopy [46].
2.4. Mitochondrial oxidative phosphorylation and enzyme activity
The rate of oxygen consumption in mitochondria was measured using a Clark-type oxygen electrode at 30°C as previously described [46]. Mitochondria were incubated in oxidative phosphorylation buffer (Composition: 80 mM KCl, 50 mM MOPS, 1 mM EGTA, 5 mM KH2PO4, and 1 mg defatted, dialyzed bovine serum albumin/ml at pH 7.4). Pyruvate (20 mM)+malate (10 mM) was used as complex I substrate. Succinate (20 mM)+rotenone (7.5 μM) was used as complex II substrate [47]. Enzyme activities of the ETC were determined in detergent-solubilized frozen-thawed mitochondria with previously published methods [47]
2.5. Cell culture and induction of the ER stress
H9c2 (ATCC® CRL-1446™) cells were grown in complete Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich, USA) including 4.5 mM glucose and supplemented with 10 % fetal bovine serum with 1 % penicillin and streptomycin. Briefly, cells (3×104 cells/ml) were seeded in 25cm2-flasks after reaching 70% confluency. THAP (1 μM) was used to induce ER stress in cultured H9c2 cells incubated in the Galaxy O2 controlled incubator at 5% CO2 and 1% O2 at 37°C for 24 hours [48]. DMSO was used as vehicle control. MDL-28170 (10 μM) was added into incubation medium to inhibit CPN1/2. Assessment of cell death was performed by measurement of dead cell protease activity according to manufacturer instruction by using the Mitochondrial ToxGlo assay (Promega, Madison, WI).
2.6. High resolution respirometry-OROBOROS
H9c2 cells were harvested after vehicle or THAP treatment and re-suspended in 2.1 ml MiR-05, pH.7.1. Cell density was counted in a hemocytometer. Glutamate-pyruvate-malate (10, 5 and 2mM respectively) were used as complex I substrates. H9c2 cells were permeabilized with digitonin (dig, 10μg/ml) and ADP (1 mM) was used to stimulate respiration. Rotenone (0.05 μM) was used to inhibit complex I before addition succinate (10mM) as complex II substrate. Thenoyltrifluoroacetone (TTFA, 40 μM) was then added to inhibit complex II respiration [48, 49].
2.7. Incubation of mouse mitochondria with H2O2 or calcium
Exogenous calcium (100 μM) treatment leads to mitochondrial swelling and MPTP opening in isolated cardiac mitochondria [7, 50]. It is difficult to assess mitochondrial function including oxidative phosphorylation due to calcium-induced mitochondrial uncoupling [7]. Although this result supports that calcium overload leads to mitochondrial damage, it is a model of relatively severe tissue injury [51]. However, in settings of chronic cardiac disease, the oxidative stress and potential calcium exposure are more subtle. Thus, we asked if a lower calcium concentration that does not result in MPTP opening or uncoupling of respiration can lead to decreases in the rate of oxidative phosphorylation in isolated cardiac mitochondria.
Mitochondrial inner membrane potential (Δψ) was used to reflect MPTP opening. The Δψ was measured using the fluorogenic indicator TMRM (tetramethylrhodamine, methyl ester)[52]. TMRM is a lipophilic cation accumulated by mitochondria in proportion to Δψ. The fluorescence intensity of TMRM is quantified using emission at 590 nm and excitation at two wavelengths: 573nm and 546nm. Freshly isolated mitochondria (0.2 mg/ml) were incubated in a single cuvette at 30 °C in the presence of 0.3 uM TMRM. The Δψ was reflected by the ADP stimulated change of fluorescence intensity of TMRM using glutamate as substrate [53].
The isolated mitochondria (0.2 mg) were incubated in 100 μl buffer (150 mM sucrose, 50 mM KCl, 2 mM KH2PO4, 20 mM Tris–HCl, pH 7.4) with 0.1% β-mercaptoethanol (to maintain the buffer in reduced state) for 30 min [27]. H2O2 (100 μM) induces oxidative stress in cardiac myocytes [54] and was used in these experiments. Exogenous calcium (25 μM Ca2+) was used. H2O2 (100 μM)/calcium (25 μM Ca2+) was used to evaluate the contributions of oxidative stress, calcium overload or their combination to mitochondrial dysfunction. Mitochondria were centrifuged at 5,000×g for 10 min at the end of the incubation. The mitochondrial pellet was resuspended in incubation buffer without 0.1% β-mercaptoethanol for functional study [45].
For the study of mitochondrial morphology, isolated mitochondria underwent the same incubation protocol. For MDL-28170 treatment, 10 uM MDL-28170 was used to inhibit calpains [27]. Mitochondria were sedimented following incubation and the supernatant was removed. The mitochondrial pellet was immersed in the 3% buffered glutaraldehyde for transmission electron microscopy and the supernatant was discarded [11, 46].
For the mCPN1-dependent study, the isolated mitochondria (0.2 mg mitochondrial protein) from wild type and CPNS1 knockout mice were incubated only with calcium (25 μM Ca2+) for 30 min [27]. Mitochondria were centrifuged at 5,000×g for 10 min at the end of the incubation. The mitochondrial pellet was used for functional measurements, proteomic analysis, and immunoblotting to confirm proteomic findings. Each sample for analysis was prepared in a separate incubation.
2.8. Mass Spectrometry (MS) Analyses with iTRAQ™ Labeling
Isolated mitochondria were reduced and tryptic digested with iTRAQ labeling followed by MudPIT analysis using a strong cation exchange to generate fractions followed by HPLC of each fraction and analysis using mass spectrometry. Mitochondria from control and CAPNS1 deletion hearts with or without calcium treatment were lysed and precipitated overnight in acetone at minus 20°C and pellets were re-suspended in 20 μL of 0.5 M triethylammonium bicarbonate (TEAB; pH 8.5) as previously described [55]. Lyophilized SCX sample fractions were reconstituted in HPLC aqueous run buffer (0.1% trifluoroacetic acid, 2% acetonitrile) and injected onto a Kinetex C18 chromatographic column, 100 × 2.1 mm (Phenomenex, Torrance, CA). The peptides were eluted from the column using an acetonitrile/formic acid gradient (2–90% acetonitrile in 120 minutes) and analyzed using a QTrap5500 mass spectrometer operated with Analyst 1.5 software. The MS acquisition was in data dependent mode. The three most intense multiply charged ions with ion intensities above a threshold of 50,000 in each regular MS scan were chosen for MS/MS analyses. Spectra achieved a S/N ≥ 70.
The resulting MS/MS spectra were analyzed using ABI Protein ProteinPilot software 4.0 (Applied Biosystems, Foster City, CA). The spectral data was searched against the mouse protein database (Uniprot_mouse_27Feb12 database customized to select for all mouse proteins) for identification of the peptides and corresponding proteins. In Protein Pilot, the sample type was selected as iTRAQ 4Plex for retrieval of the isotopic tag information from the mass spectra. After database correlation analysis, the proteins were grouped, scored, and normalized against one of four isotope correction factors. Each peptide match showed the iTRAQ isotopic labels, MMTS labeled cysteines, and other PTMs present as mass spectral shifts identified during the database correlation analysis. Each protein identified also showed the differential protein expression compared against the other iTRAQ labeled samples for relative quantization [56].
2.9. Immunoblotting
Cytosol or mitochondrial samples were solubilized in sample buffer and denatured at 95°C for 5 min. Samples were separated using 12% or 4–15% Tris-glycine gels (Bio-Rad, Hercules, CA) and transferred to PVDF membrane by semi-dry transfer (Bio-Rad). The membranes were incubated for 1 hour at room temperature in 5% (w/v) non-fat dry milk (Bio-Rad) in TBST buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween20). The membrane was washed with TBST for 5 min at room temperature. Then, the membrane was incubated with primary antibodies at 4 °C overnight (See information in Table 1). The membrane was washed with TBST buffer before addition of second antibody (HRP-conjugated anti-mouse or anti-rabbit IgG F(ab)2, 1:10,000 dilution, GE Healthcare Life Sciences, Piscataway, NJ) and incubation for 1 hour in room temperature. The blots were developed using ECL Plus Western Blotting Detection Reagents (GE Healthcare Life Sciences, Piscataway, NJ). Membranes were digitally analyzed (Bio-Rad, Hercules, CA) using Image Lab 6.0 software. Background intensity adjustment, if performed, was always adjusted for the entire membrane.
Table 1:
Antibodies used in the current manuscript.
| Antibody name | Company | Catalog number | Concentration |
|---|---|---|---|
| Subunit 4 of cytochrome oxidase | Cell signaling | 4844 | 1:10000 |
| GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) | Cell signaling | 5174 | 1:1000 |
| NDUFA4 (mitochondrial complex associated) | Abcam | ab129752 | 1:1000 |
| NDUFB1 (NADH:Ubiquinone Oxidoreductase Subunit B1) | Abcam | ab201302 | 1:1000 |
| NDUFS7 (NADH:Ubiquinone Oxidoreductase Core Subunit S7) | ThermoFisher Scientific | PA5–19343 | 1:500 |
| PDHα1 subunit (pyruvate dehydrogenase α1 subunit) | Cell signaling | 2784 | 1:1000 |
| Spectrin | Santa Cruz | csc-46696 | 1:100 |
| VDAC (Voltage-dependent anion-selective channel) | Abcam | ab14715 | 1:2500 |
2.10. Statistical analyses
Data are expressed as the mean ± standard error. Differences between groups (≥ 3 groups) were compared by one-way ANOVA. When a significant F value was obtained, means were compared using the Student-Newman-Keuls test of multiple comparisons. Differences between two groups were compared by unpaired student t-test or paired t-test where appropriate, as in incubations of mitochondria from wild type and CAPNS deletion mice with and without calcium (SigmaStat 3.5, Systat, Richmond, CA). Statistical significance was defined as a value of p<0.05. Statistical significance in proteomic studies was calculated using the software contained within the ABI Protein ProteinPilot Software 4.0 [42].
RESULTS
3.1. Mitochondrial morphology in the presence or absence of calcium exposure
Ultrastructure of normal mitochondria in situ in left ventricular myocardium using transmission electron microscopy is shown inFigure 1A. The isolated normal cardiac mitochondria are shown in Figure 1B and 1C with different magnifications. Incubation of the isolated mitochondria in non-calcium buffer did not alter mitochondrial morphology (Figure 1D). Incubation of mitochondria with calcium did not dramatically alter mitochondrial morphology, but it did leads to swelling in scatted mitochondria (Figure 1E). Inhibition of calpains with MDL-28170 (a calpain I and II inhibitor) preserved mitochondrial morphology in calcium-treated mitochondria (Figure 1F).
Figure. 1.
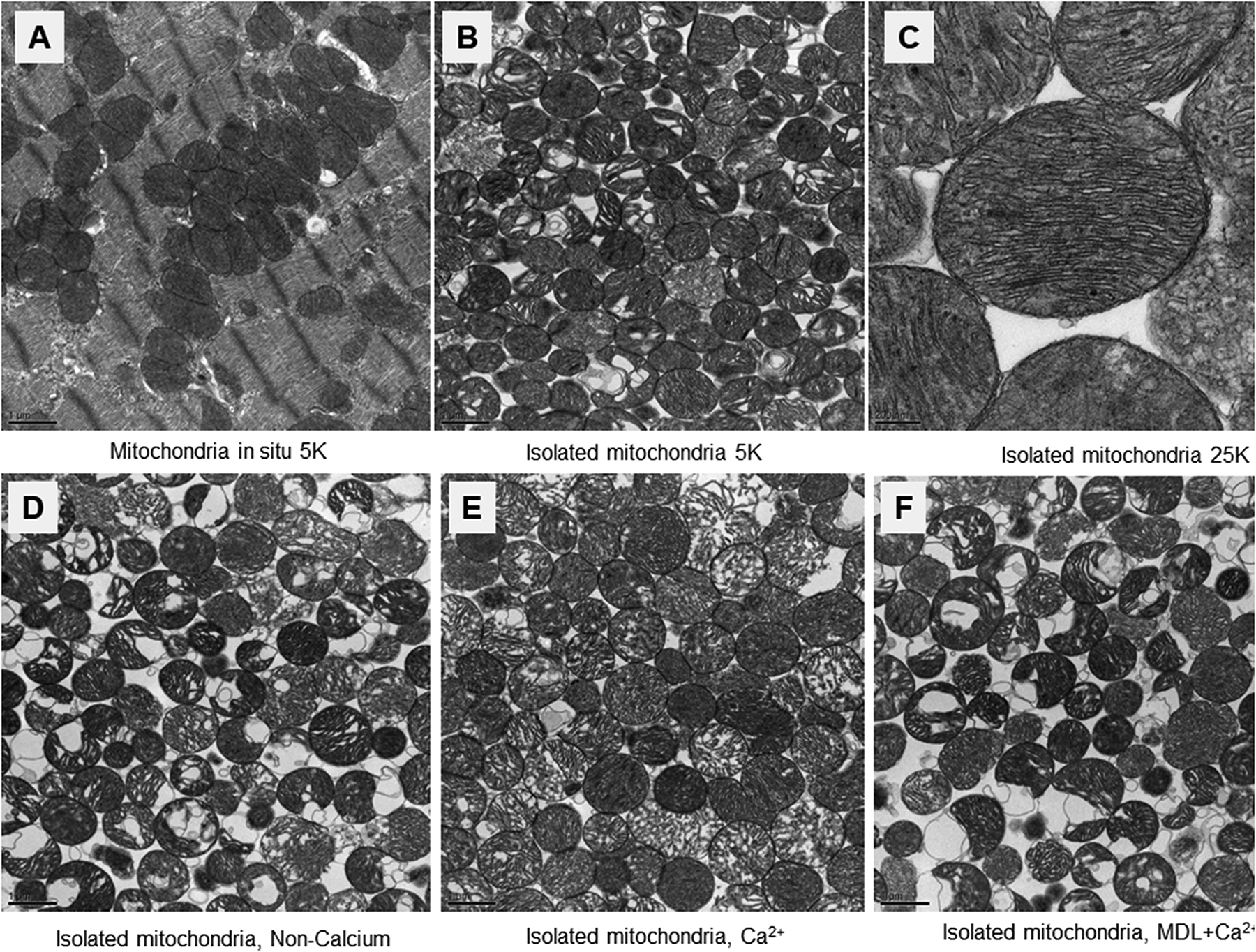
Representative electron micrographs of the mitochondria with or without calcium treatment.
Panel A: Representative cardiac mitochondrial ultra-structure in situ (Magnification x5,000).
Panel B&C: Representative ultrastructure in isolated and mixed population of cardiac mitochondria with magnification x5,000 in Panel B and x25,000 in Panel C.
Panel D: Mitochondrial morphology was well preserved after incubation with non-calcium buffer (magnification x5,000).
Panel E: Incubation of the isolated mitochondria with calcium preserved mitochondrial integrity but did result in mild to moderate mitochondrial swelling in approximately half of the organelles (magnification x5,000).
Panel F: Inhibition of calpain using MDL-28170 also led to preservation of overall mitochondrial morphology during calcium treatment (magnification x5,000).
3.2. Calcium rather than H2O2 decreased oxidative phosphorylation in cardiac mitochondria
ER stress leads to both calcium overload and ROS generation in vivo. Isolated mitochondria were used to evaluate the role of exogenous calcium and oxidative stress in mitochondrial dysfunction. Representative tracings of oxidative phosphorylation are shown in Figure 2A. Compared to vehicle, calcium treatment substantially decreased state 3 respiration using pyruvate + malate or succinate + rotenone as complex I and complex II substrates, respectively (Table 2). In contrast, H2O2 (100 μM) alone did not decrease state 3 respiration compared to vehicle treatment with either substrate. The combination of calcium and H2O2 decreased state 3 respiration with both complex I and complex II substrates (Table 2), similar to calcium treatment alone. Compared to control, calcium or calcium + H2O2 treatment also led to a decreased state 4 rate (Table 2). The respiratory control ratio (RCR, state 3/state 4) was slightly decreased in calcium or calcium+H2O2 group compared to control using complex I substrates (Table 2).
Figure. 2:
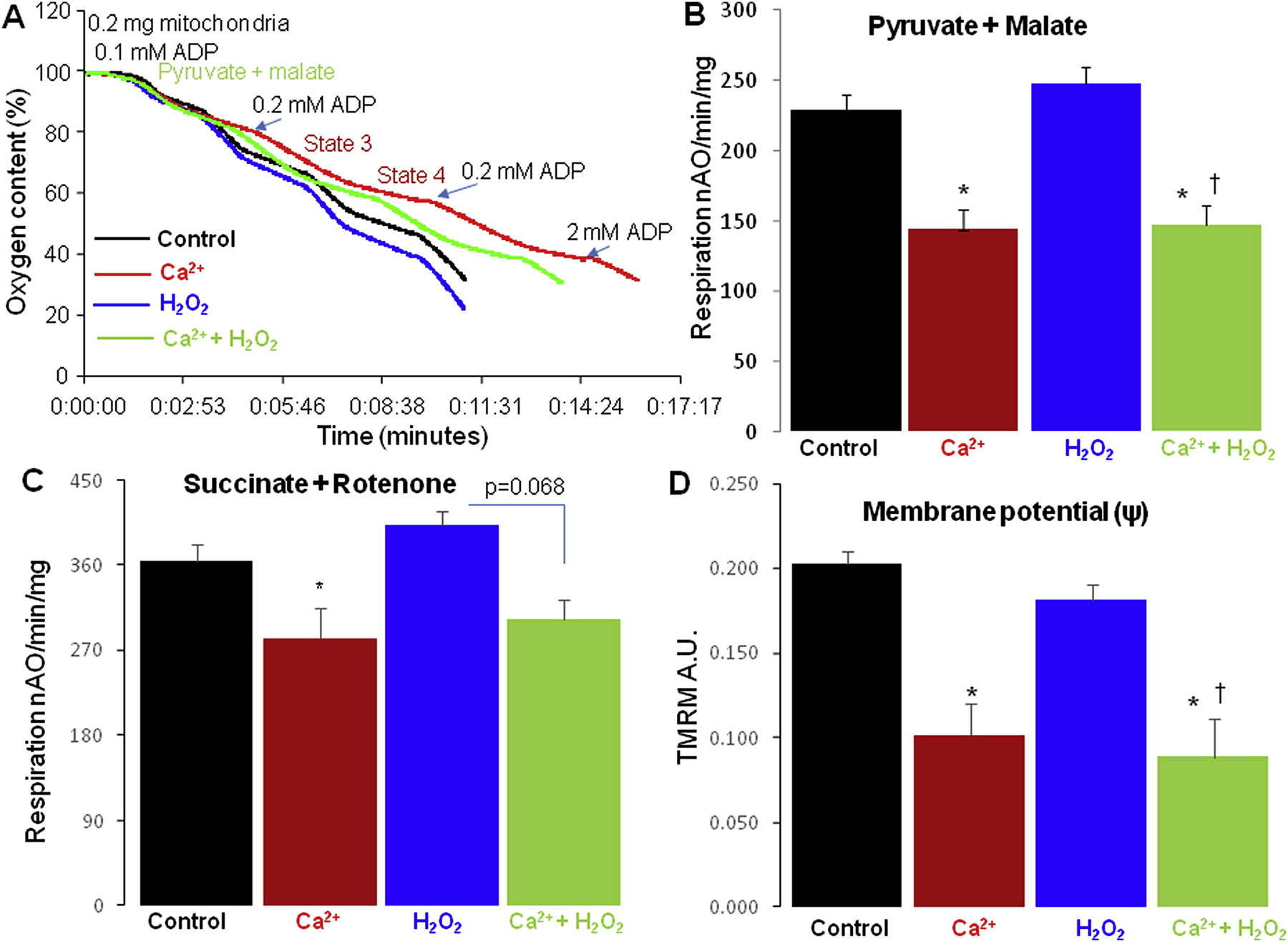
Oxidative phosphorylation in the isolated heart mitochondria.
Panel A: A representative original tracing of the oxygen consumption in isolated cardiac mitochondria is shown. Mitochondria were first added into the oxygen chamber and 0.1 mM ADP added to consume residual endogenous substrates. Then pyruvate + malate was added into the chamber followed with 0.2 mM ADP to stimulate state 3 respiration. After consumption of ADP, respiration slowed, demonstrating preserved respiratory control, leading to ADP-limited state 4 respiration. Then, 0.2 mM ADP was repeated to recheck the state 3 respiration. A saturating concentration of ADP (2mM) was finally used to stimulate maximal respiration.
Panel B: Compared to vehicle, calcium and calcium+H2O2, rather than H2O2 alone, led to the decreased maximal respiration when pyruvate + malate were used as complex I substrates.
Panel C: Compared to vehicle, calcium slightly decreased oxidative phosphorylation using succinate (plus rotenone) as a complex II substrate. H2O2 alone did not alter succinate oxidation. Calcium+H2O2 tended to decrease the maximal respiration when succinate (plus rotenone) was used as the complex II substrate.
Panel D: Compared to vehicle, calcium and calcium+H2O2, rather than H2O2 alone, led to a partially depolarized inner membrane potential (ΔΨ) when pyruvate+malate were used as complex I substrate.
Mean ± SEM. *p<0.05 vs. vehicle; † p<0.05 vs. vehicle; H2O2 treatment. Vehicle (n=9), Calcium (n=7), H2O2 (n=7), Calcium + H2O2 (n=3).
Table 2:
Oxidative phosphorylation in mitochondria from C57BL/6 mouse hearts with or without calcium and H2O2 treatment
| Vehicle N=9 |
Calcium N=6 |
H2O2 N=7 |
Calcium + H2O2 N=3 |
|
|---|---|---|---|---|
| Complex I substrate- Pyruvate + malate | ||||
| State 3 | 212±5 | 139±13 * | 216±7 | 144±10 * † |
| State 4 | 70±3 | 61±5 † | 81±3 | 62±5 † |
| RCR | 3.1±0.2 | 2.3±0.2* † | 2.7±0.1 | 2.4±0.1* † |
| Complex II substrate- Succinate + Rotenone | ||||
| State 3 | 435±17 | 308±30 * † | 451±13 | 318±24 * † |
| State 4 | 171±14 | 135±11 | 158±6 | 131±11 |
| RCR | 2.7±0.2 | 2.3±0.2 | 2.9±0.1 | 2.4±0.1 |
Mean ± SEM;
p<0.05 vs. vehicle;
p<0.05 vs. H2O2 alone;
analysis by one-way ANOVA.
A saturating concentration of ADP (2 mM) was used to measure maximal ADP-stimulated respiration. Incubation of mitochondria with 100 μM H2O2 alone did not alter the maximal rate of ADP-stimulated respiration with either substrate. In contrast, calcium exposure (25 μM) significantly decreased the maximal rate of oxidative phosphorylation in mitochondria oxidizing either complex I or complex II substrates. Likewise, the combination of H2O2 and calcium treatment led to decreased rates (Figure 2 B&C), again similar to calcium alone.
The complex I activity was decreased in calcium-treated mitochondria compared to control [Mean ± SEM, 550 ± 46 mU/mg mitochondrial protein (control, n=9) vs. 453 ± 20 (calcium treatment, n=7), p<0.05]. Calcium treatment did not alter complex II activity [Mean ± SEM, 309 ± 30 mU/mg mitochondrial protein (control, n=9) vs. 283 ± 54 (calcium treatment, n=7), p=NS]. Thus, modest exogenous calcium stress, but not oxidative stress, led to decreased respiration and complex I activity in cardiac mitochondria.
Compared to vehicle, calcium treatment led to a partially depolarized inner mitochondrial membrane potential (Figure 2D). However, state 4 respiration was not increased in calcium-treated mitochondria (Table 2), supporting that this low concentration of calcium did not lead to uncoupling of respiration or opening of the permeability transition pore. Compared to vehicle, H2O2 with calcium, rather than H2O2 alone also led to a partially depolarized inner mitochondrial membrane potential (Figure 2D).
3.3. THAP treatment decreased oxidative phosphorylation in H9c2 cells
Administration of THAP in vivo increases ER stress not only in the heart but in other organs as well. Thus, we applied THAP treatment in cells to evaluate the response of oxidative phosphorylation in cardiomyoblast H9c2 cells as a direct effect of ER stress. THAP treatment of H9c2 cells led to decreased oxidative phosphorylation in cells oxidizing complex I substrates. In contrast, the rate of oxidative phosphorylation in H9c2 cells oxidizing complex II substrates was not altered (Figure 3A). These findings support that THAP-induced ER stress leads to a complex I defect in H9c2 cells. This result is in line with the findings in mitochondria isolated from mouse hearts in vivo following the acute induction of ER stress following THAP administration [10]. THAP treatment decreased the rate of OXPHOS using complex I substrates, whereas treatment did not alter the rate of respiration using succinate (plus rotenone) as a complex II substrate.
Figure. 3:
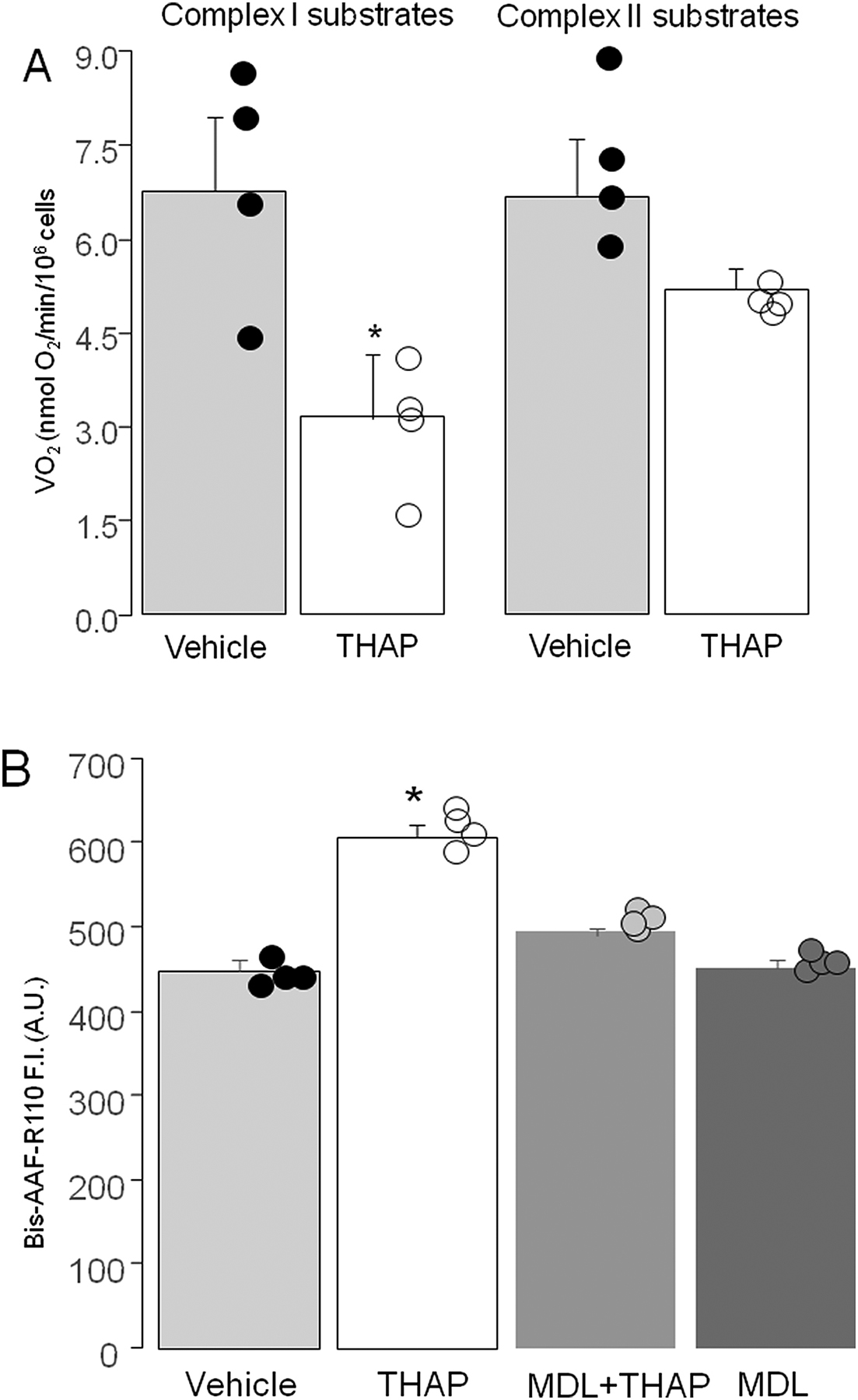
ER stress led to decreased oxidative phosphorylation and increased cell death in H9c2 cells.
Panel A: THAP treatment led to decreased oxidative phosphorylation in H9c2 cells oxidizing complex I substrates. THAP did not markedly alter oxidative phosphorylation in H9c2 cells oxidizing complex II substrates.
Panel B: THAP-induced ER stress increased cell death in H9c2 cells. Prevention of calpain activation using MDL-28170 (10 uM) led to decreased cell death in H9c2 cells, indicating that ER stress-mediated calpain activation contributes to cell death. Mean ± SEM. *p<0.05 vs. vehicle. N=4 in each group.
3.4. THAP-induced ER stress increased cell death in H9c2 cells via calpain activation
ER stress slightly increased programmed cell death in THAP-treated mouse hearts [10]. THAP treatment increased cell death in H9c2 cells (Figure 3B) compared to vehicle, consistent with the in vivo study. In addition, administration of MDL-28170 [27] decreased the THAP-induced cell death in H9c2 cells (Figure 3B), indicating that activation of calpains contributes to the cell death that occurs during ER stress.
3.5. THAP-induced ER stress activated cytosolic calpains
ER stress is increased in THAP-treated mouse hearts [10]. ER stress leads to intracellular calcium overload that favors calpain activation. Spectrin is a substrate of ubiquitous calpains including calpain 1 and calpain 2. THAP treatment decreased the content of full length spectrin in cytosol (Figure 4A), indicating that the acute induction of ER stress activated cytosolic calpain1/2 in THAP-treated hearts.
Figure. 4:
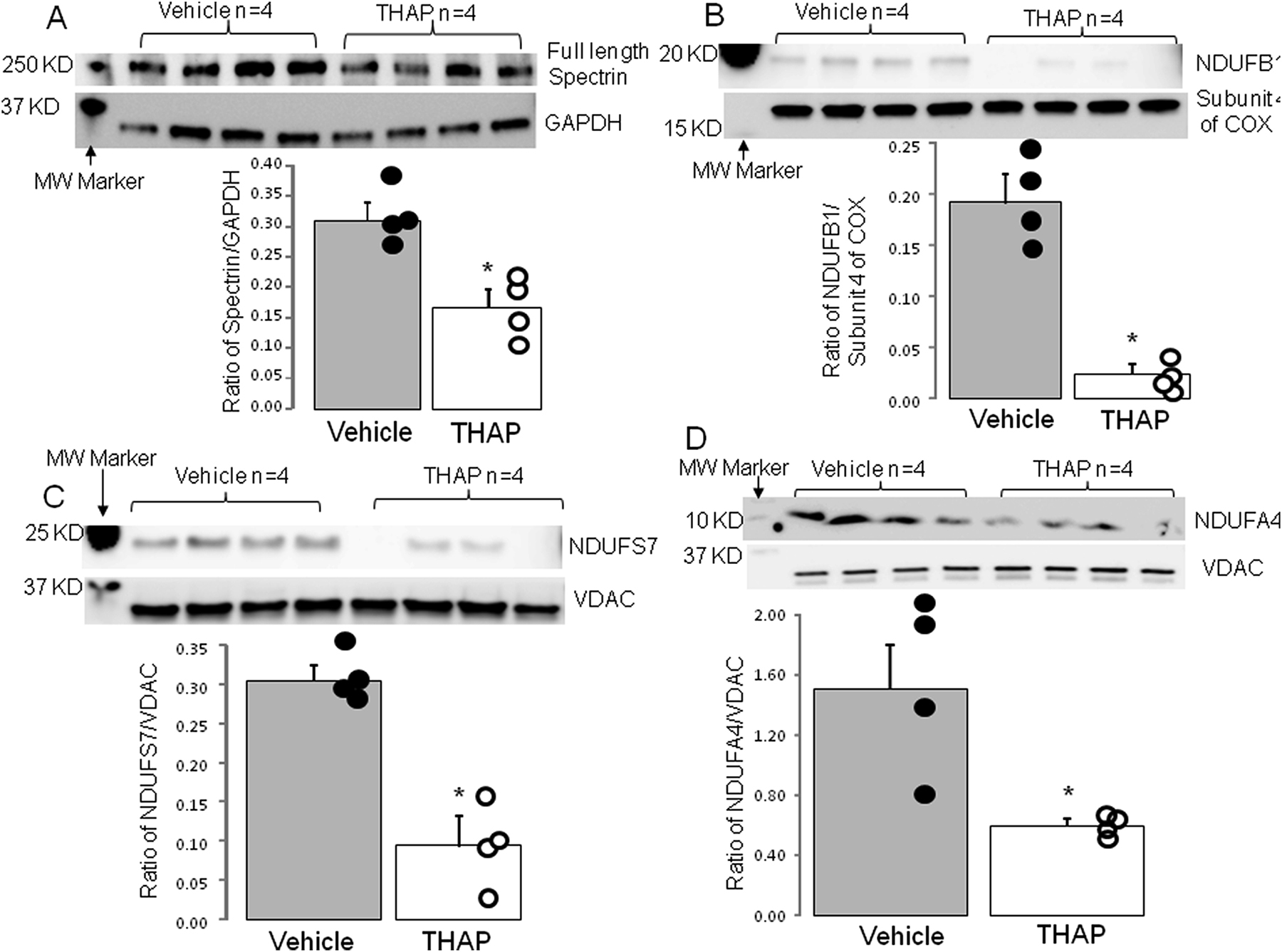
THAP-induced ER stress led to degradation of complex I subunits
Panel A: Spectrin is a substrate of ubiquitous calpains. Compared to vehicle, THAP treatment led to decreased content of full length spectrin, indicating that THAP-induced ER stress activates ubiquitous calpains. Compared to vehicle, THAP treatment led to decreased content of complex I subunit NDUFB1 (Panel B), NDUFS7 (Panel C), and NDUFA4 (Panel D). Mean ± SEM. *p<0.05 vs. vehicle. N=4 in each group. MW (molecular weight).
3.6. THAP-induced ER stress leads to degradation of complex I subunits
In vivo THAP treatment decreased complex I activity in cardiac mitochondria [11]. THAP treatment decreased the contents of complex I subunits NDUFB1 (Figure 4B), NDUFS7 (Figure 4C), and NDUFA4 (Figure 4D). These findings support that the THAP-induced complex I defect occurs due to the degradation of its protein subunits.
3.6. Elimination of mCPN1 activity improved oxidative phosphorylation in calcium-treated mitochondria
Calcium treatment decreased the ADP-stimulated respiratory rate in wild type but not in CAPNS1 deletion mitochondria oxidizing complex I substrate (Figure 5A). With succinate as substrate, calcium treatment tended to decrease maximal respiratory rate in wild type compared to vehicle treatment as shown above (p=0.062). Succinate oxidation was not altered in calcium-treated mitochondria from CAPNS1 deletion mice (Figure 5B). Thus, the genetic inactivation of mitochondria-localized calpains I and II preserves respiration with complex I substrates. Incubation of calcium with mitochondria in wild type mice led to a decreased content of the PDHα1 subunit, previously identified as a potential target of mCPN [57]. However, the content of PDHα1 subunit was preserved in the calcium-treated mitochondria from CAPNS1 deletion mice (Figure 5C&D). The calcium-mediated activation of the mCPN1 contributes to PDHα1 subunit degradation. These results in a model of genetic inactivation of mitochondrial calpain I and II reinforce previous results that identified the PDHα1 subunit as a target of mitochondrial calpains during ischemia and reperfusion [57].
Figure. 5:
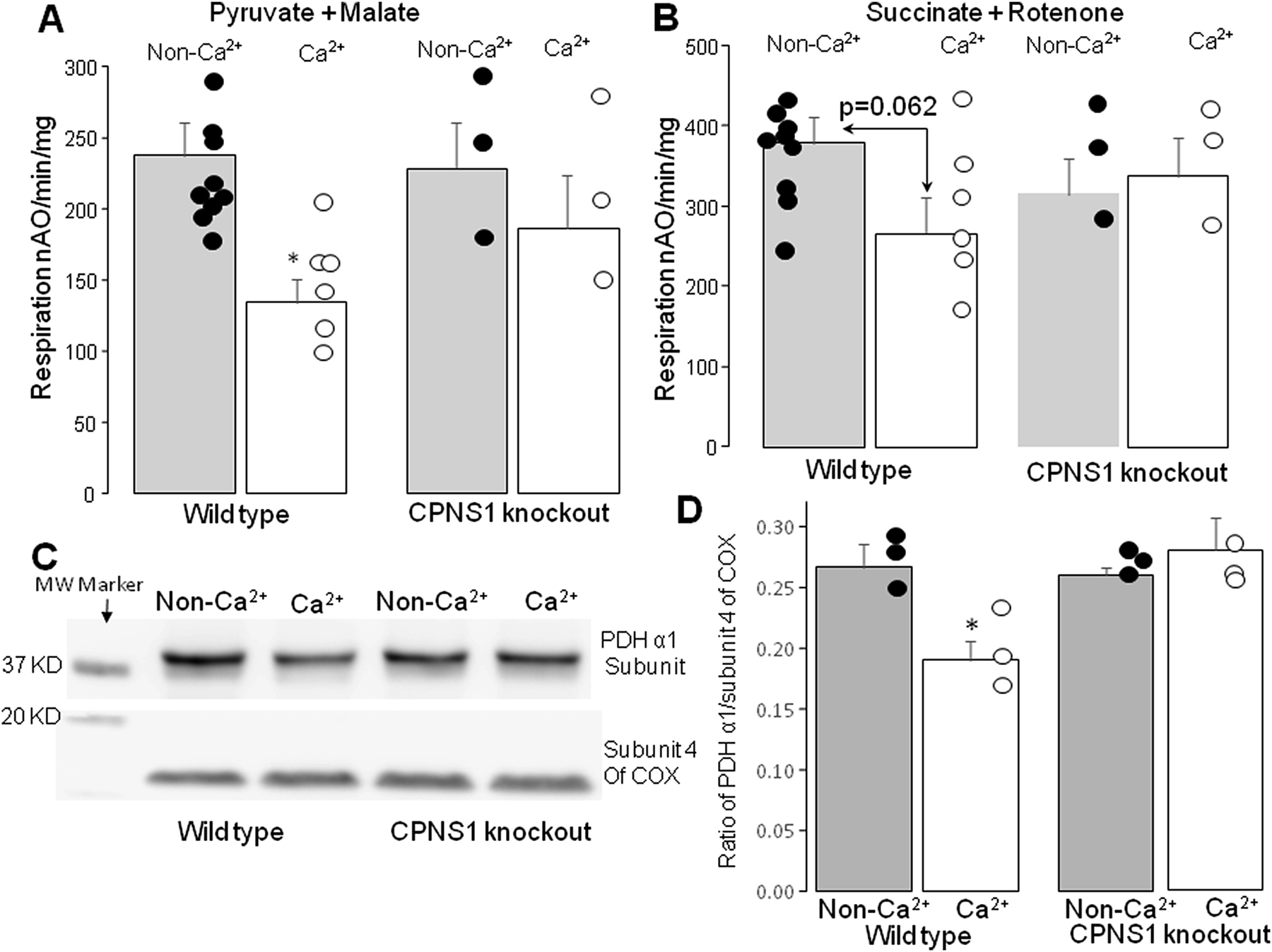
Calcium treatment led to decreased oxidative phosphorylation in wild type but not in CPNS1 knockout mice.
Panel A: Incubation of the isolated mitochondria from wild type with calcium led to decreased oxidative phosphorylation stimulated by 2 mM ADP when pyruvate + malate were used as complex I substrates. Calcium treatment did not alter oxidative phosphorylation in mitochondria from CPNS1 knockout mice.
Panel B: Calcium treatment tended to decrease oxidative phosphorylation in wild type but not in CPNS1 knockout mice when succinate was used as complex II substrate.
Panel C&D: Calcium treatment led to decreased content of PDH α1 subunit in wild type but not in CPNS1 knockout mice. Mean ± SEM. *p<0.05 vs. vehicle. N=3–9 in each group. MW (molecular weight).
3.7. Elimination of mCPN1 activity protected respiratory complexes and metabolic enzymes
A proteomic approach was used to identify other potential calpain targets in cardiac mitochondria. The primary parameter used to identify potential protein targets in mitochondria was a minimum 10% decrease in protein content measured in the calcium-treated mitochondria versus non-calcium-treated mitochondria. The percent change was used to reflect the alteration of subunit protein content.
Calcium treatment decreased the contents of NDUFS7 (complex I subunit) in mitochondria isolated from wild type mice. Calcium also led to the degradation of the subunits of ATP synthase including ATP5I and ATP5J. Enzymes involved in fatty acid metabolism including very long-chain specific acyl-CoA dehydrogenase (ACADVL), carnitine O-palmitoyltransferase 1 (CPT1B), and carnitine O-palmitoyltransferase 2 (CPT2) were decreased in calcium-treated control mitochondria. The contents of aspartate aminotransferase (GOT2), calcium-binding mitochondrial carrier protein aralar1 (SLC25A12) and branched-chain-amino-acid aminotransferase (BCAT2) were also decreased in calcium-treated control but not CAPNS1 deletion mitochondria (Table 3). Calcium treatment did not alter the proteins mentioned above in CAPNS1 deletion mice.
Table 3:
Calcium incubation-mediated mitochondrial protein degradation in wild type mice
| Protein Name | Calcium/non-calcium treated mitochondria | |
|---|---|---|
| % ↓ | P | |
| NADH dehydrogenase iron-sulfur protein 7 (NDUFS7) | 51% ↓ | 0.043 |
| ATP synthase subunit g (ATP5I) | 58% ↓ | 0.07 |
| ATP synthase-coupling factor 6 (ATP5J) | 83% ↓ | 0.0059 |
| Very long-chain specific acyl-CoA dehydrogenase (ACADVL) | 61% ↓ | 0.0169 |
| Carnitine O-palmitoyltransferase 2 (CPT2) | 62% ↓ | 0.005 |
| Carnitine O-palmitoyltransferase 1 (CPT1B) | 64% ↓ | 0.0012 |
| Isovaleryl-CoA dehydrogenase (IVD) | 46% ↓ | 0.0334 |
| Aspartate aminotransferase (GOT2) | 49% ↓ | 0.0525 |
| Calcium-binding mitochondrial carrier protein Aralar1 (SLC25A12) | 65% ↓ | 0.0156 |
| Branched-chain-amino-acid aminotransferase (BCAT2) | 84% ↓ | 0.0351 |
DISCUSSION
Our previous in vivo study found that ER stress leads to decreased complex I activity in cardiac mitochondria [11]. In the present study, we find that the ER stress-induced complex I defect is due to degradation of specific protein subunits of complex I. The in vitro study shows that a modest, exogenous H2O2-mediated oxidative stress alone does not impair function in isolated cardiac mitochondria. In contrast, exposure to a low concentration of exogenous calcium, consistent with enhanced calcium exposure of mitochondria from increased ER stress, leads to decreased oxidative phosphorylation and complex I activity. An accompanying proteomic study of calcium treated mitochondria showed that exogenous calcium treatment leads to decreased NDUFS7 content in control mitochondria. Genetic elimination of mCPN1 and 2 activities preserves oxidative phosphorylation and preserves NDUFS7 content in calcium-treated mitochondria. The current study indicates that ER stress leads to a calcium mediated complex I defect by degrading complex I protein subunits in part via activation of mitochondrial calpains.
Although the initial ER stress response is to restore ER function by slowing protein synthesis and removing unfolded proteins, severe ER stress increases cell injury and favors cell death [8, 58]. The ER contributes a key role in the regulation of intracellular calcium homeostasis [59]. ER stress leads to cytosolic and mitochondrial calcium overload that favors calpain activation. Prevention of cytosolic and mitochondrial calpain activation decreases cardiac injury during ischemia-reperfusion [27, 41, 57]. The current study shows that inhibition of calpains with MDL-28170 decreases cell death in THAP-treated H9c2 cells, supporting that activation of calpains contributes to cell death during the ER stress.
Mitochondrial dysfunction contributes a critical role in the ER stress-mediated cell injury. ER stress leads to intracellular calcium overload that can impair mitochondrial function by inducing activation of calcium-dependent proteases. Calcium can be released from the ER through the ryanodine 2 receptor or inositol 1,4,5-trisphosphate receptors (IP3R). Genetic inhibition of ryanodine 2 receptor decreases mitochondrial calcium overload and protects mitochondrial function, suggesting that the ryanodine 2 receptor is a main pathway to induce calcium overload during the ER stress [16]. The mitochondrial calcium uniporter contributes an important role in transferring calcium into the mitochondrial matrix [60, 61]. Targeting the mitochondrial calcium uniporter can also prevent calcium-induced mitochondrial damage [60–62]. In addition to calcium overload, ER stress also increases the oxidative stress that further increases mitochondrial damage [8]. The current study shows that a low concentration of exogenous calcium leads to decreased oxidative phosphorylation in mitochondria using complex I but not complex II substrates, suggesting that calcium incubation leads to decreased complex I activity. Direct enzyme activity measurement supports that calcium incubation results in a complex I defect in cardiac mitochondria. Complex I damage favors opening of the MPTP [29] that contributes to cell injury. Calcium exposure leads to a depolarized inner mitochondrial membrane potential, supporting that exogenous calcium treatment sensitizes the organelles to possible MPTP opening. Thus, exposure of mitochondria to calcium stress leads to complex I damage that may be followed by MPTP opening.
In contrast to calcium treatment, modest oxidative stress does not decrease state 3 respiration but slightly increases state 4 respiration. These results indicate that modest oxidative stress does not directly impair the ETC but may increase the proton leak through the inner mitochondrial membrane (IMM). The proton leak across the IMM is mainly regulated by mitochondrial uncoupling proteins (UCPs) [63, 64]. Isoforms of UCPs including UCP1, UCP2, and UCP3 are present in heart [65]. A decreased UCP3 content decreases state 4 respiration in aged heart mitochondria, whereas overexpression of UCP3 increases state 4 respiration [63, 64]. The UCPs are activated during oxidative stress. H2O2 treatment leads to increased state 4 respiration, supporting that notion that oxidative stress activates UCPs in the heart [66, 67].
Although modest oxidative stress alone does not damage the ETC in isolated mitochondria, it may cause mitochondrial dysfunction in vivo [18]. Oxidative stress increases calcium overload in cardiac myocytes [5, 18]. ROS generation can lead to decreased Ca2+-ATPase (SERCA) activity through prevention of ATP binding to SERCA to decrease activity [16, 68, 69] (Graphic abstract). Thus, ROS generation can increase mitochondrial calcium overload by inducing ER stress through inhibition of the Ca2+-ATPase that in turn feeds back upon the mitochondria [5, 16, 18]. In ongoing work, we found that induction of ROS generation from the ETC increases ER stress in mouse hearts [13]. Thus, modest ROS generation can trigger mitochondrial damage by inducing ER stress which then feeds back upon the mitochondria via ER-driven mitochondrial calcium overload, resulting in a vicious cycle of injury via pathologic mitochondrial-ER interactions.
THAP treatment in vivo leads to decreased oxidative phosphorylation and complex I activity in cardiac mitochondria [10, 11]. Administration of THAP in vivo increases the ER stress not only in heart, but also in other organs and vascular endothelial cells. Although we believe that THAP-induced ER stress directly contributes to mitochondrial dysfunction, cardiac mitochondria might be indirectly affected by THAP-induced dysfunction in other cell components. Thus, in addition to the in vivo study, the direct effect of THAP on in situ mitochondrial function was studied in a cardiomyoblast cell model. THAP treatment led to decreased oxidative phosphorylation in H9c2 cells, supporting that ER stress leads to a complex I defect.
Complex I is an L-shaped molecular complex including an inner mitochondrial membrane embedded arm and a matrix-oriented peripheral arm [70]. The matrix arm participates in NADH oxidation and subsequent electron transfer through the complex [70], whereas the membrane arm is responsible for vectorial H+ pumping across the inner membrane. Ischemia-reperfusion leads to decreased complex I activity by cleaving subunits of NDUFS7 [41], NDUFV1, and NDUFS3 [42]. In the current study, ER stress leads to a decreased content of the NDUFS7, NDUFB1, and NDUFA4 subunits. These results support that ER stress-mediated complex I damage is due to subunit degradation, some of the cleavages which are due to mitochondrial matrix localized calpain. In addition to CPN1/2, activation of calpain 10 has been shown to decrease complex I activity in kidney mitochondria by degrading NDUFV2 and ND6 subunits [71]. Lon is another protease located within the mitochondrial matrix. Activation of Lon protease decreases oxidative phosphorylation by impairing mitochondrial biogenesis through alteration of TFAM (mitochondrial transcription factor A) [72]. ER stress likely causes mitochondrial dysfunction at least in part by activating calcium dependent matrix enzymes. The current study shows that a modest exogenous calcium stress can impair mitochondrial function in the absence of uncoupling of respiration or MPTP opening. The extent of calcium required to activate mitochondria-localized calpains is less than that required to elicit MPTP opening [26].
Ischemia-reperfusion leads to degradation of the complex I subunit NDUFS7 in cardiac mitochondria [41]. Inhibition of CPN1 using MDL-28170 preserves NDUFS7 content in mitochondria following ischemia-reperfusion [41], indicating that NDUFS7 is a likely calpain target. The proteomic results confirm that calcium treatment leads to a decreased NDUFS7 content in wild type but not in CAPNS1 deletion mice, supporting that activation of mCPN contributes to the degradation of NDUFS7. ER stress also leads to degradation of NDUFB1 and NDUFA4 in cardiac mitochondria. The role of mitochondrial calpain activation in cleaving NDUFB1 and NDUFA4 needs to be studied further in the future. Ischemia-reperfusion leads to degradation of the PDHα1 subunit in cardiac mitochondria [57]. Pharmacologic inhibition of CPN1 using MDL-28170 protects PDHα1 in mitochondria following ischemia-reperfusion [57], indicating that PDH α1 is potential calpain target. ER stress also leads to decreased PDH α1 subunit content in cardiac mitochondria [11]. Incubation of cardiac mitochondria with exogenous calcium leads to decreased PDHα1 content in wild type but not in CPNS1 knockout mice, supporting that activation of mCPN contributes to PDH subunit degradation during ER stress. Thus, the decrease in oxidative phosphorylation with pyruvate + malate as substrate is likely due to decreases in PDH activity and complex I activity due to mCPN activation. PDH and complex I are located in series in the metabolism of glycolytic substrates that generate NADH for oxidation by the electron transport chain [73].
In addition to electron transport chain damage, activation of the mCPN leads to mitochondrial dysfunction by depleting aspartate aminotransferase, calcium-binding mitochondrial carrier, and branched-chain-amino-acid aminotransferase. Aspartate aminotransferase (AST), which is also known as glutamic oxaloacetic transaminase (GOT), is a key enzyme to catalyze the exchange of aspartate and α-ketoglutarate to oxaloacetate and glutamate [74]. Aralar1 is an isoform of calcium-binding mitochondrial carrier proteins encoded by the SLC25A12 gene. Aralar 1 is mainly expressed in heart and skeletal muscle [75]. The aralar1 shuttle is involved in the transport of cytosolic NADH into mitochondria [76]. An impairment of NADH transport into mitochondria adds another calpain-induced defect in NADH oxidation upstream of complex I. BCAT2 (branched-chain-amino-acid aminotransferase) catalyzes the metabolism of the branched chain amino acids including leucine, isoleucine, and valine. The branched chain amino acids contribute a critical role in regulating protein turnover and energy production and expenditure [77]. Thus, activation of mitochondrial calpains has a broader effect on the activities of mitochondrial metabolic enzymes and amino acid transferases.
There are several limitations to the current study. A lower exogenous calcium (25 μM) concentration was used to mainly activate mitochondrial calpain 1, but mitochondrial calpain 2 activation cannot be excluded in that knockout of CAPNS1 eliminates both CPN1 and CPN2 activities. Although a cardiac specific cre was used to generate cardiac specific CAPNS1 knockout mice, there was difficulty breeding a sufficient quantity of knockout mice which severely limited the experiments that could be performed. Thus, only limited studies could be performed using CAPNS1 knockout mice. In order to improve the mouse breeding, cardiac specific and tamoxifen-inducible cre mice are being utilized in our laboratory. Tamoxifeninducible CAPNS1 knockout decreased cardiac injury in hearts following ischemia-reperfusion [78]. These mice will be a critical tool to further evaluate the contribution of the activation of mCPN to mitochondrial damage, especially in complex I, during the ER stress. The proteomic screen for calcium dependent targets in isolated mitochondria represents an initial screen instead of a rigorously complete list, as shown by the failure to detect the decrease in PDH α1 content. Although the reason for this finding is not clear, limitations in the detection of peptides by proteomic techniques has been previously discussed [79]. Although MDL-28170 is a classic calpain inhibitor, it does have off-target effects [26]. Thus, a genetic approach is critical to evaluate the relationship between calpain activation and mitochondrial damage during the ER stress in future studies.
In summary, ER stress impairs mitochondrial function at least in part by activation of mitochondrial calpains. Attenuation of the ER stress or prevention of calpain activation is a potential approach to protect complex I from damage in the presence of cardiac disease [42].
Fig. 6.
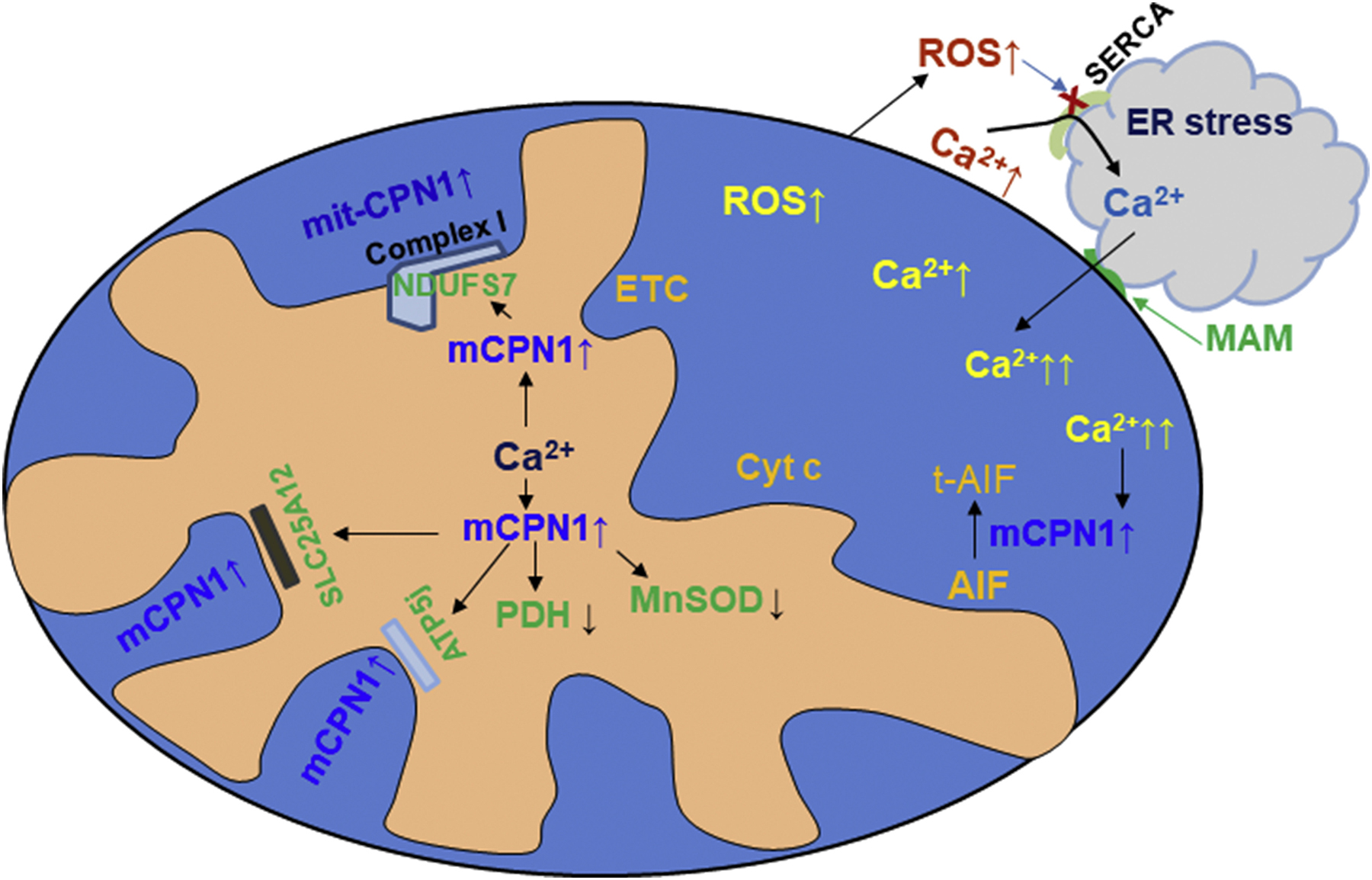
ER stress-induced mitochondrial damage.
HIGHLIGHTS:
ER stress leads to CPN1/2 activation in mitochondria and cytosol
Calcium stress contributes a key role in depression of mitochondrial function
ER stress contributes to complex I damage by activating mitochondrial CPN1/2
Inhibition of CPN1/2 is a potential approach to protect mitochondria in chronic cardiac disease
ACKNOWLEDGMENTS
This work was supported by National Institute on Aging (NIA) R21AG054975-01 (QC), the Office of Research and Development, Medical Research Service Merit Review Award (2IO1BX001355-01A2) (QC, EJL), and the Pauley Heart Center, Virginia Commonwealth University (QC, JT, YH, EJL). This work was also supported by National Institutes of Health grant R01HL128485 (JMH) and the Community Foundation for the Ohio Valley Whipkey Trust (JMH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- [1].Lesnefsky EJ, Chen Q, Hoppel CL, Mitochondrial Metabolism in Aging Heart, Circ Res, 118 (2016) 1593–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL, Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies, Annu Rev Pharmacol Toxicol, 57 (2017) 535–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Feissner RF, Skalska J, Gaum WE, Sheu SS, Crosstalk signaling between mitochondrial Ca2+ and ROS, Front Biosci (Landmark Ed), 14 (2009) 1197–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gustafsson AB, Gottlieb RA, Bcl-2 family members and apoptosis, taken to heart, Am J Physiol Cell Physiol, 292 (2007) C45–51. [DOI] [PubMed] [Google Scholar]
- [5].Wagner S, Rokita AG, Anderson ME, Maier LS, Redox regulation of sodium and calcium handling, Antioxid Redox Signal, 18 (2013) 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Adam-Vizi V, Starkov AA, Calcium and mitochondrial reactive oxygen species generation: how to read the facts, J Alzheimers Dis, 20 Suppl 2 (2010) S413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen Q, Xu H, Xu A, Ross T, Bowler E, Hu Y, Lesnefsky EJ, Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria, PLoS One, 10 (2015) e0118834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J, Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway, Circ Res, 113 (2013) 1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P, Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells, Nat Protoc, 4 (2009) 1582–1590. [DOI] [PubMed] [Google Scholar]
- [10].Chen Q, Thompson J, Hu Y, Das A, Lesnefsky EJ, Metformin attenuates ER stress-induced mitochondrial dysfunction, Transl Res, 190 (2017) 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen Q, Thompson J, Hu Y, Das A, Lesnefsky EJ, Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage, Front Cardiovasc Med, 6 (2019) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang Y, Ren J, Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: Role of Akt dephosphorylation, Free Radic Biol Med, 51 (2011) 2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [13].Chen Q, Allegood JC, Thompson J, Toldo S, Lesnefsky EJ, Increased Mitochondrial ROS Generation from Complex III Causes Mitochondrial Damage and Increases Endoplasmic Reticulum Stress, FASEB J, 33 (2019) Abstract (543.513). [Google Scholar]
- [14].Lima NCR, Melo TQ, Sakugawa AYS, Melo KP, Ferrari MFR, Restoration of Rab1 Levels Prevents Endoplasmic Reticulum Stress in Hippocampal Cells during Protein Aggregation Triggered by Rotenone, Neuroscience, 419 (2019) 5–13. [DOI] [PubMed] [Google Scholar]
- [15].Chami M, Oules B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Brechot P, Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress, Mol Cell, 32 (2008) 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Santulli G, Xie W, Reiken SR, Marks AR, Mitochondrial calcium overload is a key determinant in heart failure, Proc Natl Acad Sci U S A, 112 (2015) 11389–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang M, Sun GB, Zhang JY, Luo Y, Yu YL, Xu XD, Meng XB, Zhang MD, Lin WB, Sun XB, Elatoside C protects the heart from ischaemia/reperfusion injury through the modulation of oxidative stress and intracellular Ca(2)(+) homeostasis, Int J Cardiol, 185 (2015) 167–176. [DOI] [PubMed] [Google Scholar]
- [18].Ge C, Huang H, Huang F, Yang T, Zhang T, Wu H, Zhou H, Chen Q, Shi Y, Sun Y, Liu L, Wang X, Pearson RB, Cao Y, Kang J, Fu C, Neurokinin-1 receptor is an effective target for treating leukemia by inducing oxidative stress through mitochondrial calcium overload, Proc Natl Acad Sci U S A, 116 (2019) 19635–19645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Agip AA, Blaza JN, Bridges HR, Viscomi C, Rawson S, Muench SP, Hirst J, Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states, Nat Struct Mol Biol, 25 (2018) 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hou T, Zhang R, Jian C, Ding W, Wang Y, Ling S, Ma Q, Hu X, Cheng H, Wang X, NDUFAB1 confers cardio-protection by enhancing mitochondrial bioenergetics through coordination of respiratory complex and supercomplex assembly, Cell Res, 29 (2019) 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gomez LA, Monette JS, Chavez JD, Maier CS, Hagen TM, Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart, Arch Biochem Biophys, 490 (2009) 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ, Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion, Am J Physiol Cell Physiol, 292 (2007) C137–147. [DOI] [PubMed] [Google Scholar]
- [23].Chen Q, Lesnefsky EJ, Ischemic damage to the mitochondrial electron transport chain favors opening of the permeability transition pore., FASEB J, 22 (2008) E345 (abstract 750.346). [Google Scholar]
- [24].Nadtochiy SM, Burwell LS, Brookes PS, Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemiareperfusion injury, J Mol Cell Cardiol, 42 (2007) 812–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rosca M, Minkler P, Hoppel CL, Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV, Biochim Biophys Acta, 1807 (2011) 1373–1382. [DOI] [PubMed] [Google Scholar]
- [26].Chen Q, Lesnefsky EJ, Heart mitochondria and calpain 1: Location, function, and targets, Biochim Biophys Acta, 1852 (2015) 2372–2378. [DOI] [PubMed] [Google Scholar]
- [27].Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ, Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion, Biochem Biophys Res Commun, 415 (2011) 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ozaki T, Tomita H, Tamai M, Ishiguro S, Characteristics of mitochondrial calpains, J Biochem, 142 (2007) 365–376. [DOI] [PubMed] [Google Scholar]
- [29].Shintani-Ishida K, Yoshida K, Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion, Int J Cardiol, 197 (2015) 26–32. [DOI] [PubMed] [Google Scholar]
- [30].Yamada KH, Kozlowski DA, Seidl SE, Lance S, Wieschhaus AJ, Sundivakkam P, Tiruppathi C, Chishti I, Herman IM, Kuchay SM, Chishti AH, Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress, J Biol Chem, 287 (2012) 13182–13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Goll DE, Thompson VF, Li H, Wei W, Cong J, The calpain system, Physiol Rev, 83 (2003) 731–801. [DOI] [PubMed] [Google Scholar]
- [32].Vosler PS, Brennan CS, Chen J, Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration, Mol Neurobiol, 38 (2008) 78–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen J, Henderson GI, Freeman GL, Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart, J Mol Cell Cardiol, 33 (2001) 1919–1927. [DOI] [PubMed] [Google Scholar]
- [34].Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA, Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion, J Biol Chem, 276 (2001) 30724–30728. [DOI] [PubMed] [Google Scholar]
- [35].Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D, Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion, J Mol Cell Cardiol, 49 (2010) 271–279. [DOI] [PubMed] [Google Scholar]
- [36].Li S, Ma J, Li JB, Lacefield JC, Jones DL, Peng TQ, Wei M, Over-expression of calpastatin attenuates myocardial injury following myocardial infarction by inhibiting endoplasmic reticulum stress, J Thorac Dis, 10 (2018) 5283–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Teng X, Ji C, Zhong H, Zheng D, Ni R, Hill DJ, Xiong S, Fan GC, Greer PA, Shen Z, Peng T, Selective deletion of endothelial cell calpain in mice reduces diabetic cardiomyopathy by improving angiogenesis, Diabetologia, 62 (2019) 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang Y, Zheng D, Wei M, Ma J, Yu Y, Chen R, Lacefield JC, Xu H, Peng T, Overexpression of calpastatin aggravates cardiotoxicity induced by doxorubicin, Cardiovasc Res, 98 (2013) 381–390. [DOI] [PubMed] [Google Scholar]
- [39].Zheng D, Su Z, Zhang Y, Ni R, Fan GC, Robbins J, Song LS, Li J, Peng T, Calpain-2 promotes MKP-1 expression protecting cardiomyocytes in both in vitro and in vivo mouse models of doxorubicin-induced cardiotoxicity, Arch Toxicol, 93 (2019) 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cao T, Fan S, Zheng D, Wang G, Yu Y, Chen R, Song LS, Fan GC, Zhang Z, Peng T, Increased calpain-1 in mitochondria induces dilated heart failure in mice: role of mitochondrial superoxide anion, Basic Res Cardiol, 114 (2019) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen Q, Thompson J, Hu Y, Dean J, Lesnefsky EJ, Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in hearts following ischemiareperfusion, Am J Physiol Cell Physiol, 317 (2019) C910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen Q, Younus M, Thompson J, Hu Y, Hollander JM, Lesnefsky EJ, Intermediary metabolism and fatty acid oxidation: novel targets of electron transport chain-driven injury during ischemia and reperfusion, Am J Physiol Heart Circ Physiol, 314 (2018) H787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA, Conditional disruption of ubiquitous calpains in the mouse, Genesis, 44 (2006) 297–303. [DOI] [PubMed] [Google Scholar]
- [44].Ma J, Wei M, Wang Q, Li J, Wang H, Liu W, Lacefield JC, Greer PA, Karmazyn M, Fan GC, Peng T, Deficiency of Capn4 gene inhibits nuclear factor-kappaB (NF-kappaB) protein signaling/inflammation and reduces remodeling after myocardial infarction, J Biol Chem, 287 (2012) 27480–27489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC, Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species, J Biol Chem, 286 (2011) 29610–29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL, Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria, Am J Physiol, 273 (1997) H1544–1554. [DOI] [PubMed] [Google Scholar]
- [47].Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ, Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion, J Pharmacol Exp Ther, 319 (2006) 1405–1412. [DOI] [PubMed] [Google Scholar]
- [48].Mohsin AA, Chen Q, Quan N, Rousselle T, Maceyka MW, Samidurai A, Thompson J, Hu Y, Li J, Lesnefsky EJ, Mitochondrial Complex I Inhibition by Metformin Limits Reperfusion Injury, J Pharmacol Exp Ther, 369 (2019) 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, Kordula T, Milstien S, Lesnefsky EJ, Spiegel S, Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration, Faseb J, 25 (2011) 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Paillard M, Gomez L, Augeul L, Loufouat J, Lesnefsky EJ, Ovize M, Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential, J Mol Cell Cardiol, 46 (2009) 902–909. [DOI] [PubMed] [Google Scholar]
- [51].An J, Varadarajan SG, Novalija E, Stowe DF, Ischemic and anesthetic preconditioning reduces cytosolic [Ca2+] and improves Ca2+ responses in intact hearts, Am J Physiol Heart Circ Physiol, 281 (2001) H1508–1523. [DOI] [PubMed] [Google Scholar]
- [52].Scaduto RC Jr., Grotyohann LW, Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives, Biophys J, 76 (1999) 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ, Postconditioning modulates ischemia-damaged mitochondria during reperfusion, J Cardiovasc Pharmacol, 59 (2012) 101–108. [DOI] [PubMed] [Google Scholar]
- [54].Jones SP, Teshima Y, Akao M, Marban E, Simvastatin attenuates oxidant-induced mitochondrial dysfunction in cardiac myocytes, Circ Res, 93 (2003) 697–699. [DOI] [PubMed] [Google Scholar]
- [55].Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM, Mitochondrial Dysfunction in the Type 2 Diabetic Heart is Associated with Alterations in Spatially-Distinct Mitochondrial Proteomes, Am J Physiol Heart Circ Physiol, 299 (2010) H529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Baseler WA, Dabkowski ER, Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Croston TL, Powell M, Razunguzwa TT, Lewis SE, Schnell DM, Hollander JM, Reversal of mitochondrial proteomic loss in Type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase, Am J Physiol Regul Integr Comp Physiol, 304 (2013) R553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Thompson J, Hu Y, Lesnefsky EJ, Chen Q, Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release, Am J Physiol Heart Circ Physiol, 310 (2016) H376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liu K, Shi Y, Guo X, Wang S, Ouyang Y, Hao M, Liu D, Qiao L, Li N, Zheng J, Chen D, CHOP mediates ASPP2-induced autophagic apoptosis in hepatoma cells by releasing Beclin-1 from Bcl-2 and inducing nuclear translocation of Bcl-2, Cell Death Dis, 5 (2014) e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gorlach A, Klappa P, Kietzmann T, The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control, Antioxid Redox Signal, 8 (2006) 1391–1418. [DOI] [PubMed] [Google Scholar]
- [60].Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW, MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress, Circulation, 140 (2019) 1720–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gao P, Jiang Y, Wu H, Sun F, Li Y, He H, Wang B, Lu Z, Hu Y, Wei X, Cui Y, He C, Wang L, Zheng H, Yang G, Liu D, Yan Z, Zhu Z, Inhibition of Mitochondrial Calcium Overload by SIRT3 Prevents Obesity- or Age-Related Whitening of Brown Adipose Tissue, Diabetes, 69 (2020) 165–180. [DOI] [PubMed] [Google Scholar]
- [62].Pan M, Han Y, Basu A, Dai A, Si R, Willson C, Balistrieri A, Scott BT, Makino A, Overexpression of hexokinase 2 reduces mitochondrial calcium overload in coronary endothelial cells of type 2 diabetic mice, Am J Physiol Cell Physiol, 314 (2018) C732–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bosetti F, Baracca A, Lenaz G, Solaini G, Increased state 4 mitochondrial respiration and swelling in early post-ischemic reperfusion of rat heart, FEBS Lett, 563 (2004) 161–164. [DOI] [PubMed] [Google Scholar]
- [64].Kerner J, Turkaly PJ, Minkler PE, Hoppel CL, Aging skeletal muscle mitochondria in the rat: decreased uncoupling protein-3 content, Am J Physiol Endocrinol Metab, 281 (2001) E1054–1062. [DOI] [PubMed] [Google Scholar]
- [65].Akhmedov AT, Rybin V, Marin-Garcia J, Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart, Heart Fail Rev, 20 (2015) 227–249. [DOI] [PubMed] [Google Scholar]
- [66].Talbot DA, Hanuise N, Rey B, Rouanet JL, Duchamp C, Brand MD, Superoxide activates a GDP-sensitive proton conductance in skeletal muscle mitochondria from king penguin (Aptenodytes patagonicus), Biochem Biophys Res Commun, 312 (2003) 983–988. [DOI] [PubMed] [Google Scholar]
- [67].Aguirre E, Cadenas S, GDP and carboxyatractylate inhibit 4-hydroxynonenal-activated proton conductance to differing degrees in mitochondria from skeletal muscle and heart, Biochim Biophys Acta, 1797 (2010) 1716–1726. [DOI] [PubMed] [Google Scholar]
- [68].Kukreja RC, Okabe E, Schrier GM, Hess ML, Oxygen radical-mediated lipid peroxidation and inhibition of Ca2+-ATPase activity of cardiac sarcoplasmic reticulum, Arch Biochem Biophys, 261 (1988) 447–457. [DOI] [PubMed] [Google Scholar]
- [69].Xu KY, Zweier JL, Becker LC, Hydroxyl radical inhibits sarcoplasmic reticulum Ca2+-ATPase function by direct attack on the ATP binding site, Circ Res, 80 (1997) 76–81. [DOI] [PubMed] [Google Scholar]
- [70].Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT, Understanding mitochondrial complex I assembly in health and disease, Biochim Biophys Acta, 1817 (2012) 851–862. [DOI] [PubMed] [Google Scholar]
- [71].Arrington DD, Van Vleet TR, Schnellmann RG, Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction, Am J Physiol Cell Physiol, 291 (2006) C1159–1171. [DOI] [PubMed] [Google Scholar]
- [72].Kunkel GH, Chaturvedi P, Tyagi SC, Mitochondrial pathways to cardiac recovery: TFAM, Heart Fail Rev, 21 (2016) 499–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER, The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility, Nutr Metab (Lond), 11 (2014) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kuznetsov AV, Schneeberger S, Seiler R, Brandacher G, Mark W, Steurer W, Saks V, Usson Y, Margreiter R, Gnaiger E, Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion, Am J Physiol Heart Circ Physiol, 286 (2004) H1633–1641. [DOI] [PubMed] [Google Scholar]
- [75].del Arco A, Morcillo J, Martinez-Morales JR, Galian C, Martos V, Bovolenta P, Satrustegui J, Expression of the aspartate/glutamate mitochondrial carriers aralar1 and citrin during development and in adult rat tissues, Eur J Biochem, 269 (2002) 3313–3320. [DOI] [PubMed] [Google Scholar]
- [76].Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, Palmieri F, Citrin and aralar1 are Ca2+-stimulated aspartate/glutamate transporters in mitochondria, Embo J, 20 (2001) 5060–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, Hutson SM, Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle, Cell Metab, 6 (2007) 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Chen Q, Thompson J, Hu Y, Lesnefsky EJ, Genetic Elimination of Calpain 4 Decreases Cardiac Injury by Improving Mitochondrial Function, Circulation (2019) Abstract (15954) [Google Scholar]
- [79].Radko SP, Poverennaya EV, Kurbatov LK, Ponomarenko EA, Lisitsa AV, Archakov AI, The “Missing” Proteome: Undetected Proteins, Not-Translated Transcripts, and Untranscribed Genes, J Proteome Res, 18 (2019) 4273–4276. [DOI] [PubMed] [Google Scholar]