

Abstract
Severe acute respiratory syndrome (SARS) main protease or 3C-like protease (3CLpro) is essential for the propagation of the coronaviral life cycle and is regarded as one of the main targets for structure-based anti-SARS drug design. It is an attractive approach to find new uses for old drugs as they have already been through extensive clinical testing and could easily be accelerated for clinical approval. Briefly, we performed virtual screening of a database of small molecules against SARS 3CLpro, analyzed inhibitor–protease complexes, and identified several covalent and non-covalent inhibitors. Several old drugs that bind to SARS 3CLpro active site were selected and in silico derivatized to generate covalent irreversible inhibitors with enhanced affinity. Furthermore, we show that pharmacophores derived from clusters of compounds resulting out of virtual screening could be useful probes for future structure–activity relationship studies (SARs) and fine-tune the lead molecules identified.
Keywords: Severe acute respiratory syndrome, Cysteine protease, Virtual screening, Protease inhibitors, 3CL protease
Severe acute respiratory syndrome (SARS) is a life-threatening viral respiratory illness caused by a new coronavirus (CoV). The virus induces symptoms of atypical pneumonia, clinically indistinguishable from similar syndromes and is thought to be of animal origin [1]. In the course of a few months, SARS had spread rapidly from its likely origin in Guangdong Province, China, to 32 countries. World Health Organization (WHO) reported that a total of 8098 people worldwide became sick with SARS that was accompanied by either pneumonia or respiratory distress syndrome and of these 774 patients died. National laboratories, several biotechnology, and diagnostics firms have joined the global rush to combat the infectious disease [2], [3]. The scientific community has already learnt many important lessons from HIV, which could accelerate anti-SARS drug/vaccine development. WHO declared the global outbreak of SARS was contained, as no new cases were reported by August 2003. However, the virus is not yet eradicated.
The important proteins associated with the SARS CoV infection include the polymerase, the spike (S) glycoprotein, the envelope (E) protein, the membrane (M) protein, the nucleocapsid (N) protein, and the 3C-like protease (3CLpro) [4]. SARS 3CLpro, which plays pivotal role in the viral replication, is one of the potential targets for structure-based drug design. SARS 3CLpro has three domains: I (residues 8–101), II (102–184), and III (201–301). Domains I and II, which contains the active site region, are β-barrel domains and III is an α-helical domain. SARS 3CLpro folds similar to generic serine protease but with a catalytic Cys-His dyad playing critical role in the active site. The protease reaction is conjured by the active nucleophile Cys-145 and the acid–base catalyst His-41. Structural conclusions from active site similarity within the coronavirus family and virtual screening on homology models have provided some clues regarding the class of compounds that could interact with SARS protease. Rhinovirus 3Cpro inhibitors like AG7088 fit into the active site pocket of SARS 3CLpro and their derivatives could be potential inhibitors of SARS [5]. The X-ray crystal structure of AG7088-bound HRV protease formed the basis for early anti-SARS drug design (PDB: 1cqq). Further, this is supported by the observation that KZ7088, a derivative of AG7088, could interact with the active site of the SARS protease through six hydrogen bonds [6]. Virtual screening on a 3D model of the SARS 3CLpro against a database of 73 protease inhibitors shows that available protease inhibitors could provide clues toward anti-SARS drug design [7]. Molecular dynamics and docking studies using a database of 29 FDA approved compounds suggested that L-700417, a pseudo C2 symmetric HIV protease inhibitor, fits well into the active site compared to AG7088 [8]. Toney et al. [9] have reported that Sanabandine, a compound from the NCI diversity set, could inhibit SARS protease. The first crystal structure of the SARS 3CLpro was reported in July 2003 (PDB: 1q2w) [10] and a set of crystal structures including SARS 3CLpro in complex with a specific hexa-peptide inhibitor have been reported recently (PDB: 1UJ1, 1UK2, 1UK3, and 1UK4) [11]. Bifunctional aryl boronic acid compounds targeting the cluster of serine residues (Ser139, Ser144, and Ser147) near the active site cavity were found to be effective protease inhibitors [12]. At this juncture, researchers revisited the old adage “old drugs for new bugs” [13], [14] and started investigating old drugs that show potential of inhibiting the replication of SARS-CoV. They have shown that a few old drugs could be used as templates for designing SARS 3CLpro inhibitors [15]. Since these old drugs are originally not designed to inhibit SARS 3CLpro, they have to be fine-tuned to interact with the new target and “taught” how to kill new bugs! The present study employs in silico derivatization as a method to “teach old drugs to kill new bugs.” We have designed irreversible covalent inhibitors by selective derivatization of top non-covalent leads, which includes several old drugs especially a class of HIV inhibitors identified from virtual screening. Our study has resulted in identification of several peptidomimetics and small molecule candidates as potential non-covalent/covalent inhibitors of SARS 3CL protease.
Materials and methods
The catalytically active chain A of the SARS 3CLpro X-ray crystal structure (PDB ID: 1UK4) without the bound CMK peptide and water molecules was used in the study from the PDB structure. The resultant structure was energy minimized using the DISCOVER module of Insight II (Accelrys) and used as the initial structure. A database of more than 15,000 compounds comprising of protease inhibitors (aspartyl, cysteine, serine, and metallo-proteases), HETATM records extracted from PDB (http://www.rcsb.org/) [30], HIV inhibitors (polymerase, integrase, and reverse transcriptase), and a set of thiol reactive compounds filtered from Maybridge (http://www.maybridge.com), Leadquest (http://www.leadquest.com/), ACD laboratories (http://www.acdlabschem.com), and NCI small-molecule databases (http://dtp.nci.nih.gov) was used in the study. Virtual screening of the small molecule inhibitors against SARS 3Clpro was performed using the FlexX module in SYBYL6.9 (Tripos) set at default parameters unless otherwise indicated in the text. We utilized multiple well-known scoring functions: a GOLD-like function [16], a DOCK-like function [17], ChemScore [18], a PMF function [19], and FlexX [20] to rank order the complexes resulting from virtual screening. A list of top 200 compounds was selected from each scoring function, generated and purged into a dataset of 760 unique compounds. These compounds were subjected to physicochemical filters and merged into single database of 330 compounds. Identified lead molecules were in silico derivatized with suitable thiol reactive warheads. The resultant complexes were subjected to Molecular Dynamics simulations and energy minimization using DISCOVER module of Insight II. Molecular dynamics simulations consisted of an initial equilibration of 5 pico seconds (ps) and followed by 100 ps dynamics at 300 K. The final complex structure at the end of the MD simulation was subjected to 5000 steps of steepest descent energy minimization followed by conjugate gradient energy minimization. For all the above calculations, a distance-dependent dielectric constant and non-bonded distance cutoff of 20 Å were used. Molecular graphics images were produced using SYBYL6.9 and the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (http://www.cgl.ucsf.edu/chimera; [21]). Inhibitor–protease interactions were analyzed using occluded surface (OS) [22], [23].
Results and discussion
Design principle
Designing specific inhibitors to block SARS 3CLpro requires a clear understanding of the successful inhibitors designed against class of cysteine proteases. The first set of specific covalent inhibitors for proteases has been designed by adding reactive warheads such as diazo compounds or haloketones to a good substrate of the protease under concern. One of the major disadvantages in using early covalent inhibitors such as haloketones arises due to the inherent reactivity towards non-target molecules. This compromises their stability/selectivity and in turn makes them unsuitable for in vivo studies. The discovery of E64, potent natural epoxysuccinyl inhibitor and Michael acceptors such as AG7088, has shown that lowering the reactivity of the warhead essentially increases the stability, inhibitory potency, and in turn makes them viable for in vivo studies. The entry of AG7088 into clinical trials [24], [25], [26] has rejuvenated the interest in developing irreversible covalent inhibitors for cysteine proteases. Essentially the first step is the subsite mapping with a library of peptide substrates [27]. In lieu of such studies, it is often easier to identify the potential substrate peptide from closer homologues within the family of the protease. A closer examination of the substrate specificity profile of 3C-like proteases of coronaviruses reveals that P1′ position of the substrate is usually small (Gly, Ala or Ser), a conserved Gln at the P1 position, and the P2 position of the substrates seems to favor large hydrophobic residues. The side chains of His163 and Phe140 and the main-chain atoms of Met165, Glu166, and His172 form the S1 subsite, which confers specificity towards Gln. Thus, specific covalent inhibitors of SARS 3CLpro could be designed by substituting the amino acid at the P1′ position with a thiol specific reactive organic moiety like chloromethyl ketone. The affinity for the peptide with correct Pi–Pi′ (where i = 1, 2, 3, etc.) amino acid arrangement has always been the highest. The criteria for any small molecule mimic would be to span the critical length required for the inhibition and make critical interactions with the binding site residues. Secondary structure studies using peptide substrates demonstrate that substrates with more β-sheet like structure tend to react fast [28].
Comparison of SARS 3CLpro homology models with X-ray crystal structures
The availability of X-ray crystal structures with bound ligands facilitates computer-assisted design of structural analogues with increased potency. Due to the lack of X-ray crystal structures before July 2003, we constructed several homology models based on TGEV Mpro (PDB ID: 1lvo). Though there were significant differences between the homology models and the available crystal structures, by comparison of RMS deviations of the binding sites of all the structures alone with respect to that of 1uk4, it could be observed that the differences in the binding site are localized to ‘minor’ loop reorganization and side chain orientations (Fig. 1A ). Our homology models were comparable with other models available at that time (Fig. 1B, marked I). Early structures 1p9s [5], 1p9t [5], and 1p9u [5] provided structural basis for initial virtual screening efforts. These were based on the assumption that the substrate peptide binds in the normal mode i.e., conserved Si–Pi and Si′–Pi′ interactions (where i = 1, 2, 3, etc.). However, in the reported CMK-peptide bound crystal structure, Leu-P2 is partially solvent accessible and does not interact with the S2 subsite. This results in a shift in subsite interaction; Thr-P3 and Asn-P5 occupy the S2 and S4 subsites, respectively. This unusual mode of binding could attribute to lower specificity of the P2-amino acid in comparison with other coronaviruses [11]. The authors have also reported large cooperative movements of the side chains of Glu166, Phe140, Leu141, and Tyr118, and the N terminus of the partner protomer in the dimer as a function of pH. Especially there is a marked difference amongst the structures crystallized at different pH (PDB: 1uj1, 1uk2, 1uk3, and 1uk4; Fig. 1B, marked II). Our earlier virtual screening studies with homology models of the SARS 3CLpro provided clues about potential protease inhibitors.
Fig. 1.
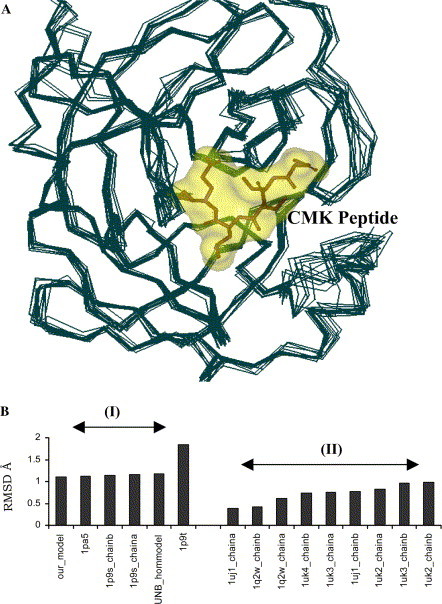
(A) Superposition of SARS 3CLpro homology models and structure homologues with X-ray crystal structure (PDB: 1UK4, bound CMK peptide is shown as a stick model). (B) Plot of the RMS deviations of the homology models with respect to the X-ray crystal structure (PDB: 1UK4 chain A): (I) structure homologues (1pa5, 1p9s, and 1p9t) and homology models (UNB model [29] and our model) and (II) SARS 3CLpro crystal structures.
Virtual screening of potential small molecule inhibitors of SARS 3Clpro
We eliminated molecules with poor scores based on our early virtual screening studies with SARS 3CLpro homology models. This not only reduced about 40% of unwanted molecules but also reduced the computer time taken for virtual screening against the X-ray crystal structure as outlined in Materials and methods. Virtual screening resulted in 330 unique compounds with 157 small molecule hits containing at least one thiol reactive functional group. These compounds could potentially serve as covalent inhibitors of SARS 3CLpro. Analysis of the docked complexes reveals that the thiol reactive functional groups are not properly oriented towards catalytic Cys145 in most of the docked complexes. Out of the 157 complexes only 17 compounds including KZ7088 were oriented properly towards Cys145 but still were 4–5 Å away to induce possible nucleophilic attack. Reorganization of the ligand fragments to provide proper orientation for protease reaction disrupted critical interactions with the binding site residues. In the process, we learnt that it was easier to modify small molecules that do not contain thiol functionality but bind well to the protease pocket.
Apart from the 157 potential covalent inhibitors, virtual screening has also resulted in 173 molecules that did not contain any thiol reactive functionality. Based on visual examination, we selected 58 small molecules that bind to the substrate-binding pocket by mimicking several critical hydrogen bonding interactions similar to the CMK peptide. The identified non-covalent inhibitors were composed of several popular HIV protease inhibitors (as shown in Fig. 2 , structures #1–15): Amprenavir (141W94; VX-478, agenerase; #4); DMP-323 (XM323; #5), Nelfinavir (Viracept, AG1343, #6), A-98881 (#7), U89360 (#8), Indinavir (L-735524; Crixivan, #9), L700417 (#10), Lopinavir (ABT378, Aluviran, #12), A80987 (Ritonavir derivative, #13), BMS-186318 (#14), and Saquinavir (Invirase, Fortovase, Ro 31-8959, #15). It is noteworthy that 33 of the 58 non-covalent inhibitors identified in the study were potent HIV protease inhibitors.
Fig. 2.
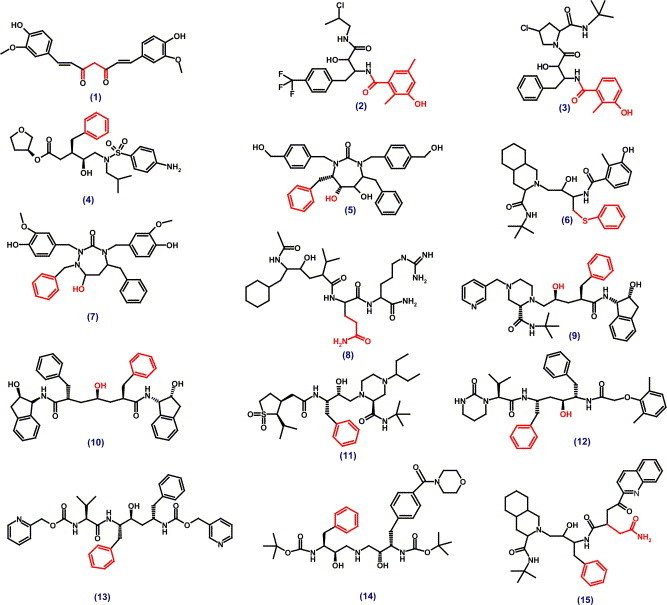
2D chemical structures of non-covalent inhibitors of SARS 3CLpro (#1–15). Red highlights indicate the moiety selected for in silico derivatization. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
SARs on SARS?
Pharmacophores derived from clusters of compounds resulting from virtual screening form excellent data set for future structure–activity relationship studies (SARs). We observed that 9 out of the 33 HIV protease inhibitors shared similar pharmocophoric features (Fig. 2, structure #3). A 2D structure-based search of the pharmacophore using CrossFire Commander V6 (MDL) resulted in 64 molecules with various functional groups at R1-11 positions. Fig. 3 shows the docked pose of 53 successful hits out of the 64 identified compounds in SARS 3CLpro-binding site. Steric substituents at R1-5 (highlighted in Red, Fig. 3) alter the scores drastically when compared to functional groups at R6-10 positions. Molecules with a hydroxyl- or an amino-group at the R2 position are favored. These molecules could potentially serve directly as non-covalent inhibitors of SARS 3CLpro or provide templates for designing covalent inhibitors as described in the following section.
Fig. 3.
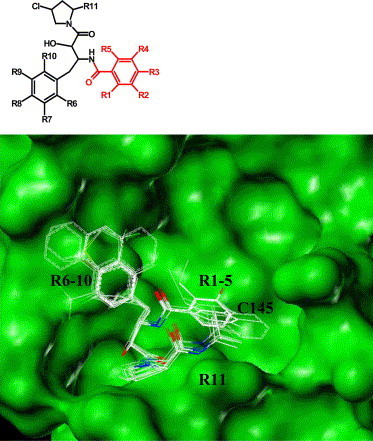
Illustration of the binding modes of 53 successful ligands selected from pharmacophore search in SARS 3CLpro-binding site. Functional group variations with respect to the pharmacophore are indicated by R1-11.
Teaching old drugs to kill new bugs
We reexamined the set of non-covalent inhibitor bound complexes and observed the class of HIV inhibitors that fit well in SARS 3CLpro active site. In an attempt to design irreversible covalent inhibitors, the top candidates from the virtual screening have been subjected to a rule-based secondary screening to select the small molecules that lie within 2–3 Å away from the Sγ of Cys-145. The resultant candidates were subjected to in silico derivatization and thiol reactive organic warheads had been incorporated at appropriate chemically viable positions as shown in Fig. 4A . The warheads were covalently ligated to Sγ of Cys-145 and the structures were re-minimized and those with bumps and structural deformity arising out of the new linkage were carefully eliminated by visual examination. In this study, we have incorporated thiol-reactive organic moieties or “warheads,” extracted from the 157 covalent inhibitors identified in the study, with both fast and slow reactivity as shown in Fig. 4B. Analyses of the inhibitor-bound complexes reveal that covalent inhibitor picks up more interactions compared to its non-covalent analogue. A list of top non-covalent inhibitors selected after secondary rule-based screen with functional groups selected for in silico derivatization is highlighted in red (Fig. 2, Fig. 3, Fig. 4). Graphical illustration of cyclic urea-based non-covalent inhibitor (colored in orange) and its covalent analogue (colored in cornflower blue) bound to SARS 3CLpro active site is shown in Fig. 5 . Visual examination of the interacting residues shows that the covalent inhibitor interacts with the binding site residues much better than the non-covalent analogue.
Fig. 4.
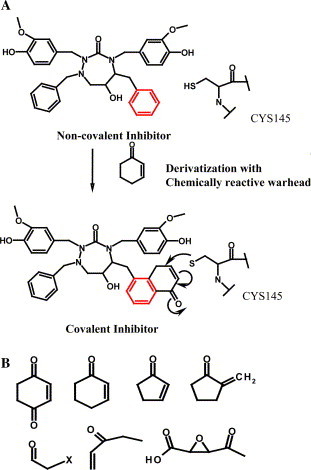
(A) Graphical illustration of in silico derivatization or the “Teaching” strategy; (B) List of organic warheads used in the study.
Fig. 5.
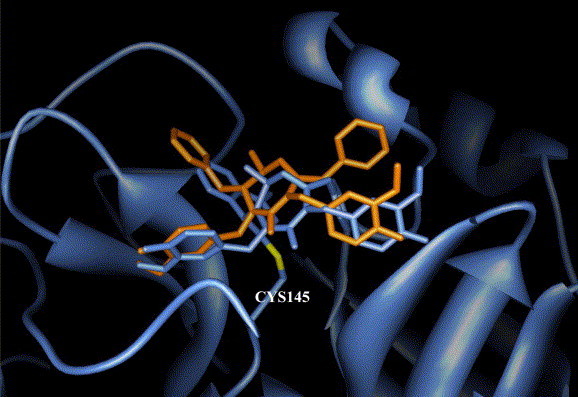
Comparison of binding modes of the cyclic urea-based HIV protease inhibitor (colored in orange) and its covalent analogue (colored in cornflower blue) to SARS 3CLpro active site. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Analysis of inhibitor–protease interactions
The CMK peptide inhibitor forms six hydrogen bonds with 3CLpro active site residues Phe140, Ser144, Cys145, His163, Glu166, and Gln189 (PDB: 1uk4). Most of the top ranked inhibitors picked in our study form at least 4 hydrogen bonds and the corresponding interacting residues are as follows: Thr26, Asn119, Phe140, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, and Gln189. Figs. 6A–C illustrate the docked poses of the CMK peptide, HIV inhibitor (non-covalent inhibitor) and covalent irreversible analogue of the HIV inhibitor, respectively, in SARS 3CLpro-binding pocket.
Fig. 6.
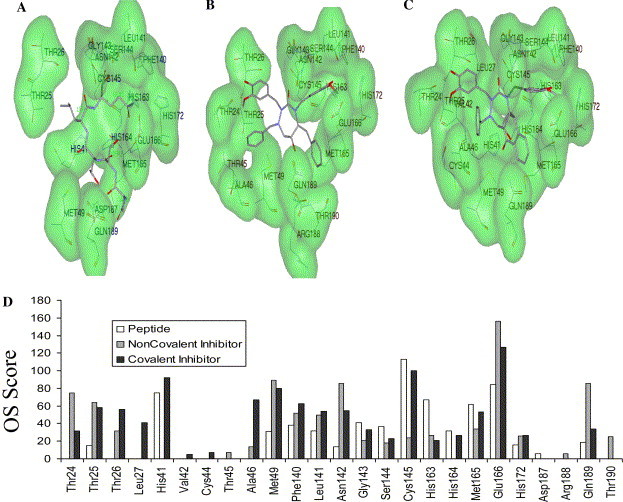
Occluded surface-based comparison of protease–inhibitor interactions. SARS 3CLpro bound to (A) CMK peptide, (B) non-covalent inhibitor, and (C) covalent analogue of B. Surfaces of OS-identified interacting residues are colored green. (D) Quantitative graphical analyses of interacting residues using OS. The interacting residues are plotted in the x-axis and the corresponding OS scores in y-axis for the protease–inhibitor complexes A–C. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Occluded surface program (OS), a package of programs to calculate the occluded surface and atomic packing of protein structures developed by Pattabiraman et al., is used to analyze inhibitor–protease interactions. Occluded surface is defined as the molecular surface that is less than 2.8 Å from the surface of neighboring non-bonded atoms. That is, if a water molecule cannot fit between two atoms they occlude each other. Occluded surface is similar to buried surface but is more sensitive to packing geometry than buried surface using a rolling probe. To calculate occluded surface, normals at the molecular surface are extended outward until they intersect neighboring van der Waals surface. The collection of extended normals, and their respective lengths, defines the packing of each atom in a structural model. A combination of occluded surface area and average length of the normals was used to obtain the occluded surface packing (OSP) value for each residue and the analysis of inter-chain occluded surface allows a detailed calculation of protein–protein interactions.
Surfaces of OS-identified interacting residues of SARS 3CLpro active site are highlighted in Figs. 6A–C. Occluded surface scores for each ligand atom with corresponding atoms from the binding site were generated. The OS scores averaged per amino acid give a quantitative measure of the protein–inhibitor interactions. The non-covalent inhibitor does not interact with the catalytic residue His41. The corresponding covalent analogue interacts with His41 better than the CMK peptide (Fig. 6D). Both the CMK peptide and the covalent inhibitor bound to Cys145 have higher OS score for this residue compared to the corresponding non-covalent analogue (Fig. 6D). However, the non-covalent inhibitor interacts with the residues Glu166 and Gln189 better than the covalent and the CMK peptide. It is evident that OS scores could quantitatively differentiate the interactions of covalent and non-covalent analogue of the HIV inhibitors with binding site residues of SARS 3CLpro.
We are in the process of in vitro biological testing of top non-covalent inhibitors using cloned SARS protease and have initiated the ex silico derivatization of top covalent irreversible inhibitors identified in the study. These results would help focus the substrate-optimization and lead discovery.
Conclusions
We have used structure-based screening to identify compounds that bind to the SARS 3CLpro-binding site. These molecules could potentially retard the proteolytic action of the SARS protease and be used in combination with other anti-viral therapeutics. Protease inhibitor design has evolved beyond mere addition of reactive warheads to cognate protease substrates. In silico derivatization of viable functional groups as identified by the secondary rule-based screen enables the old drugs to react with Cys145 by serving as centers of nucleophilic attack. The use of low and high reactive organic warheads provides means to control the reactivity of the inhibitor, its stability, inhibitory potency and in turn helps in designing inhibitors viable for in vivo studies. Thus, our strategy not only educates old drugs to kill new bugs but also teaches them to behave according to the needs. Occluded Surfaces generated scores provide unique and novel method for quantitative comparison of non-covalent and covalent inhibitor bound complexes. Unlike with a new drug, old drugs or drugs with minimal modifications do not have to undergo extensive pre-clinical testing to prove their safety, efficacy and have the possibility of gaining accelerated approval by US Food and Drug Administration. Our strategy could be extended to identify potent inhibitors and fine-tune old drugs against other disease targets that are cysteine proteases such as cathepsins, caspases, calpains, and papain.
Acknowledgment
We acknowledge the National Cancer Institute (NCI) for allocation of computing time and staff support at the Advanced Biomedical Computing Center of the National Cancer Institute, Frederick.
References
- 1.Peiris J.S. Severe acute respiratory syndrome (SARS) J. Clin. Virol. 2003;28:245–247. doi: 10.1016/j.jcv.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu P. Biotech firms jump on SARS bandwagon. Nat. Biotechnol. 2003;21:720. doi: 10.1038/nbt0703-720a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbott A. US Army joins hunt for SARS drug. Nature. 2003;423:103. doi: 10.1038/423103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 5.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 6.Chou K.C., Wei D.Q., Zhong W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y., Yue L.D., Duan W.H., Shen J.K., Qin L., Shi T.L., Li Y.X., Chen K.X., Luo X.M., Shen X., Shen J.H., Jiang H.L. A 3D model of SARS_CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 8.Jenwitheesuk E., Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003;13:3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toney J.H., Navas-Martin S., Weiss S.R., Koeller A. Sabadinine: a potential non-peptide anti-severe acute-respiratory-syndrome agent identified using structure-aided design. J. Med. Chem. 2004;47:1079–1080. doi: 10.1021/jm034137m. [DOI] [PubMed] [Google Scholar]
- 10.J.B. Bonanno, R. Fowler, S. Gupta, J. Hendle, D. Lorimer, R. Romero, M. Sauder, C.L. Wei, E.T. Liu, S.K. Burley, T. Harris, X-Ray Crystal Structure of the Sars Coronavirus Main Protease. Available from: <http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1q2w> (2003)
- 11.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CL(pro) Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 13.Vastag B. Old drugs for a new bug: influenza, HIV drugs enlisted to fight SARS. JAMA. 2003;290:1695–1696. doi: 10.1001/jama.290.13.1695. [DOI] [PubMed] [Google Scholar]
- 14.Koren G., King S., Knowles S., Phillips E. Ribavirin in the treatment of SARS: A new trick for an old drug? CMAJ. 2003;168:1289–1292. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X.W., Yap Y.L. Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main proteinase with HIV, psychotic and parasite drugs. Bioorg. Med. Chem. 2004;12:2517–2521. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones G., Willett P., Glen R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 17.Meng E.C., Shoichet B.K., Kuntz I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992;13:505–524. [Google Scholar]
- 18.Eldridge M.D., Murray C.W., Auton T.R., Paolini G.V., Mee R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 19.Muegge I., Martin Y.C. A general and fast scoring function for protein–ligand interactions: a simplified potential approach. J. Med. Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 20.Rarey M., Kramer B., Lengauer T., Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 21.Huang C.C., Couch G.S., Pettersen E.F., Ferrin T.E. Chimera: an extensible molecular modeling application constructed using standard components. Pac. Symp. Biocomput. 1996;1:724. [Google Scholar]
- 22.Pattabiraman N. Occluded molecular surface analysis of ligand-macromolecule contacts: application to HIV-1 protease–inhibitor complexes. J. Med. Chem. 1999;42:3821–3834. doi: 10.1021/jm980512c. [DOI] [PubMed] [Google Scholar]
- 23.Pattabiraman N., Ward K.B., Fleming P.J. Occluded molecular surface: analysis of protein packing. J. Mol. Recognit. 1995;8:334–344. doi: 10.1002/jmr.300080603. [DOI] [PubMed] [Google Scholar]
- 24.Witherell G. AG-7088 Pfizer. Curr. Opin. Investig. Drugs. 2000;1:297–302. [PubMed] [Google Scholar]
- 25.Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T., Zhou R., Tikhe J., Ford C.E., Meador J.W., Ferre R.A., Brown E.L., Binford S.L., Brothers M.A., DeLisle D.M., Worland S.T. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dragovich P.S., Prins T.J., Zhou R., Webber S.E., Marakovits J.T., Fuhrman S.A., Patick A.K., Matthews D.A., Lee C.A., Ford C.E., Burke B.J., Rejto P.A., Hendrickson T.F., Tuntland T., Brown E.L., Meador 3rd J.W., Ferre R.A., Harr J.E., Kosa M.B., Worland S.T. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 1999;42:1213–1224. doi: 10.1021/jm9805384. [DOI] [PubMed] [Google Scholar]
- 27.Thornberry N.A., Rano T.A., Peterson E.P., Rasper D.M., Timkey T., Garcia-Calvo M., Houtzager V.M., Nordstrom P.A., Roy S., Vaillancourt J.P., Chapman K.T., Nicholson D.W. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 28.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.A. Wiley, G. Deslongchamps, Homology Model of SARS-CoV Mpro Protease. Available from: <http://biocomp.chem.unb.ca:8080/GD/SARS/> (2003)
- 30.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]