

Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV) is the etiological agent of SARS. Analysis of SARS-CoV spike glycoprotein (S) using recombinant plasmid and virus infections demonstrated that the S-precursor (proS) exists as a ∼190 kDa endoplasmic reticulum form and a ∼210 kDa Golgi-modified form. ProS is subsequently processed into two C-terminal proteins of ∼110 and ∼80 kDa. The membrane-bound proprotein convertases (PCs) furin, PC7 or PC5B enhanced the production of the ∼80 kDa protein. In agreement, proS processing, cytopathic effects, and viral titers were enhanced in recombinant Vero E6 cells overexpressing furin, PC7 or PC5B. The convertase inhibitor dec-RVKR-cmk significantly reduced proS cleavage and viral titers of SARS-CoV infected cells. In addition, inhibition of processing by dec-RVKR-cmk completely abrogated the virus-induced cellular cytopathicity. A fluorogenically quenched synthetic peptide encompassing Arg761 of the spike glycoprotein was efficiently cleaved by furin and the cleavage was inhibited by EDTA and dec-RVKR-cmk. Taken together, our data indicate that furin or PC-mediated processing plays a critical role in SARS-CoV spread and cytopathicity, and inhibitors of the PCs represent potential therapeutic anti-SARS-CoV agents.
Keywords: SARS-CoV, Proprotein convertases, Furin, Inhibitor, Viral infection, Viral spread, Biosynthesis, Spike glycoprotein processing
Severe acute respiratory syndrome (SARS) is an emerging disease which was first reported in the Guangdong province of China in November 2002. Worldwide efforts led to the rapid isolation of the etiological agent as SARS-CoV, a novel member of the family Coronaviridae [1]. Complete genome sequencing of the SARS-CoV [2], [3] confirmed that this pathogen is not closely related to any previously known coronavirus serogroups and was therefore classified in a new group IV, although it shares similarity with group II. Budding of the SARS-CoV occurs in the Golgi [4], resulting in the viral incorporation of the envelope spike glycoprotein (S). The S-protein of SARS-CoV is a type I membrane-bound protein essential for host cell receptor–viral attachment and target cell fusion. Cell surface expression of S may lead to a receptor mediated cell–cell fusion resulting in multinucleated giant cell formation, as observed in the lungs of SARS patients [5]. Angiotensin converting enzyme-2 (ACE2) was recently identified as a functional receptor of SARS-CoV S-glycoprotein [6].
The precursor proS of group II and III coronaviruses, but not of group I, is cleaved at basic residues into S1 and S2 subunits [7]. This cleavage occurs in the trans Golgi network (TGN) by specific protease(s) likely to be one or more of the basic amino acid (aa)-specific proprotein convertases (PCs) that recognize an RXXR↓ motif [8], [9]. These convertases cleave secretory precursors at the general motif (K/R)–(X)n–(K/R)↓, where n = 0, 2, 4, or 6, and are responsible for the tissue-specific limited proteolysis of multiple polypeptide precursors [9], [10]. Of the seven basic aa-specific PCs, only three are membrane-bound: furin, PC7, and the isoform PC5B, and two others are widely expressed as soluble proteins: PACE4 and PC5A [9].
Alignment of SARS-CoV S-sequence with representative group II coronavirus S-glycoproteins (Fig. 1 A) suggested that basic aa-specific PCs are likely to be their convertases. Indeed, it was recently shown in cell culture that a specific PC inhibitor, the membrane-permeable peptide decanoyl-RVKR-chloromethylketone (dec-RVKR-cmk) [11], reduced the processing of the murine hepatitis coronavirus (MHV) and its cell–cell but not its virus–cell fusion [8]. Despite the conservation of 19 out of 20 cysteines within the S1 subunit of group II with those of the S-protein of SARS-CoV [12], the latter seems to have a distinct sequence lacking a PC-like motif (site A; Fig. 1A). In contrast, a candidate PC-like cleavage site (RNTR 761↓) is located C-terminal to site A (site B; Fig. 1A). We therefore hypothesized that SARS-CoV S-glycoprotein could be processed by PCs and that PC-specific inhibitors, like dec-RVKR-cmk, might inhibit the processing. Herein, we demonstrate that overexpression of membrane-bound PC-like enzymes enhances the processing of an ∼80 kDa protein and inhibition of processing leads to a dramatic decrease in SARS-CoV titer, cellular spread, and abrogation of cytopathic effects (CPE).
Fig. 1.
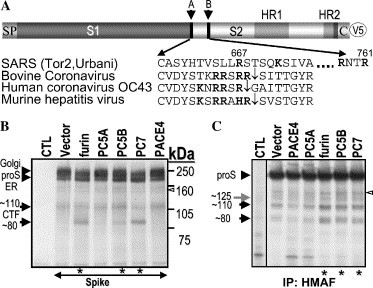
Processing of the SARS-CoV spike glycoprotein precursor. (A) Schematic representation of the S-protein. Arrows point to potential cleavage sites (sites A and B). The signal peptide (SP), S1/S2 domains, heptad repeats (HR1, HR2), cytosolic tail (CT), and V5-epitope (V5) are illustrated. Underneath the spike scheme is an amino acid (aa) alignment of group II coronavirus cleavage sites with the SARS-CoV S. Basic aa are emphasized in bold. Downward arrows indicate group II proS identified cleavage sites. (B) FD11 cells stably expressing either the vector alone or a defined convertase were transiently transfected with the spike glycoprotein cDNA. At 48 h post-transfection with the designated constructs, cell lysates were V5-immunoprecipitated, proteins were resolved by 7% SDS–PAGE, and analyzed by Western blot with a V5 antibody. (C) Vero E6 cells stably expressing the PCs were infected with SARS-CoV, control (CTL) representing uninfected cells. After virus adsorption at a multiplicity of infection (MOI) of one for 1 h, inoculums were removed. Pulse-labeled cell lysate proteins were HMAF–immunoprecipitated and resolved by 7% NuPAGE. Proteins were visualized by autoradiography. Asterisk (∗) denotes increased spike processing.
Materials and methods
Plasmids and expression vector. Three Tor2 SARS-CoV cDNA clones (B21, H21, and N08) were purchased from the British Columbia Cancer Agency (BC, Canada). These clones were assembled by polymerase chain reaction (PCR) to get the full length SARS-CoV proS cDNA fused to a C-terminal V5 epitope in the bicistronic mammalian expression vector pIRES2-EGFP (Invitrogen, Burlington, ON, Canada) between NheI and BamHI sites (pIR-proS-V5). Recombinant proS-V5 DNA sequence was confirmed by DNA sequencing. The R667A, K672A, R758A, R797A, and RRKR667 mutations were introduced into the pIR-proS-V5 by PCR.
Cells and transfection. African green monkey Vero E6 kidney cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. Furin deficient FD11 Chinese hamster ovary (CHO) derived cells were generously given by Dr. Stephen Leppla (National Institutes of Health, MD, USA) and cultured as reported [13]. The cDNAs of PCs (hfurin, mPC5A, mPC5B, rPC7, and hPACE4) were cloned into the pIRES2-EGFP vector. FD11 cells (furin deficient) were transfected with individual PCs using Lipofectamine 2000 (Invitrogen) and stable cells were selected for 14 days with 800 μg/mL Geneticin (Invitrogen), followed by a single round of FACS selection for EGFP using a method described before [14]. Vero E6 cells overexpressing the above-mentioned PCs were produced in a similar manner and were selected after 48 h post-transfection with 500 μg/mL Geneticin for 14 days. A 100 mm dish of each FD11 cell line stably overexpressing a PC was transfected with 24 μg of the pIR-proS-V5 DNA. FD11-furin cell line was also transfected by the same technique with pIR-proS-V5 mutant forms of vectors (R667A, K672A, R761Q, R761K, R758A + R761K, R758A, R797A, and RRKR667).
Antibodies and Western blot. Forty-eight hours (h) after transfections, cells were washed twice with PBS and lysed in RIPA buffer [50 mM Tris/HCl, pH 8.0, 1% (v/v) Nonidet P40, 0.5% sodium deoxycholate, 150 mM NaCl, and 0.1% (v/v) sodium dodecyl sulfate (SDS)] with a Complete Protease Inhibitor Cocktail (Roche Diagnostics GmbH). Clarified lysates were immunoprecipitated with a monoclonal anti-V5 antibody (Invitrogen). Proteins were precipitated with protein A/G plus agarose beads (Santa Cruz). Immunoprecipitated proteins were separated by SDS–polyacrylamide gel electrophoresis (SDS–PAGE, 7% gel) and transferred to polyvinylidene fluoride membranes (Schleicher and Schuell). Membranes were then incubated with horseradish peroxidase coupled V5-antibody (Invitrogen) for 16 h at 4 °C. Proteins were revealed with the chemiluminescent ECL plus reagent (Amersham Biosciences). SARS-CoV specific hyperimmune mouse ascitic fluid (HMAF) was kindly provided by Dr. Pierre Rollin and Thomas G. Ksiazek (Centers for Disease Control and Prevention, Atlanta, GA). HMAF for SARS-CoV was made by a previously described protocol [15].
Peptides and in vitro assay. The intramolecularly quenched fluorogenic spike peptides (QS1 = Abz-E755-Q-D-R-N-T-R761-E-V-F-A- Q-3-Nitro-Y-CONH2, QS2 = Abz-P-D-P-L-K-P-T-K-R797-S-F-I-E-D-3-Nitro-Y-Ala- CONH2, and QS3 = Abz-S-Y-H-T-V-S-L-L-R667-S-T-S-3-Nitro-Y-CONH2, where Abz = 2-amino benzoic acid) were synthesized by solid phase Fmoc chemistry using Pioneer instrument (ABI) as described earlier [16], [17]. Crude peptides were purified and characterized by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (ABI). In vitro digestion of each peptide (20 μg) was carried out at 37 °C for 8 h in a total volume of 50 μL buffer consisting of 25 mM Tris + 25 mM 2-morpholinoethanesulfonic acid + 2.5 mM CaCl2, pH 7.4, with recombinant furin (New England Biolab, 2 μL) exhibiting an enzymatic activity of 2 U/μL, 1 U representing the release of 1 pmol/minute (pmol/min) of free 7-amino-4-methylcoumarin from 100 μM Boc-RVRR-4-methylcoumarin-7-amide. In addition, similar digestions were also performed in the presence of either 10 μM dec-RVKR-cmk or 20 mM EDTA following a 20 min pre-incubation with the enzyme before the quench peptide substrate was added. Each digestion was monitored for 8 h by progress curves and end time assay (excitation and emission wavelengths fixed at 320 and 420 nm, respectively), following which the reaction was stopped with glacial acetic acid (2 μL) and the resultant crude digests were analyzed by reverse phase high pressure chromatography (RP-HPLC) with online dual (UV and fluorescence) detection systems followed by MALDI-TOF mass spectrometry analysis of each collected peptide.
SARS-CoV infection, metabolic labeling, and immunofluorescence. The proprotein convertase inhibitor dec-RVKR-cmk was purchased from Biomol (PA, USA). Vero E6 cells stably expressing the PCs (see text above) were infected with SARS-CoV at a multiplicity of infection (MOI) of one for 1 h. After removal of the inoculums, cells were extensively washed with PBS. Eighteen hours post-infection, prior to labeling with [35S]cysteine, the virus containing supernatants was removed, subjected to centrifugation at 12,000 revolutions per minute, and stored at −80 °C until titrations were performed. Cells were pulsed with [35S]cysteine for 30 min and lysed in RIPA. Clarified cell lysates were incubated with HMAF and immunoprecipitated proteins were separated by NuPAGE (7% gel, Invitrogen). Gels were dried and proteins were revealed by autoradiography. For dec-RVKR-cmk pulse labeling experiments, Vero E6 cells were infected for 1 h with SARS-CoV at MOI = 0.5 (see text above). After virus removal, cells were incubated in 1 mL OPTI-MEM medium (Invitrogen) containing 60 μM dec-RVKR-cmk for 18 h. The onset of CPE was monitored by light microscopy. Immunofluorescence staining was performed with HMAF. Briefly, cells were fixed with a mix an equimolar solution of methanol and acetone for 15 at −20 °C. SARS-CoV reactive protein(s) were revealed with an anti-mouse fluorescein coupled antibody (ICN).
Treatments of SARS-CoV infected Vero E6 cells with dec-RVKR-cmk were achieved with the following protocol. Upon removal of the inoculums and extensive washing, cells were maintained in fresh OPTI-MEM medium with 15, 30, or 60 μM concentration of dec-RVKR-cmk for 15–18 h. Cells were then labeled for 30 min with [35S]cysteine in the presence of appropriate quantities of dec-RVKR-cmk. In some experiments, cells were chased for 3 h with cold medium. Clarified cell supernatants were also immunoprecipitated with HMAF, and in parallel-unlabeled media were stored at −80 °C for viral titer determination. Titration was performed on a fresh monolayer of Vero E6 cells. Serial dilutions of the virus containing supernatants were added to cells followed by agar overlay. The plaques were visualized by neutral red staining and counted.
Results
Processing of recombinant SARS-CoV proS
In order to address the significance of the PCs in proS processing, we cloned the S gene in a pIRES2-EGFP bicistronic vector and added a V5-tag at its C-terminus for immunological detection (pIR-proS-V5; Fig. 1A). Addition of such an epitope in C-terminal was reported not to interfere with the SARS-CoV spike glycoprotein mediated fusion [18]. We first obtained furin-deficient FD11 cells [19] stably expressing either the vector alone or one of the PCs of the constitutive secretory pathway, namely: furin, PC5A, PC5B, PC7, and PACE4. A functional proprotein convertase activity assay showed that these enzymes induced complete processing of pro-platelet derived growth factor-A (proPDGF-A) [20] into PDGF-A (not shown). The expression and processing of the spike protein was analyzed by transient transfection of the cDNA encoding S, followed by Western blotting using a V5 reactive monoclonal antibody. In absence of overexpressed convertase (vector) we observed the presence of a ∼190 kDa and a ∼210 kDa proteins (Fig. 1B, lane 2). Endoglycosidase treatment indicated that the ∼190 kDa protein was endoH sensitive, possibly of an endoplasmic reticulum (ER) form, and the ∼210 kDa protein was endoH resistant, likely representing a Golgi-modified form (not shown). A very small amount of non-glycosylated proS was also detected (∼150 kDa). In addition to the ER and Golgi forms, we also observed two V5-immunoreactive C-terminal fragments (CTFs): a ∼110 kDa protein and a very small amount of a ∼80 kDa protein (Fig. 1B, lane 2). Interestingly, the level of the ∼80 kDa CTF was enhanced by overexpression of the membrane-bound furin or PC7, and least by PC5B (Fig. 1B, lanes 3, 5, and 6), but not by the soluble PC5A or PACE4 (lanes 4 and 7), nor by a soluble form of furin lacking the transmembrane domain (not shown). We also noticed that whenever processing took place, the level of the ∼210 kDa proS was significantly reduced. This agrees with the known zymogen activation of furin, PC7, and PC5B in the trans Golgi network (TGN). The ∼110 kDa product was least affected by the overexpression of the PCs. The enhancement in the appearance of the ∼80 kDa CTF in membrane-bound PC-overexpressing cells suggests that the spike protein of SARS-CoV is a potential substrate for PC-dependent cleavage. It remains to be seen whether the cleavage is a result of direct action of the PCs or indirect via the activation by PCs of other cellular proteases to effect the generation of the ∼80 kDa CTF.
ProS is cleaved in SARS-CoV infected cells
In order to verify the relevance and authenticity of the observed processing products of recombinant proS, we analyzed the biosynthesis of proS produced following SARS-CoV infection of Vero E6 cells stably expressing either an empty vector or a recombinant of each of the PCs. Sixteen hours post-infection, the cells were pulse-labeled with [35S]cysteine for 30 min and the cell lysates were immunoprecipitated with the spike reactive polyclonal antibody HMAF. In the vector co-expressing cells, a ∼190 kDa protein was visualized together with significant amounts of ∼110 and ∼80 kDa proteins, and low levels of ∼150 and ∼125 kDa proteins (Fig. 1C, vector lane). Interestingly, the levels of the ∼110 and ∼80 kDa products were significantly increased upon overexpression of furin, PC7, and PC5B (Fig. 1C, lanes 5–7), the same convertases that enhanced the production of the ∼80 kDa CTF from recombinant proS (Fig. 1B). The ∼125 kDa product, which was not observed in the V5 antibody experiments (Fig. 1B), might represent an N-terminal S1 fragment, only seen with the HMAF antibody. Thus, apart from the ∼125 kDa protein, the SDS–polyacrylamide gel electrophoresis (SDS–PAGE) profile of SARS-CoV infected cells closely resembled that observed with the recombinant proS in furin-expressing FD11 cells (Fig. 1B). The low levels of the processed proteins precluded microsequencing analysis, and hence the N-terminal sequences of the cleavage products could not be determined.
Enhanced virus release in Vero E6 cells stably expressing furin, PC7 or PC5B
Since membrane-bound PCs increased the processing of proS (Figs. 1B and C), we measured the cytopathic effect (CPE) (Table 1 ) and the SARS-CoV titers (Fig. 2 ) from those cells 24 h post-infection. We observed a correlation between the efficiency of processing with both the extent of CPE (Table 1) and titer of the virus recovered from the media (Fig. 2A). Accordingly, PC7, furin, and PC5B released more viruses resulting in more extensive CPE than either vector alone or PC5A expressing Vero E6 cells. Although the increase in virus titer is modest (∼2- to 3-fold), it concurs with the notion that PC-mediated processing of the spike protein enhances virus release.
Table 1.
CPE evaluation of SARS-CoV infected cells
| CPE | |
|---|---|
| Cell line | |
| Vector | +++ |
| PACE4 | +++ |
| PC5A | +++ |
| PC5B | +++++ |
| Furin | ++++++ |
| PC7 | +++++++ |
| dec-RVKR-cmk | |
| None | +++++++ |
| 15 μM | +++++ |
| 30 μM | +++ |
| 60 μM | − |
Vero E6 overexpressing PCs (cell line) or incubated with dec-RVKR-cmk. Vero E6 were infected at a MOI of 0.5 for 1 h. Inoculums were removed and medium containing or not different doses of dec-RVKR-cmk were added to the cells. Twenty-four hours later cytopathic effects (CPE) were monitored by microscopy.
Fig. 2.
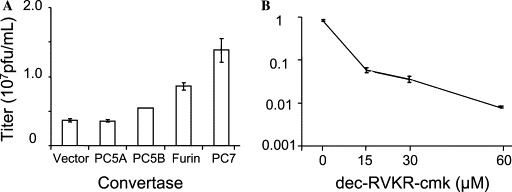
The membrane-bound PCs increase SARS-CoV titers and their inhibition by dec-RVKR-cmk reduces SARS-CoV titers. (A) Cells were infected as in Fig. 1C. Twenty-four hours post-infection, clarified cell supernatants were titrated. Serial dilutions of the virus containing supernatant were added to a fresh monolayer of Vero E6 cells followed by agar overlay. The plaques were visualized by neutral red staining. (B) Vero E6 cells were infected as in Fig. 1C, except that different doses of dec-RVKR-cmk were added to the media after inoculum removal. Viruses were titrated as described in (A). These data are representative of at least four independent experiments and are presented as average with the corresponding standard deviation.
PC inhibition reduce, SARS-CoV spread and abrogate, CPE
Since membrane-bound PCs increased both spike processing and viral titers, we investigated whether the membrane-permeable PC-inhibitor, decanoyl-RVKR-chloromethylketone (dec-RVKR-cmk), could interfere with CPE, viral spread, and proS processing. This type of PC-inhibitor has been widely used to block glycoprotein processing and infectivity of several viruses including HIV-1 [11], respiratory syncytial virus [21], and MHV [8]. Thus, Vero E6 cells were infected with SARS-CoV for 1 h without inhibitor; the inoculums were then changed with fresh media supplemented with 60 μM dec-RVKR-cmk and incubated for 15–24 h. Microscopic examination of the cell monolayer indicated that the infected cells showed extensive CPE, while virtually no CPE was observed with the dec-RVKR-cmk treated cells (left panels in Fig. 3 ). This demonstrates that the extracellularly added PC-inhibitor abrogated the CPE caused by SARS-CoV infection. In an attempt to gain more insight into the infection process, we performed immunofluorescence of dec-RVKR-cmk treated and untreated infected cells (right panels in Fig. 3). Infected cells showed extensive antigen staining while dec-RVKR-cmk treated cells showed a modest number of cells infected with greatly reduced spreading evident throughout the intact monolayer. The concentration dependence of the dec-RVKR-cmk effect revealed that at 60 μM the viral titer was reduced by a factor of ∼100 (Fig. 2B) and that the CPE was abolished (Table 1). These results strongly suggested the possibility that the inhibitor dec-RVKR-cmk blocks a PC-mediated processing step required for extensive virus spread and ensuing CPE.
Fig. 3.
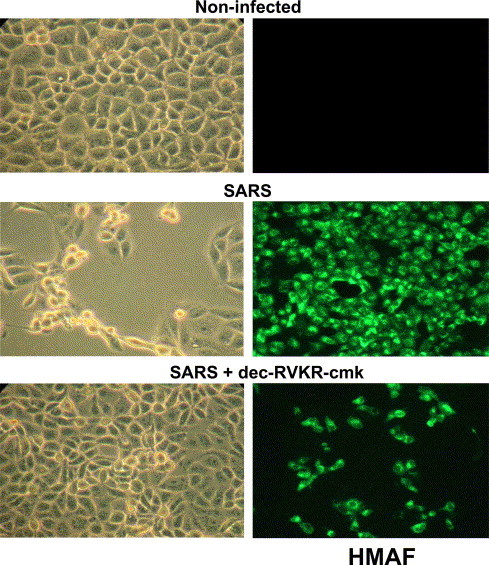
PC-CMK inhibits SARS-CoV spread and CPE. Vero E6 cells were infected with SARS-CoV (MOI = 0.5) as in Fig. 1C. Upon removal of the virus fresh media containing 60 μM dec-RVKR-cmk were added to the cells. The onset of CPE was monitored by microscopic observation. Left panel pictures were taken 24 h post-infection. At 15 h post-infection, another set of cells were permeabilized and viral antigens were probed with HMAF and stained with a fluorescein-conjugated secondary antibody (right panel).
Dec-RVKR-cmk inhibition of proS processing
We next analyzed whether dec-RVKR-cmk could inhibit SARS-CoV proS processing. Accordingly, we incubated infected Vero E6 cells in the presence of increasing doses of this inhibitor for 1 h, and then pulse-labeled the cells for 30 min, followed by immunoprecipitation of the lysates with the HMAF antibody (Fig. 4 A). The data revealed a dose-dependent inhibition of the formation of the ∼80 kDa CTF (∼86% inhibition at 60 μM; Fig. 4B) and at most only a ∼40% effect on the ∼110 kDa CTF (Fig. 4B). This suggests that the dec-RVKR-cmk exerts a dominant inhibitory effect on the generation of the ∼80 kDa CTF. In addition, it may also exert a partial, possibly non-specific, inhibitory effect on the enzymes generating the ∼100–110 kDa CTF. Such dominant inhibitory effect of dec-RVKR-cmk has been previously reported for β-secretase [22] and Adamalysins (ADAMs) [10]. Since viral titer and CPE were maximally reduced at 60 μM dec-RVKR-cmk (Fig. 2, Fig. 4B, and Table 1), we propose that the inhibition of the ∼80 kDa CTF production best correlates with these observations.
Fig. 4.
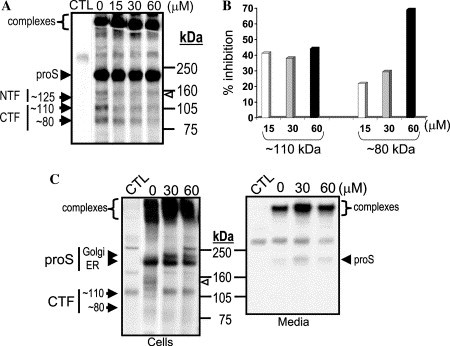
Biosynthetic analysis of PC-inhibition of SARS-CoV S-processing. (A) Vero E6 cells were infected as described in Fig. 1C. Cells were then incubated with media containing the indicated concentrations of dec-RVKR-cmk for 16 h. Labeling and electrophoresis were performed as in Fig. 1D, except that starvation and pulse labeling were performed in the presence of indicated amounts of dec-RVKR-cmk. (B) Percentage inhibition of ∼110 and ∼80 kDa CTF levels were quantified from the Phosphor Imager acquired data of (A). (C) Infections and dec-RVKR-cmk treatments were done as in (A), except that the samples were chased with cold media for 3 h. CTL lane indicates an uninfected control treated with 60 μM dec-RVKR-cmk. At the end of the 3 h chase period, media were immunoprecipitated with the antiserum HMAF.
Pulse-chase analysis revealed S-immunoreactive high molecular weight SDS resistant complexes (>500 kDa, Fig. 4C). These complexes were only observed in SARS-CoV infected cells, but not in cells transfected with recombinant proS, possibly due to the different heating and gel conditions used. Indeed, similar large molecular weight proteins were recently observed by Song et al. [23] that disappear upon boiling at 100 °C, a similar temperature used in our transfection experiments. In contrast, our protein analysis involving SARS-CoV infection was performed using NuPAGE gel system which requires heating of the samples at 70 °C for 10 min. Size exclusion chromatography suggested that the ∼570 kDa complex likely represents a homotrimer of proS [23]. In the media of infected Vero E6 cells, S-containing complexes were largely dominant over proS (Fig. 4C), and no processed products were detected. This raises the possibility that either budding virions selectively incorporated proS or that proS-cleavage products are tightly bound to the high molecular weight complexes. It should be noted that no accumulation of processed spike glycoproteins was observed upon chase periods. However, incubation with dec-RVKR-cmk increased the cellular level of the Golgi-modified ∼210 kDa proS form in parallel with the inhibition of its cleavage (Fig. 4C). Taken together, our data demonstrate that dec-RVKR-cmk exerts a dose-dependent inhibitory effect on the generation of ∼80 kDa CTF with a concomitant increase in the levels of the Golgi-modified proS form, clearly demonstrating an alteration in the SARS-CoV spike biosynthesis.
Mutagenesis of the potential processing sites
We next attempted to identify the cleavage positions by site-directed mutagenesis of selected basic amino acids in and around sites A and B (Fig. 1A). While the mutant K672A did not abrogate processing, the R667A variant diminished the formation of the ∼80 kDa product (Fig. 5 A). These data suggest that while Lys672 is not critical for the generation of the ∼110 or ∼80 kDa CTF-products, Arg667 influences the level of the ∼80 kDa product, possibly via a conformational effect. Since all the mutations tested around the potential sites A and B (Fig. 5A) did not block the processing and in some cases resulted in major retention in the ER (R761A) (not shown), we are unable to precisely map by mutagenesis the processing sites. However, to better define if the generation of the ∼110 kDa CTF results from the a processing around site A, we tried to enhance proS processing at the potential site A through replacement of the SLLR667 sequence (Fig. 1A) by a furin-favored RRKR667 motif [9]. Compared to control (WT), furin caused a drastic reduction of this mutant ∼210 kDa Golgi form at the expense of an enhanced ∼105–110 kDa protein, without significantly increasing the levels of the ∼80 kDa product (Fig. 5B). Based on the mobility in SDS–PAGE, we suggest that the ∼110 kDa CTF is produced by processing of proS at a site close to the aligned cleavage sites in other coronaviruses (site A; Fig. 1A), and that the ∼80 kDa CTF is generated by cleavage at a site downstream from Arg667 by either PC-like enzymes or proteinases activated by PCs. The presence of the RNTR 761 and PTKR 797 motifs within the S-protein suggested that one of them might represent the sought PC-like cleavage site responsible for the generation of the ∼80 kDa CTF. However, neither of the mutants R758A (P4 mutant), R761K or R761Q (P1 mutants), or the double mutant R758A + R761K (P1 + P4 mutant), nor the R797A (P1 mutant; data not shown) eliminated the ∼80 kDa CTF (Fig. 5A). This suggests that none of them are candidate PC-sites, or that another compensating enzyme(s) could cleave these mutagenized sites, as reported for Ebola virus [24].
Fig. 5.
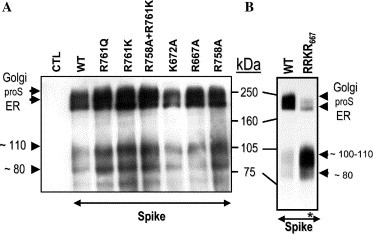
Processing of proS mutants. (A) Wild type (WT) and mutant proS were transfected in FD11 cells stably expressing furin. (B) Stable FD11-furin cells were transfected with either proS-V5 or proS-(RRKR667)-V5 mutant. Immunoprecipitation and Western blot were performed as in Fig. 1B.
Furin cleavage of a proS site B synthetic internally quenched fluorogenic peptide
Since the mutagenesis studies were inconclusive, we used an alternative approach whereby we tested the in vitro cleavage by recombinant furin of synthetic internally quenched fluorogenic peptides [17] mimicking the candidate sites A and B. Site B encompassing peptide (QS1; Abz-E-Q-D-R-N-T-R 761 ↓ E-V-F-A-Q-3-nitrotyrosine amide; Abz = 2 amino benzoic acid) was incubated with purified furin in the presence or absence of inhibitors (EDTA and dec-RVKR-cmk) and analyzed by RP-HPLC. The RP-HPLC chromatogram of the furin digestion showed the disappearance of the QS1 peptide (retention time: R t = 34.3 min, Fig. 6 A, UV panel) and the appearance of two cleavage products (R t = 19.4 and 31.2 min). In contrast, when EDTA and dec-RVKR-cmk (CMK) were added to the peptide digestions, such a cleavage was prevented as expected. Detection using the fluorescence channel indicated the appearance of the N terminal cleaved product eluting at 19.6 min. To confirm the identity of the location of the peptide bond cleavage, we analyzed the collected peptides by MALDI-TOF mass spectrometry. Mass spectral data showed that the RNTR 761 containing peptide was indeed cleaved by furin C-terminal to R761 (Fig. 6B). In contrast, the PTKR 797 (QS2) and SLLR 667 (QS3) containing peptides were not cleaved at all (not shown). This may be rationalized by molecular modeling of S, whereby the RNTR 761 sequence is in turn sandwiched between two α-helices, while the PTKR 797 is itself within an α-helix [25], a secondary structure that usually does not favor cleavage [26]. Although the above in vitro data suggested that furin is able to process a peptide encompassing the RNTR 761 site, and the inhibitor dec-RVKR-cmk reduced the generation of the ∼80 kDa, ex vivo processing was not affected by mutations around this sequence (Fig. 5A). We conclude that it is likely that the ∼80 kDa CTF is generated indirectly by a PC-activated enzyme.
Fig. 6.
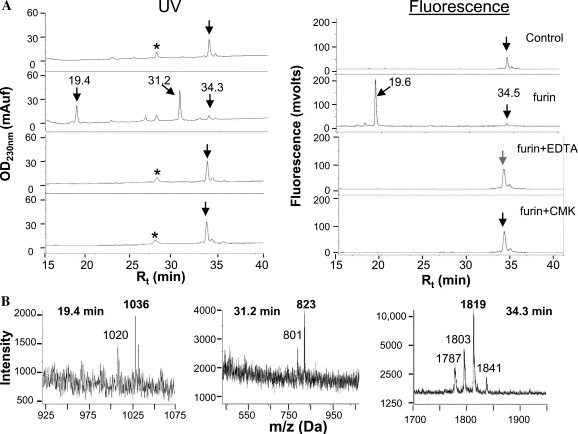
In vitro digestion of proS QS1 peptide by recombinant furin. (A) The internally quenched fluorogenic peptide QS1 was incubated with recombinant furin in the absence or presence of EDTA and dec-RVKR-cmk (CMK) as indicated. Crude reactions were submitted to RP-HPLC equipped with consecutive online monitoring of ultraviolet (UV) absorbance followed by fluorescence emission. Chromatograms of QS1 were produced by the simultaneous monitoring of UV absorbance (left panel) and fluorescence emission (right panel). Control indicates undigested QS1 that showed a single major peak with a retention time (Rt) of 34.3 min. The asterisk marks a peak not specific to furin digestion. Note in the furin digest chromatogram (furin) the disappearance of the 34.3 min peak and appearance of a fluorescent N-terminal (Rt = 19.6 min) and a non-fluorescent C-terminal (Rt = 31.2 min) products. These processing peptides were not detected with dec-RVKR-cmk or EDTA. (B) Mass spectrometry identified each peptide: undigested peptide showed peaks at an m/z of 1841 (M + Na)+, 1819 (M + H)+, 1803 (M + H–NH2)+, and 1787 (M + H–2NH2)+. The N-terminal product exhibited an m/z of 1036 (M + H)+ and 1020 (M + H–NH2)+. The C-terminal product displayed an m/z of 801 (M + H)+ and 823 (M + Na)+, indicating cleavage after Arg761.
Discussion
The present report demonstrated that both recombinant proS and SARS-CoV infected cells produce a ∼80 kDa CTF. Although other investigators reported the lack of proS processing [27], [28], [29], [30], this report and others [31], [32] detected CTFs by transient transfection of recombinant spike glycoprotein. The inconsistency between these observations may perhaps be due to the recombinant virus expression systems chosen, levels of expression, and the antibodies used. Pulse labeling analysis of infected Vero E6 lysates with a commercially available S-specific antibody and with sera of convalescent patients (not shown) also detected proS processing products. In Vero E6 cells or Vero E6 cells expressing the empty vector alone, the levels of the ∼80 kDa product were very modest (less than 10% of proS). This would mean that the processing is very inefficient, but that possibly such low level of cleavage may be sufficient to support virus assembly and spread. On the other hand, in cells overexpressing membrane-bound PCs (furin, PC7, and PC5B), we observed an enhancement of CPE and viral titers, indicating a positive correlation between those parameters and the increased appearance of the ∼80 kDa protein. Interestingly, a soluble form of furin lacking its transmembrane domain was not able to mediate the processing to the ∼80 kDa protein (not shown). The significance of proS cleavage in SARS-CoV infection was further emphasized by the complete abrogation of CPE with a PC specific inhibitor (dec-RVKR-cmk) (Table 1).
Even though no basic-amino acid stretch aligned with the S-sequences of group II coronaviruses, at least one of the cleavages of the SARS-CoV proS is likely to occur within or close to aa 667, as demonstrated by the enhancement of the ∼110 kDa CTF production with the RRKR667 mutant. The enzyme responsible for this cleavage appears to be active early in the secretory pathway, since the fungal metabolite brefeldin A [33] does not affect its activity (not shown). It is possible that the cognate protease could be related to the one responsible for the early cleavage of the Crimean Congo Hemorrhagic fever Gc glycoprotein [34]. Moreover, peptides encompassing this region were not cleaved in vitro by furin, supporting the conclusion that furin does not directly generate the ∼110 kDa CTF. In contrast, the generation of the ∼80 kDa CTF was sensitive to dec-RVKR-cmk in a dose-dependent manner. Additionally, based on in vitro and ex vivo data, furin or membrane-bound PCs are likely to process proS into a ∼80 kDa CTF. Whether the proS cleavage is effected directly by the PCs or indirectly by PCs activating another cellular protease remains to be elucidated. The fact that the fluorogenic peptide containing RNTR761 is efficiently cleaved by furin and the cleavage is inhibited by EDTA or dec-RVKR-cmk raised the possibility for a direct role for PCs in proS processing. If that was true, then our extensive mutagenesis analyses should have enabled us to identify the potential processing site(s). Since none of the mutations of the candidate cleavage sites have resulted in the lack of processing, we suggest that the role played by PCs is indirect. In the case of Nipah virus, the F0 protein is cleaved ubiquitously into F1 and F2, and here also none of the mutations of potential cleavage sites were able to inhibit the processing [35]. The authors concluded that Nipah virus fusion protein is processed by a ubiquitous protease which is different from the PCs since it does not require the presence of basic residues at the cleavage site. It is possible that SARS-CoV spike protein is processed by similar proteases, which may require PCs for activation.
Interestingly, it was reported that addition of trypsin augmented proS cleavage into a ∼85 kDa CTF (similar to the ∼80 kDa CTF described in this work), which resulted in efficient cell–cell fusion (syncytia) [36]. This result raised the possibility that the kinetics of SARS-CoV syncytia formation or cell to cell spread is affected by proS processing. Furthermore, a recent report demonstrated the significance of dec-RVKR-cmk in the prevention of murine hepatitis coronavirus (MHV) spike protein processing and the subsequent effect on cell–cell fusion and not virus–cell fusion as supported by dec-RVKR-cmk treatment [8]. Similarly, in our experiments, dec-RVKR-cmk directly or indirectly reduced the appearance of the ∼80 kDa protein, which in turn may have resulted in the abrogation of the cytopathicity associated with virus spread. The concentration of dec-RVKR-cmk used in these experiments did not result in microscopically identifiable toxicity effects to the cells and is well below the concentration used in other reports. Future studies should address if the replication of virus was affected by dec-RVKR-cmk. Taken together, our work strongly suggests that proS processing and viral spread are interconnected, and that processed proS may be required for direct cell–cell spread and/or for CPE initiation resulting in enhanced secretion of mature virions [4].
In conclusion, this work showed that SARS-CoV S-protein processing is upregulated by the overexpression of membrane-bound PCs and suppression of the processing by dec-RVKR-cmk results in abrogation of CPE and decline in virus titer. Inhibition of the PCs may represent an alternative strategy beneficial to patients infected with SARS-CoV, as already suggested for other infectious pathogens requiring host cell PC-processing of their surface proteins [10]. A PC-inhibitor could possibly be used along with vaccination [37] and/or in conjunction with interferon treatment [13] or with the novel compounds targeting the SARS-CoV proteins recently reported [38]. In view of the widespread implication of the PCs in the processing of various surface glycoproteins of infectious viruses, development of a small molecule inhibitor of the convertases is urgently needed, as its usefulness may also extend to too many emerging deadly infectious agents.
Acknowledgments
We thank S. Basak for peptide synthesis and in vitro furin analysis, M.-C. Asselin for help in cell biology. We thank Dr. J. Towner and J. Dover for their suggestions for the plaque assay and Dr. P. Rollin and Dr. T. Ksiazek for generously providing the SARS-CoV stocks, antibodies, and support. This work was supported by a Canadian PENCE grant (T3), CIHR group Grant #MGC–64518 (to N.G.S. and M.C.), and CIHR Grant #MGP-44363 (to N.G.S.). The secretarial help of Brigitte Mary was greatly appreciated.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E, Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 3.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 4.Ng M.L., Tan S.H., See E.E., Ooi E.E., Ling A.E. J. Gen. Virol. 2003;84:3291–3303. doi: 10.1099/vir.0.19505-0. [DOI] [PubMed] [Google Scholar]
- 5.Nicholls J.M., Poon L.L., Lee K.C., Ng W.F., Lai S.T., Leung C.Y., Chu C.M., Hui P.K., Mak K.L., Lim W., Yan K.W., Chan K.H., Tsang N.C., Guan Y., Yuen K.Y., Peiris J.S. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Haan C.A., Stadler K., Godeke G.J., Bosch B.J., Rottier P.J. J. Virol. 2004;78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidah N.G., Chretien M. Brain Res. 1999;848:45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- 10.Thomas G. Nat. Rev. Mol. Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallenberger S., Bosch V., Angliker H., Shaw E., Klenk H.D., Garten W. Nature. 1992;360:358–361. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 12.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haagmans B.L., Kuiken T., Martina B.E., Fouchier R.A., Rimmelzwaan G.F., Van Amerongen G., van Riel D., de Jong T., Itamura S., Chan K.H., Tashiro M., Osterhaus A.D. Nat. Med. 2004;10:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergeron E., Basak A., Decroly E., Seidah N.G. Biochem. J. 2003;373:475–484. doi: 10.1042/BJ20021630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandt W.E., Buescher E.L., Hetrick F.M. Am. J. Trop. Med. Hyg. 1967;16:339–347. doi: 10.4269/ajtmh.1967.16.339. [DOI] [PubMed] [Google Scholar]
- 16.Basak A., Chretien M., Seidah N.G. FEBS Lett. 2002;514:333–339. doi: 10.1016/s0014-5793(02)02394-3. [DOI] [PubMed] [Google Scholar]
- 17.Basak A., Zhong M., Munzer J.S., Chretien M., Seidah N.G. Biochem. J. 2001;353:537–545. doi: 10.1042/0264-6021:3530537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofmann H., Hattermann K., Marzi A., Gramberg T., Geier M., Krumbiegel M., Kuate S., Uberla K., Niedrig M., Pohlmann S. J. Virol. 2004;78:6134–6142. doi: 10.1128/JVI.78.12.6134-6142.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon V.M., Klimpel K.R., Arora N., Henderson M.A., Leppla S.H. Infect. Immun. 1995;63:82–87. doi: 10.1128/iai.63.1.82-87.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siegfried G., Khatib A.M., Benjannet S., Chretien M., Seidah N.G. Cancer Res. 2003;63:1458–1463. [PubMed] [Google Scholar]
- 21.Sugrue R.J., Brown C., Brown G., Aitken J., McL R.H. J. Gen. Virol. 2001;82:1375–1386. doi: 10.1099/0022-1317-82-6-1375. [DOI] [PubMed] [Google Scholar]
- 22.Benjannet S., Elagoz A., Wickham L., Mamarbachi M., Munzer J.S., Basak A., Lazure C., Cromlish J.A., Sisodia S., Checler F., Chretien M., Seidah N.G. J. Biol. Chem. 2001;276:10879–10887. doi: 10.1074/jbc.M009899200. [DOI] [PubMed] [Google Scholar]
- 23.Song H.C., Seo M.Y., Stadler K., Yoo B.J., Choo Q.L., Coates S.R., Uematsu Y., Harada T., Greer C.E., Polo J.M., Pileri P., Eickmann M., Rappuoli R., Abrignani S., Houghton M., Han J.H. J. Virol. 2004;78:10328–10335. doi: 10.1128/JVI.78.19.10328-10335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wool-Lewis R.J., Bates P. J. Virol. 1999;73:1419–1426. doi: 10.1128/jvi.73.2.1419-1426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spiga O., Bernini A., Ciutti A., Chiellini S., Menciassi N., Finetti F., Causarono V., Anselmi F., Prischi F., Niccolai N. Biochem. Biophys. Res. Commun. 2003;310:78–83. doi: 10.1016/j.bbrc.2003.08.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brakch N., Rholam M., Boussetta H., Cohen P. Biochemistry. 1993;32:4925–4930. doi: 10.1021/bi00069a029. [DOI] [PubMed] [Google Scholar]
- 27.Bisht H., Roberts A., Vogel L., Bukreyev A., Collins P.L., Murphy B.R., Subbarao K., Moss B. Proc. Natl. Acad. Sci. USA. 2004;101:6641–6646. doi: 10.1073/pnas.0401939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bukreyev A., Lamirande E.W., Buchholz U.J., Vogel L.N., Elkins W.R., St Claire M., Murphy B.R., Subbarao K., Collins P.L. Lancet. 2004;363:2122–2127. doi: 10.1016/S0140-6736(04)16501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao X., Chakraborti S., Dimitrov A.S., Gramatikoff K., Dimitrov D.S. Biochem. Biophys. Res. Commun. 2003;312:1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao Y.X., Ren J., Heinen P., Zambon M., Jones I.M. J. Infect. Dis. 2004;190:91–98. doi: 10.1086/421280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying W., Hao Y., Zhang Y., Peng W., Qin E., Cai Y., Wei K., Wang J., Chang G., Sun W., Dai S., Li X., Zhu Y., Li J., Wu S., Guo L., Dai J., Wang J., Wan P., Chen T., Du C., Li D., Wan J., Kuai X., Li W., Shi R., Wei H., Cao C., Yu M., Liu H., Dong F., Wang D., Zhang X., Qian X., Zhu Q., He F. Proteomics. 2004;4:492–504. doi: 10.1002/pmic.200300676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X.D., Shang B., Yang R.F., Yu H., Ma Z.H., Shen X., Ji Y.Y., Lin Y., Wu Y.D., Lin G.M., Tian L., Gan X.Q., Yang S., Jiang W.H., Dai E.H., Wang X.Y., Jiang H.L., Xie Y.H., Zhu X.L., Pei G., Li L., Wu J.R., Sun B. Cell Res. 2004 doi: 10.1038/sj.cr.7290240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lippincott-Schwartz J., Yuan L., Tipper C., Amherdt M., Orci L., Klausner R.D. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- 34.Vincent M.J., Sanchez A.J., Erickson B.R., Basak A., Chretien M., Seidah N.G., Nichol S.T. J. Virol. 2003;77:8640–8649. doi: 10.1128/JVI.77.16.8640-8649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moll M., Diederich S., Klenk H.D., Czub M., Maisner A. J. Virol. 2004;78:9705–9712. doi: 10.1128/JVI.78.18.9705-9712.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Proc. Natl. Acad. Sci. USA. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z.Y., Kong W.P., Huang Y., Roberts A., Murphy B.R., Subbarao K., Nabel G.J. Nature. 2004;428:561–564. doi: 10.1038/nature02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kao R.Y., Tsui W.H., Lee T.S., Tanner J.A., Watt R.M., Huang J.D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.H., Tse H., To A.P., Ng L.W., Wong B.C., Tsoi H.W., Yang D., Ho D.D., Yuen K.Y. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]