

Graphical abstract
Abbreviations: ARS, aminoacyl-tRNA synthetase; TLR, toll-like receptor; RIG-I, retinoic acid-inducible gene-I; MAVS, mitochondrial antiviral signaling protein; IFN, interferon; ITCH, E3 ubiquitin-protein ligase Itchy homolog; MSC, multi-tRNA synthetase complex; AIMP, ARS-interacting multifunctional protein; WT, wild-type; BMDC, bone marrow-derived dendritic cell; Oas, 2′-5′-oligoadenylate synthase; HCV, hepatitis C virus; PTM, post-translational modification; GAIT, IFN-γ-activated inhibition of translation; MAPK, mitogen-activated protein kinase; MEK, MAPK kinase kinase; ERK, extracellular-related kinase; MITF, microphthalmia-associated transcription factor; PCBP2, poly(rC)-binding protein 2; TH1, T-helper type 1; LPS, lipopolysaccharide; TCID50, a median tissue culture infective dose; PBMC, peripheral blood mononuclear cell; BMDM, bone marrow-derived macrophage; TLS, tRNA-like structure; eEF1A, eukaryotic elongation factor 1 alpha; MDA5, melanoma differentiation-associated protein 5; HIV, human immunodeficiency virus; TGF-β, tumor growth factor-β; IFN-α, interferon-α; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; EMAPII, endothelial monocyte-activating polypeptide II; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; IP-10, interferon-inducible protein 10; CRP, C-reactive protein; HMGB1, high mobility group box 1; AA-AMP, aminoacyl-adenylate
Keywords: Aminoacyl-tRNA synthetase, Multi-tRNA synthetase complex, Infection, Antiviral immunity, Antibiotics
Abstract
Despite remarkable advances in medical science, infection-associated diseases remain among the leading causes of death worldwide. There is a great deal of interest and concern at the rate at which new pathogens are emerging and causing significant human health problems. Expanding our understanding of how cells regulate signaling networks to defend against invaders and retain cell homeostasis will reveal promising strategies against infection. It has taken scientists decades to appreciate that eukaryotic aminoacyl-tRNA synthetases (ARSs) play a role as global cell signaling mediators to regulate cell homeostasis, beyond their intrinsic function as protein synthesis enzymes. Recent discoveries revealed that ubiquitously expressed standby cytoplasmic ARSs sense and respond to danger signals and regulate immunity against infections, indicating their potential as therapeutic targets for infectious diseases. In this review, we discuss ARS-mediated anti-infectious signaling and the emerging role of ARSs in antimicrobial immunity. In contrast to their ability to defend against infection, host ARSs are inevitably co-opted by viruses for survival and propagation. We therefore provide a brief overview of the communication between viruses and the ARS system. Finally, we discuss encouraging new approaches to develop ARSs as therapeutics for infectious diseases.
1. Introduction
Humans come into contact with a range of infectious microbes, including bacteria, viruses, and parasites. Diverse safeguard systems are available to detect and defend against invading pathogens. Upon recognition of pathogen-associated molecular patterns, cells transmit intracellular signals to trigger immediate proinflammatory and antimicrobial responses [1]. These innate immune responses are the first line of defense in the early phase of infection. Toll-like receptors (TLRs), Nod-like receptors, and retinoic acid-inducible gene-I (RIG-I)-like receptors are representative families of innate immune receptors that distinguish microbial products as ‘non-self’ molecules [2].
TLRs can be grouped into two types depending on their cellular locations: cell surface TLRs (1, 2, 4, 5, and 6) and endosomal TLRs (3, 7, 8, and 9) [3], [4]. Surface-exposed TLRs mainly detect bacterial products, while TLRs located in intracellular compartments recognize nucleic acids [1]. RIG-I mediates antiviral signaling by sensing viral RNA and subsequently interacting with the central antiviral signaling protein, mitochondrial antiviral signaling protein (MAVS) [5]. This signaling cascade ultimately leads to the transcription of type I interferons (IFNs) and other antiviral molecules [5], [6]. All these innate immune responses orchestrate the early host response to infection, which subsequently activates and shapes the adaptive immune response [1], [7]. The adaptive immune system controls the late phase of infection and generates immunological memory through T cell- or B cell-mediated (humoral) mechanisms [8], [9].
Well-integrated defense systems will provide effective and long-lasting protective immunity [2]. The induction, maintenance, and termination of host immune responses to infection must be tightly controlled [10]. Turning off the immune system is as important as turning it on to avoid unwanted immune responses. Several positive and negative regulatory mechanisms control the immune response through a complex signaling network [11], [12], [13]. A number of endogenous proteins that either enhance or limit the magnitude and duration of the inflammatory response regulate this system. These non-immune proteins serve as a second safeguard system by fine-tuning host immune responses and ensuring cellular homeostasis. For example, a mitochondrial import receptor protein, Tom7, positively regulates MAVS signaling by serving as a critical adaptor linking MAVS to downstream signaling molecules that activate antiviral immunity [14]. Alternatively, cells have developed diverse strategies to halt antiviral signaling through MAVS [15]. Several E3 ligases, including E3 ubiquitin-protein ligase Itchy homolog (ITCH), Smurf1, Gp78, and TRIM25, have been shown to accelerate MAVS degradation after viral infection [16], [17], [18]. Collectively, cells employ multiple mechanisms to regulate immunity against microbial invaders, with the help of non-immunogenic proteins to prevent uncontrolled cellular damage.
Aminoacyl-tRNA synthetases (ARSs) are essential components for translation in every living species. Although the role of ARSs in protein synthesis is well recognized, it has taken scientists decades to acknowledge their roles beyond translation. There is an apparent propensity for new sequences and domains to be added to ARSs during evolution. The emergence of such new domains is consistent with the involvement of ARSs in a broad range of biological functions in addition to protein synthesis and correlates with the increasing biological complexity of higher organisms.
Interestingly, the activity of many ARSs in higher eukaryotes appears to be regulated by their presence in the cytoplasmic multi-tRNA synthetase complex (MSC), which is assembled in most cases via the appended domains and consists of nine ARSs [EPRS (which consists of ERS and PRS), DRS, IRS, KRS, LRS, MRS, QRS, and RRS, according to the standard nomenclature in which the ARSs are designated by the single-letter amino acid code followed by “RS”] and three auxiliary ARS-interacting multifunctional proteins (AIMPs), AIMP1, AIMP2, and AIMP3 [19]. MSC has been proposed to function as a depot system to perform specific regulatory functions via release of its components [19].
Recent studies identified novel molecular and cellular processes through which ARSs control host immune responses against infections [20], [21], [22]. EPRS contributes to innate antiviral immunity and promotes viral clearance [20]. The MSC protein AIMP1 also has a critical role in innate and adaptive antiviral immunity against influenza A virus infection [21]. In addition, secreted WRS acts as an early warning signal of host infections [22]. Together, these findings suggest that ARSs may act as cellular sensors as well as immunoregulators in the context of infection.
In this review, we will provide an overview of the current understanding of the mechanisms through which the ubiquitous, non-immunogenic ARSs are appropriated to regulate cell signaling upon infection. We also briefly discuss current strategies targeting pathogenic ARSs for antibiotics development. We hope that this review will stimulate emerging research in the field of ARSs in infectious diseases, including clinical applications for ARSs in anti-infective pharmacology.
2. Immunoregulatory roles of mammalian ARSs against infection
2.1. Multi-omics profiles reveal that ARSs respond to infection
Profiles of the transcriptional and proteomic changes in infected cells provide key information about cellular factors that may be central to host immune responses [23]. Multiple lines of evidence support a role for ARSs in the infected host. Our group conducted an RNA sequencing analysis of human bronchial epithelial cells infected with influenza A virus at 8 h and 24 h post-infection [20]. We particularly noted that the MSC components IRS, RRS, EPRS, AIMP1, and AIMP3 were up-regulated at 8 h, while the expression of KRS gradually increased up to 24 h [20]. With respect to non-MSC ARS genes, early induction was observed for YRS, TRS, and SRS, whereas the expression of the WRS gene was increased at both 8 h and 24 h post-infection (GEO accession code for RNA-Seq data: GSE75699).
In agreement with our data, another group showed that AIMP1 is up-regulated after influenza A infection. This transcriptome analysis revealed that AIMP1 is closely related to antiviral immunity [21]. Microarray analysis of antigen-loaded wild-type (WT) or AIMP1−/− mouse bone marrow-derived dendritic cells (BMDCs) showed that AIMP1 regulates a variety of immune processes through important antiviral genes including IFN-activated genes, IFN regulatory factor genes, and innate immune sensors such as 2′-5′-oligoadenylate synthase (Oas) family genes and Ddx58.
An integrated analysis of microarray and proteomics data showed that hepatitis C virus (HCV)-infected hepatocytes up-regulate the WRS, SRS, AlaRS, TRS, and GRS genes and express higher levels of YRS and SRS proteins [24]. In A549 cells inoculated with swine influenza virus, WRS was up-regulated at both the mRNA and protein levels [25]. Increased expression of WRS was also observed in cells infected with Vibrio cholera, human cytomegalovirus, and human hepatitis B virus [22].
A microarray analysis revealed upregulation of ARS genes (including the MSC members Irs, Lrs, Mrs, Rrs, and Eprs, as well as the free ARSs Alars, Crs, Grs, Nrs, Srs, Trs, Vrs, Wrs, and Yrs) in the brains of mice infected with flaviviruses, including Japanese encephalitis virus and West Nile virus, which are important causes of central nervous system diseases [26]. Note that this study performed the RNA analysis at 5–8 days post-infection, when clinical symptoms of encephalitis are already apparent [26]. All these data indicate that ARSs are responsive to infection and may be involved in immune regulation against infection.
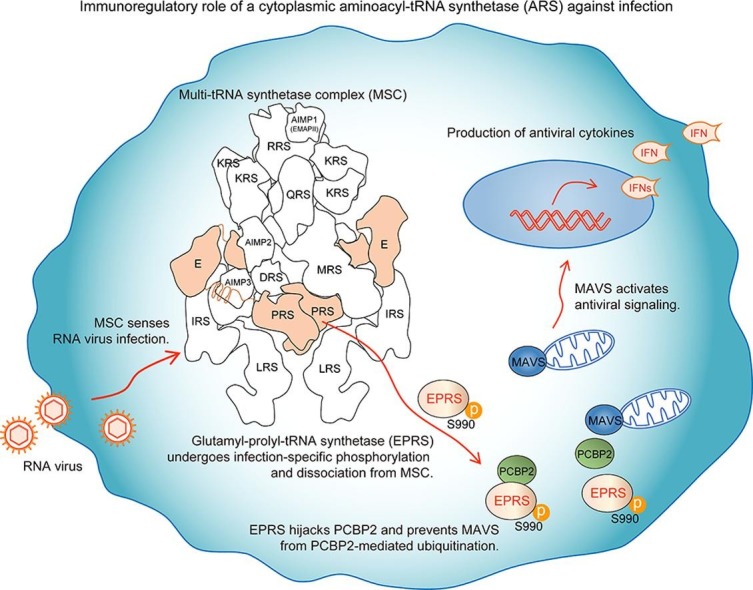
In addition to these transcriptional and proteomic analyses, post-translational modifications (PTMs) in ARSs have been characterized in response to various stimuli [27]. These studies revealed their diverse and extended functions in biological and pathological processes [28], [29]. Phosphorylation is the most frequent PTM occurring in ARS proteins, and it is thought to induce structural changes that may drive their dissociation from the MSC and/or allow for new interacting partners [30]. It is well established that the Ser886 and Ser999 residues of the EPRS WHEP domains are sequentially phosphorylated in response to IFN-γ stimulation. IFN-γ induces phosphorylation of EPRS at Ser886 by activating cyclin-dependent kinase 5 and indirectly induces phosphorylation at Ser999 via a distinct kinase [31]. This promotes the dissociation of EPRS from the MSC, and subsequently promotes its association with the IFN-γ-activated inhibition of translation (GAIT) complex [32]. The heterotetrameric GAIT complex, including EPRS, binds to distinct 3′-untranslated regions of mRNAs involved in chronic inflammation to suppress their translation [33]. RNA virus infection-specific phosphorylation was also identified at the EPRS Ser990 residue [20]. This modification was critical for dissociation of EPRS from the MSC and activation of antiviral signaling [20].
Because infections cause massive production of diverse cytokines and activate multiple cellular signaling pathways, including the mitogen-activated protein kinase (MAPK) cascade, infectious signals may also indirectly induce PTMs of ARS proteins [5], [34]. In the case of KRS, allergen stimulation of activated mast cells results in phosphorylation of KRS at Ser207 in a MAPK-dependent manner [35]. Signaling through MAPK kinase kinase (MEK)/extracellular-related kinase (ERK), but not p38 MAPK, was critical for the KRS phosphorylation. Ultimately, this phosphorylation triggered dissociation of KRS from the MSC and its translocation into the nucleus, where it controls the level of diadenosine tetraphosphate [36], an important signaling molecule in microphthalmia-associated transcription factor (MITF) transcriptional activity [37]. Therefore, KRS phosphorylation might be induced directly by infection-related MAPK signaling or indirectly by infection-induced products like cytokines, which can activate multiple regulatory pathways.
Collectively, these changes in the expression and regulation of ARSs by microbial infections suggest that ARSs have substantial potential to regulate pathogenesis and anti-infective immunological responses. ARSs are ubiquitous and abundant proteins; therefore, transient changes in their expression may not significantly affect global protein synthesis. Alteration of TRS and AlaRS expression does not markedly alter global protein synthesis [38]. PTMs located in the additional domains of vertebrate ARSs do not affect aminoacylation of these enzymes [20], [33]. However, PTMs of residues located in the catalytic core domain can influence the enzymatic activities of ARSs. Phosphorylation of MRS at Ser662, which is located in the critical tRNA-binding site, significantly down-regulates global translation by inactivating methionine-charging activity until damaged DNA is repaired [39]. By looking at the distinct transcriptional and protein expression profiles of ARSs, as well as the dynamics of their regulation by PTMs, novel, infection-specific roles for individual ARSs will be elucidated. Future studies should investigate whether changes in the expression and PTMs of each ARS upon infection correlate with its canonical tRNA aminoacylation activity and global protein synthesis as well as with its non-canonical functions.
2.2. The role of EPRS in antiviral immunity
Human EPRS, one of the members of the MSC, is the only bifunctional enzyme for protein synthesis. It comprises ERS and PRS, which are linked together by three WHEP domains [40]. During the past two decades, EPRS has become known for its non-canonical function as a translational silencer by forming the IFN-γ-dependent GAIT complex [32], [40], [41].
Our group hypothesized that the cytoplasmic MSC may function as a surveillance system for infection. We found that the MSC immediately senses invasion by RNA viruses and activates EPRS to promote antiviral immunity via its infection-specific phosphorylation and release from the MSC [20]. EPRS knockdown in macrophages increased the replication of the RNA viruses influenza A virus and vesicular stomatitis virus, and reduced the production of antiviral cytokines. Conversely, ectopically expressed EPRS in macrophages significantly attenuated viral replication and increased antiviral IFN-β production. Moreover, RNA viruses were much more lethal in EPRS+/− haploid mice due to the high rates of viral replication and weak antiviral cytokine responses. Intriguingly, infection with RNA viruses led to a previously unreported phosphorylation of EPRS at Ser990 that triggers the release of EPRS from the MSC. This phosphorylation was not induced by IFN-γ, and thus the released EPRS was not involved in the GAIT system. Instead, the EPRS associated with poly(rC)-binding protein 2 (PCBP2), a well-known negative regulator of MAVS that mediates its degradation via ubiquitin ligase ITCH [13] after release from the MSC. Ultimately, the phosphorylated EPRS protected MAVS from PCBP2-mediated ubiquitination and degradation, and the stabilized MAVS activated antiviral immunity.
These data indicate that EPRS displays a phospho-code that controls its function in response to different stimuli. Furthermore, the role of EPRS in antiviral immune responses demonstrates the functional significance of the MSC as an immune regulatory system in the early stages of infection.
2.3. The role of AIMP1 in innate and adaptive antiviral immunity
AIMP1 is a non-enzymatic component of the MSC that mainly associates with RRS, QRS, and another non-enzymatic component, AIMP2, within the complex [42]. Extracellularly, AIMP1 activates innate immune cells such as macrophages and monocytes to produce inflammatory cytokines through MAPK signaling [43] and induces maturation of BMDCs [44]. AIMP1 potentiates the link between innate and adaptive immunity by activating NK cells [45] and B cells [46] and by promoting IL-12-mediated T-helper type 1 (TH1) cell immunity [47]. AIMP1 expression in BMDCs directly promoted TH1 cell polarization, which is characterized by the secretion of IL-12 and IFN-γ from antigen-presenting cells and T cells, respectively, and is associated with the generation of cell-mediated adaptive immune responses [48]. These adaptive responses were critical for effective clearance of intracellular infections as well as antitumor immunity [49], [50]. Lipopolysaccharide (LPS) treatment of AIMP1−/− BMDCs impaired the secretion of the TH1 polarizing cytokine IL-12p70, but secretion of the proinflammatory cytokines IL-6 and IL-1β was not changed. AIMP1−/− BMDCs also expressed lower levels of costimulatory molecules and MHC class II following LPS treatment. The p38 MAPK signaling pathway was also significantly impaired in AIMP1−/− BMDCs which reduced TH1 polarization [21].
AIMP1 is also critical for innate and adaptive antiviral immunity against influenza A virus infection [21]. Microarray analysis of WT and AIMP1−/− BMDCs suggested its close correlation to the expression of antiviral genes. Challenge of AIMP1−/− mice with influenza A virus (H3N2) by aerosol spray resulted in 100% lethality, whereas WT mice showed only 20% lethality at a high infectious dose [a median tissue culture infective dose (TCID50) of 20] per mouse at 2 weeks post-infection. A sub-lethal dose of 7.50 TCID50 per mouse still caused 60% mortality among AIMP1−/− mice, while all WT mice survived. Evaluation of survivors indicated that the levels of infiltrating neutrophils and macrophages in bronchoalveolar lavage fluid were significantly reduced in AIMP1−/− mice on day 7 post-infection. Moreover, IFN-γ+ T cells in the lung and levels of IgG2a (the TH1-specific antibody isotype) in the serum were highly reduced in AIMP1 knockout mice on day 15 post-infection. By varying the titers of infectious virus and investigating different time-points, this study revealed that AIMP1 deficiency can lead to insufficient innate and adaptive antiviral immune responses in vivo [21]. This was the first report on the role of MSC components in adaptive immunity during sequential phases of virus infection. However, further studies will be necessary to determine whether dissociation of AIMP1 from the MSC and its secretion by cells are required for its function against viral infection. Whether AIMP1 functions differently in intracellular and extracellular environments in infection should also be addressed by future studies. Such molecular details will strengthen the interrelated roles of AIMP1 in innate and adaptive immunity in infectious disease.
2.4. WRS as a primary responsive molecule to infection
In addition to its enzymatic activity, human WRS contains an N-terminal extension WHEP domain. WRS exists as two different forms; the major form is the full-length WRS (fWRS, aa 1–471), and the other form is a truncated version (mini-WRS, aa 48–471) in which the N-terminal WHEP domain is deleted by alternative splicing [51]. Both forms of WRS are secreted by various cells, and the fWRS is further cleaved into T1 (aa 71–471) or T2 (aa 94–471), which possess angiostatic activity, by leukocyte elastase or plasmin in the extracellular space [52]. Secreted WRS also plays an important role in high-affinity Trp uptake by human cells, which is linked with immune regulation [53].
Ahn et al. reported a critical role for human WRS as a primary responsive molecule to activate innate immune defense signaling against bacterial infection [22]. They showed that fWRS primes innate immunity upon bacterial infection, although infected cells secreted both fWRS and mini-WRS, suggesting that the N-terminal WHEP domain is responsible for control of infection [22].
These authors further showed that the secretion of WRS is significantly increased in sepsis patients infected with pathogens, including Gram-negative bacteria, Gram-positive bacteria, and fungi, but not in sepsis patients with sterile inflammation. WRS was secreted from monocytes upon infection and directly bound to the TLR4-MD2 complex on macrophages to activate phagocytosis and chemokine production. WRS seems to function prior to the full activation of the immune response in infected cells. Its secretion occurred relatively early, at around 15 min post-infection, which is much earlier than TNF-α production, which could be first detected at 120 min [22]. These results suggest that WRS acts as a warning molecule to prime the first line of defensive signaling against invading microbes.
Interestingly, treatment of human peripheral blood mononuclear cells (PBMCs) with WRS induced their production of the chemokines CCL3 and CCL4, which recruit inflammatory cells, particularly neutrophils and macrophages. Moreover, secreted fWRS increased phagocytic cell numbers in mice and enhanced the phagocytic activity of mouse bone marrow-derived macrophages (BMDMs). Furthermore, administration of WRS to Salmonella typhimurium-infected mice increased neutrophil infiltration and decreased the bacterial load in the spleen and liver, which ultimately improved the survival of the infected mice [22].
It would be very interesting to see the molecular details underlying the rapid and infection-specific secretion of WRS. Further studies will also be required to identify the mechanisms through which WRS secretion is tightly controlled by different signals. Fig. 1 and Table 1 summarize the immunoregulatory roles of ARSs.
Fig. 1.

Immune functions of ARSs. Viral infection releases EPRS from the MSC to exert antiviral activity by inhibiting MAVS degradation by PCBP2. AIMP1 also promotes antiviral immunity by potentiating the link between innate and adaptive immunity. WRS is secreted after bacteria or virus infection that subsequently primes innate immunity via binding to TLR4. Secreted cytokine-like ARSs are displayed with cognate stimuli. Each ARS function is represented by arrows in different colors. See text for details.
Table 1.
Immunoregulatory roles of mammalian ARSs.
| ARS | Stimulus | PTM | Interactor | Function | Cell | Refs. |
|---|---|---|---|---|---|---|
| EPRS |
|
Phosphorylation at Ser990 | PCBP2 | Activation of MAVS-mediated antiviral signaling | U937 Raw264.7 |
[20] |
| EPRS | IFN-γ | Phosphorylation at Ser886 and Ser999 | NASP1 GAPDH L13a |
Inhibition of inflammatory gene translation by heterotetrameric GAIT complex formation | U937 | [32] |
| EPRS | IFN-γ | Phosphorylation at Ser999 | GAPDH L13a |
Inhibition of inflammatory gene translation by heterotrimeric GAIT complex formation | Raw264.7 | [41] |
| WRS |
|
Not identified | TLR4 | Priming the first line of defensive signaling | PBMC HEK293-PRRs |
[22] |
| AIMP1 |
|
Not identified | Not identified |
|
BMDC | [21] |
| KRS | IgE-Ag | Phosphorylation at Ser207 | MITF |
|
RBL mast cell | [35] |
PTM, post-translational modification; VSV, vesicular stomatitis virus; GAIT, IFN-γ-activated inhibition of translation; RSV, respiratory syncytia virus; PBMC, peripheral blood mononuclear cell; PRR, pattern recognition receptor; MAPK, mitogen-activated protein kinase; TH1, T-helper type 1; BMDC, bone marrow-derived dendritic cell; IgE-Ag, immunoglobulin E-antigen; MITF, microphthalmia-associated transcription factor; Ap4A, diadenosine tetraphosphate.
3. Communication between the mammalian ARS system and viruses
In general, RNA viruses have very small genomes, which limits their own protein synthesis [54]. Accordingly, viruses have to rely on the host translational system for their survival and replication [55]. Unavoidably, the ARS system, including ARSs and tRNAs, is co-opted by viruses, which lies in contrast to its immunoregulatory role in defense against infection.
Viral hijacking of the host translational machinery, particularly the ribosomes and the ARS system, consequently reduces host translational fidelity [56]. Methionine misacylation was increased up to 10-fold in cells exposed to various viruses, including adenovirus and vaccinia virus [56]. In the case of vaccinia virus, its replication and progeny assembly take place exclusively in the cytoplasm of infected cells at distinct foci termed viral factories or virosomes, which are enriched in viral DNA and proteins [57]. Interestingly, MSC was recruited to these active viral translation sites in vaccinia virus-infected cells [58]. Immunofluorescence analysis indicated that MSC proteins (KRS, MRS, and EPRS) and some free ARSs (SRS and YRS) are largely recruited to the viral factories [58]. Moreover, upregulation of diverse ARS genes after flavivirus infection in mice brain implies that host ARSs may be used by viruses for their propagation, based on the observation that flavivirus does not shut off host protein synthesis and therefore must compete with the cellular translational machinery for limiting factors [26].
Although the substrate specificity of ARS is sequence-dependent, several studies revealed that ARSs can recognize plant viral RNAs with different sequences from those of tRNAs [59]. Some viral RNAs have adopted structures that mimic those of tRNA, which increases the efficiency of their genomic RNA translation. tRNA-like structures (TLSs) have been found at the 3′ end of RNAs from eight genera of plant viruses [60], including turnip yellow mosaic virus [61], brome mosaic virus [62], and tobacco mosaic virus [63]. These TLSs can be aminoacylated specifically on valine, tyrosine, and histidine [64]. This mimetic activity is strategically valuable to viruses for evading the host’s immune system as well as increasing their translational efficiency [65]. Structural analysis showed that the viral TLS can bind to ARS, eukaryotic elongation factor 1 alpha (eEF1A), and the ribosome, implying that the tRNA mimicry functions as a translation enhancer and a means of protecting the 3′ end of viral genomic RNA [66]. Similarly, a 32-nucleotide RNA motif in the genome of the transmissible gastroenteritis coronavirus formed a similar structure with the GAIT element that can selectively interact with EPRS [67], [68]. The sequence of this viral RNA motif showed a high identity with a known GAIT element that interacts with the first and second linker regions in the WHEP domains of EPRS [40]. Direct binding of EPRS to the viral GAIT-like RNA motif inhibited the translation of this viral RNA motif in vitro [68]. Interestingly, the viral GAIT-like RNA motif affected the host melanoma differentiation-associated protein 5 (MDA5)-mediated antiviral signaling pathway. Although disruption of this GAIT-like RNA motif did not affect viral growth, the innate immune response was exacerbated through MDA5 activation, indicating that this motif interferes with the host defense system by blocking the accessibility of EPRS to host sensors of viral RNA [68].
All retroviruses utilize host cellular tRNA isoacceptors as primers for reverse transcription [69]. Human immunodeficiency virus-1 (HIV-1) uses the specific tRNALys3 to initiate reverse transcriptase-catalyzed synthesis of minus-strand DNA [70]. During viral assembly, human KRS was found to be selectively packaged into HIV-1 virions via direct interaction with the viral Gag protein, and a truncated KRS associated with the tRNALys packaging was found within the virion [70], [71], [72]. Gag codes for core structural proteins of the virus, which are the fundamental building blocks of the retrovirus particle [73]. The source of viral KRS has been a topic of significant investigation. One study showed that the incorporation of KRS into HIV-1 virions occurs rapidly and is sensitive to the inhibition of newly synthesized KRS [72]. Another study suggested that HIV-1 uses host mitochondrial KRS during the packaging process [74]. Because KRS is normally associated with the MSC, HIV-1 could exploit the dynamic nature of the MSC to redirect and co-opt cellular translation factors [75]. Indeed, HIV-1 infection triggers release of KRS from the MSC, possibly through phosphorylation at Ser207 [75]. This PTM is important for KRS packaging into virions and progeny virus infectivity [75]. The free pool of KRS translocates to the nucleus in a phosphorylation-dependent manner, although the relevance of nuclear KRS for viral infectivity is not yet entirely clear [75].
HCV is a major cause of chronic liver disease characterized by persistent infection and immune escape [76]. HCV E2 is an envelope protein that plays an important role in viral entry [77]. HCV E2 directly interacted with AIMP1 and led to its degradation [78]. Because AIMP1 is a negative regulator of tumor growth factor-β (TGF-β) [79], the decreased cellular levels of AIMP1 in HCV infection caused liver fibrosis by increasing TGF-β signaling. Examining whether this interaction is involved in the immune evasion strategy of HCV will be valuable, as AIMP1 was recently shown to exert antiviral activity by enhancing TH1 polarization [21].
Influenza A virus encodes NS2, a nuclear export protein that plays an important role in overcoming host restriction [80]. The NS2 protein interacted directly with AIMP2 to protect it from ubiquitin-mediated degradation [81]. AIMP2 promotes virus replication by enhancing the stability of the virus M1 protein, facilitating the switch from ubiquitination to SUMOylation of M1 [81].
Collectively, these data demonstrate that viruses do not interact with the ARS system solely to co-opt the host translational machinery for their own protein synthesis. Instead, viruses also act through the ARS to regulate diverse cellular processes involved in viral propagation and replication.
4. Therapeutic potential of mammalian ARSs
4.1. Secreted ARSs acting as cytokines and chemokines
As key players in the innate and adaptive immune system, cytokines can be used as therapeutic agents for immune-related diseases [82]. The immune-modulatory activity of cytokines represents a promising feature of anti-infective therapeutics and immune adjuvants for vaccines directed against pathogens [83]. For example, interferon-α (IFN-α) has been proven to be clinically useful in virus infection by inducing IFN-stimulated genes that limit virus replication and other genes involved in apoptosis of infected cells [84], [85]. Several cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), are produced as precursor proteins and cleaved into active mature forms by proteases, which are subsequently secreted into the extracellular space [86].
In the last few decades, it has been reported that several ARSs are secreted by intact cells and function like cytokines [51], [87], [88], [89], [90]. ARSs are secreted in response to extracellular stimuli and are also secreted continuously, suggesting they are constitutively and actively secreted [43], [89]. They are secreted as either full-length or truncated forms generated by alternative splicing or proteolytic cleavage and exhibit activity in cytokine signaling pathways [91], [92]. For instance, under apoptotic conditions, YRS is secreted and cleaved into two distinct cytokines by an elastase, yielding an N-terminal catalytic domain (mini-YRS) and a C-terminal endothelial monocyte-activating polypeptide II (EMAPII)-like domain [87], [93]. The latter showed potent chemotactic activity for leukocytes and monocytes and stimulated the production of myeloperoxidase, TNF-α, and tissue factor, whereas mini-YRS functioned as an IL-8-like cytokine via its Glu-Leu-Arg (ELR) motif, which is critical for polymorphonuclear leukocyte receptor binding [87].
WRS has strong structural homology to YRS [92]. As discussed above, WRS can be secreted in either a full-length or a truncated form. Based on a structural comparison, the ELR motif in YRS seems to be replaced with a Tyr-Gly-Tyr (YGY) motif in mini-WRS [94]. However, the YGY sequence of WRS was not essential for its angiostatic cytokine activity, but instead was important for maintaining structural stability. Alternatively, the Glu-His-Arg (EHR)-containing eight-residue motif (aa 382–389) in mini-WRS was directly involved in its angiostatic activity [94]. Whether the EHR motif of WRS affects binding to chemokine receptors and activates signal transduction should be investigated.
The extracellular signaling activity of KRS has been described [89]. KRS was secreted from intact human cells in its full-length form, which functioned as an active cytokine. The secreted KRS bound to monocytes and macrophages to induce immune responses through the ERK and p38 MAPK signaling pathways. TNF-α but not TGF-β induced the release of endogenous KRS from cells [89]. Interestingly, Shiga toxins produced by Shigella dysenteriae and a subset of diarrheagenic Escherichia coli [95] triggered the dissociation of KRS from the MSC and its secretion by macrophages [96]. The secreted KRS contributed to Shiga toxin-mediated inflammatory signals by increasing the production of proinflammatory cytokines and chemokines from target cells. This study showed that a bacteria-derived virulence factor could use KRS as a cytokine to synergistically exacerbate inflammatory responses in the host. HRS secreted in its full-length form [97] induced lymphocyte migration and activated monocytes and immature dendritic cells through CCR5 binding [98].
AIMP1 is also secreted as a full-length form or as a 22 kDa C-terminal EMAPII domain [99]. Secreted AIMP1 displayed diverse biological functions, including upregulation of proinflammatory genes [43] and activation of immune cells [46], [100]. The EMAPII domain activated host immune responses by acting as a proinflammatory cytokine or as a chemoattractant to induce endothelial/mononuclear cell migration [99], [101], [102].
Due to their specific cytokine-like and chemotactic activities, the secreted ARSs have great potential for boosting immunity against primary infections. Alternatively, blocking the release of ARSs from cells or targeting the secreted ARSs with specific antibodies or inhibitors could be used to alleviate severe inflammation during infections, similar to anti-TNF-α therapy in sepsis [103]. Although the mechanisms underlying ARS secretion have not been fully elucidated, recent studies showed that GRS and KRS are released via exosomes [104], [105]. Application of an exosome pathway inhibitor (e.g., GW4869) or an ER-Golgi pathway inhibitor (e.g., brefeldin A) is a potential strategy to control ARS secretion and attenuate excessive inflammation induced by infection. Extracellular functions of ARSs in eliciting cytokine signaling responses are summarized in Fig. 1 and Table 2 .
Table 2.
Cytokine-like functions of ARSs.
| ARS | Stimulus | Active form | Target cell | Receptor | Function | Refs. |
|---|---|---|---|---|---|---|
| YRS | Apoptotic condition |
|
Leukocyte | CXCR1/2 |
|
[87] |
| WRS | IFN-γ |
|
Endothelial cell | VE-cadherin | Angiostatic activity with EHR motif | [52], [94] |
| KRS |
|
Full-length (exosome-associated) |
|
Not identified |
|
[89], [96], [105] |
| HRS | Inflammatory myositis |
(internal deletion of catalytic domain) |
|
CCR5 |
|
[97], [98] |
| AIMP1 |
|
|
|
CD23 |
|
[99], [101], [102] |
EMAPII, endothelial monocyte-activating polypeptide II; ELR, Glu-Leu-Arg; EHR, Glu-His-Arg; DC, dendritic cell.
4.2. ARS-derived peptides as potential anti-infective drugs
Development of human ARS-derived peptides is an attractive pharmacological approach in that they are relatively safe and well tolerated [106]. Currently, clinical approaches to treat acute infections depend mainly on the use of antibiotics or antiviral drugs. Host-directed therapies to augment anti-infective responses are greatly needed [107]. ARSs that have bioactive roles in anti-infectious signaling represent potential candidates to enhance host defense. Our group showed that an EPRS-derived peptide has potential as a therapeutic agent against a broad range of RNA viruses [20]. A domain mapping study of EPRS showed that the EPRS L1 region (aa 168–196) was critical for protection of MAVS from PCBP2-mediated ubiquitination and activation of antiviral type I IFN signaling [20]. Based on this observation, an antiviral peptide called Tat-Epep was designed by fusing the EPRS L1 region to the cell-permeable HIV-1 Tat tag. Treatment of macrophages with Tat-Epep increased their production of antiviral cytokines and reduced RNA viral replication in vitro. Tat-Epep was shown to increase MAVS stability by inhibiting the negative regulation of PCBP2 in cells. To further examine the antiviral mechanism of Tat-Epep, structure-based modifications of the peptide should be introduced for optimization, and its activity should be functionally validated in vivo.
The ELR motif in human YRS is a representative leukocyte chemoattractant. The ELR peptide binds to host macrophages and is internalized, leading to enhanced secretion of the proinflammatory cytokines TNF-α and IL-6 [108]. Introduction of the YRS ELR motif into yeast cells, which lack this protein, led to a gain of cytokine function [109], [110]. A structural analysis of the embedded ELR motif in YRS to investigate how it interacts with neighboring residues and binds with high affinity to the chemokine receptor CXCR1 only in the case of mini-YRS will support the development of this motif as a therapeutic agent to regulate inflammation as well as angiogenesis.
The WRS N-terminal region (aa 1–154), which acts as a danger signal early in infections, may also be a very promising target for an antimicrobial peptide [22]. Direct binding between the N-terminal region of WRS and TLRs stimulates anti-infective immune responses in the host. Identification of a functional motif required for this binding and annotation of specific residues by structural analysis of the WRS N-terminal domain will help us design potent drug candidates to treat infectious disease.
To overcome the general weakness of peptide drugs, including their short half-life, fast elimination, and low membrane permeability [106], small, synthetic compounds designed based on the identified ARS peptides may offer an effective solution for the development of anti-infectious drugs.
4.3. ARSs as diagnostic biomarkers
Infectious diseases can be monitored by direct detection of pathogens. However, antigenic variation from a broad diversity of microbial pathogens and the presence of unknown antigens can make it difficult to diagnose the exact type of pathogen [111]. An approach that has the potential to address these challenges relies on monitoring the expression of host genes or proteins [112]. Analyzing gene expression profiles from virus-infected human blood showed that different pathogens elicited distinct host RNA signatures [113]. Host proteins involved in immune responses can also be an indicator of infection, and elevated levels of these factors can provide useful clinical information in the context of infectious disease. For example, detection of the concentrations of three proteins, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), interferon-inducible protein 10 (IP-10), and C-reactive protein (CRP), could be used to discriminate between bacterial and viral diseases and reduce antibiotic misuse [114]. High mobility group box 1 (HMGB1) protein is also an important mediator of inflammation, and elevated plasma levels are significantly associated with infection [115].
A close correlation between the secretion of ARSs and the presence of infection was observed in a previous study [22]. WRS was detected in supernatants from PBMC cultures 2 h after bacterial infection, while HMGB1 was not detected during this period [22]. This result suggested that, compared with HMGB1, which is secreted at a relatively late stage (at 12 h) after exposure to pathogens [116], WRS may be used as an early biomarker of infection [22]. Moreover, while HMGB1 is released in response to numerous inflammatory signals in addition to infection [115], WRS secretion is dependent on the presence of infection, which differentiates sepsis from systemic inflammatory response syndrome. Therefore, WRS may be a useful diagnostic tool for infectious diseases including sepsis without the necessity of detecting pathogenic antigens in patient serum.
KRS was also secreted by intact human cells in response to proinflammatory cytokines and stimulated TNF-α production by immune cells [89]. Bacterial toxins also induced KRS secretion, which in turn increased cytokine production [96]. This positive feedback loop augments the KRS signal during the infection, and may be utilized as a detection tool for infectious disease. Because the types and amounts of secreted ARSs might differ depending on the pathogen and the duration of the infection, the classification of detectable ARSs from human samples can provide valuable information about disease status [22], [89].
Another strategy to detect infection is measuring ARS antibodies. Specific auto-antibodies are found in the sera of patients with autoimmune disease. Antisynthetase syndrome is a chronic autoimmune condition caused by the production of auto-antibodies against eight different ARSs, of which anti-HRS is the most prevalent [117], [118]. Antisynthetase syndrome is a form of idiopathic inflammatory myopathy characterized by myositis, interstitial lung disease, and arthritis [119]. Although the exact underlying mechanism for this syndrome is unknown, it seems to occur after certain viral infections or exposure to certain drugs [119]. Indeed, infectious agents are thought to induce autoimmunity once self-tolerance is broken [120]. For example, infectious myositis, one of the symptoms of antisynthetase syndrome, is an acute, subacute, or chronic infection of skeletal muscle caused by viruses including HIV, bacteria, fungi, and parasites [121]. Therefore, ARS auto-antibodies may be clinically useful as biomarkers of infection.
Finally, a robust and specific ARS detection system is required for accurate prediction of infectious diseases. For purification of human ARSs as antigens, specific design is required to minimize cross-reactivity with pathogenic ARSs that contain conserved catalytic regions. Determination of antigenicity and specificity based on the host ARS sequence and structure should improve ARS detection [30]. Several ARS antibodies and sandwich ELISA systems are commercially available, but most utilize polyclonal antibodies and are quite expensive, and the sensitivities are not sufficient for use with human serum samples, which have high levels of background [122]. To overcome this low resolution, new diagnostic tools such as protein chips should be developed to identify multiple ARSs simultaneously [123]. PCR-based methods to detect host ARSs can also be used due to their high sensitivity and specificity [124]. The rational design of discriminative primers for human ARSs will promote the timely diagnosis of infections. Moreover, in addition to secreted ARSs, examining the host ARS proteome or genome signature in cells isolated from patient peripheral blood or tissues will help correlate the abnormal expression of ARSs with infections [125], [126], [127].
Collectively, ARSs are novel biomarkers that may be used to identify early or secondary infections. The combined detection of pathogenic antigens and host ARSs that are specifically involved in infections will improve current diagnosis systems for infections.
5. Bacterial ARSs as targets for antimicrobial drug development
5.1. Representative compounds inhibiting essential functions of bacterial ARSs
There is a great deal of interest and concern about the rate at which new pathogens are emerging and causing significant human health problems. Among a wide variety of factors that predispose pathogens to invade host populations, the spread of antibiotic-resistant bacteria is a serious challenge in medical research.
Inhibition of protein synthesis by targeting tRNA aminoacylation has proven to be an effective strategy for anti-pathogenic drug development [128]. The catalytic domain of ARS has three distinct binding pockets responsible for recognition of amino acids, adenylate, and tRNA moieties [129]. Based on these substrate-binding sites, three classes of ARS inhibitors have been explored. While some inhibitors target a single specific pocket, many bind to one or more sites within the catalytic domain or the separate editing domain. Firstly, amino acid analogs can act as competitors for particular amino acids in ARS domains. Halofuginone binds to the proline- and tRNA-binding sites of PRS, and thereby inhibits its enzymatic activity and has been used as an herbal medicine to treat malaria [130], [131], [132]. Other biosynthetic products include indolmycin/chuangxinmycin (tryptophan analogs), cispentacin/icofungipen (an isoleucine analog), ochratoxin A (a phenylalanine analog), and SB219383 (a tyrosine analog), although they have not been used as therapeutic agents because of their poor antibacterial activity and low specificity [133]. Developing compounds that mimic aminoacyl-adenylate (AA-AMP) intermediate or bind to AA-AMP pockets may also be a powerful strategy [134]. Mupirocin is a mimetic of isoleucyl-AMP, which occupies the AA-AMP-binding site [135]. It is a very successful ARS inhibitor, exhibiting a high degree of specificity to several species of bacteria, including methicillin-resistant Staphylococcus aureus, E. coli, and Bacillus subtilis [135]. The clinical use of mupirocin validates the idea that ARSs can be effective antibiotic targets in human patients. Lastly, AN2690 inhibits fungal LRS by forming a tRNALeu-AN2690 adduct in the editing site [136]. It traps the 3′-end A76 nucleotide of tRNA, thus inhibiting subsequent rounds of aminoacylation. Interestingly, borrelidin acts as a triple competitive inhibitor [137]. Co-crystal structures of borrelidin with TRS showed that borrelidin simultaneously occupied four distinct subsites, including substrate-binding sites for amino acids, ATP, and tRNAThr, and an additional binding site [137].
5.2. Developing bacterial ARS inhibitors
The full set of 20 ARSs present in most bacterial pathogens offers great opportunities for the development of diverse antibacterial target-based drugs [133]. The considerable evolutionary divergence between prokaryotic and eukaryotic ARSs may reduce potential off-target effects of ARS inhibitors. Bacterial ARSs have many advantages as druggable targets. These enzymes are relatively easy to purify as recombinant proteins. Typical aminoacylation assays measuring the rate of AA-tRNAAA formation and kinetic assays to measure the rate of conversion of PPi into ATP using radioisotopes or specific organic compounds like malachite green are available for early characterization of ARS inhibitors [128], [138].
The traditional approach to identify ARS-targeted drugs would include high-throughput screening with large libraries of chemicals or bacteria-derived natural products. Technological advances in automated high-throughput screening will enable the rapid, efficient, and inexpensive discovery of low molecular weight compounds that perturb bacterial ARS functions [139]. Potent inhibitors of S. aureus MRS were identified from a high-throughput screen by GlaxoSmithKline [140]. The compound was later characterized as a quinolinone derivative that acts by competition with a methionine substrate [141]. Anacor also used a combinatorial screening of a boron-containing small molecule library and discovered a novel synthetic inhibitor, AN2690, that inhibited fungal LRS [142]. However, a serendipitous discovery from a large chemical library would be like looking for a needle in a haystack, and such compounds may have a high risk of toxicity because they would work through unknown mechanisms.
Structure-based drug design is the most powerful approach for drug development. This technique could especially be applied to ARS inhibitors since the architecture of ARS catalytic domains is structurally conserved throughout evolution. The low amount of variation in the ARS active domains suggests that targeting bacterial ARSs may be a broad strategy for use against a range of pathogens, but selectivity between eukaryotic and prokaryotic ARSs might be difficult to achieve. As enzymes, the core structures of ARSs without additional domains (which are usually present in higher eukaryotes) are readily soluble, and many of their unique structures are publicly available in the Protein Data Bank. Atomic-level characterization with structure-guided drug design is critical for developing and modifying ARS-based drugs. Co-crystallization of ARSs and inhibitors clearly shows how small compounds can potently control ARS function across a variety of species. The detailed structural and functional analysis of binding between the natural product borrelidin and TRS is a good example [137]. The authors determined the crystal structures of both human and E. coli TRS in complex with borrelidin and found that borrelidin has potent and redundant mechanisms to inhibit TRS during protein synthesis. This study revealed that borrelidin acted as a quadrivalent inhibitor by filling three substrate-binding sites and an extra space through its extended macrolide ring [137].
Growing reports on the crystal structures of ARSs showing the detailed catalytic sites and their availability provide valuable information for drug design and docking. Instead of resolving the co-crystal of an ARS and its inhibitor experimentally, new computational algorithms and approaches for modeling ARS drug interactions have become indispensable and time-saving tools [143]. Several bioinformatics tools are available to explore sequence and structure-derived enzyme active site residues to annotate drug inserts [143]. In silico methods including virtual screening and structure-based drug design have been used to design ARS inhibitors targeting LRS, WRS, NRS, MRS, IRS, and TRS [144], [145], [146]. The inhibitory activities of these compounds must be confirmed. Moreover, ARS substrate-based modification such as exploiting the structure of tRNA can be considered. Trana Discovery has developed a modern bioinformatics tool to conduct detailed analyses of tRNAs and employed a high-throughput screening system with a fluorescent probe mimicking the T-loop region to identify novel drugs that bind at the anticodon stem loop region of tRNAs to inhibit pathogen growth [147]. In addition, despite the structural homology, specific sequence differences between prokaryotic and eukaryotic ARSs can be used to design discriminating and specific ARS-targeted drugs. For example, the successful application of mupirocin is derived from a mere two-amino-acid difference between the prokaryotic and eukaryotic ARS [148]. Mutational analysis of amino acid residues will further characterize the critical sites involved in the interactions between ARSs and their inhibitors.
Taken together, the integration of diverse methods will greatly enhance the rational development of more optimized and sophisticated ARS inhibitors that will meet the need for new antimicrobial, antifungal, and antiparasitic drugs.
5.3. Bacterial resistance against ARS-targeted antibiotics
Unfortunately, the use of antibiotics has inevitably led to bacterial resistance. ARS inhibitors are no exception. S. aureus is an extraordinarily adaptable Gram-positive pathogen with a proven ability to develop drug resistance [149]. Although mupirocin has been used to treat methicillin-resistant S. aureus clinically at a MIC of ≤4 μg/ml, this strain appears to have evolved two types of resistance to mupirocin [150]. First, point mutations in the targeted S. aureus IRS led to the development of low-level (mupirocin MIC ≤ 8–64 μg/ml) antibiotic resistance, while horizontal gene transfer of resistance genes (mupA, which encodes an alternate IRS, or mupB, which contains common IRS motifs) from either eukaryotes or other bacteria caused high-level resistance (mupirocin MIC ≥ 512 μg/ml) [151]. Additionally, although the MRS inhibitors were potent against a large number of pathogenic strains such as S. aureus and Enterococcus, Streptococcus pneumoniae exhibited a wide range of susceptibility to these compounds [140]. S. pneumoniae strains encoding a second, distantly related MRS enzyme called MRS2 are resistant to synthetically derived inhibitors, including REP8839 [141]. S. pneumoniae acquired the mrs2 gene by horizontal transfer from Gram-positive pathogens or close relatives [141].
Many ARS inhibitors are naturally produced by several microbes. Because biosynthesized ARS inhibitors can be toxic to their sources by targeting their own ARSs, bacteria that produce ARS inhibitors require self-immunity. Here, self-immunity is defined as a resistance system against antimicrobial compounds that can kill related bacteria. Ironically, plasmids encoding these resistance genes (usually genes encoding secondary ARSs) are readily transmissible by conjugation and confer polymicrobial resistance. For example, Pseudomonas fluorescens produces mupirocin by quorum sensing to kill competitor bacteria in the local environment [152]. P. fluorescens protects itself by expressing the mupM gene, which encodes a second IRS. Transfer of P. fluorescens mupM to E. coli induced resistance to mupirocin [153]. Similarly, the Agrobacterium radiobacter strain K84 produces agrocin 84, which inhibits a plant pathogen, A. tumefaciens C58 [154]. Immunity to agrocin 84 is conferred by the agnB2 gene, which encodes an active LRS. Introduction of this gene to pathogenic A. tumefaciens imparted antibiotic resistance, suggesting that the biocontrol organism carries a self-protected ARS to avoid cell death [155]. Thus, although ARSs are good drug targets due to their high selectivity and potency toward microbes, sharing of ARS resistance genes not only locally, among neighboring bacteria, but also across long distances by selective pressure is a major obstacle for ARS drug application [148]. Identification of ARS inhibitors that are not associated with self-immunity and that do not transfer natural resistance genes should be identified. Finally, based on the different chemical properties of the 20 ARSs, targeting multiple ARSs simultaneously may be a safe way to overcome the emergence of resistance.
6. Conclusions and outlook
Infectious diseases remain an ever-growing risk to human health due to increasing antibiotic resistance, immune-compromised populations, and the emergence of new viruses that spread rapidly through the population due to globalization. Identification of novel drug targets to control these complex infective situations is inevitably of great importance.
Beyond their roles in protein synthesis, ubiquitously existed ARSs can sense and immediately respond to danger signals. MSC, a complex consisting of ARSs and non-enzymatic proteins, has been regarded as a hub for the cellular regulatory functions of ARSs [19]. The anti-infective functions of EPRS, which positively regulates antiviral signaling [20], and AIMP1, which increases the expression of antiviral genes [21], provide further evidence that the MSC serves as an immune regulatory system against infection. In addition, the non-MSC protein WRS is considered a primary defense molecule that helps cells of the immune system to respond rapidly to infectious signals [22]. Moreover, some secreted ARSs have evolved activities similar to those of classical proinflammatory cytokines [35], [87]. All these functions demonstrate that ARSs are promising therapeutic targets that can boost defensive immune responses and control infectious pathogens. Additionally, because the duration of infection can range from days to years, screening for ARS expression and secretion could provide a potential prognostic maker for acute and chronic infections.
The emergence of multidrug-resistant microbes is a substantial threat to human health. Therapeutic applications of ARS-targeted inhibitors will offer the alternative drugs to overcome the inevitable bacterial resistance to currently used antibiotics. Although there are no known antiviral drugs targeting ARSs, identification of a GAIT-like RNA motif in the viral genome [68] or discovery of host ARS-RNA intermediates that are packaged into retroviruses may offer a new opportunities for the development of antiviral agents [148].
Still, limited studies are available on the efficacy of targeting ARSs against infections. Recent studies including our findings provide new insights on the roles of ARSs in infection and anti-infective immunity. We hope to stimulate further in-depth research in this field to advance both basic science and clinical applications of ARS.
7. Conflicts of interest
The authors declare no conflicts of interest related to this manuscript.
Acknowledgments
This work was supported by the National Research Foundation of Korea, which is funded by the Ministry of Science and Communications Technology (NRF-2010-0029767 and 2015M3C9A4053394 to M.H.K.; NRF-M3A6A4-2010-0029785 to S.K.), and the Korea Research Institute of Biology and Biotechnology (KRIBB) Initiative Program (to M.H.K.).
References
- 1.Mogensen T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009;22:240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoshi T., Koyama S., Kobiyama K., Akira S., Ishii K.J. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011;1:226–232. doi: 10.1016/j.coviro.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Chaplin D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010;125:S3–S23. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 5.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.McWhirter S.M., Tenoever B.R., Maniatis T. Connecting mitochondria and innate immunity. Cell. 2005;122:645–647. doi: 10.1016/j.cell.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 7.Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 8.Reis e Sousa C. Activation of dendritic cells: translating innate into adaptive immunity. Curr. Opin. Immunol. 2004;(16):21–25. doi: 10.1016/j.coi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Caramalho I., Lopes-Carvalho T., Ostler D., Zelenay S., Haury M., Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J. Exp. Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marrack P., Scott-Browne J., MacLeod M.K. Terminating the immune response. Immunol. Rev. 2010;236:5–10. doi: 10.1111/j.1600-065X.2010.00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gack M.U., Shin Y.C., Joo C.H., Urano T., Liang C., Sun L. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 12.Belgnaoui S.M., Paz S., Samuel S., Goulet M.L., Sun Q., Kikkert M. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe. 2012;12:211–222. doi: 10.1016/j.chom.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 13.You F., Sun H., Zhou X., Sun W., Liang S., Zhai Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009;10:1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 14.Liu X.Y., Wei B., Shi H.X., Shan Y.F., Wang C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 2010;20:994–1011. doi: 10.1038/cr.2010.103. [DOI] [PubMed] [Google Scholar]
- 15.Papatriantafyllou M. Innate immunity: MAVS build-ups for defence. Nat. Rev. Immunol. 2011;11:570. doi: 10.1038/nri3050. [DOI] [PubMed] [Google Scholar]
- 16.Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol. Cell Biol. 2015;93:3–10. doi: 10.1038/icb.2014.75. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y., Tong X., Ye X. Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 2012;189:5304–5313. doi: 10.4049/jimmunol.1201445. [DOI] [PubMed] [Google Scholar]
- 18.Castanier C., Zemirli N., Portier A., Garcin D., Bidere N., Vazquez A. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray P.S., Arif A., Fox P.L. Macromolecular complexes as depots for releasable regulatory proteins. Trends Biochem. Sci. 2007;32:158–164. doi: 10.1016/j.tibs.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Lee E.Y., Lee H.C., Kim H.K., Jang S.Y., Park S.J., Kim Y.H. Infection-specific phosphorylation of glutamyl-prolyl tRNA synthetase induces antiviral immunity. Nat. Immunol. 2016;17:1252–1262. doi: 10.1038/ni.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang D., Tian L., You R., Halpert M.M., Konduri V., Baig Y.C. AIMp1 potentiates TH1 polarization and is critical for effective antitumor and antiviral immunity. Front. Immunol. 2017;8:1801. doi: 10.3389/fimmu.2017.01801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn Y.H., Park S., Choi J.J., Park B.K., Rhee K.H., Kang E. Secreted tryptophanyl-tRNA synthetase as a primary defence system against infection. Nat. Microbiol. 2016;2:16191. doi: 10.1038/nmicrobiol.2016.191. [DOI] [PubMed] [Google Scholar]
- 23.Baddal B., Muzzi A., Censini S., Calogero R.A., Torricelli G., Guidotti S. Dual RNA-seq of nontypeable haemophilus influenzae and host cell transcriptomes reveals novel insights into host-pathogen cross talk. MBio. 2015;6:e01765–e1815. doi: 10.1128/mBio.01765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodhouse S.D., Narayan R., Latham S., Lee S., Antrobus R., Gangadharan B. Transcriptome sequencing, microarray, and proteomic analyses reveal cellular and metabolic impact of hepatitis C virus infection in vitro. Hepatology. 2010;52:443–453. doi: 10.1002/hep.23733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X., Wang S., Yu Y., Zhang J., Sun Z., Yan Y. Subcellular proteomic analysis of human host cells infected with H3N2 swine influenza virus. Proteomics. 2013;13:3309–3326. doi: 10.1002/pmic.201300180. [DOI] [PubMed] [Google Scholar]
- 26.Clarke P., Leser J.S., Bowen R.A., Tyler K.L. Virus-induced transcriptional changes in the brain include the differential expression of genes associated with interferon, apoptosis, interleukin 17 receptor A, and glutamate signaling as well as flavivirus-specific upregulation of tRNA synthetases. MBio. 2014;5:e00902–e914. doi: 10.1128/mBio.00902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clemens M.J. Does protein phosphorylation play a role in translational control by eukaryotic aminoacyl-tRNA synthetases? Trends Biochem. Sci. 1990;15:172–175. doi: 10.1016/0968-0004(90)90153-3. [DOI] [PubMed] [Google Scholar]
- 28.Guo M., Yang X.L., Schimmel P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010;11:668–674. doi: 10.1038/nrm2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo M., Schimmel P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013;9:145–153. doi: 10.1038/nchembio.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo M., Schimmel P., Yang X.L. Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett. 2010;584:434–442. doi: 10.1016/j.febslet.2009.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arif A., Jia J., Moodt R.A., DiCorleto P.E., Fox P.L. Phosphorylation of glutamyl-prolyl tRNA synthetase by cyclin-dependent kinase 5 dictates transcript-selective translational control. Proc. Natl. Acad. Sci. U.S.A. 2011;(108):1415–1420. doi: 10.1073/pnas.1011275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arif A., Jia J., Mukhopadhyay R., Willard B., Kinter M., Fox P.L. Two-site phosphorylation of EPRS coordinates multimodal regulation of noncanonical translational control activity. Mol. Cell. 2009;35:164–180. doi: 10.1016/j.molcel.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arif A., Yao P., Terenzi F., Jia J., Ray P.S., Fox P.L. The GAIT translational control system. Wiley Interdiscip. Rev. RNA. 2017 doi: 10.1002/wrna.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guha M., Mackman N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 35.Yannay-Cohen N., Carmi-Levy I., Kay G., Yang C.M., Han J.M., Kemeny D.M. LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol. Cell. 2009;34:603–611. doi: 10.1016/j.molcel.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Ofir-Birin Y., Fang P., Bennett S.P., Zhang H.M., Wang J., Rachmin I. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell. 2013;49:30–42. doi: 10.1016/j.molcel.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y.N., Nechushtan H., Figov N., Razin E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity. 2004;20:145–151. doi: 10.1016/s1074-7613(04)00020-2. [DOI] [PubMed] [Google Scholar]
- 38.Jeong S.J., Kim J.H., Lim B.J., Yoon I., Song J.A., Moon H.S. Inhibition of MUC1 biosynthesis via threonyl-tRNA synthetase suppresses pancreatic cancer cell migration. Exp. Mol. Med. 2018;50:e424. doi: 10.1038/emm.2017.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwon N.H., Kang T., Lee J.Y., Kim H.H., Kim H.R., Hong J. Dual role of methionyl-tRNA synthetase in the regulation of translation and tumor suppressor activity of aminoacyl-tRNA synthetase-interacting multifunctional protein-3. Proc. Natl. Acad. Sci. U.S.A. 2011;108:19635–19640. doi: 10.1073/pnas.1103922108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia J., Arif A., Ray P.S., Fox P.L. WHEP domains direct noncanonical function of glutamyl-Prolyl tRNA synthetase in translational control of gene expression. Mol. Cell. 2008;29:679–690. doi: 10.1016/j.molcel.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arif A., Chatterjee P., Moodt R.A., Fox P.L. Heterotrimeric GAIT complex drives transcript-selective translation inhibition in murine macrophages. Mol. Cell. Biol. 2012;32:5046–5055. doi: 10.1128/MCB.01168-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y., Kim Y., Jin K.S., Kim H.S., Kim J.H., Wang D. Structure of the ArgRS-GlnRS-AIMP1 complex and its implications for mammalian translation. Proc. Natl. Acad. Sci. U S A. 2014;111:15084–15089. doi: 10.1073/pnas.1408836111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ko Y.G., Park H., Kim T., Lee J.W., Park S.G., Seol W. A cofactor of tRNA synthetase, p43, is secreted to up-regulate proinflammatory genes. J. Biol. Chem. 2001;276:23028–23033. doi: 10.1074/jbc.M101544200. [DOI] [PubMed] [Google Scholar]
- 44.Kim E., Kim S.H., Kim S., Cho D., Kim T.S. AIMP1/p43 protein induces the maturation of bone marrow-derived dendritic cells with T helper type 1-polarizing ability. J. Immunol. 2008;180:2894–2902. doi: 10.4049/jimmunol.180.5.2894. [DOI] [PubMed] [Google Scholar]
- 45.Kim M.S., Song J.H., Cohen E.P., Cho D., Kim T.S. Aminoacyl tRNA synthetase-interacting multifunctional protein 1 activates NK cells via macrophages in vitro and in vivo. J. Immunol. 2017;198:4140–4147. doi: 10.4049/jimmunol.1601558. [DOI] [PubMed] [Google Scholar]
- 46.Kim M.S., Kim T.S. Aminoacyl tRNA synthetase-interacting multifunctional protein 1 acts as a novel B cell-activating factor in vitro and in vivo. J. Immunol. 2015;194:4729–4736. doi: 10.4049/jimmunol.1401352. [DOI] [PubMed] [Google Scholar]
- 47.Liang D., Halpert M.M., Konduri V., Decker W.K. Stepping out of the cytosol: AIMp1/p43 potentiates the link between innate and adaptive immunity. Int. Rev. Immunol. 2015;34:367–381. doi: 10.3109/08830185.2015.1077829. [DOI] [PubMed] [Google Scholar]
- 48.Steinman R.M., Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr. Top. Microbiol. Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 49.Mlecnik B., Bindea G., Angell H.K., Maby P., Angelova M., Tougeron D. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 50.Kranz L.M., Diken M., Haas H., Kreiter S., Loquai C., Reuter K.C. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 51.Wakasugi K., Slike B.M., Hood J., Otani A., Ewalt K.L., Friedlander M. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:173–177. doi: 10.1073/pnas.012602099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otani A., Slike B.M., Dorrell M.I., Hood J., Kinder K., Ewalt K.L. A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:178–183. doi: 10.1073/pnas.012601899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miyanokoshi M., Yokosawa T., Wakasugi K. Tryptophanyl-tRNA synthetase mediates high-affinity tryptophan uptake into human cells. J. Biol. Chem. 2018 doi: 10.1074/jbc.RA117.001247. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holland J., Spindler K., Horodyski F., Grabau E., Nichol S., VandePol S. Rapid evolution of RNA genomes. Science. 1982;215:1577–1585. doi: 10.1126/science.7041255. [DOI] [PubMed] [Google Scholar]
- 55.Pavon-Eternod M., David A., Dittmar K., Berglund P., Pan T., Bennink J.R. Vaccinia and influenza A viruses select rather than adjust tRNAs to optimize translation. Nucleic Acids Res. 2013;41:1914–1921. doi: 10.1093/nar/gks986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Netzer N., Goodenbour J.M., David A., Dittmar K.A., Jones R.B., Schneider J.R. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009;462:522–526. doi: 10.1038/nature08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castro A.P., Carvalho T.M., Moussatche N., Damaso C.R. Redistribution of cyclophilin A to viral factories during vaccinia virus infection and its incorporation into mature particles. J. Virol. 2003;77:9052–9068. doi: 10.1128/JVI.77.16.9052-9068.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.David A., Netzer N., Strader M.B., Das S.R., Chen C.Y., Gibbs J. RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J. Biol. Chem. 2011;286:20688–20700. doi: 10.1074/jbc.M110.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schimmel P., Giege R., Moras D., Yokoyama S. An operational RNA code for amino acids and possible relationship to genetic code. Proc. Natl. Acad. Sci. U S A. 1993;90:8763–8768. doi: 10.1073/pnas.90.19.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dreher T.W. Viral tRNAs and tRNA-like structures. Wiley Interdiscip. Rev. RNA. 2010;1:402–414. doi: 10.1002/wrna.42. [DOI] [PubMed] [Google Scholar]
- 61.Pinck M., Yot P., Chapeville F., Duranton H.M. Enzymatic binding of valine to the 3' end of TYMV-RNA. Nature. 1970;226:954–956. doi: 10.1038/226954a0. [DOI] [PubMed] [Google Scholar]
- 62.Hall T.C., Shih D.S., Kaesberg P. Enzyme-mediated binding of tyrosine to brome-mosaic-virus ribonucleic acid. Biochem. J. 1972;129:969–976. doi: 10.1042/bj1290969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sela I. Tobacco enzyme-cleaved fragments of TMV-RNA specifically accepting serine and methionine. Virology. 1972;49:90–94. doi: 10.1016/s0042-6822(72)80009-6. [DOI] [PubMed] [Google Scholar]
- 64.Dreher T.W. Role of tRNA-like structures in controlling plant virus replication. Virus Res. 2009;139:217–229. doi: 10.1016/j.virusres.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ariza-Mateos A., Gomez J. Viral tRNA mimicry from a biocommunicative perspective. Front. Microbiol. 2017;8:2395. doi: 10.3389/fmicb.2017.02395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colussi T.M., Costantino D.A., Hammond J.A., Ruehle G.M., Nix J.C., Kieft J.S. The structural basis of transfer RNA mimicry and conformational plasticity by a viral RNA. Nature. 2014;511:366–369. doi: 10.1038/nature13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galan C., Sola I., Nogales A., Thomas B., Akoulitchev A., Enjuanes L. Host cell proteins interacting with the 3' end of TGEV coronavirus genome influence virus replication. Virology. 2009;391:304–314. doi: 10.1016/j.virol.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marquez-Jurado S., Nogales A., Zuniga S., Enjuanes L., Almazan F. Identification of a gamma interferon-activated inhibitor of translation-like RNA motif at the 3' end of the transmissible gastroenteritis coronavirus genome modulating innate immune response. MBio. 2015;6:e00105. doi: 10.1128/mBio.00105-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rein A., Datta S.A., Jones C.P., Musier-Forsyth K. Diverse interactions of retroviral Gag proteins with RNAs. Trends Biochem. Sci. 2011;36:373–380. doi: 10.1016/j.tibs.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cen S., Khorchid A., Javanbakht H., Gabor J., Stello T., Shiba K. Incorporation of lysyl-tRNA synthetase into human immunodeficiency virus type 1. J. Virol. 2001;75:5043–5048. doi: 10.1128/JVI.75.11.5043-5048.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Javanbakht H., Halwani R., Cen S., Saadatmand J., Musier-Forsyth K., Gottlinger H. The interaction between HIV-1 Gag and human lysyl-tRNA synthetase during viral assembly. J. Biol. Chem. 2003;278:27644–27651. doi: 10.1074/jbc.M301840200. [DOI] [PubMed] [Google Scholar]
- 72.Halwani R., Cen S., Javanbakht H., Saadatmand J., Kim S., Shiba K. Cellular distribution of Lysyl-tRNA synthetase and its interaction with Gag during human immunodeficiency virus type 1 assembly. J. Virol. 2004;78:7553–7564. doi: 10.1128/JVI.78.14.7553-7564.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freed E.O. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 74.Kaminska M., Shalak V., Francin M., Mirande M. Viral hijacking of mitochondrial lysyl-tRNA synthetase. J. Virol. 2007;81:68–73. doi: 10.1128/JVI.01267-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duchon A.A., St Gelais C., Titkemeier N., Hatterschide J., Wu L., Musier-Forsyth K. HIV-1 exploits a dynamic multi-aminoacyl-tRNA synthetase complex to enhance viral replication. J. Virol. 2017;91 doi: 10.1128/JVI.01240-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burke K.P., Cox A.L. Hepatitis C virus evasion of adaptive immune responses: a model for viral persistence. Immunol. Res. 2010;47:216–227. doi: 10.1007/s12026-009-8152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gale M., Jr., Foy E.M. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 78.Kim M.S., Kim S., Myung H. Degradation of AIMP1/p43 induced by hepatitis C virus E2 leads to upregulation of TGF-beta signaling and increase in surface expression of gp96. PLoS One. 2014;9:e96302. doi: 10.1371/journal.pone.0096302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee Y.S., Han J.M., Son S.H., Choi J.W., Jeon E.J., Bae S.C. AIMP1/p43 downregulates TGF-beta signaling via stabilization of smurf2. Biochem. Biophys. Res. Commun. 2008;371:395–400. doi: 10.1016/j.bbrc.2008.04.099. [DOI] [PubMed] [Google Scholar]
- 80.Manz B., Brunotte L., Reuther P., Schwemmle M. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun. 2012;3:802. doi: 10.1038/ncomms1804. [DOI] [PubMed] [Google Scholar]
- 81.Gao S., Wu J., Liu R.Y., Li J., Song L., Teng Y. Interaction of NS2 with AIMP2 facilitates the switch from ubiquitination to SUMOylation of M1 in influenza A virus-infected cells. J. Virol. 2015;89:300–311. doi: 10.1128/JVI.02170-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kwak J.Y., Mamura M., Barlic-Dicen J., Grage-Griebenow E. Pathophysiological roles of cytokine-chemokine immune network. J. Immunol. Res. 2014;2014:615130. doi: 10.1155/2014/615130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aoki N., Xing Z. Use of cytokines in infection. Expert Opin. Emerg. Drugs. 2004;9:223–236. doi: 10.1517/14728214.9.2.223. [DOI] [PubMed] [Google Scholar]
- 84.Friedman R.M. Clinical uses of interferons. Br. J. Clin. Pharmacol. 2008;65:158–162. doi: 10.1111/j.1365-2125.2007.03055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoofnagle J.H., Seeff L.B. Peginterferon and ribavirin for chronic hepatitis C. N. Engl. J. Med. 2006;355:2444–2451. doi: 10.1056/NEJMct061675. [DOI] [PubMed] [Google Scholar]
- 86.Dinarello C.A., Margolis N.H. Cytokine-processing enzymes. Stopping the cuts. Curr. Biol. 1995;5:587–590. doi: 10.1016/s0960-9822(95)00116-3. [DOI] [PubMed] [Google Scholar]
- 87.Wakasugi K., Schimmel P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science. 1999;284:147–151. doi: 10.1126/science.284.5411.147. [DOI] [PubMed] [Google Scholar]
- 88.van Horssen R., Eggermont A.M., ten Hagen T.L. Endothelial monocyte-activating polypeptide-II and its functions in (patho)physiological processes. Cytokine Growth Factor Rev. 2006;17:339–348. doi: 10.1016/j.cytogfr.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Park S.G., Kim H.J., Min Y.H., Choi E.C., Shin Y.K., Park B.J. Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. U S A. 2005;102:6356–6361. doi: 10.1073/pnas.0500226102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Park M.C., Kang T., Jin D., Han J.M., Kim S.B., Park Y.J. Secreted human glycyl-tRNA synthetase implicated in defense against ERK-activated tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E640–E647. doi: 10.1073/pnas.1200194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kleeman T.A., Wei D., Simpson K.L., First E.A. Human tyrosyl-tRNA synthetase shares amino acid sequence homology with a putative cytokine. J. Biol. Chem. 1997;272:14420–14425. doi: 10.1074/jbc.272.22.14420. [DOI] [PubMed] [Google Scholar]
- 92.Doublie S., Bricogne G., Gilmore C., Carter C.W., Jr. Tryptophanyl-tRNA synthetase crystal structure reveals an unexpected homology to tyrosyl-tRNA synthetase. Structure. 1995;3:17–31. doi: 10.1016/s0969-2126(01)00132-0. [DOI] [PubMed] [Google Scholar]
- 93.Casas-Tinto S., Lolo F.N., Moreno E. Active JNK-dependent secretion of Drosophila Tyrosyl-tRNA synthetase by loser cells recruits haemocytes during cell competition. Nat. Commun. 2015;6:10022. doi: 10.1038/ncomms10022. [DOI] [PubMed] [Google Scholar]
- 94.Kise Y., Lee S.W., Park S.G., Fukai S., Sengoku T., Ishii R. A short peptide insertion crucial for angiostatic activity of human tryptophanyl-tRNA synthetase. Nat. Struct. Mol. Biol. 2004;11:149–156. doi: 10.1038/nsmb722. [DOI] [PubMed] [Google Scholar]
- 95.Johannes L., Romer W. Shiga toxins–from cell biology to biomedical applications. Nat. Rev. Microbiol. 2010;8:105–116. doi: 10.1038/nrmicro2279. [DOI] [PubMed] [Google Scholar]
- 96.Lee M.S., Kwon H., Nguyen L.T., Lee E.Y., Lee C.Y., Choi S.H. Shiga toxins trigger the secretion of lysyl-tRNA synthetase to enhance proinflammatory responses. J. Microbiol. Biotechnol. 2016;26:432–439. doi: 10.4014/jmb.1511.11056. [DOI] [PubMed] [Google Scholar]
- 97.Zhou J.J., Wang F., Xu Z., Lo W.S., Lau C.F., Chiang K.P. Secreted histidyl-tRNA synthetase splice variants elaborate major epitopes for autoantibodies in inflammatory myositis. J. Biol. Chem. 2014;289:19269–19275. doi: 10.1074/jbc.C114.571026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Howard O.M., Dong H.F., Yang D., Raben N., Nagaraju K., Rosen A. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J. Exp. Med. 2002;196:781–791. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kao J., Ryan J., Brett G., Chen J., Shen H., Fan Y.G. Endothelial monocyte-activating polypeptide II. A novel tumor-derived polypeptide that activates host-response mechanisms. J. Biol. Chem. 1992;267:20239–20247. [PubMed] [Google Scholar]
- 100.Kim E., Kim S.H., Kim S., Kim T.S. The novel cytokine p43 induces IL-12 production in macrophages via NF-kappaB activation, leading to enhanced IFN-gamma production in CD4+ T cells. J. Immunol. 2006;176:256–264. doi: 10.4049/jimmunol.176.1.256. [DOI] [PubMed] [Google Scholar]
- 101.Barnett G., Jakobsen A.M., Tas M., Rice K., Carmichael J., Murray J.C. Prostate adenocarcinoma cells release the novel proinflammatory polypeptide EMAP-II in response to stress. Cancer Res. 2000;60:2850–2857. [PubMed] [Google Scholar]
- 102.Park H., Park S.G., Lee J.W., Kim T., Kim G., Ko Y.G. Monocyte cell adhesion induced by a human aminoacyl-tRNA synthetase-associated factor, p43: identification of the related adhesion molecules and signal pathways. J. Leukoc. Biol. 2002;71:223–230. [PubMed] [Google Scholar]
- 103.Ghezzi P., Cerami A. Tumor necrosis factor as a pharmacological target. Mol. Biotechnol. 2005;31:239–244. doi: 10.1385/MB:31:3:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.He W., Bai G., Zhou H., Wei N., White N.M., Lauer J. CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature. 2015;526:710–714. doi: 10.1038/nature15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim S.B., Kim H.R., Park M.C., Cho S., Goughnour P.C., Han D. Caspase-8 controls the secretion of inflammatory lysyl-tRNA synthetase in exosomes from cancer cells. J. Cell Biol. 2017;216:2201–2216. doi: 10.1083/jcb.201605118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fosgerau K., Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov. Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 107.Basil M.C., Levy B.D. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016;16:51–67. doi: 10.1038/nri.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bhatt T.K., Khan S., Dwivedi V.P., Banday M.M., Sharma A., Chandele A. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat. Commun. 2011;2:530. doi: 10.1038/ncomms1522. [DOI] [PubMed] [Google Scholar]
- 109.Liu J., Yang X.L., Ewalt K.L., Schimmel P. Mutational switching of a yeast tRNA synthetase into a mammalian-like synthetase cytokine. Biochemistry. 2002;41:14232–14237. doi: 10.1021/bi0205395. [DOI] [PubMed] [Google Scholar]
- 110.Kapoor M., Otero F.J., Slike B.M., Ewalt K.L., Yang X.L. Mutational separation of aminoacylation and cytokine activities of human tyrosyl-tRNA synthetase. Chem. Biol. 2009;16:531–539. doi: 10.1016/j.chembiol.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lange A., Ferguson N.M. Antigenic diversity, transmission mechanisms, and the evolution of pathogens. PLoS Comput. Biol. 2009;5:e1000536. doi: 10.1371/journal.pcbi.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ramilo O., Mejias A. Shifting the paradigm: host gene signatures for diagnosis of infectious diseases. Cell Host Microbe. 2009;6:199–200. doi: 10.1016/j.chom.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 113.Zaas A.K., Chen M., Varkey J., Veldman T., Hero A.O., 3rd, Lucas J. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe. 2009;6:207–217. doi: 10.1016/j.chom.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.van Houten C.B., de Groot J.A.H., Klein A., Srugo I., Chistyakov I., de Waal W. A host-protein based assay to differentiate between bacterial and viral infections in preschool children (OPPORTUNITY): a double-blind, multicentre, validation study. Lancet Infect. Dis. 2017;17:431–440. doi: 10.1016/S1473-3099(16)30519-9. [DOI] [PubMed] [Google Scholar]
- 115.Harris H.E., Andersson U., Pisetsky D.S. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 116.Wang H., Bloom O., Zhang M., Vishnubhakat J.M., Ombrellino M., Che J. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 117.Witt L.J., Curran J.J., Strek M.E. The diagnosis and treatment of antisynthetase syndrome. Clin. Pulm. Med. 2016;23:218–226. doi: 10.1097/CPM.0000000000000171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hervier B., Benveniste O. Clinical phenotypes and prognosis of antisynthetase syndrome. Rev. Med. Interne. 2014;35:453–460. doi: 10.1016/j.revmed.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 119.Hervier B., Benveniste O. Clinical heterogeneity and outcomes of antisynthetase syndrome. Curr. Rheumatol. Rep. 2013;15:349. doi: 10.1007/s11926-013-0349-8. [DOI] [PubMed] [Google Scholar]
- 120.Zampieri S., Ghirardello A., Iaccarino L., Briani C., Sarzi-Puttini P., Atzeni F. Polymyositis-dermatomyositis and infections. Autoimmunity. 2006;39:191–196. doi: 10.1080/08916930600622348. [DOI] [PubMed] [Google Scholar]
- 121.Satoh M., Tanaka S., Ceribelli A., Calise S.J., Chan E.K. A Comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin. Rev. Allergy Immunol. 2017;52:1–19. doi: 10.1007/s12016-015-8510-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park S.G., Park H.S., Jeong I.K., Cho Y.M., Lee H.K., Kang Y.S. Autoantibodies against aminoacyl-tRNA synthetase: novel diagnostic marker for type 1 diabetes mellitus. Biomarkers. 2010;15:358–366. doi: 10.3109/13547501003777823. [DOI] [PubMed] [Google Scholar]
- 123.Natesan M., Ulrich R.G. Protein microarrays and biomarkers of infectious disease. Int. J. Mol. Sci. 2010;11:5165–5183. doi: 10.3390/ijms11125165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stahlberg A., Krzyzanowski P.M., Jackson J.B., Egyud M., Stein L., Godfrey T.E. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 2016;44:e105. doi: 10.1093/nar/gkw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schwartz M.H., Pan T. Determining the fidelity of tRNA aminoacylation via microarrays. Methods. 2017;113:27–33. doi: 10.1016/j.ymeth.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim S.S., Hur S.Y., Kim Y.R., Yoo N.J., Lee S.H. Expression of AIMP1, 2 and 3, the scaffolds for the multi-tRNA synthetase complex, is downregulated in gastric and colorectal cancer. Tumori. 2011;97:380–385. doi: 10.1177/030089161109700321. [DOI] [PubMed] [Google Scholar]
- 127.Hause R.J., Stark A.L., Antao N.N., Gorsic L.K., Chung S.H., Brown C.D. Identification and validation of genetic variants that influence transcription factor and cell signaling protein levels. Am. J. Hum. Genet. 2014;95:194–208. doi: 10.1016/j.ajhg.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ho J.M., Bakkalbasi E., Soll D., Miller C.A. Drugging tRNA aminoacylation. RNA Biol. 2018:1–11. doi: 10.1080/15476286.2018.1429879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Woese C.R., Olsen G.J., Ibba M., Soll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou H., Sun L., Yang X.L., Schimmel P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2013;494:121–124. doi: 10.1038/nature11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jain V., Yogavel M., Oshima Y., Kikuchi H., Touquet B., Hakimi M.A. Structure of prolyl-tRNA synthetase-halofuginone complex provides basis for development of drugs against malaria and Toxoplasmosis. Structure. 2015;23:819–829. doi: 10.1016/j.str.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 132.Keller T.L., Zocco D., Sundrud M.S., Hendrick M., Edenius M., Yum J. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012;8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hurdle J.G., O'Neill A.J., Chopra I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005;49:4821–4833. doi: 10.1128/AAC.49.12.4821-4833.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Belrhali H., Yaremchuk A., Tukalo M., Larsen K., Berthet-Colominas C., Leberman R. Crystal structures at 2.5 angstrom resolution of seryl-tRNA synthetase complexed with two analogs of seryl adenylate. Science. 1994;263:1432–1436. doi: 10.1126/science.8128224. [DOI] [PubMed] [Google Scholar]
- 135.Silvian L.F., Wang J., Steitz T.A. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- 136.Rock F.L., Mao W., Yaremchuk A., Tukalo M., Crepin T., Zhou H. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 137.Fang P., Yu X., Jeong S.J., Mirando A., Chen K., Chen X. Structural basis for full-spectrum inhibition of translational functions on a tRNA synthetase. Nat. Commun. 2015;6:6402. doi: 10.1038/ncomms7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Francklyn C.S., First E.A., Perona J.J., Hou Y.M. Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods. 2008;44:100–118. doi: 10.1016/j.ymeth.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu B., Li S., Hu J. Technological advances in high-throughput screening. Am. J. Pharmacogenomics. 2004;4:263–276. doi: 10.2165/00129785-200404040-00006. [DOI] [PubMed] [Google Scholar]
- 140.Jarvest R.L., Berge J.M., Berry V., Boyd H.F., Brown M.J., Elder J.S. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against gram-positive pathogens. J. Med. Chem. 2002;45:1959–1962. doi: 10.1021/jm025502x. [DOI] [PubMed] [Google Scholar]
- 141.Gentry D.R., Ingraham K.A., Stanhope M.J., Rittenhouse S., Jarvest R.L., O'Hanlon P.J. Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae with two distinct methionyl-tRNA synthetase genes. Antimicrob. Agents Chemother. 2003;47:1784–1789. doi: 10.1128/AAC.47.6.1784-1789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baker S.J., Zhang Y.K., Akama T., Lau A., Zhou H., Hernandez V. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690), for the potential treatment of onychomycosis. J. Med. Chem. 2006;49:4447–4450. doi: 10.1021/jm0603724. [DOI] [PubMed] [Google Scholar]
- 143.Sliwoski G., Kothiwale S., Meiler J., Lowe E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhao Y., Meng Q., Bai L., Zhou H. In silico discovery of aminoacyl-tRNA synthetase inhibitors. Int. J. Mol. Sci. 2014;15:1358–1373. doi: 10.3390/ijms15011358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu Y., Yu K., Xu B., Chen L., Chen X., Mao J. Potent and selective inhibitors of Staphylococcus epidermidis tryptophanyl-tRNA synthetase. J. Antimicrob. Chemother. 2007;60:502–509. doi: 10.1093/jac/dkm229. [DOI] [PubMed] [Google Scholar]
- 146.Zhao Y., Wang Q., Meng Q., Ding D., Yang H., Gao G. Identification of Trypanosoma brucei leucyl-tRNA synthetase inhibitors by pharmacophore- and docking-based virtual screening and synthesis. Bioorg. Med. Chem. 2012;20:1240–1250. doi: 10.1016/j.bmc.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 147.Chopra S., Reader J. tRNAs as antibiotic targets. Int. J. Mol. Sci. 2014;16:321–349. doi: 10.3390/ijms16010321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dewan V., Reader J., Forsyth K.M. Role of aminoacyl-tRNA synthetases in infectious diseases and targets for therapeutic development. Top. Curr. Chem. 2014;344:293–329. doi: 10.1007/128_2013_425. [DOI] [PubMed] [Google Scholar]
- 149.Chambers H.F., Deleo F.R. Waves of resistance: staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009;7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Patel J.B., Gorwitz R.J., Jernigan J.A. Mupirocin resistance. Clin. Infect. Dis. 2009;49:935–941. doi: 10.1086/605495. [DOI] [PubMed] [Google Scholar]
- 151.Seah C., Alexander D.C., Louie L., Simor A., Low D.E., Longtin J. MupB, a new high-level mupirocin resistance mechanism in Staphylococcus aureus. Antimicrob. Agents Chemother. 2012;56:1916–1920. doi: 10.1128/AAC.05325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Thomas C.M., Hothersall J., Willis C.L., Simpson T.J. Resistance to and synthesis of the antibiotic mupirocin. Nat. Rev. Microbiol. 2010;8:281–289. doi: 10.1038/nrmicro2278. [DOI] [PubMed] [Google Scholar]
- 153.Yanagisawa T., Lee J.T., Wu H.C., Kawakami M. Relationship of protein structure of isoleucyl-tRNA synthetase with pseudomonic acid resistance of Escherichia coli. A proposed mode of action of pseudomonic acid as an inhibitor of isoleucyl-tRNA synthetase. J. Biol. Chem. 1994;269:24304–24309. [PubMed] [Google Scholar]
- 154.Kim J.G., Park B.K., Kim S.U., Choi D., Nahm B.H., Moon J.S. Bases of biocontrol: sequence predicts synthesis and mode of action of agrocin 84, the Trojan horse antibiotic that controls crown gall. Proc. Natl. Acad. Sci. U.S.A. 2006;103:8846–8851. doi: 10.1073/pnas.0602965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ryder M.H., Slota J.E., Scarim A., Farrand S.K. Genetic analysis of agrocin 84 production and immunity in Agrobacterium spp. J. Bacteriol. 1987;169:4184–4189. doi: 10.1128/jb.169.9.4184-4189.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]