

Abstract
SARS coronavirus, SCV, has been recently responsible of a sudden and widespread infection which caused almost 800 victims. The limited amount of SCV protein structural information is partially responsible of the lack of specific drugs against the virus. Coronavirus helicases are very conserved and peculiar proteins which have been proposed as suitable targets for antiviral drugs, such as bananins, which have been recently shown to inhibit the SCV helicase in vitro. Here, the quaternary structure of SCV helicase has been predicted, which will provide a solid foundation for the rational design of other antiviral helicase inhibitors.
Keywords: SARS coronavirus, Protein structure, Structure prediction, Molecular modeling, Helicase structure, Drug design
Severe acute respiratory syndrome coronavirus (SCV) was the protagonist of a recent severe world-wide spread infection which caused almost 800 deaths within few months between 2002 and 2003 [1]. Genomic investigations have yielded a wealth of information on SCV evolution in terms of sequence mutations [2], [3], [4] to understand mechanism of viral transmission between animals and humans. Since SCV is still circulating, so far almost asymptomatically in southern China, our preparedness against new dangerous coronavirus outbreaks should include the development of specific antiviral drugs which, at the moment, remain unavailable. In this respect, structural genomics can play an important role in gaining insight into viral protein targets. Inhibition of these targets may interfere with the metabolism of the infecting virus without strong side effects on patients.
Helicase have been targeted for therapy in other viruses, suggesting that the SCV helicase may be a suitable target for new anti-SCV drugs [5], [6], [7], [8], [9]. The SCV helicase tertiary structure, not yet experimentally solved, would be useful for designing specific inhibitors of this SCV enzyme. Here, we report a structure prediction procedure to obtain a reliable molecular model of this viral protein.
Materials and methods
The sequence of SCV helicase (576 aminoacid residues with NCBI Accession No. NP_828870) [10] was aligned with that of representatives of the other groups of coronavirus. Sequence alignment of SCV helicase with other coronavirus helicases were obtained using program ClustalW v.1.8 [11]. The ATP binding site motif was predicted with MotifScan software available at the Expasy web-site [12]. Secondary structure prediction and fold recognition studies were performed by using PsiPred v.2.1 [13] and Pyre v.0.2 [14], respectively. Using the fold recognition method the helicase of SARS Coronavirus (SwissProt Accession ID P59641) [15] identifies the 1UAA pdb entry [16]. Model building of the major domain (494 aminoacids) of SARS helicase was carried out using ClustalW v.1.8 [11] alignment between the target sequence and that of the template structure. Models were subsequently optimized according to secondary structure predictions. Substitution of aminoacid residues and modeling of insertions and deletions in the target structure were performed using DeepView v.3.7 software [17]. The 3D models were optimized by a 900 step minimization run with AMBER [18] and finally validated with the PROCHECK v.3.5.4 procedure [19] and PROSAII v.4.0 software [20].
A model of the metal binding domain (MBD) was obtained using DYANA v.1.5 software [21]. The insertion of zinc atoms was carried out using GROMACS v.3.2 [22] software using the force field ffgmx43a2 that allows a molecular minimization of this domain with the bound metal atoms. To investigate the structure of MBD and helicase domain, a docking simulation was performed using ESCHER software [23]. A first coarse run with a rotation step of 10° was carried out and approximately 500 results were collected. Only those models that were consistent with the available experimental data were subjected to a second run with a rotation step of 2° between ± 20° of the starting position in order to refine the most probable structures. The major side-chain clashes were removed by a 900 cycles minimization in the AMBER force Field [18].
Results and discussion
A set of 35 different amino acid sequences of SCV helicase proteins, collected from the NCBI protein database, was aligned using the ClustalW v.1.8 software [11] and among all the considered sequences pairwise identities of 99% were observed. Thus, a representative consensus sequence was obtained for the SCV helicase, suggesting the presence of two separate domains, i.e., the helicase domain (Hel) and a metal binding domain (MBD). Such a combination of a putative MBD and a Hel domain in the same protein, as in Equine Arterivirus nsp10, has been found in a number of other viral and cellular proteins [24], [25], [26]. Accordingly, the nsp13 model building was performed in two separate steps, one for each domain of the enzyme, and a hybrid protein structure was assembled from the two modeled domains.
A 3D model of the SCV helicase Hel domain, spanning residues 80–568, was made on the basis of the crystal structure of the Escherichia coli Rep helicase. Both helicases belong to the same Superfamily 1 helicase classification on the basis of seven conserved sequence motifs [27]. Therefore, the Protein Data Bank entry 1UAA [16], corresponding to E. coli Rep helicase crystal structure, was used as a suitable template for a model building of the Hel domain using the threading program Pyre v.0.2 [14]. A manual alignment between the secondary structure elements predicted by shuffled PsiPred runs [13] and the ones observed in the model enhanced the correspondence of identical and positively conserved residues, respectively to 11% and 23%.
As far as the MBD structure is concerned, it should be noted that all coronaviruses exhibit very different amino acid sequences. However, coronavirus MBDs, as well as all the other nidoviruses, have a Cys and His rich motif which can orient the overall modeling procedure of this domain. In fact, arterivirus helicases have been shown to possess MBD domains which coordinate four Zn2+ [28], [29] in a way that could be similarly adopted by coronaviruses. In this Zn2+ binding domain, known also as binuclear cluster, a Cys residue coordinates two closely spaced Zn2+ ions bridging a Zn2Cys6 group, typically found in GAL4-like proteins [30] with Zn2Cys4His2, found in RAG1 domain [31]. The SCV MBD structure has been reconstructed using tetrahedral geometry characterization, distance constraints, and orientations typical of Cys and His residues of the latter clusters, suggested by the crystal structures of GAL4-like and RAG1. The corresponding Protein Data Bank files, 1PYI, and 1RMD, respectively [30], [31], provided accurate distance constraints for the two components of the MBD.
The reliability of the predicted tertiary structures of both SCV helicase domains was tested on the basis of simple physico-chemical rules, such as the ones suggested by Ramachandran plots, and the agreement between hydropathy profile and residue accessibility in the two modeled structures. Additional features which support the predicted structure of the Hel domain are given by the fact that 10 out of a total 15 cysteine residues, which are predicted to form cystine bonds, are involved in disulphide bridges. After refinements of loop regions and manual tracing of side-chains for both Hel and MBD models no severe disallowed atomic contact was detected with PROCHECK v.3.5.4 [19], suggesting an essentially good stereochemistry, with 63% and 29% of the residues in the most favored and additional allowed regions, respectively, and with 4.8% and 3.2% residues in generously allowed and disallowed regions of the Ramachandran plot.
Once the models of the Hel and MBD domains had been successfully predicted, a molecular docking simulation was carried out to obtain the complete nsp13 tertiary structure in an unbiased way. Among the lowest energy solutions, only the ones consistent with the overall protein sequence were taken into account and manually refined before a final energy minimization. The theoretical structure of SCV nsp13, shown in Fig. 1 , was again confirmed by PROCHECK v.3.5.4 [19] and deposited in the Protein Data Bank with the ID code 2G1F.
Fig. 1.
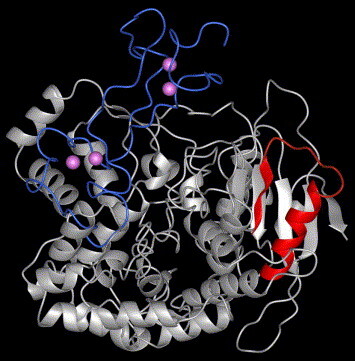
Ribbon representations of SCV nsp13 tertiary structure. In blue the MBD domains is shown and in red and violet the P-loop and zinc atoms are highlighted. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Reliability of protein structures may be conveniently assessed by PROSAII program package [20] and, accordingly, the predicted single components of the SCV helicase as well as the final tertiary structure have been considered by this program package. In Fig. 2 , we compared the energy profiles obtained with PROSAII program package [20] for the isolated Hel and MBD domains with the one calculated for the entire SCV helicase structure. From the negative values of the energy, which have been obtained by subtracting the energy profile of the two separate domains from the one calculated for the final protein structure, see Fig. 2, it is apparent that higher stability is achieved when the two domains are assembled. Negative peaks in the latter profiles, indeed, are found for the interface regions of the two domains, i.e., 42–48 residues for MBD and 198–204, 325–335, and 353–360 residues for the Hel domain, respectively.
Fig. 2.
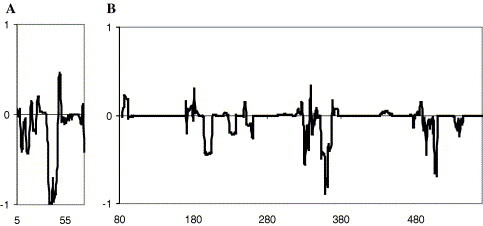
The difference of the energy profile obtained by using ProSaII program package for the entire nsp13 proteins and MBD is shown in (A). (B) The same difference is calculated for the He1 domain.
Since a typical ATP binding site motif between residues 282 and 289 of the helicase sequence may be predicted, it is noteworthy that in the obtained nsp13 model the corresponding fragment, the so-called P-loop, is surface exposed and located between a β strand and a short α helix, exactly as this active site should be [32], see Fig. 1.
We conclude that a theoretical model of SCV helicase is here proposed which will provide a foundation for the rational design of helicase inhibitors that could potentially act as anti-SCV drugs.
References
- 1.World Health Organization. Summary of probable SARS cases with onset of illness from 1st November 2002 to 31st July 2003. Based on data as of the 31st December 2003. Available from: http://www.who.int/csr/sars/country/table2004_04_21/en/.
- 2.Song H.D., Tu C.C., Zhang G.W., Wang S.Y., Zheng K., Lei L.C., Chen Q.X., Gao Y.W., Zhou H.Q., Xiang H., Zheng H.J., Chern S.W., Cheng F., Pan C.M., Xuan H., Chen S.J., Luo H.M., Zhou D.H., Liu Y.F., He J.F., Qin P.Z., Li L.H., Ren Y.Q., Liang W.J., Yu Y.D., Anderson L., Wang M., Xu R.H., Wu X.W., Zheng H.Y., Chen J.D., Liang G., Gao Y., Liao M., Fang L., Jiang L.Y., Li H., Chen F., Di B., He L.J., Lin J.Y., Tong S., Kong X., Du L., Hao P., Tang H., Bernini A., Yu X.J., Spiga O., Guo Z.M., Pan H.Y., He W.Z., Manuguerra J.C., Fontanet A., Danchin A., Niccolai N., Li Y.X., Wu C.I., Zhao G.P. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA. 2005;102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivanov K.A., Thiel V., Dobbe J.C., van der Meer Y., Snijder E.J., Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 5.Kleymann G., Fischer R., Betz U.A., Hendrix M., Bender W., Schneider U., Handke G., Eckenberg P., Hewlett G., Pevzner V., et al. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 2002;8:392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 6.Crute J.J., Grygon C.A., Hargrave K.D., Simoneau B., Faucher A.M., Bolger G., Kibler P., Liuzzi M., Cordingley M.G. Herpes simplex virus helicase-primase inhibitors are active in animal models of human disease. Nat. Med. 2002;8:386–391. doi: 10.1038/nm0402-386. [DOI] [PubMed] [Google Scholar]
- 7.Borowski P., Schalinski S., Schmitz H. Nucleotide triphosphatase/helicase of hepatitis C virus as a target for antiviral therapy. Antiviral Res. 2002;55:397–412. doi: 10.1016/s0166-3542(02)00096-7. [DOI] [PubMed] [Google Scholar]
- 8.Tanner J.A., Watt R.M., Chai Y.B., Lu L.Y., Lin M.C., Peiris J.S.M., Poon L.L.M., Kung H.F., Huang J.D. The severe acute respiratory syndrome (SARS) coronavirus NTP/helicase belongs to a distinct class of 5′ to 3′ viral helicase. J. Biol. Chem. 2003;278:39578–39582. doi: 10.1074/jbc.C300328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kao R.Y., Tsui W.H., Lee T.S., Tanner J.A., Watt R.M., Huang J.D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.H., Tse H., To A.P., Ng L.W., Wong B.C., Tsoi H.W., Yang D., Ho D.D., Yuen K.Y. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.http://www.ncbi.nlm.nih.gov/.
- 11.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.http://myhits.isb-sib.ch/cgi-bin/motif_scan.
- 13.Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 14.http://www.sbg.bio.ic.ac.uk/~phyre/.
- 15.http://www.expasy.ch/sprot/.
- 16.Korolev S., Hsieh J., Gauss G.H., Lohman T.M., Waksman G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 17.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 18.Case D.A., Cheatham T.E., III, Darden T., Gohlke H., Luo R., Merz K.M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. The Amber biomolecular simulation programs. J. Computat. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993;26:283–291. [Google Scholar]
- 20.Sippl M.J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 21.Güntert P., Wüthrich K. Improved efficiency of protein structure calculations from NMR data using the program DIANA with redundant dihedral angle constraints. J. Biomol. NMR. 1991;1(4):447–456. doi: 10.1007/BF02192866. [DOI] [PubMed] [Google Scholar]
- 22.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 23.Ausiello G., Cesareni G., Helmer-Citterich M. ESCHER: a new docking procedure applied to the reconstruction of protein tertiary structure. Proteins. 1997;28(4):556–567. [PubMed] [Google Scholar]
- 24.Dracheva S., Koonin E.V., Crute J.J. Identification of the primase active site of the herpes simplex virus type 1 helicase-primase. J. Biol. Chem. 1995;270(23):14148–14153. doi: 10.1074/jbc.270.23.14148. [DOI] [PubMed] [Google Scholar]
- 25.Johnson R.E., Henderson S.T., Petes T.D., Prakash S., Bankmann M., Prakash L. Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol. Cell. Biol. 1992;12(9):3807–3818. doi: 10.1128/mcb.12.9.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kusakabe T. The role of the zinc motif. J. Biol. Chem. 1996;271:19563–19570. doi: 10.1074/jbc.271.32.19563. [DOI] [PubMed] [Google Scholar]
- 27.Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Dinten L.C., van Tol H., Gorbalenya A.E., Snijder E.J. The predicted metal-binding region of the arterivirus helicase protein is involved in subgenomic mRNA synthesis, genome replication, and virion biogenesis. J. Virol. 2000;74(11):5213–5223. doi: 10.1128/jvi.74.11.5213-5223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seybert A., Posthuma C.C., van Dinten L.C., Snijder E.J., Gorbalenya A.E., Ziebuhur J. A complex zinc finger controls the enzymatic activities of nidovirus helicases. J. Virol. 2005;79(2):696–704. doi: 10.1128/JVI.79.2.696-704.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marmorstein R., Carey M., Ptashne M., Harrison S.C. DANN recognition by GAL4: structure of a protein–DNA complex. Nature. 1992;356:379–380. doi: 10.1038/356408a0. [DOI] [PubMed] [Google Scholar]
- 31.Bellon S.F., Rodgers K.K., Schatz D.G., Coleman J.E., Steitz T.A. Crystal structure of the RAG1 dimerization domain reveals multiple znc-binding motifs including a novel zinc binuclear cluster. Nat. Struct. Biol. 1997;4:586–591. doi: 10.1038/nsb0797-586. [DOI] [PubMed] [Google Scholar]
- 32.Saraste M., Sibbald P.R., Wittinghofer A. The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]