

Abstract
Anion redox in lithium transition metal oxides such as Li2RuO3 and Li2MnO3, has catalyzed intensive research efforts to find transition metal oxides with anion redox that may boost the energy density of lithium-ion batteries. The physical origin of observed anion redox remains debated, and more direct experimental evidence is needed. In this work, we have shown electronic signatures of oxygen-oxygen coupling, direct evidence central to lattice oxygen redox (O2−/(O2)n−), in charged Li2-xRuO3 after Ru oxidation (Ru4+/Ru5+) upon first-electron removal with lithium de-intercalation. Experimental Ru L3-edge high-energy-resolution fluorescence detected X-ray absorption spectra (HERFD-XAS), supported by ab-initio simulations, revealed that the increased intensity in the high-energy shoulder upon lithium de-intercalation resulted from increased O-O coupling, inducing (O-O) σ*-like states with π overlap with Ru d-manifolds, in agreement with O K-edge XAS spectra. Experimental and simulated O K-edge X-ray emission spectra (XES) further supported this observation with the broadening of the oxygen non-bonding feature upon charging, also originated from (O-O) σ* states. This lattice oxygen redox of Li2-xRuO3 was accompanied by a small amount of O2 evolution in the first charge from differential electrochemistry mass spectrometry (DEMS) but diminished in the subsequent cycles, in agreement with the more reduced states of Ru in later cycles from Ru L3-edge HERFD-XAS. These observations indicated that Ru redox contributed more to discharge capacities after the first cycle. This study has pinpointed the key spectral fingerprints related to lattice oxygen redox from a molecular level and constructed a transferrable framework to rationally interpret the spectroscopic features by combining advanced experiments and theoretical calculations to design materials for Li-ion batteries and electrocatalysis applications.
Graphical Abstract
1. Introduction
The redox process of positive electrode materials largely dictates the energy density and cycling stability of the Li-ion batteries. Conventional positive electrode materials including LiCoO2 and LiNixMnyCozO2, with layered structures, hinge on the reversible redox couples of 3d transition metals including Co3+/Co4+,1–2 Ni2+/Ni4+ (or Ni3+/Ni4+)3–4 and Mn3+/Mn4+,5–6 accompanied by lithium (de-)intercalation, delivering a capacity of around 140 mAh/g,1–6 upon charging to 4.3 V with respect to Li metal (VLi). This redox process is rooted in the electronic structure of lithium early transition metal oxides. Due to their large electronegativity difference, oxygen 2p-states lie well below the transition metal d-states, hence, the Fermi level has a predominantly metallic character, and the transition metals mainly undertake the burden of the redox reaction.7–9 For most transition metals, this cationic redox scenario usually provides one electron per metal site and poses significant limitations on the energy density of positive electrode materials for mobile devices and automotive applications.9 However, as we move from early to late transition metals, or to higher valence metal states via lithium de-intercalation, we increase the electronegativity of the transition metal species.10 This results in a larger degree of energetic overlap between the metal d-states and the oxygen p-states, and therefore a more covalent interaction between the transition metal and oxygen.8–9, 11 In this scenario a direct extraction/replenishment of the electron density from the oxygen density of states can occur.7–8, 12 This scenario is usually referred to as anionic redox, in contrast with conventional cationic redox. The notion of anion redox arose for the first time in LiCoO2 in the 1990s, where oxygen release was observed upon charging up to 4.4 VLi.13 Ceder et al.12 explained this finding using density functional theory (DFT) calculations in which both metal and oxygen contribute to the redox processes at high potentials.12 Within the 3d metals family, going from early transition metals to late transition metals, this oxygen charge compensation mechanism is augmented due to a more covalent metal-oxygen framework12.
Li-rich layered oxides, Li2MO3 (M = Ru, Ir, Mn),8, 11, 14–22 have been studied extensively and offer much higher energy density than LiMO2 by providing more lithium for (de-)intercalation. These oxides can be written generally as Li(Li1/3M2/3)O2, with 1/3 of the transition metal sites within the transition metal layer occupied by Li. This Li excess allows a higher transition metal valence (4+) , compared to LiMO2 (3+), and therefore leads to more overlap between transition metal and oxygen states, resulting in a highly covalent MO bonding.7, 10, 12 These reported Li-rich metal oxides can be divided into two classes. The first class is represented by oxides derived from Li2MnO3,17–18, 21, 23–37 which exhibits a relatively poor reversibility attributed to oxygen redox (oxidation) into molecular oxygen, leading to oxygen loss. Although Li2MnO3 can yield a first cycle discharge capacity higher than 200 mAh/g, upon charging to 4.8 VLi,25, 29–32, 34–35 it suffers from a poor cycling performance.23, 29–31 Considerable oxygen loss occurs upon charging Li2MnO317,32, where 0.125 oxygens can be released per unit of extracted Li30. The Li2MnO3 derived compounds such as Li(LixMnyNizCo1-x-y-z)O2 (Li-rich Mn-rich NMC) introduce additional redox couples (Ni2+/Ni4+ and Co3+/Co4+) that may boost both cationic and lattice oxygen redox, reaching a reversible capacity of around 300 mAh/g.36–38 Although their cycling stability is much improved compared to pure Li2MnO3 electrodes, they still suffer from a significant voltage decay over subsequent cycles37, and the oxygen redox contribution to the redox process is diminished upon extended cycling.37 The second group of Li-rich metal oxides is derived from noble metals including Li2RuO311, 39–41 and Li2IrO3,42–43 which show reversible capacities with a two-electron transfer process and a high cycle life. For example, Tarascon and co-workers15–16, 39–40 have shown that Li2Ru1-xMnxO3 and Li2Ru1-xSnxO3 deliver first discharge capacities as high as 250 mAh/g,16, 39–40 and capacity retention of higher than 80 % after 100 cycles.16, 39 It has been proposed that the capacity corresponding to the second electron transfer results from reversible lattice oxygen oxidation into peroxo- and superoxo- like species in the bulk.11, 16, 39–40
The mechanism giving rise to the reversible capacities associated with the lattice oxygen redox is not well understood. Unambiguous experimental evidence or an electronic structure signature for lattice oxygen redox for these Mn-, Ir- and Ru- based oxides is missing. There are primarily two schools of thought on the physical origin of lattice oxygen redox in Li-rich oxides. The first class of proposed mechanisms argues that anion redox induces the formation of (O2)n− peroxo-like species,11, 39–41, 43 and the redox couple O2−/(O2)n− can proceed reversibly upon cycling. A certain M-O bond covalency is required for reversible oxidation of the anion as proposed by Doublet, Tarascon and coworkers.11, 41 Tarascon et al. have proposed the existence of peroxo-like O2n− species for Li2-xRuO3 at 4.6 VLi during the first cycle via X-ray photoelectron spectroscopy (XPS) studies.16, 39 The formation of such peroxo-like species is accompanied by Ru reduction which is referred to as a reductive coupling mechanism.11, 40 The Ru6+ species are stabilized by modifying the coordination sphere of Ru through oxygen-oxygen bond formation and the transfer of extra electrons to the transition metal for reduction, eventually forming Ru5+-(O2)2− peroxo-species or Ru4+-(O2)− superoxo-species. However, we should note that the XPS measurement has a penetration depth of only 5 nm to 10 nm44. Therefore, the evidence provided by XPS is from the surface and subsurface of the materials which is likely not representative of the bulk. Moreover, the XPS fingerprint is not clear and cannot be unequivocally assigned to peroxo-like species, but might also come from surface electrolyte decomposition products. Hence, there is no unambiguous experimental evidence from an electronic structure perspective to support this proposed redox process. The formation of peroxo-like species is further demonstrated and visualized by McCalla et al.43 using high-resolution transmission electron microscopy and neutron diffraction in delithiated Li2-xIrO3, where an O-O bond distance of around 2.4 Å has been shown.43 In addition, Saubanère et al.11 have reported from crystal orbital overlap population (COOP) analysis that the covalent bonding between Ru-O stabilizes the peroxo-like species in Li2-xRuO3 upon oxidation. In contrast, the Mn-O bond has less covalency, resulting in further oxidation of peroxo-like species and eventually evolving molecular oxygen. This idea has also been further explored by Bruce and coworkers.36 Their O K-edge resonant inelastic X-ray scattering (RIXS) study on Li-rich Mn-rich NMC showed an increased intensity of the elastic peak (around 531 eV) upon charging to around 4.4 VLi, a fingerprint they assigned to formation of oxygen-localized electron holes.36 However, the rationale for such an assignment remains unclear. Moreover, there is no strong indication of true peroxide or superoxide species formation since there is no clear Raman signal from 800 cm−1 to 1100 cm−1 in ex-situ charged Li-rich NMC samples,36 the characteristic wavenumber range for metal peroxide and superoxides.36 Bruce and coworkers have concluded that the reversibility of anionic redox relies on more delocalized O holes, which require more delocalized M-O bonds from higher M-O covalency.
In the second school of thought, the oxygen redox of Li-rich oxides is attributed to the reversible oxidation and reduction of the non-bonding oxygen density of states created by Li-O-Li configuration,45–46 unique to Li-excess or disordered oxides. Due to the high ionicity of Li-O interaction, the oxygen 2p orbital bond with lithium resembles a lone-pair non-bonding oxygen, and its density of states lies at a higher electron energy level compared to the corresponding ones of transition metal bonded oxygen (bonding oxygen), thus providing a more facile electron transfer from oxygen.45 Therefore, oxygen redox can be represented as a redox couple of O2−/O−, where electron density is directly taken from non-bonding oxygen 2p states. However, there is no unequivocal experimental evidence that oxygen redox proceeds as a redox couple of O2−/O− without oxygen sub-lattice distortion. This mechanism is further extended by Gent and Chueh38, 47, where they proposed that anionic redox is a dynamic process, requiring synergistic transition metal migration and O2−/O− redox couple. In-situ X-ray diffraction (XRD) and RIXS38 on Li-rich and Mn-rich NMC (Li1.2Mn0.54Ni0.13Co0.13O2) demonstrated that the oxygen redox and the transition metal migration are coupled. The oxygen-bonded transition metal migrates to a tetrahedral site, creating non-bonding oxygen 2p states on the de-coordinated oxygen. Similar to what Seo et al.45 have proposed, this non-bonding oxygen can perform O2−/O− redox, coupled with continuous transition metal migration. In particular, a RIXS feature with an emission energy of 523.7 eV, different from the 525 eV emission feature for conventional O2− species, could be assigned to the anionic redox state in both Li-ion and Na-ion battery electrodes.38, 48 However, although further theoretical study shows that this specific RIXS feature corresponds to excitations into unoccupied O-2p states, e.g., oxidized oxygen because 2p orbitals of O2− are full occupied, a simple extension of a peroxide model into transition-metal oxide material remains questionable49.
In this study, we find unambiguous evidence of lattice oxygen redox upon lithium deintercalation from Li2RuO3. By coupling hard and soft X-ray absorption (XAS) and X-ray emission spectroscopy (XES) experimental studies with ab-initio Bethe-Salpeter equation (BSE) spectroscopy calculations50, we capture the redox processes of both oxygen and ruthenium in Li2-xRuO3 upon the first charge, which contrasts the redox process of conventional battery materials including LiCoO2 and LiNi1/3Mn1/3Co1/3O2. This detailed characterization of the redox process allows us to qualify different previously proposed hypotheses and experimental studies regarding lattice oxygen redox. Furthermore, combining the redox process with differential mass spectrometry (DEMS) measurements, we evaluate the reversibility of the anionic redox of Li2RuO3 and discuss the implications for the design of high-energy positive electrode materials leveraging anionic redox.
2. Result and Discussion
Electronic signatures of oxygen-oxygen coupling in Li2-xRuO3
XAS and XES on the charged oxide-only pellet electrodes, without conductive carbon and binder, revealed electronic structure evidence of oxidation of non-bonding oxygen in Li2-xRuO3. Both O K-edge XAS total fluorescence yield (TFY) and Ru L3-edge high-energyresolution fluorescence detected (HERFD)-XAS of the pristine electrode (Li2RuO3) were composed of two major peaks (c.a. 528.8 eV and 530.8 eV for the O K-edge in Figure 1(a) and 2841.1 eV and 2843.6 eV for the Ru L3-edge in Figure 1(b)). Ab-initio theoretical calculations with OCEAN50–51 using the pristine Li2RuO3 structure (C2/c)52–53 resulted in simulated O K-edge (Figure 1(a)) and Ru L3-edge (Figure 1(b)) spectra that agree well with the experimental spectral fingerprints of the pristine electrode. Furthermore, these calculations also enabled us to visualize the electron densities corresponding to specific excited state features. The low-energy peak can be attributed to a t2g feature, having a metal 4dxy (degenerate with 4dyz and 4dzx) orbital with a π overlap with O 2p states, whereas the high-energy peak can be attributed to the eg* states, comprising the σ* overlap between transition metal 4dx2-y2 states and O 2p states, as shown in Figure 1(c). Here we should note that the computed spectra have slightly larger t2g and eg splitting compared to the experimental spectra, which indicates we have an overestimation of the ligand field. Moreover, as we might note that in the pristine experimental Ru L3-edge XAS spectra of Li2RuO3 (Figure 1(b)), there is a small high-energy shoulder which is not understood, which might potentially come from mixed oxidation states54–56, and/or surface reacting with moisture undergoing proton and lithium ions exchange.57 Worth notably, this feature has never been reported by previous Ru L3-edge measurements of Ru-based oxides58 and complexes54–55 since they were limited by the significant core-hole lifetime broadening and therefore such small feature was not resolved, which requires further studies.
Figure 1:
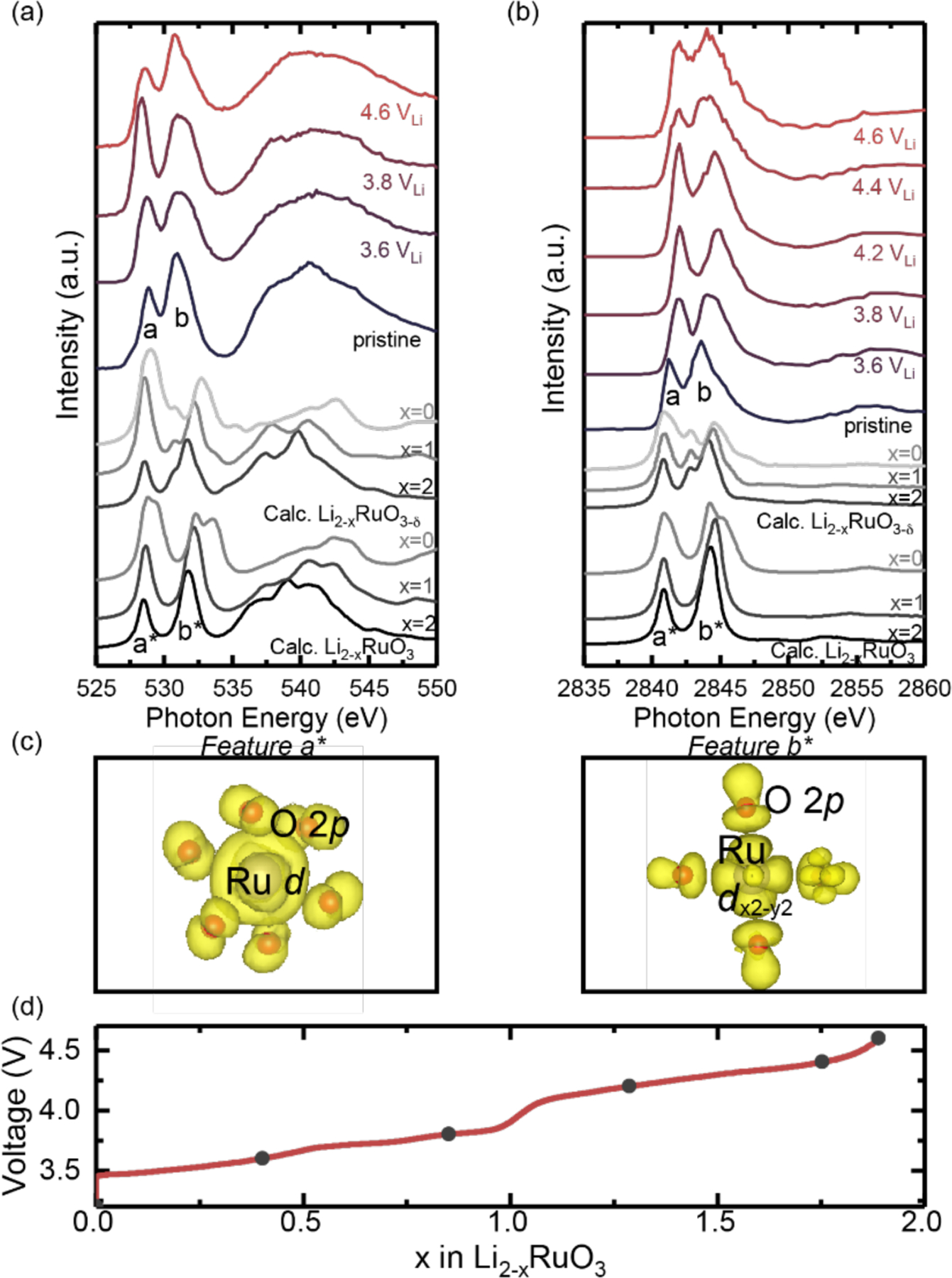
(a) O K-edge XAS collected in TFY mode, and (b) Ru L3-edge HERFD-XAS spectra of Li2-xRuO3 pellets as a function of charging potential (3.6 VLi, 3.8 VLi, 4.2 VLi, 4.4 VLi, and 4.6 VLi). Corresponding simulated spectra of the Li2RuO3, Li1RuO3, Li0RuO3 as well as Li2RuO3-δ, Li1RuO3-δ, Li0RuO3-δ (δ =1/4), are also shown for comparison, representing the structures with oxygen vacancies. The calculated O K-edge and Ru L3-edge spectra are shifted by 528 eV and 2884 eV, respectively, to allow for a direct comparison with the experimental spectra. The computed features labeled “a*” and “b*” match with the experimental features “a” and “b” in the pristine Li2RuO3. (c) Visualization of charge densities that corresponds to a* and b* of the computed spectra of pristine Li2RuO3; a* can be assigned to the t2g feature comprised of Ru d- manifolds and O 2p state with a π overlap, and b* can be assigned to eg* feature composed of Ru 4dx2-y2 and O 2p orbitals with a σ* overlap. (d) The voltage profile of Li2-xRuO3 pellets in the first cycle show two major plateaus at 3.6 VLi and 4.2 VLi in line with previous works.16, 39–40 The closed black circles indicate the voltages where the ex-situ samples were collected. The fitted peak intensities of t2g and eg peak at each state of charge is shown in Figure S1, and the fitted peaks for O K-edge and Ru L-edge are shown in Figure S2 and S3, respectively.
Upon charging, the low-energy t2g peak intensity (integrated area) gradually increased up to 3.8 VLi, and then decreased, relative to that of the high-energy eg feature, in both O K-edge XAS (Figure 1(a) and Figure S1(a)) and Ru L-edge HERFD XAS (Figure 1(b) and Figure S1(b)). The increase of the t2g peak integrated area up to 3.8 VLi, which corresponds to the first lithium de-intercalation per formula from Li2RuO3 in the low-voltage plateau as shown in Figure 1(d), can be attributed to the removal of approximately one t2g electron upon oxidation of Ru4+ to Ru5+ in Li2-xRuO3. Similar spectroscopic changes have been reported for oxides relying predominantly on cationic redox including LiCoO259–60 and LiNi1/3Mn1/3Co1/3O261, where the metal L-edge shifts monotonically and the pre-peak area in O K-edge XAS increased,59–61 marking the removal of the electron from transition metal d states upon lithium de-intercalation. On the other hand, upon charging to voltages greater than 3.8 VLi, the eg feature in both Ru L3-edge and O K-edge spectra were found to grow relative to the low-energy t2g peak and broaden considerably on the high-energy side (Figure 1(a), (b), and Figure S1). Through DFT calculations, we determined the most energetically favorable structures at different lithium contents of Li2-xRuO3 by enumerating all possible configurations (computational details shown in Methods section). Simulated Ru L3-edge (Figure 2(a) and (b)) and O K-edge (Figure S4) spectra of Li2-xRuO3 revealed the appearance of a high-energy shoulder in the vicinity of the eg feature for Li0.5RuO3 and Li0RuO3, which could potentially account for the broadening observed in the experimental spectra Aided by a charge density visualization at the shoulder peak energy, we assigned this feature to a stronger oxygen-oxygen (O-O) σ* − like antibonding states with a π* overlap with transition metal d-manifolds (Figure 2(b)). This combined experimental and simulated approach for the Li2-xRuO3 revealed electronic structure evidence for the antibonding state of oxygen-oxygen coupling associated with lattice oxygen redox in bulk proposed previously from DFT studies.11
Figure 2:
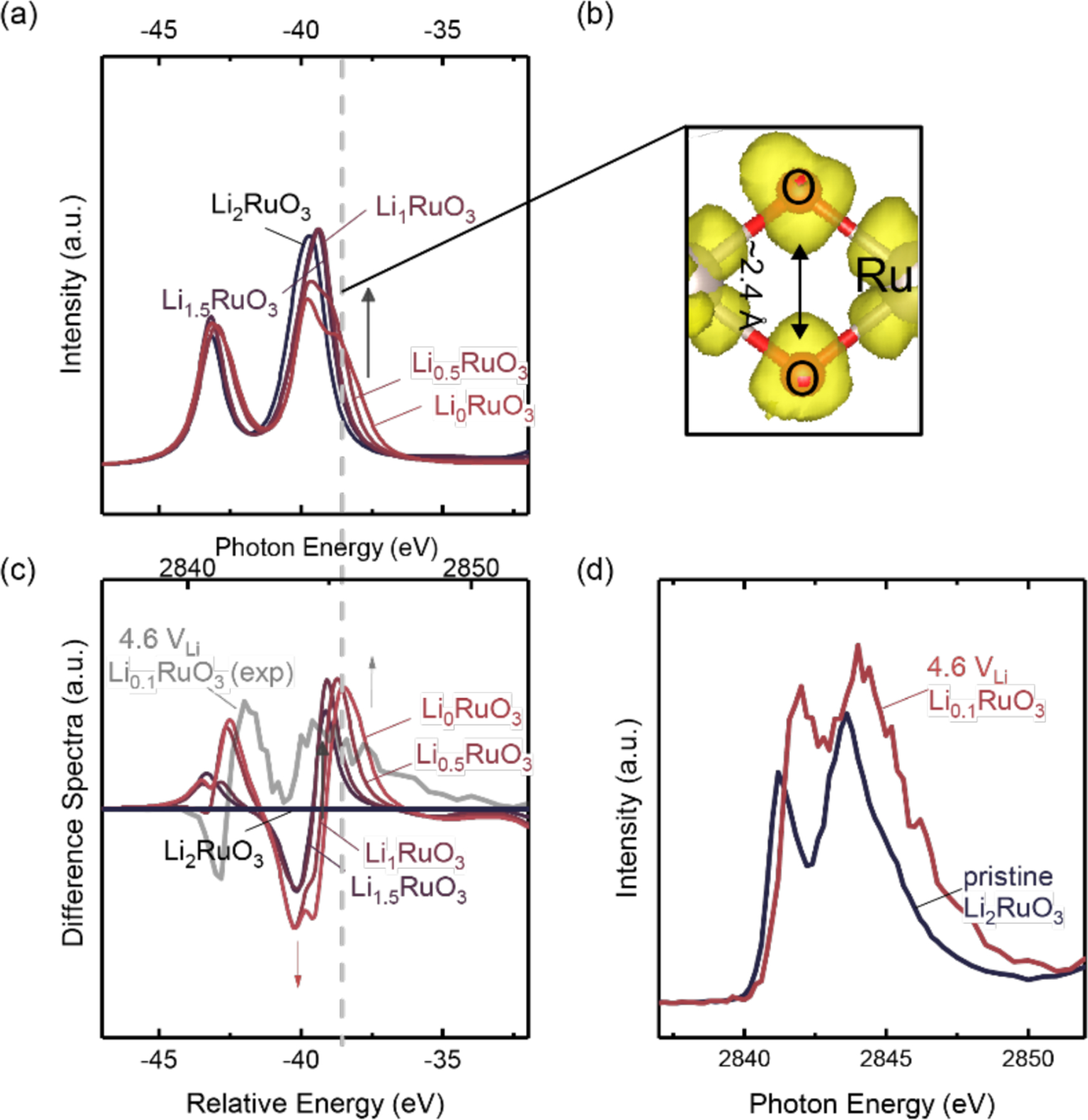
(a) Calculated Ru L3-edge HERFD XAS and (c) calculated and experimental difference spectra of Li2-xRuO3. Upon delithiation, an additional peak appears at a photon energy just above the eg feature. The electron density of this excitation shows it to be an (O-O) σ*-like feature hybridized with Ru d-manifolds due to oxygen cage distortion and shortening of the O-O bond from 2.8 Å to 2.4 Å upon delithiation, as shown in the schematic in (b). (d) Experimental Ru L3-edge HERFD XAS of Li2-xRuO3 charged to 4.6 VLi (red) and pristine Li2-xRuO3 (black), where a broadening of eg peak was observed with higher intensity at higher photon energy, matching with the observation in the computed spectra, and can be attributed to the creation of (O-O) σ*-like feature. The voltage profile determined from the DFT-calculated structure of Li2-xRuO3 is shown in comparison with the experimental profile in Figure S5.
Theoretical spectra simulations of other candidate structures show that the experimentally observed changes in the O K- and Ru L-edge XAS spectra cannot be solely explained by introducing oxygen vacancies in Li2-xRuO3, which is known to evolve oxygen gas at voltages around 4.2 VLi,40 nor by true peroxo (dO-O ≈1.5 Å )62 or superoxo- (dO-O ≈1.2 Å – 1.3 Å)63 species of oxygen with bond length of less than 1.8 Å . The high-energy peak shoulder observed in the experimental XAS spectra of Li2-xRuO3 obtained from the 4.2 VLi plateau cannot be explained by the presence of lattice oxygen vacancies. The simulated O K-edge (Figure 1(a) and Figure S6) and Ru L3-edge (Figure 1(b) and Figure S7) of Li2-xRuO3-δ with δ = 0.25 oxygen vacancies per formula unit in the bulk led to additional features between the t2g and eg peaks, as well as a significant decrease in the intensity of the eg relative to the t2g feature, which were not clearly experimentally observed (Figure 1(a), (b) and Figure S1). Moreover, we have also performed a test calculation on Ru L-edge XAS of a surface Ru atom surrounded by surface oxygen vacancies as in Figure S7(d), and a similar trend as the bulk oxygen vacancy calculations was also observed. Here, we saw a more intense feature between t2g and eg peaks and more depressed eg peaks due to larger degree of Ru reduction, compared to the cases with bulk oxygen vacancies. (Figure S7(a)) However, we cannot rule out the presence of oxygen vacancy formation as we have also observed a slight rise of the feature between eg and t2g, and this observation could potentially coming from oxygen vacancies formed in the oxides upon charging, but the additional features at high photon energy cannot be solely explained by oxygen vacancy formation. Previous computational work on Li2-xRuO3 and Li2-xIrO3 showed the presence of distorted oxygen cages with an oxygen distance around 2.4 Å,40, 43 and refined neutron diffraction data in chemically delithiated Li2-xIrO3 samples also confirmed an oxygen-oxygen distance of around 2.4 Å.43 This O-O bond distance does not support the presence of true peroxide or superoxide species. We performed Ru L3-edge and O K-edge spectra simulations by reducing the oxygen-oxygen bond distance from 2.4 Å to 1.8 Å in Li0RuO3 where we forced two oxygen atoms closer together while keeping the position of other atoms the same. This led to increased high-energy shoulder intensities for both Ru L3-edge and O K-edge, indicating a stronger oxygen-oxygen coupling, as shown in Figure S8 and Figure S9(a). However, a further reduction of the O-O bond distance from 1.8 Å to 1.2 Å, so to form a true superoxide, led to a drastic reduction in the eg intensity compared to the t2g peak. This could be the result of reduced σ overlap between Ru 4d and O 2p states coming from a smaller O-Ru-O bond angle associated with shorter O-O bonds. As such changes were not observed in the experimental XAS data in Figure 1, the formation of peroxo-like and superoxo-like species with an O-O bond length less than 1.8 Å in bulk Li2-xRuO3 (1≤x≤2) is also not likely based on our combined experimental and simulated study. Moreover, as we force the O-O bond distance from 2.4 Å to 1.8 Å and 1.2 Å, we induced an energy penalty of above 3 eV respectively, which also indicates that from an energetic standpoint, forming such short O-O bond distance in Li2-xRuO3 is not possible.
The oxygen-oxygen coupling in Li2-xRuO3 (1≤x≤2) is further supported by O K-edge valence XES, which probes the projected occupied local density of state (DOS) with O-2p symmetry. The XES spectra of the pristine electrode in Figure 3(a) and Figure 3(b) consisted of three major features: one major peak centered at ≈523 eV and two shoulders located at ≈521 eV and ≈526 eV, respectively. When comparing these, on a binding energy scale, with valence band XPS data from Li2RuO3, similar features are observed with different relative intensities due to different selection rules dictating valence XES and XPS (Figure 3(a)).64 In line with previous work on perovskite materials that have comparable O K-edge XES spectra64, we assign the major XES peak to non-bonding oxygen, and the high- and low-energy shoulders to anti-bonding and bonding Ru-O bonds, respectively.64 This assignment is supported by the DFT projected DOS of Ru 4d and oxygen 2p in Figure 3(a), where the high-energy shoulder reflects Ru-O bonding states (≈ 6 eV in the DOS) with a predominantly oxygen character, the low-energy shoulder reflects anti-bonding Ru-O states with mostly metal character (≈1 eV in the DOS), and the center peak indicates non-bonding oxygen states without significant contribution of Ru d-electrons (≈2.5 eV in the DOS). Here, we should note that the center non-bonding peak position of computed ground-state DOS had some discrepancies with experimental O K-edge XES spectra (Figure 3(a)). When charging Li2-xRuO3 up to 3.8 VLi, there was no significant change observed for the O K-edge XES spectra (Figure 3(b)), in agreement with corresponding simulated O K-edge XES spectra of Li2-xRuO3 (0 ≤ x ≤ 1) in Figure 3(c). The O K-edge XES indicates that there is no oxygen participation upon first lithium removal. In contrast, upon charging Li2-xRuO3 to 4.2 VLi and beyond a spectral broadening was observed (Figure 3(b)), indicative of changes to the probed oxygen states. This broadening was also found in the ab-initio calculated XES spectra of Li0.5RuO3 and Li0RuO3 (Figure 3(c)), especially for the oxygen non-bonding peak. Here we should note that for OCEAN simulation of O K-XES, since no core-hole effect is considered, the energy is referenced to the Fermi level, and the relative energy scale is the same as E-Ef scale. Using the crystal orbital overlap population (COOP), in line with previous work,11, 41, 65–66 the broadening in the oxygen non-bonding feature can be attributed to an increased O-O antibonding character (by showing negative COOP intensity) in the energy range where we observed the broadening in computed spectra (from −2 eV to 0 eV vs. the Fermi level) in Figure 4(d). Furthermore, by visualizing the charge density in the same energy range, we observed that the charge density also resembled a (O-O) σ* interaction with a nodal plane in the middle as shown in Figure 3(d). This finding further supports the formation of oxygen-oxygen coupling upon charging Li2-xRuO3 in the high-voltage plateau of 4.2 VLi. The peak broadening in the XES data most likely came from the (O-O) σ* antibonding states (Figure 3(d)). These states are different from those with an additional π* participation from Ru 4d electrons which are responsible for the increased intensity at the high-energy shoulder found in the XAS spectra (Figure 2). Here, we captured the creation of both “non-bonding” (O-O) σ* in the O K-edge XES (Figure 3(d)) in contrast with “anti-bonding” (O-O) σ* with metal (Ru) π* contribution in XAS, shown in the charge density in Figure 2(a). Furthermore, such broadening in the experimental XES spectra was not observed in charged Li1-xCoO2 (Figure S12) and Li1-xNi1/3Mn1/3Co1/3O2 (Figure S12). Their largely unchanged peak widths indicate the absence of lattice oxygen-oxygen coupling (Figure 4(e)). However, broadening in the XES spectra was observed in substituted Li2-xRu1-yMyO3 such as Li2-xRu0.μmn0.5O3 (Figure 4(e) and Figure S12) as well as Li2-xMnO3 derived materials including Li-rich Mn-rich NMC probed by RIXS,36 where the central peak of the RIXS spectra broadened upon charging. This broadening in O K-edge XES appears concurrently with changes in the O K-edge absorption, including broadening and the creation of an additional feature around 531 eV, but not in metal L-edges as in Ru cases.38 This observation is in agreement with previous COOP analysis performed by Saubanère et al. on Li2MnO3,11 where upon lithium deintercalation, a stronger O-O coupling exists, inducing (O-O) π and σ states. However, contrary to the present Ru case, the (O-O) σ* states of Li2MnO3 have no Mn contribution,11 and therefore the additional states did not appear in Mn L-edge spectra of the charged electrodes from previous experiemtnal measurement, where upon charging, the Mn L-edge spectra of Li2-xMnO3 did not show significant change.67 This suggested that for the Ru-based material, the (O-O) σ* is stabilized through π overlap with Ru d-manifolds because of the much more delocalized d states. In contrast, for the Mn scenario, this (O-O) σ* is not coordinated with any transition metal, and oxygen is released due to more weakened metal oxygen coordination. This observation suggests that strongly covalent Ru-O bonds is critical to enable lattice oxygen-oxygen coupling.11, 41
Figure 3:
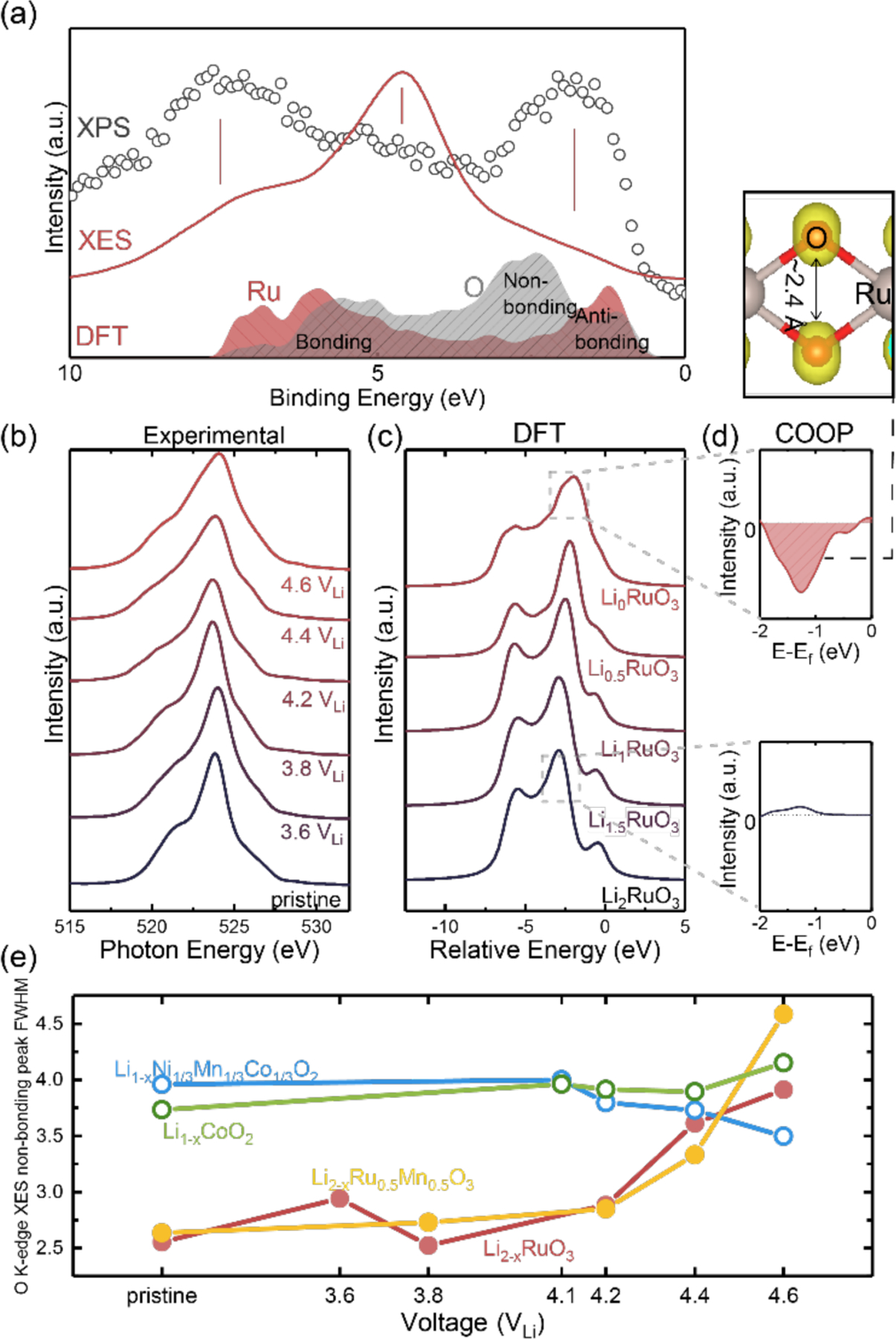
(a) Alignment of the O K-edge XES to XPS valence band measurement on the binding energy scale and comparison of ground-state DFT projected orbital-wise density of state calculation for Ru 4d and O 2p. In the XES spectra the three features can be assigned to bonding, non-bonding, anti-bonding oxygen states from low to high photon energy, respectively. (b) O K-edge XES of Li2-xRuO3 as a function of state of charge. (c) Simulated O K-edge XES of Li2-xRuO3 as a function of lithium content, where peak broadening was noted for Li0.5RuO3 and Li0RuO3. (d) Crystal orbital overlap population (COOP) of Li2-xRuO3 (bottom x = 0 and top x = 2), where, upon delithiation, Li0RuO3 showed a stronger O-O σ* feature around the non-bonding region in the experimental and computed spectra compared to the fully-lithiated case of Li2RuO3. (e) Non-bonding peak full width at half maximum (FWHM) as a function of charging potential of different positive electrode materials: LiCoO2 (LCO), LiNi1/3Mn1/3Co1/3O2 (NMC), Li2RuO3 (LRO) and Li2Ru0.5mMn0.5O3(LRMO). The materials claimed to have anionic redox (closed symbol) were observed to have significant peak broadening upon charging in contrast with LCO and NMC (open symbol), which utilize predominantly cationic redox activity upon oxidation. Here, the FWHM was extracted based on the width at the half height of the most predominant peak (non-bonding peak) as the other shoulder peaks are much smaller than non-bonding peak, thus we assume that the shoulder peaks evolution will not have significant effect on the FWHM of the center non-bonding peak, the FWHM extraction procedure is also shown schematically in Figure S10. The ground-state projected DOS of the fully lithiated and delithiated Li2-xRuO3 is shown in Figure S11 (a) and (b), respectively, and the COOP analysis of full energy range of Li2RuO3 and Li0RuO3 is shown in Figure S11(c). Raw XES data of LCO, NMC and LRMO is shown in Figure S12 and more detailed analysis is shown in Figure S13.
Figure 4.
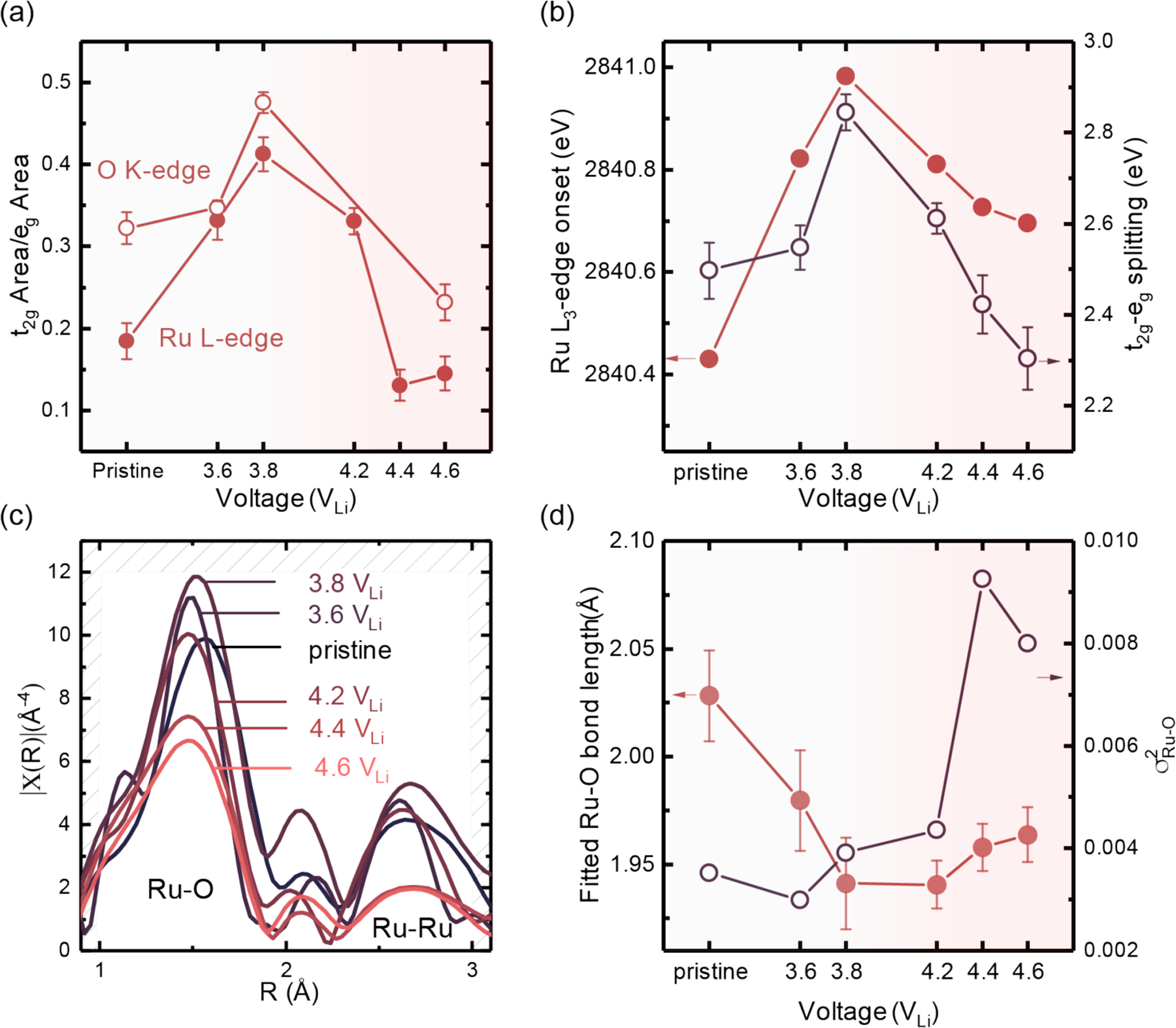
(a) Ratio of integrated peak area of the t2g peak (feature a in Figure 1) and eg peak (feature b in Figure 1) of O K-edge (open circle) and Ru L-edge (closed circle) from XAS spectra. Detailed fitting results are shown in Figure S1–S3. (b) Ru L-edge edge shift (closed circle) and t2g (feature a in Figure 1) and eg (feature b in Figure 1) features splitting (open circle). (c) Raw EXAFS data, Fourier-transformed χ(R) as a function of R (reduced distance) of Li2-xRuO3 in the first charge as a function of state of charge including 3.6 VLi, 3.8 VLi, 4.2 VLi, 4.4 VLi, and 4.6 VLi, showing two major features, one centered around 1.5 Å (reduced distance), representing Ru-O interaction, and another centered around 2.6 Å (reduced distance), representing Ru-Ru interaction. The k-range for Fourier transformation is 1 Å −1 to 11 Å −1. (d) Fitted Ru-O bond length (closed circle) and Debye-Waller factor from Ru K-edge EXAFS shown in Figure 2(c). In the first plateau, Ru is monotonically oxidized with increasing t2g and eg splitting, accompanied by Ru-O bond contraction. Upon charging to 4.2 VLi, Ru metal is slightly reduced with less t2g and eg splitting, and there is an increase in the Ru-O bond length and disorder in the system. The raw EXAFS data in k-space and fitting results are shown in Figure S15 and S16, and the summary of the EXAFS fitting parameter is shown in Table S1. Summary of alternative EXAFS fitting parameter by only using the first coordination shell is shown in Table S2.
Moreover, the broadening observed in the O K-edge XES spectra cannot be explained by the creation of oxygen vacancies in the structure as shown by the simulated O K-XES spectra of Li2-xRuO3-δ (δ=1/4) in Figure 3(c) and Figure S6, where only a small shift of the nonbonding peak towards lower energy was observed, whereas in the experimental spectra a positive shift was observed for Li2-xRuO3-δ upon charging. In addition, such broadening also cannot come from the formation of true peroxo- or superoxo- species with O-O bond length less than 1.8 Å. Simulated XES spectra of Li0RuO3 with equilibrium O-O bond distance of 2.4 Å and shortened distance of 1.8 Å and 1.2 Å in Figure S9(b) reveal distinctive new peak features in the simulated XES spectra for the 1.2 Å case, potentially coming from fingerprints of σ, π, π * of the O-O bonding, which were clearly not observed experimentally in this case. Our experimental and simulated XAS and XES revealed unambiguous electronic structure signatures for lattice oxygen redox in form of oxygen-oxygen coupling upon first charge of Li2-xRuO3 on the 4.2 VLi plateau, which include 1) the growth of the eg peak shoulder in the O K-edge and Ru L3-edge XAS spectra originated from the (O-O) σ*-like states with π overlap with Ru d-manifolds and 2) the broadening of non-bonding oxygen originated from the (O-O) σ* states (without Ru d hybridization). Simulated XAS data further revealed that such O-O coupling in Li2-xRuO3 has an oxygen-oxygen distance not smaller than 1.8 Å, namely much longer than those in true molecular oxygen dimers, peroxide and superoxide, which is in agreement with previous proposal by Doublet et al.41 that the fully de-coordinated oxygen couples can escape from the lattice.
XAS Spectroscopy evidence of reductive coupling in Li2-xRuO3
We further examined the electronic structure signature for the reductive coupling in Li2-xRuO3,11, 39, 41 in which the reduction of Ru is accompanied by the oxidation of lattice oxygen (or lattice oxygen-oxygen coupling) in the 4.2 VLi plateau. Upon charging Li2-xRuO3 to 3.8 VLi, the ratio of the integrated peak intensities between the t2g and eg features, in both Ru L3-edge and O K-edge XAS spectra (Figure 4(a)), indicated electron removal from the t2g states upon oxidation of Ru. This assignment is supported by simulated Ru L3-edge (Figure 2(b)) and O K-edge XAS spectra (Figure S4) of Li2-xRuO3 obtained from BSE calculations, which show the increase of the low-energy t2g peak relative to the high-energy eg peak with de-intercalation of one lithium per formula unit from Li2RuO3 to LiRuO3. Ru oxidation within the 3.8 VLi plateau is further evidenced by positive shifts in the edge photon energy of the Ru L3-edge (Figure 4(b)) and Ru K-edge XAS (Figure S14), greater energy splitting between t2g and eg features in Ru L3-edge XAS (Figure 4(b)) coming from larger ligand fields associated with higher oxidation states54–56, and shortened Ru-O bonds obtained from the extended X-ray absorption fine structure (EXAFS) analysis of the Ru K edge in Figure 4(c), matching with previous observations from Mori et al.58 Upon further charging within the 4.2 VLi plateau, the ratio of integrated peak intensities of t2g to eg features in both Ru L3-edge and O K-edge XAS spectra began to decrease (Figure 4(a)), accompanied by negative shifts in the onset energy of the Ru L3 edge (Figure 4(b)) and K edge (Figure S14), as well as a smaller energy splitting between t2g and eg features in Ru L-edge XAS(Figure 4(b)), indicative of Ru reduction. The trend observed in the Ru L3-edge shift (Figure 4(b)) is consistent with other metrics to estimate Ru oxidation states used in previous work68. For example, the centroid and the pre-peak position of Ru L3-edge shifted towards higher photon energies during charging up to 3.8 VLi, and towards lower energies at 4.2 VLi and above (Figure S17). Such trend of energy splitting between t2g and eg is also evidenced from the computed Ru L3-edge XAS spectra (Figure 2(a)) where as we remove the first Li+, the crystal field splitting increases slightly, and then decreases after removing the second Li+ per formula unit. Therefore, experimental Ru L3- and O K-edge XAS data of Li2-xRuO3 during the first charge along with simulated XAS spectra support the simultaneous Ru reduction and oxygen-oxygen coupling in the 4.2 VLi plateau reported previously as reductive coupling.11, 39–40, 43 Similarly, slight reduction of Ir has been reported by Hong et al.47 using Ir L-edge XAS, accompanied by proposed lattice oxygen oxidation in fully de-intercalated Sn-substituted LixIrySn1-yO3 (x = 0, y = 0.5 and 0.75).47 While the Ru oxidation state reduction in the 4.2 VLi plateau did not yield a statistically significant increase in the Ru-O bond length from Ru K-edge EXAFS (Figure 4(d)), the Debye-Waller factor associated with the Ru-O bond was increased markedly at 4.2 VLi and above, indicating increased variances of Ru-O bond length and angles as well as a distorted Ru-O cage, which requires further study.
The XAS spectral evidence of reductive coupling at the 4.2 VLi plateau during the first charge was diminished in subsequent cycles. The voltage profile of Li2-xRuO3 in the subsequent charges was also markedly different from the first cycle. In fact, the first charge distinct voltage plateaus at 3.8 VLi and 4.2 VLi were replaced by a sloppy voltage profile with an average potential of around 3.6 VLi in subsequent cycles, as shown in Figure 5(a). During the third charge, the Ru L3-edge onset energy was found to increase monotonically with increasing lithium de-intercalation. This is in contrast to behavior during the first charge with first an increase and then a decrease in the edge onset energy (Figure 5(a) and Figure 4(b)), characteristic of reductive coupling (reduction of Ru) with an enhanced oxygen-oxygen interaction. This observation suggests that reductive coupling, and potentially oxygen-oxygen coupling, is substantially diminished in subsequent cycles, and Ru redox was largely responsible for reversible capacities. This postulate is supported by a much lower photon energy onset at comparable lithium content in Li2-xRuO3 in the third charge (Figure 5(a) bottom panel) compared to the first charge (Figure 5(a) top panel). Further support comes from the DEMS in Figure 5(b), where an irreversible oxygen redox (oxygen oxidation) generated oxygen in the first charge at around 4.2 VLi, but in subsequent cycles oxygen evolution fell below the detection limit. The amount of oxygen evolution can only be translated to a molar oxygen deficiency fraction of 2 %. From the DEMS measurement, we can also capture the first onset of CO2 evolution at ≈3.9 VLi, potentially due to residual Li2CO3 from the synthesis precursor.69–70 Upon charging to 4.2 VLi, lattice oxygen redox was accompanied by pronounced electrolyte oxidation to generate CO2 evolution (2.0 % mole fraction), which was also reduced considerably in subsequent cycles. Previous online electrochemistry mass spectrometry (OEMS)4, 71 on NMC and Li-rich NMC has attributed part of the CO2 evolution to singlet oxygen evolved from electrolyte decomposition at the cathode surface. Even by attributing all the CO2 evolution to the oxide lattice (although this is unlikely), we would only create around 0.4 mol of oxygen vacancies (0.8 mol of electron transfer) per formula unit of oxide during the 4.2 VLi plateau. Accounting for the charge associated with the simultaneous Li-removal in this stage (≈ 0.8 mol Li+ and e−), oxygen and CO2 evolution alone cannot explain the reduction of transition metal within the high-voltage plateau observed in Ru L3- and O K-edge XAS in Figure 4 (a) and (b), and Figure 5(a) in the first charge. As a result, during the first cycle, the redox process proceeded with Ru4+/5+ accompanied by oxygen-oxygen coupling and transition metal reduction, whereas in subsequent cycles the high reversibility of Li2-xRuO3 most likely relied heavily on reversible Ru redox of Ru3.x+/Ru5+ (where x is closer to 1 than 0), which surprisingly resembles the trends found in 3d transition metal systems in both Li-rich and Na-ion compounds37, 48
Figure 5:
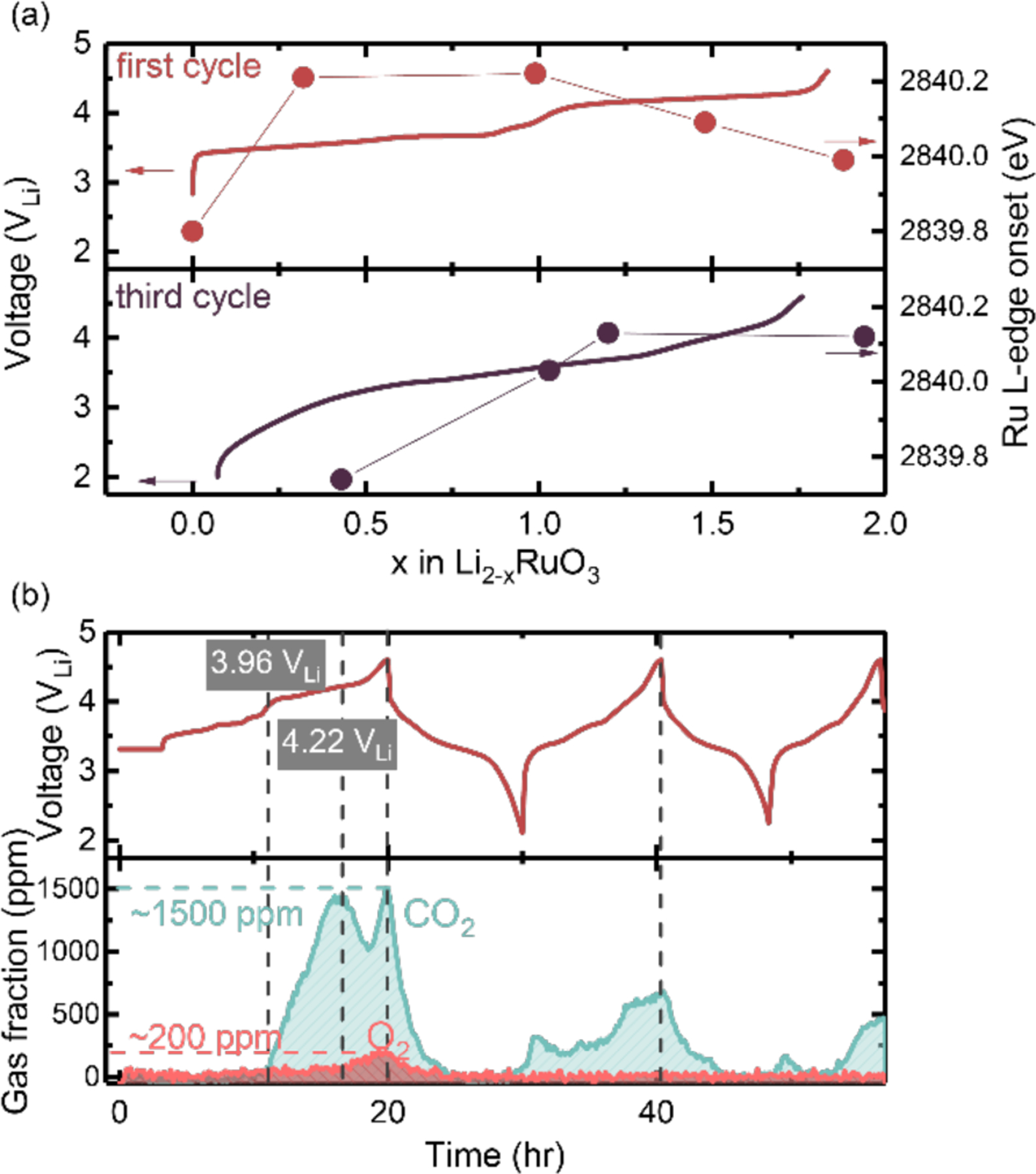
(a) Voltage profiles of Li2RuO3 composite electrodes during the first (dark red) and third (purple) charge cycle with current density of 16.35 mA per gram of fully lithiated oxide (goxides) or 0.088 mA per cm2 of the electrode cross-section (cmgeo2), and the Ru-L3 HERFD XAS edge onset of the corresponding composite electrodes as a function of lithium content indicated in filled circle (Figure S18). The lithium content was calculated based on the current applied and time of charge/discharge process and the charge transferred during the 5 h potential holding at the end-of-charge, which can be translate to how many lithium ions per formula unit (de-)intercalate upon cycling. (b) Differential electrochemistry mass spectrometry (DEMS) of Li2RuO3 during the first three cycles, where on the top panel is the voltage profile with a current density of 16.35 mA/goxides (0.049 mA/cmgeo2) and the bottom panel is the CO2 and O2 evolution as a function of time during the first three cycles.
Implications in oxygen redox reported for other oxides
The electronic structure signature of oxygen-oxygen coupling associated with lattice oxygen oxidation in Li2-xRuO3 revealed that non-bonding oxygen is primarily being oxidized,45 giving rise to characteristic features in the O K-edge and Ru L3-edge XAS as well as to the O K-edge XES. The oxidized non-bonding species can be formed through oxygen sublattice distortion which also has a transition metal (Ru) coordination, where this intermediate O-O dimer-like species can be stabilized through metal oxygen covalency,11, 40 in contrast with Mn-based oxides exhibiting anionic redox.47, 67 The electronic fingerprints and the physical origin proposed in this work can be applied to explain spectroscopic data of Li-excess materials reported previously. For charged LixIrySn1-yO3, the appearance of an additional spectral feature, i.e. at ≈523 eV in O K-edge XES and at ≈531 eV in O K-edge XAS, as well as the broadening of the eg feature (≈531 eV) for charged Li2IrO3 have been previously attributed to oxygen redox without any specific electronic structure details. Similarly, for Li2MnO3 and its derivative materials including Li-rich Mn-rich NMC, researchers have observed similar increased intensities at the high-energy peak of ≈531 eV of O K-edge36, 38. This peak has been proposed to be the fingerprint of oxygen redox, but no specific physical origin has been assigned to it. Based on this work, this feature can be assigned to the π/σ states of (O-O) bonding from enhanced oxygen coupling, providing electronic structure details for oxygen redox. While spectral changes of Ir L-edge XAS and O K-edge XAS spectra of LixIrySn1-yO3 are expected to be similar to those of Ru found in this work (Figure 1), previous Ir L-edge measurements47, 72 suffered from significant peak broadening due to the much shorter core-hole lifetime of Ir73 and failed to detect any spectroscopic changes. High-energy resolution L-edge XAS is critical to follow any changes in such 4d and 5d transition metal oxides. Importantly, in Li2MnO367 and Li-rich Mn-rich NMC38, even though similar changes have been observed in O K-edge spectra, no simultaneous broadening or additional peak is reported in the Mn L-edge upon charging,67 which suggest that this (O-O) peroxo-like state did not have any metallic contribution, and therefore is more prone to oxygen evolution compared to metal-hybridized peroxo-like species as in Li2RuO3. Similarly, an emerging peak at ≈531 eV was observed in the O K-edge XAS of charged Li1.2Ti0.4M0.4O2 (M = Fe, Mn)74 and Li1.3Nb0.3M0.4O2 (M = Fe, Mn)75, while minimal differences were noted for transition metal L edges,74–75 indicating the (O-O) coupled states in these compounds are not hybridized with the transition metal, resulting in lower capacity retention compared to Ru and Ir-based materials.
The observed electronic structure fingerprints for oxygen redox in Li2RuO3 and implications are discussed with respect to different proposed mechanisms for oxygen redox in the literature. The finding on oxidation of non-bonding oxygen can be related to the DFT studies of oxygen redox in Li2MnO3, Li2Ru0.5Sn0.5O3 and Li-excess disordered rocksalt reported by Seo and Ceder,45 where the oxidation of non-bonding oxygen (the oxygen bound to Li) was proposed. However, similar to what Tarascon, Doublet and coworkers claim,11, 40 we argue that the strong hybridization between Ru and O2n− is critical to stabilize the oxidized oxygen species otherwise oxygen-oxygen coupling would form molecular oxygen, causing capacity loss (irreversible oxygen redox). In addition, transition metal migration can create under-coordinated oxygen, which can facilitate the coupling between transition metal migration and oxygen redox in Li-rich Mn-rich NMC (Li1.2Mn0.54Ni0.13Co0.13O2) observed by Gent and Chueh.38 Nevertheless, such oxygen, without transition metal coordination, is prone to be released and would not contribute to capacities in later cycles, as shown by hard XAS data from Hu et al.37, where the capacity contribution from oxygen redox of such materials is diminished to 50 mAh/g in the 83rd cycle compared to 120 mAh/g in the first cycle.37 Moreover, we show that in such covalent oxides with a 4d transition metal, even though stable (O-O) coupling can be formed in the first charge, the capacity attributed to oxygen redox can be overwhelmed by a more significant contribution from cation redox in subsequent cycles.
3. Conclusions
In this work, by combining O K-edge XAS, Ru L3-edge HERFD-XAS and O K-edge XES spectroscopy with ab-initio theoretically calculated spectra, we have captured and rationalized electronic structure fingerprints that reveal an enhanced O-O coupling in Li2-xRuO3 in the 4.2 VLi plateau during the first charge after removing ≈ 50 % of the lithium. This observation provides direct evidence central to lattice oxygen redox (O2−/(O2)n−) in charged Li2-xRuO3 after Ru oxidation (Ru4+/Ru5+). Such oxygen-oxygen coupling (lattice oxygen redox) of Li2-xRuO3 was also accompanied with simultaneous Ru reduction that cannot be solely explained by oxygen vacancy formation in the oxides at high potentials. Upon the first charge, the lattice oxygen redox is accompanied by a small amount of O2 evolution in the first charge (captured by DEMS), which is diminished in subsequent cycles. This observation is in agreement with the changes in Ru L3-edge HERFD-XAS. Ru was found to be in a more reduced state in later cycles despite similar lithium content, indicating that the Ru redox contributed more to discharge capacities in later cycles than in the first cycle. This study has provided theoretical insights on previously reported spectroscopic features in resonant inelastic X-ray scattering spectroscopy and O K-edge XAS across different materials families exhibiting anionic redox. This work highlights the importance of combining with the state-of-the-art core-level measurements and ab-initio excited-state Bethe-Salpeter equation calculations in order to interpret the spectroscopic features. The presented approach can be used as a transferable framework to monitor the spectroscopy changes of metal oxides undergoing redox/electrochemical reactions for Li-ion and electrocatalysis applications.
4. Methods
Experimental methods
Solid-state synthesis
The pristine Li2RuO3 was synthesized through the solid-state route previously undertaken by Goodenough et al.76 and Tarascon et al.40 Li2CO3 and RuO2 precursors are first dried under air flow at 300 °C for 4 h. The precursors were then mixed in stoichiometric amounts, but with a Li excess of 10 %, with a mortar and a pestle for 1 h in a argon-filled glovebox (H2O, O2 < 0.1ppm). The resultant powder was pelletized under 1 MPa and calcined and sintered at 1000 °C and 900°C under air with heating rate of 2 °C/min for 24 h and 48 h, respectively, with intermediate grinding. The phase purity of Li2RuO3 is confirmed through X-ray diffraction (XRD) with PANalyt’cal X’Pert PRO (Figure S19).
Positive electrode preparation
The pellet electrode was prepared by pelletizing around 48 mg of active material using 6 mm diameter pressing die set (Across International) for 15 mins. The pellets were then sintered under air at 900 °C for 6 hours. The cooling and heating rates were 2 °C/min. The pellet electrodes were then broken into pieces of around 3 mg each and dried in vacuum under 120 °C overnight before being transferred into an Argon-filled glovebox (<0.5 ppm of H2O and O2). The pellet electrodes were used for the X-ray measurements to remove any ambiguities introduced by conductive carbon and binder.
Composite electrodes were prepared by mixing active material (80 % mass fraction), 10 % acetylene black (AB) and 10 % polyvinylidene fluoride (PVDF) (Kynar) dispersed in N-Methyl-2-pyrrolidone (NMP) with Fritsch Minimill 23 homogenizer. The slurry was then blade-coated onto an aluminum sheet with a gap of 5 μm. The composite electrodes were punched into 1.27 cm disc and pressed at 5.7×104 kg/cm2 under a hydraulic press, to embed the powder into the aluminum disc. Finally, the electrodes were completely dried at 120 °C under vacuum for 12 h before being transferred into Argon-filled glovebox (<0.5 ppm of H2O and O2). The active loading of the composite electrodes for the cycling experiments is around 3 mg/cm2. Composite electrodes for DEMS experiments were prepared with a slurry dispersed in less NMP to make a more viscous slurry. The resultant slurry was then blade-coated onto a stainless steel 304 mesh with an opening of 20 μm with a wire diameter of 0.2 mm with an aluminum sheet underneath, and with a gap of 15 μm. The electrodes were then dried and separated from the aluminum sheet underneath and punched into 15 mm diameter discs. The DEMS electrodes were pressed at 4.11×104 kg/cm2 under a hydraulic press. Finally, the electrodes were completely dried at 120 °C under vacuum for 12 h before being transferred into an Argon-filled glovebox (<0.5 ppm of H2O and O2). The active loading of the composite electrodes for the DEMS experiments was around 5.27 mg/cm2, which is higher than the loading of composite electrodes for XAS measurements to ensure that enough gaseous species were evolved to be detected during cycling.
Electrochemical measurements
Electrochemical behavior of the pellet electrodes was confirmed by galvanostatic measurements in two-electrode cells (Tomcell type TJ-AC). Cells were assembled in an argon-filled glovebox (<0.5 ppm of H2O and O2) and comprised a lithium metal foil as the negative electrode and the positive electrode, separated by two pieces of polypropylene separator (2500 Celgard), impregnated with 150 μL of LP57 (1M LiPF6 in a 3:7 ethylene carbonate (EC): ethylmethyl carbonate (EMC) electrolyte (BASF)). After assembly, the cells rested for 6 h prior to measurement and then were charged with different end-of-charge potentials (3.6 VLi, 3.8 VLi, 4.2 VLi, 4.4 VLi, and 4.6 VLi) at a rate of ≈1.64 mA/g. This rate of charge corresponds to a rate of C/100 based on the theoretical capacity calculated assuming one lithium extraction per Li2RuO3. The cell was held at the end-of-charge potential for 5 h before disassembly inside the glovebox. The composite electrode was charged in a two-electrode coin cell (Hohsen CR2016) with Li metal foil as the negative electrode. The positive and negative electrodes were separated by two pieces of polypropylene separator (2500 Celgard), impregnated with 100 μL of LP57. The coin cells were charged at a rate of C/10 (16.35 mA/g) with different end-of-charge potential (first cycle: 3.6 VLi, 3.8 VLi, 4.2 VLi and 4.6 VLi, third cycle: 3 VLi, 3.4 VLi, 4 VLi, and 4.6 VLi). The cells were held at the end-of-charge potential for 5 h before being disassembled inside the glovebox. The collected pellets and composite electrodes were rinsed in 1 mL EMC solution with stirring for 24 h to remove the residual electrolyte and electrolyte decomposition products on the surface. The pellets and composite electrodes were then dried in vacuum for 4 h before further characterization using XAS and XES.
Differential electrochemistry mass spectrometry measurements
The DEMS setup was constructed in-house based on the design reported by McCloskey et al.77 and used for the detection of gas evolution including CO2 and O2 in the head space of the electrochemistry cell along with pressure monitoring. Details on the DEMS setup and cell design are documented in Harding et al.78 The cell volume is around 6.7 cm3 for the measurement and Argon (Airgas, 99.999 % pure, O2, H2O, CO2 < 1 ppm ) was used as a carrier gas. The DEMS cells were prepared inside an argon-filled glovebox (<0.5 ppm of H2O and O2) and comprised a lithium metal foil as the negative electrode and the DEMS positive electrodes, separated by two pieces of polypropylene separator (2500 Celgard), impregnated with 150 μL of LP57. The cell was first pressurized with argon to around 150 kPa and allowed to rest for 8 h prior to electrochemical measurements to check for any potential leakage. DEMS measurement started 4 h after pressurization with a time interval of 10 min of collecting the gas from the headspace to obtain a CO2 evolution background from evaporated EMC fragments. The gas evolution profile upon charge/discharge as a function of time presented in this work were already background subtracted by fitting the gas evolution profile collected in the open circuit voltage stage after pressurizing with a polynomial function. The electrochemical measurements were carried out at a current density of 16.35 mA/g (0.088 mA/cm2) for charge and discharge (corresponding to a C/10 rate), and a time interval of 10 min was set between each DEMS sequence.
Synchrotron measurements
After rinsing and drying, the dried pellet electrodes were mixed with sucrose or BN powder to make 10 mm pellets. The pellets were wrapped around with Kapton tape for protection during transportation for hard X-ray absorption measurements.
High-energy resolution fluorescence detected Ru L3-edge XAS was conducted at the Stanford Synchrotron Radiation Lightsource (SSRL) at SLAC National Accelerator Laboratory. The measurements were performed with a high-resolution tender x-ray spectrometer developed on a dispersive Johansson geometry in a 500 mm Rowland circle using a Si(111) analyzer.79 The spectrometer recorded in dispersive mode the overall Ru Lα emission line; for extracting the HERFD spectrum, the peak intensity of the Lα emission line is plotted as a function of the incident photon energy. By using the Si(111) monochromator of SSRL beamline 6–2, an overall energy resolution of 0.6 eV was achieved enabling the sharpening of the Ru-L3 XAS features when compared to conventional x-ray absorption spectroscopy, i.e., total fluorescence yield or transmission mode XAS (comparison shown in Figure S20). The incident beam was about 400 μm × 800 μm (FWHM) and the photon flux ≈ 1012 sec−1. The dried electrode material was mixed with sucrose to minimize any self-absorption effects across the very intense Ru L3-edge resonances, resulting in pressed pellets with an active material mass fraction concentration of 0.5 %.
Ruthenium K-edge EXAFS was conducted at beamline 20-BM-B of the Advanced Photon Source (APS) at Argonne National Laboratory in transmission mode with an active materials mass fraction of 10 %, diluted with boron nitride. A Ru metal foil was used as reference. O K-edge XAS and XES were conducted at the Advanced Light Source (ALS) on beamline 8.0.2. All spectroscopy data were normalized and processed using the ATHENA software package.80
Computational Methods
DFT calculations
We used the Perdew-Burke-Enzerhof (PBE)81 formulation of the density functional and projected augmented wave (PAW) potentials implemented in Vienna Ab-initio Simulation Package (VASP)82–84, with Dudarev’s rotationally invariant Hubbard-type U85 applied on the transition metal where we applied a Ueff = 4 eV on Ru 4d orbital as reported from previous work.11, 45 A k-point per reciprocal atom of 8000 was used. The delithiated structures were generated with a primitive cell with 4 formula unit of Li2RuO3, where for each different lithium content, the most energetically favorable configuration was taken. The voltage profile calculated using the energetics from the delithiated structures is shown in Figure S5, where the computed voltage profile matched relatively well with experimental charging curve. However, we should note that to compute the true ground-state at each lithium content along the convex hull, a cluster expansion approach should be employed.86 The surface calculations in Figure S7(d) was conducted with VASP using a stoichiometric slab model with slab thickness of 5 Å and a vacuum thickness of 10 Å to avoid any spurious interactions across the periodic boundary. For simplicity, a surface orientation of (001) was chosen for the test, and a k-point grid of 7 × 7 × 1 was used. We fully relaxed the surface structure before the OCEAN calculations.
The XAS and XES simulations were performed with the OCEAN 2.5.2 code50,51, which combines ground-state density-functional theory and the Bethe-Salpeter equation (BSE) to account for the core-hole presence and to properly capture the spin-orbit interactions of transition metals. The ground-state charge density and wave functions used in the core-hole screening and BSE calculations were obtained using Quantum Espresso.87 Norm-conserving pseudopotentials from the ABINIT88 distribution were used. The k-point grid used to solve the Kohn-Sham states was typically 6×6×3 for the pristine Li2RuO3 (24 atom cell). Similar values were used for all structures. The screening calculations used a 2×2×2 k-point grid. The number of unoccupied bands used for the BSE calculation was 250, and the screened core-hole potential calculation included 750 bands. A spectral broadening of 0.5 eV was applied and dipole and quadrupole transitions were considered. To enable easy spectroscopic feature comparison, the core-level shift was not turned on for the OCEAN calculations. Given the polycrystalline type of our samples, all simulated spectra were obtained by averaging the contribution from all photon polarizations.
Supplementary Material
ACKNOWLEDGMENT
This work made use of the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-1419807. Research at MIT related to this work was supported financially by BMW. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work also used resources of the Extreme Science and Engineering Discovery Environment (XSEDE)119, which is supported by National Science Foundation grant number ACI-1548562. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences (contract No. DE-AC02-76SF00515). Part of the instrument used for this study was supported by U.S. Department of Energy, Office of Energy Efficiency & Renewable Energy, Solar Energy Technology Office BRIDGE Program. This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, and was supported by the U.S.DOE under Contract No. DE-AC02-06CH11357, and the Canadian Light Source and its funding partners. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. Certain commercial equipment, instruments, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by NIST, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Reference
- 1.Mizushima K; Jones P; Wiseman P; Goodenough JB, LixCoO2 (0< x<−1): A new cathode material for batteries of high energy density. Materials Research Bulletin 1980, 15 (6), 783–789. [Google Scholar]
- 2.Amatucci G; Tarascon J; Klein L, Cobalt dissolution in LiCoO2-based non-aqueous rechargeable batteries. Solid State Ionics 1996, 83 (1–2), 167–173. [Google Scholar]
- 3.Broussely M; Perton F; Biensan P; Bodet J; Labat J; Lecerf A; Delmas C; Rougier A; Peres J, LixNiO2, a promising cathode for rechargeable lithium batteries. Journal of power sources 1995, 54 (1), 109–114. [Google Scholar]
- 4.Jung R; Metzger M; Maglia F; Stinner C; Gasteiger HA, Oxygen Release and Its Effect on the Cycling Stability of LiNixMnyCozO2 (NMC) Cathode Materials for Li-Ion Batteries. Journal of The Electrochemical Society 2017, 164 (7), A1361–A1377. [Google Scholar]
- 5.Koetschau I; Richard M; Dahn J; Soupart J; Rousche J, Orthorhombic LiMnO2 as a High Capacity Cathode for Li-Ion Cells. Journal of the Electrochemical Society 1995, 142 (9), 2906–2910. [Google Scholar]
- 6.Vitins G; West K, Lithium intercalation into layered LiMnO2. Journal of the Electrochemical Society 1997, 144 (8), 2587–2592. [Google Scholar]
- 7.Goodenough JB; Kim Y, Challenges for rechargeable Li batteries. Chemistry of materials 2009, 22 (3), 587–603. [Google Scholar]
- 8.Grimaud A; Hong W; Shao-Horn Y; Tarascon J-M, Anionic redox processes for electrochemical devices. Nature materials 2016, 15 (2), 121. [DOI] [PubMed] [Google Scholar]
- 9.Assat G; Tarascon J-M, Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nature Energy 2018, 1. [Google Scholar]
- 10.Li K; Xue D, Estimation of electronegativity values of elements in different valence states. The Journal of Physical Chemistry A 2006, 110 (39), 11332–11337. [DOI] [PubMed] [Google Scholar]
- 11.Saubanère M; McCalla E; Tarascon J-M; Doublet M-L, The intriguing question of anionic redox in high-energy density cathodes for Li-ion batteries. Energy & Environmental Science 2016, 9 (3), 984–991. [Google Scholar]
- 12.Ceder G; Aydinol M; Kohan A, Application of first-principles calculations to the design of rechargeable Li-batteries. Computational materials science 1997, 8 (1–2), 161–169. [Google Scholar]
- 13.Jansen A; Kahaian A; Kepler K; Nelson P; Amine K; Dees D; Vissers D; Thackeray M, Development of a high-power lithium-ion battery. Journal of power sources 1999, 81, 902–905. [Google Scholar]
- 14.Perez AJ; Jacquet Q; Batuk D; Iadecola A; Saubanère M; Rousse G; Larcher D; Vezin H; Doublet M-L; Tarascon J-M, Approaching the limits of cationic and anionic electrochemical activity with the Li-rich layered rocksalt Li 3 IrO 4. Nature Energy 2017, 2 (12), 954. [Google Scholar]
- 15.Sathiya M; Leriche J-B; Salager E; Gourier D; Tarascon J-M; Vezin H, Electron paramagnetic resonance imaging for real-time monitoring of Li-ion batteries. Nature communications 2015, 6, 6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sathiya M; Ramesha K; Rousse G; Foix D; Gonbeau D; Prakash A; Doublet M; Hemalatha K; Tarascon J-M, High Performance Li2Ru1–y Mn y O3 (0.2≤ y≤ 0.8) Cathode Materials for Rechargeable Lithium-Ion Batteries: Their Understanding. Chemistry of Materials 2013, 25 (7), 1121–1131. [Google Scholar]
- 17.Armstrong AR; Holzapfel M; Novák P; Johnson CS; Kang S-H; Thackeray MM; Bruce PG, Demonstrating oxygen loss and associated structural reorganization in the lithium battery cathode Li [Ni0. 2Li0. 2Mn0. 6] O2. Journal of the American Chemical Society 2006, 128 (26), 8694–8698. [DOI] [PubMed] [Google Scholar]
- 18.Johnson C; Kim J; Lefief C; Li N; Vaughey J; Thackeray M, The significance of the Li 2 MnO 3 component in ‘composite’xLi 2 MnO 3·(1− x) LiMn 0.5 Ni 0.5 O 2 electrodes. Electrochemistry Communications 2004, 6 (10), 1085–1091. [Google Scholar]
- 19.Johnson CS; Li N; Lefief C; Vaughey JT; Thackeray MM, Synthesis, Characterization and Electrochemistry of Lithium Battery Electrodes: x Li2MnO3·(1− x) LiMn0. 333Ni0. 333Co0. 333O2 (0≤ x≤ 0.7). Chemistry of Materials 2008, 20 (19), 6095–6106. [Google Scholar]
- 20.Kim S; Aykol M; Hegde VI; Lu Z; Kirklin S; Croy JR; Thackeray MM; Wolverton C, Material design of high-capacity Li-rich layered-oxide electrodes: Li 2 MnO 3 and beyond. Energy & Environmental Science 2017, 10 (10), 2201–2211. [Google Scholar]
- 21.Thackeray MM; Johnson CS; Vaughey JT; Li N; Hackney SA, Advances in manganese-oxide ‘composite’electrodes for lithium-ion batteries. Journal of Materials Chemistry 2005, 15 (23), 2257–2267. [Google Scholar]
- 22.Thackeray MM; Wolverton C; Isaacs ED, Electrical energy storage for transportation—approaching the limits of, and going beyond, lithium-ion batteries. Energy & Environmental Science 2012, 5 (7), 7854–7863. [Google Scholar]
- 23.Dong X; Xu Y; Yan S; Mao S; Xiong L; Sun X, Towards low-cost, high energy density Li 2 MnO 3 cathode materials. Journal of Materials Chemistry A 2015, 3 (2), 670–679. [Google Scholar]
- 24.Lee E; Blauwkamp J; Castro FC; Wu J; Dravid VP; Yan P; Wang C; Kim S; Wolverton C; Benedek R, Exploring Lithium-Cobalt-Nickel Oxide Spinel Electrodes for≥ 3.5 V Li-Ion Cells. ACS applied materials & interfaces 2016, 8 (41), 27720–27729. [DOI] [PubMed] [Google Scholar]
- 25.Croy JR; Park JS; Dogan F; Johnson CS; Key B; Balasubramanian M, First-cycle evolution of local structure in electrochemically activated li2mno3. Chemistry of Materials 2014, 26 (24), 7091–7098. [Google Scholar]
- 26.Croy JR; Balasubramanian M; Gallagher KG; Burrell AK, Review of the US department of energy’s “deep dive” effort to understand voltage fade in Li-and Mn-rich cathodes. Accounts of chemical research 2015, 48 (11), 2813–2821. [DOI] [PubMed] [Google Scholar]
- 27.Croy JR; Iddir H; Gallagher K; Johnson CS; Benedek R; Balasubramanian M, Firstcharge instabilities of layered-layered lithium-ion-battery materials. Physical Chemistry Chemical Physics 2015, 17 (37), 24382–24391. [DOI] [PubMed] [Google Scholar]
- 28.Kim S; Kim C; Jhon Y-I; Noh J-K; Vemuri SH; Smith R; Chung KY; Jhon MS; Cho B-W, Synthesis of layered–layered 0.5 Li 2 MnO 3· 0.5 LiCoO 2 nanocomposite electrode materials by the mechanochemical process and first principles study. Journal of Materials Chemistry 2012, 22 (48), 25418–25426. [Google Scholar]
- 29.Kalyani P; Chitra S; Mohan T; Gopukumar S, Lithium metal rechargeable cells using Li 2 MnO 3 as the positive electrode. Journal of power sources 1999, 80 (1), 103–106. [Google Scholar]
- 30.Denis Y; Yanagida K; Kato Y; Nakamura H, Electrochemical activities in Li2MnO3. Journal of The Electrochemical Society 2009, 156 (6), A417–A424. [Google Scholar]
- 31.Kim S; Noh J-K; Yu S; Chang W; Chung KY; Cho B-W, Effects of transition metal doping and surface treatment to improve the electrochemical performance of Li2MnO3. Journal of Electroceramics 2013, 30 (3), 159–165. [Google Scholar]
- 32.Robertson AD; Bruce PG, Mechanism of electrochemical activity in Li2MnO3. Chemistry of Materials 2003, 15 (10), 1984–1992. [Google Scholar]
- 33.Johnson CS, Development and utility of manganese oxides as cathodes in lithium batteries. Journal of Power Sources 2007, 165 (2), 559–565. [Google Scholar]
- 34.Park S; Ahn H; Park G; Kim J; Lee Y, Cycle mechanism and electrochemical properties of lithium manganese oxide prepared using different Mn sources. Materials Chemistry and Physics 2008, 112 (2), 696–701. [Google Scholar]
- 35.Park S-H; Sato Y; Kim J-K; Lee Y-S, Powder property and electrochemical characterization of Li 2 MnO 3 material. Materials chemistry and physics 2007, 102 (2), 225–230. [Google Scholar]
- 36.Luo K; Roberts MR; Hao R; Guerrini N; Pickup DM; Liu Y-S; Edström K; Guo J; Chadwick AV; Duda LC, Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen. Nature chemistry 2016, 8 (7), 684. [DOI] [PubMed] [Google Scholar]
- 37.Hu E; Yu X; Lin R; Bi X; Lu J; Bak S; Nam K-W; Xin HL; Jaye C; Fischer DA, Evolution of redox couples in Li-and Mn-rich cathode materials and mitigation of voltage fade by reducing oxygen release. Nature Energy 2018, 3 (8), 690. [Google Scholar]
- 38.Gent WE; Lim K; Liang Y; Li Q; Barnes T; Ahn S-J; Stone KH; McIntire M; Hong J; Song JH, Coupling between oxygen redox and cation migration explains unusual electrochemistry in lithium-rich layered oxides. Nature communications 2017, 8 (1), 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sathiya M; Abakumov AM; Foix D; Rousse G; Ramesha K; Saubanère M; Doublet M; Vezin H; Laisa C; Prakash A, Origin of voltage decay in high-capacity layered oxide electrodes. Nature materials 2015, 14 (2), 230–238. [DOI] [PubMed] [Google Scholar]
- 40.Sathiya M; Rousse G; Ramesha K; Laisa C; Vezin H; Sougrati MT; Doublet M-L; Foix D; Gonbeau D; Walker W, Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nature materials 2013, 12 (9), 827–835. [DOI] [PubMed] [Google Scholar]
- 41.Xie Y; Saubanere M; Doublet M-L, Requirements for reversible extra-capacity in Li-rich layered oxides for Li-ion batteries. Energy & Environmental Science 2017, 10 (1), 266–274. [Google Scholar]
- 42.Pearce PE; Perez AJ; Rousse G; Saubanère M; Batuk D; Foix D; McCalla E; Abakumov AM; Van Tendeloo G; Doublet M-L, Evidence for anionic redox activity in a tridimensional-ordered Li-rich positive electrode β-Li 2 IrO 3. Nature materials 2017, 16 (5), 580. [DOI] [PubMed] [Google Scholar]
- 43.McCalla E; Abakumov AM; Saubanère M; Foix D; Berg EJ; Rousse G; Doublet M-L; Gonbeau D; Novák P; Van Tendeloo G, Visualization of OO peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 2015, 350 (6267), 1516–1521. [DOI] [PubMed] [Google Scholar]
- 44.Matthew J, Surface analysis by Auger and x-ray photoelectron spectroscopy Briggs D and Grant JT (eds). IMPublications, Chichester, UK: and SurfaceSpectra, Manchester, UK, 2003. 900 pp., ISBN 1-901019-04-7, 900 pp. Surface and Interface Analysis: An International Journal devoted to the development and application of techniques for the analysis of surfaces, interfaces and thin films 2004, 36 (13), 1647–1647. [Google Scholar]
- 45.Seo D-H; Lee J; Urban A; Malik R; Kang S; Ceder G, The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nature chemistry 2016, 8 (7), 692–697. [DOI] [PubMed] [Google Scholar]
- 46.Zhan C; Yao Z; Lu J; Ma L; Maroni VA; Li L; Lee E; Alp EE; Wu T; Wen J, Enabling the high capacity of lithium-rich anti-fluorite lithium iron oxide by simultaneous anionic and cationic redox. Nature Energy 2017, 2 (12), 963. [Google Scholar]
- 47.Hong J; Gent WE; Xiao P; Lim K; Seo D-H; Wu J; Csernica PM; Takacs CJ; Nordlund D; Sun C-J, Metal–oxygen decoordination stabilizes anion redox in Li-rich oxides. Nature materials 2019, 1. [DOI] [PubMed] [Google Scholar]
- 48.Dai K; Wu J; Zhuo Z; Li Q; Sallis S; Mao J; Ai G; Sun C; Li Z; Gent WE; Chueh WC; Chuang Y.-d.; Zeng R; Shen Z.-x.; Pan F; Yan S; Piper LFJ; Hussain Z; Liu G; Yang W, High Reversibility of Lattice Oxygen Redox Quantified by Direct Bulk Probes of Both Anionic and Cationic Redox Reactions. Joule 2019, 3 (2), 518–541. [Google Scholar]
- 49.Zhuo Z; Pemmaraju CD; Vinson J; Jia C; Moritz B; Lee I; Sallies S; Li Q; Wu J; Dai K; Chuang YD; Hussain Z; Pan F; Devereaux TP; Yang W, Spectroscopic Signature of Oxidized Oxygen States in Peroxides. J Phys Chem Lett 2018, 9 (21), 6378–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilmore K; Vinson J; Shirley EL; Prendergast D; Pemmaraju CD; Kas JJ; Vila FD; Rehr JJ, Efficient implementation of core-excitation Bethe–Salpeter equation calculations. Computer Physics Communications 2015, 197, 109–117. [Google Scholar]
- 51.Vinson J; Rehr J; Kas J; Shirley E, Bethe-Salpeter equation calculations of core excitation spectra. Physical Review B 2011, 83 (11), 115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jimenez-Segura M-P; Ikeda A; Yonezawa S; Maeno Y, Effect of disorder on the dimer transition of the honeycomb-lattice compound Li 2 RuO 3. Physical Review B 2016, 93 (7), 075133. [Google Scholar]
- 53.Li B; Shao R; Yan H; An L; Zhang B; Wei H; Ma J; Xia D; Han X, Lithium-Ion Batteries: Understanding the Stability for Li-Rich Layered Oxide Li2RuO3 Cathode (Adv. Funct. Mater. 9/2016). Advanced Functional Materials 2016, 26 (9), 1306–1306. [Google Scholar]
- 54.Sham T, X-ray absorption spectra of ruthenium L edges in hexaammineruthenium trichloride. Journal of the American Chemical Society 1983, 105 (8), 2269–2273. [Google Scholar]
- 55.Alperovich I; Smolentsev G; Moonshiram D; Jurss JW; Concepcion JJ; Meyer TJ; Soldatov A; Pushkar Y, Understanding the electronic structure of 4d metal complexes: From molecular spinors to L-edge spectra of a di-Ru catalyst. Journal of the American Chemical Society 2011, 133 (39), 15786–15794. [DOI] [PubMed] [Google Scholar]
- 56.Cramer S; DeGroot F; Ma Y; Chen C; Sette F; Kipke C; Eichhorn D; Chan M; Armstrong W, Ligand field strengths and oxidation states from manganese L-edge spectroscopy. Journal of the American Chemical Society 1991, 113 (21), 7937–7940. [Google Scholar]
- 57.Perez AJ; Beer R; Lin Z; Salager E; Taberna PL; Abakumov AM; Simon P; Tarascon JM, Proton Ion Exchange Reaction in Li3IrO4: A Way to New H3+ xIrO4 Phases Electrochemically Active in Both Aqueous and Nonaqueous Electrolytes. Advanced Energy Materials 2018, 8 (13), 1702855. [Google Scholar]
- 58.Mori D; Kobayashi H; Okumura T; Nitani H; Ogawa M; Inaguma Y, XRD and XAFS study on structure and cation valence state of layered ruthenium oxide electrodes, Li2RuO3 and Li2Mn0. 4Ru0. 6O3, upon electrochemical cycling. Solid State Ionics 2016, 285, 66–74. [Google Scholar]
- 59.Yoon W-S; Kim K-B; Kim M-G; Lee M-K; Shin H-J; Lee J-M; Lee J-S; Yo C-H, Oxygen contribution on Li-ion intercalation− deintercalation in LiCoO2 investigated by O K-edge and Co L-edge X-ray absorption spectroscopy. The Journal of Physical Chemistry B 2002, 106 (10), 2526–2532. [Google Scholar]
- 60.Montoro L; Abbate M; Rosolen JM, Changes in the electronic structure of chemically deintercalated LiCoO2. Electrochemical and Solid-State Letters 2000, 3 (9), 410–412. [Google Scholar]
- 61.Nam KW; Bak SM; Hu E; Yu X; Zhou Y; Wang X; Wu L; Zhu Y; Chung KY; Yang XQ, Combining in situ synchrotron X-ray diffraction and absorption techniques with transmission electron microscopy to study the origin of thermal instability in overcharged cathode materials for lithium-ion batteries. Advanced Functional Materials 2013, 23 (8), 1047–1063. [Google Scholar]
- 62.Hayyan M; Hashim MA; AlNashef IM, Superoxide ion: generation and chemical implications. Chemical reviews 2016, 116 (5), 3029–3085. [DOI] [PubMed] [Google Scholar]
- 63.Koput J, An ab initio study on the equilibrium structure and torsional potential energy function of hydrogen peroxide. Chemical physics letters 1995, 236 (4–5), 516–520. [Google Scholar]
- 64.Hong WT; Stoerzinger KA; Moritz B; Devereaux TP; Yang W; Shao-Horn Y, Probing LaMO3 Metal and Oxygen Partial Density of States Using X-ray Emission, Absorption, and Photoelectron Spectroscopy. The Journal of Physical Chemistry C 2015, 119 (4), 2063–2072. [Google Scholar]
- 65.Dronskowski R; Blöchl PE, Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. The Journal of Physical Chemistry 1993, 97 (33), 8617–8624. [Google Scholar]
- 66.Deringer VL; Tchougréeff AL; Dronskowski R, Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. The journal of physical chemistry A 2011, 115 (21), 5461–5466. [DOI] [PubMed] [Google Scholar]
- 67.Oishi M; Yamanaka K; Watanabe I; Shimoda K; Matsunaga T; Arai H; Ukyo Y; Uchimoto Y; Ogumi Z; Ohta T, Direct observation of reversible oxygen anion redox reaction in Li-rich manganese oxide, Li 2 MnO 3, studied by soft X-ray absorption spectroscopy. Journal of Materials Chemistry A 2016, 4 (23), 9293–9302. [Google Scholar]
- 68.Risch M; Stoerzinger KA; Han B; Regier TZ; Peak D; Sayed SY; Wei C; Xu Z; Shao-Horn Y, Redox processes of manganese oxide in catalyzing oxygen evolution and reduction: An in situ soft X-ray absorption spectroscopy study. The Journal of Physical Chemistry C 2017, 121 (33), 17682–17692. [Google Scholar]
- 69.Renfrew SE; McCloskey BD, Residual lithium carbonate predominantly accounts for first cycle CO2 and CO outgassing of Li-stoichiometric and Li-rich layered transition-metal oxides. Journal of the American Chemical Society 2017, 139 (49), 17853–17860. [DOI] [PubMed] [Google Scholar]
- 70.Mahne N; Renfrew SE; McCloskey BD; Freunberger SA, Electrochemical oxidation of lithium carbonate generates singlet oxygen. Angewandte Chemie International Edition 2018, 57 (19), 5529–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wandt J; Freiberg AT; Ogrodnik A; Gasteiger HA, Singlet oxygen evolution from layered transition metal oxide cathode materials and its implications for lithium-ion batteries. Materials Today 2018, 21 (8), 825–833. [Google Scholar]
- 72.Li L; Castro FC; Park JS; Li H; Lee E; Boyko T; Freeland JW; Yao Z; Fister TT; Vinson J, Probing Electrochemically-Induced Structural Evolution and Oxygen Redox Reactions in Layered Lithium Iridate. Chemistry of Materials 2019, 31 (12), 4341–4352. [Google Scholar]
- 73.Horsley J, Relationship between the area of L 2, 3 x-ray absorption edge resonances and the d orbital occupancy in compounds of platinum and iridium. The Journal of Chemical Physics 1982, 76 (3), 1451–1458. [Google Scholar]
- 74.Yabuuchi N; Nakayama M; Takeuchi M; Komaba S; Hashimoto Y; Mukai T; Shiiba H; Sato K; Kobayashi Y; Nakao A, Origin of stabilization and destabilization in solid-state redox reaction of oxide ions for lithium-ion batteries. Nature communications 2016, 7, 13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yabuuchi N; Takeuchi M; Nakayama M; Shiiba H; Ogawa M; Nakayama K; Ohta T; Endo D; Ozaki T; Inamasu T, High-capacity electrode materials for rechargeable lithium batteries: Li3NbO4-based system with cation-disordered rocksalt structure. Proceedings of the National Academy of Sciences 2015, 112 (25), 7650–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.James A; Goodenough J, Structure and bonding in lithium ruthenate, Li2RuO3. Journal of Solid State Chemistry 1988, 74 (2), 287–294. [Google Scholar]
- 77.McCloskey BD; Bethune DS; Shelby RM; Girishkumar G; Luntz AC, Solvents’ critical role in nonaqueous lithium–oxygen battery electrochemistry. The Journal of Physical Chemistry Letters 2011, 2 (10), 1161–1166. [DOI] [PubMed] [Google Scholar]
- 78.Harding JR Investigation of oxidation in nonaqueous lithium-air batteries. Massachusetts Institute of Technology, 2015. [Google Scholar]
- 79.Nowak SHA,R; Schwartz CP; Gallo A; Abraham B; Garcia-Esparaza AT; Biasin E; Prado A; Maciel A; Zhang D; Day D; Christensen S; Alonso-Mori R; Kroll T; Nordlund D; Weng T-C; Sokaras D , A versatile Johansson-type tender X-ray emission spectrometer. under review 2019. [DOI] [PubMed] [Google Scholar]
- 80.Ravel B; Newville M, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. Journal of synchrotron radiation 2005, 12 (4), 537–541. [DOI] [PubMed] [Google Scholar]
- 81.Perdew JP; Burke K; Ernzerhof M, Generalized gradient approximation made simple. Physical review letters 1996, 77 (18), 3865. [DOI] [PubMed] [Google Scholar]
- 82.Kresse G; Furthmüller J, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational materials science 1996, 6 (1), 15–50. [Google Scholar]
- 83.Kresse G; Furthmüller J, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical review B 1996, 54 (16), 11169. [DOI] [PubMed] [Google Scholar]
- 84.Kresse G; Hafner J, Ab initio molecular dynamics for liquid metals. Physical Review B 1993, 47 (1), 558. [DOI] [PubMed] [Google Scholar]
- 85.Dudarev S; Botton G; Savrasov S; Humphreys C; Sutton A, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study. Physical Review B 1998, 57 (3), 1505. [Google Scholar]
- 86.Wolverton C; Zunger A, First-principles prediction of vacancy order-disorder and intercalation battery voltages in Li x CoO 2. Physical review letters 1998, 81 (3), 606. [Google Scholar]
- 87.Giannozzi P; Baroni S; Bonini N; Calandra M; Car R; Cavazzoni C; Ceresoli D; Chiarotti GL; Cococcioni M; Dabo I, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. Journal of physics: Condensed matter 2009, 21 (39), 395502. [DOI] [PubMed] [Google Scholar]
- 88.Gonze X; Beuken J-M; Caracas R; Detraux F; Fuchs M; Rignanese G-M; Sindic L; Verstraete M; Zerah G; Jollet F, First-principles computation of material properties: the ABINIT software project. Computational Materials Science 2002, 25 (3), 478–492. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.