

Abstract
SARS-CoV 3CL protease is essential for viral protein processing and is regarded as a good drug target to prevent SARS-CoV replication. In the present study, we established a high-throughput FRET technique for screening for anti-SARS-CoV 3CL protease drugs. Of a thousand existing drugs examined, hexachlorophene was identified as the most potent in inhibiting SARS-CoV 3CL protease. Further characterization showed that it was effective at micromolar concentrations (Ki = 4 μM). The binding mode was competitive, and the inhibitory effect was dependent on preincubation time. Two other drugs, triclosan and nelfinavir, were about 10 times less potent. The structure-based search and biological evaluation of various hexachlorophene analogues were described. These analogues gave optimal inhibitory activity against SARS-CoV 3CL protease with IC50 values ranging from 7.6 to 84.5 μM. Optimization of hexachlorophene analogues was shown to provide several active 3CL protease inhibitors that function as potential anti-SARS agents.
Keywords: SARS, SARS-CoV, 3C-like protease, SARS-CoV 3CL protease, Fluorescence resonance energy transfer, Hexachlorophene, Molecular docking, Inhibitors, HPLC, Kinetic analysis
Severe acute respiratory syndrome (SARS) is a new disease that emerged in 2002 and, by 2003, had spread to 30 countries and areas, including China, Hong Kong, Singapore, Canada, and Taiwan. Epidemiological evidence suggests that the pathogen involved spreads by contact with infected patients and contaminated objects. SARS is characterized by high fever, malaise, rigor, headache, and nonproductive cough or dyspnea, and may progress to generalized interstitial infiltrates in the lung, requiring intubation and mechanical ventilation. The fatality rate in subjects diagnosed as having WHO-defined SARS is about 15%. In 2003, the coronavirus causing SARS (SARS-CoV) was identified [1], [2]. Currently, there is no effective drug for the treatment of SARS-CoV infection.
SARS-CoV is a positive strand RNA virus with approximately 29,700 nucleotides. After SARS-CoV infection, the open reading frame, ORF1ab, of the viral genome is translated into a polyprotein, from which functional proteins, including replicase and RNA-dependent RNA polymerase, are released by extensive proteolytic processing [3], [4]. This is primarily achieved by the 33 kDa 3C-like protease (3CLpro), a Cys protease [5], [6]. The functional importance of 3CLpro in the viral life cycle makes it an attractive target for the development of drugs directed against SARS and other coronavirus infections [7]. Recently, a recombinant 3CLpro from another coronavirus was successfully expressed as a maltose-binding protein (MBP) fusion protein, allowing the production of large amounts of active enzyme [8], [9]. In addition, the high conservation of substrate specificities of different 3CLpros makes it possible to design substrates by comparison with previously known coronavirus cleavage sites [10]. An intramolecularly quenched fluorogenic substrate containing a donor and an acceptor chromophore in the same molecule can be used to provide a rapid simple method of measuring enzyme activity [11] and the availability of a fluorescence resonance energy transfer (FRET) synthetic peptide allows the use of high-throughput screening to identify lead candidates in compound libraries.
Previous studies have shown that existing drugs might have unknown potential to treat diseases [12], [13]. Following this logic, we have now screened a thousand existing drugs for activity as 3CLpro inhibitors using our well-established high-throughput screening system and examined the mechanism of inhibition of those drugs which were found to be effective.
Materials and methods
Expression of SARS-CoV 3CL pro in Escherichia coli and its purification. SARS-CoV 3CLpro was expressed in E. coli TB1 cells. The coding sequence of SARS-CoV 3CLpro was amplified by PCR using the forward primer, 5′-AGTGGTTTTAGGAAAATGGCATTCCCGTCAGGC-3′, and the reverse primer, 5′-GTGACAAGCTTCTATTGGAAGGTAACACCAGAGC-3′. After amplification, the PCR product was digested by HindIII and ligated into XmnI/HindIII-digested pMAL-c2x DNA (New England Biolabs, Beverly, MA) to generate plasmid pMAL-3CL, which was transformed into TB1 cells. To induce expression of the fusion protein, E. coli maltose-binding protein (MBP)–3CLpro, 0.3 mM isopropyl thio-β-d-galactoside was added and incubation was continued for 3 h, and then the transformed cells were harvested by centrifugation and stored at −20 °C. 3CLpro was purified essentially as described previously for 3CLpro from another coronavirus [14], [15]. Briefly, the cells were sonicated in buffer A (20 mM Tris–HCl, pH 7.4, 200 mM NaCl, and 1 mM EDTA), then the homogenate was centrifuged (4000g, 20 min, 4 °C) and the pellet was discarded. The supernatant was loaded onto an amylose affinity column (New England Biolabs) and the fusion protein was eluted with buffer A containing 10 mM maltose. Following Factor Xa cleavage of the eluted fusion protein, the released 3CLpro was isolated by applying the material to a phenyl Sepharose HP column (Amersham Biosciences, Uppsala, Sweden) and collecting the 3CLpro fraction, which was dialyzed against buffer A, concentrated (YM-10, Millipore, Bedford, WA), and stored at −20 °C. The concentration of the protein was determined using bicinchoninic acid reagent (Pierce, Rockford, IL) with bovine serum albumin as the standard.
Measurement of SARS-CoV 3CL pro activity using HPLC. The enzyme activity of SARS-CoV 3CLpro was measured by cleavage of the synthetic 15-mer peptide, (H2N–SITSAVLQSGFRKMA–COOH), the cleavage site in the SARS-CoV polyprotein. This peptide was synthesized by United Biochemical Research (Seattle, WA) to about 75% purity and was purified to 95% purity in our laboratory by HPLC on a reverse-phase C18 column. The identity and purity of the peptide was confirmed by MALDI-TOF mass spectrometry (DE-STR, Applied Biosystem, Foster City, CA). Cleavage was assayed by incubating 50 nM SARS-CoV 3CLpro and 15 nmoles of synthetic 15-mer peptide for 30 min at 37 °C in a total volume of 200 μl in 20 mM Tris–HCl (pH 7.4), 200 mM NaCl, 1 mM EDTA, and 1 mM DTT (buffer B). After incubation, the products were separated by HPLC on a Beckman ODS column (Beckman, Fullerton, CA) using a 30 min 0–60% linear gradient of acetonitrile in 0.1% trifluoroacetic acid and the predicted cleaved peptides identified by MALDI-TOF.
Measurement of SARS-CoV 3CL pro activity using FRET. To establish a high-throughput screening method, we designed a FRET peptide, Abz-SAVLQSGFRK-Dnp, as 3CLpro substrate. The FRET peptide was synthesized by Open Biosystems (Huntsville, AL). Inhibition of enzyme activity was assayed by preincubating the enzyme (20 nM) with the test compound (10 μM) in buffer B for 15 min at 25 °C in 96-well plates, then adding 30 μM FRET peptide, incubating for another 15 min at 25 °C, and measuring the cleaved product on a 96-well plate spectrophotometer (Safire, Tecan, Salzburg, Austria) using an excitation wavelength of 320 nm and an emission wavelength of 425 nm. Control reactions were carried out using the same reaction mixture without the test compound (no inhibition) or without the enzyme (blank). This method can also be used in an automatic robotic system for a high-throughput screen platform. The assay performance was evaluated using the following equation [16]:
where Z′ is the Z factor (0–1, with 1 being a perfect system and 0.5 excellent), X is the mean value for the sample, and σ is the standard deviation.
Preparation of test compounds. For the assessment of inhibitory effect, a chemical library of 1000 molecules was purchased from MDS Pharma Services (Taipei, Taiwan). All compounds to be screened were dissolved in dimethyl sulfoxide (DMSO) and tested in duplicate at a concentration of 10 μM in 1% DMSO.
Determination of kinetic parameters and the K i of effective compounds using the FRET peptide. Kinetic parameters were obtained using various concentrations of FRET peptide in the fluorescent assay. The maximal velocity (V max) and Michaelis–Menten constant (K m) were calculated from the Eadie–Hofstee plot. If the type of inhibition was found to be competitive using a Lineweaver–Burk double reciprocal plot, then the inhibitory constant (K i) for 3CLpro was estimated using the equation:
Plots were performed, and kinetic parameters were calculated, using Prism software (Graphpad Software, San Diego, CA).
Hexachlorophene docking and data analysis. The 3D model of the SARS-CoV 3CLpro was retrieved from the Protein data bank (code 1UK4). PRODRG program (http://davapc1.bioch.dundee.ac.uk/programs/prodrg/) was used to generate the coordinate and topology files of hexachlorophene. Hexachlorophene was computationally docked into the 3D structure of SARS-CoV 3CLpro using the docking program GOLD (version 2.1.2; Genetic Optimization for Ligand Docking, CCDC, Cambridge, UK).
GOLD operates with a genetic search algorithm and allows for complete ligand and partial-binding site flexibility [17]. Because hexachlorophene is a competitive inhibitor of SARS-CoV 3CLpro, we defined the binding site to the inhibitor as the putative hexachlorophene-binding pocket. The active site radius was set to be 10 Å from the midpoint between the Sγ atom of the catalytic residue Cys-145 and Nε2 atom of His-41. GOLD generated a maximum of ten docking results for hexachlorophene. The results were ranked by GOLD’s scoring function which is a molecular mechanics-like function with four terms based on protein–ligand hydrogen bond energy, protein–ligand van der Waals energy, ligand internal van der Waals energy, and ligand torsional strain. The 100 steps of energy minimization of the best docking structure was carried out using the CNS programs [18]. The interactions between hexachlorophene and SARS-CoV 3CLpro were analyzed and illustrated by LIGPLOT [19].
Results and discussion
SARS-CoV 3CLpro preparation and activity
SARS-CoV 3CLpro was expressed as an MBP fusion protein in E. coli, resulting in the generation of a prominent protein band with an apparent molecular mass of 75 kDa. The fusion protein was purified and cleaved by Factor Xa, then the released recombinant 3CLpro was further purified and obtained to have the same amino acid composition as native SARS-CoV 3CLpro and to contain 306 amino acid residues. Enzyme activity was checked by cleavage of the 15-mer synthetic substrate, H2N–SITSAVLQSGFRKMA–COOH, using HPLC to monitor the cleavage of the substrate into the two peptides, SITSAVLQ and SGFRKMA (data not shown). The results showed that the SARS-CoV 3CLpro was pure and active, and suitable for establishing the high-throughput system.
Establishment of the FRET-based high-throughput assay for SARS-CoV 3CLpro
Peptide cleavage by SARS-CoV 3CLpro can be easily monitored using a fluorogenic substrate. We used Abz and DNP as fluorescent and quench molecules, whereas other groups have used Dabcyl–Edens pairs [20], [21]. The present FRET peptide (MW 1490 Da) is the smallest to be used in such studies, but still large enough to be recognized by SARS-CoV 3CLpro. The purified enzyme was assayed using the FRET technique described under Materials and methods; an evaluation of the performance of the assay is shown in Fig. 1 , which shows 48 readings for the basal and maximal signals. The basal signals (signal in the absence of enzyme) for the 48 replicates were quite low and similar. The maximal signals (signal after enzyme reaction) for the 48 replicates showed a little variation, but were quite high, and the detection window (difference between the maximal and basal readings) was good and the signal-to-noise ratio about 20. The Z′-factor value for the assay was 0.81, which corresponds to a stable and excellent system [16]. Thus, this approach was suitable for a high-throughput screening method. We concluded that this system could be used as a simple, continuous, and rapid screening assay for identifying candidates for SARS-CoV 3CLpro inhibitors in compound libraries.
Fig. 1.
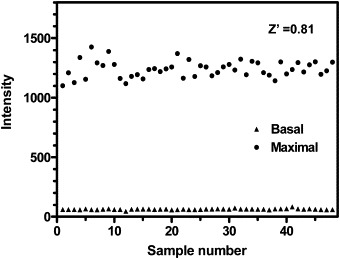
Detection window for the high-throughput FRET SARS-CoV 3CLpro activity assay in the 96-well plate format. Basal and maximal signals corresponding to protease activity measured for 48 samples in the presence of vehicle (1% DMSO). The reaction was performed as described under Materials and methods.
Determination of the Km of SARS-CoV 3CLpro using the FRET peptide for use in a primary screen for SARS-CoV 3CLpro inhibition
To identify anti-SARS drugs available for clinical use as quickly as possible, we screened a collection of a thousand existing drugs. Before screening these drugs, the K m of the FRET peptide was determined in order to optimize the assay conditions, since a concentration close to the K m is usually assumed to be optimal by considering enzyme activity and screening sensitivity. An Eadie–Hofstee plot of the initial velocities gave a K m value of 16 ± 1.1 μM and a V max value of 6.8 ± 0.2 μmol/min/mg using the FRET peptide.
Hexachlorophene, triclosan, and nelfinavir inhibit 3CLpro
Screening a thousand existing drugs, we found that hexachlorophene exhibited a half-inhibitory concentration (IC50) of 5 μM for 3CLpro using this method. The triclosan and nelfinavir, reported to be potential SARS-CoV 3CLpro inhibitors [22], inhibited 3CLpro activity, although with a potency lower than hexachlorophene; the respective IC50s were 75 and 46 μM.
Hexachlorophene is a competitive inhibitor and the inhibition depends on the preincubation time
We examined the inhibition mechanism of hexachlorophene using Eadie–Hofstee plot. As shown in Fig. 2 A, as the hexachlorophene concentration was increased (3, 10, or 30 μM), the respective apparent K m value increased (20 ± 1.6, 35 ± 3.4, or 121 ± 4.4 μM), whereas the apparent V max value was fairly constant (6.98 ± 0.16 μmol/min/mg). We concluded that hexachlorophene is a classic competitive inhibitor of SARS-CoV 3CLpro and its K i value is estimated to be 4 μM. Fig. 2B shows that the inhibitory effect of hexachlorophene depended on the preincubation time, showing that it is a slowly binding inhibitor.
Fig. 2.
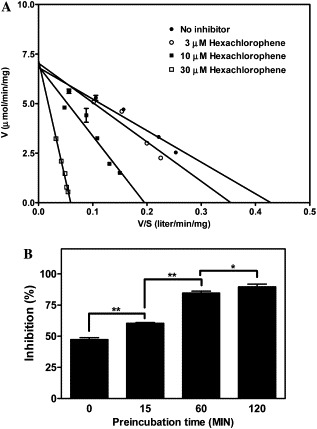
Kinetic analysis of the inhibition of SARS-CoV 3CLpro by hexachlorophene. (A) Lineweaver–Burk plot. The assay was performed for 15 min at 25 °C in buffer B using 20 nM enzyme and different concentrations of the FRET peptide (Abz-SAVLQSGFRK-DNP) in the presence of 0, 3, 10, or 30 μM hexachlorophene. (B) Effect of preincubation. The mixture containing 20 nM SARS-CoV 3CLpro and 10 μM hexachlorophene in buffer B was preincubated for different times at 25 °C, then 30 μM FRET peptide was added and incubation was continued for a further 15 min at 25 °C, and then the percentage inhibition was calculated. The data are means ± SD for three separate experiments. The linear regression curves were plotted using GraphPad Prism. The P values were derived using a two-tailed t test; *P < 0.05, **P < 0.01.
Docking of hexachlorophene
The docking simulation reveals that the drug, hexachlorophene, partially blocks the active site (Fig. 3 A). As shown in Fig. 3B, we found that the hexachlorophene forms hydrogen bonds with the side chains of Glu-166, His-163, Cys-145, Ser-144, and Asn-142, the oxygen on the main chains of Phe-140 and Thr-26 of SARS-CoV 3CLpro. In addition, His-41 donates hydrophobic interaction to hexachlorophene. We also found that the Cys-145 donates two hydrogen bonds to the ClαP and OαA atoms of hexachlorophene in SARS-CoV 3CLpro. Thus, 3D modeling data indicated that hexachlorophene might be a lead compound for the design of anti-SARS drugs.
Fig. 3.
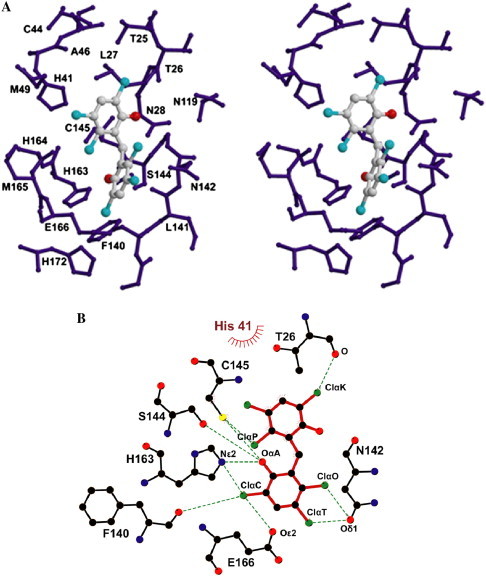
Molecular docking of hexachlorophene in the active site of SARS-CoV 3CLpro. (A) A stereo view of the substrate-binding site. The hexachlorophene was docked onto the SARS-CoV 3CLpro. These residues are within a radius set to be 8 Å from the hexachlorophene. (B) Illustration of amino-acid contacts to the hexachlorophene in the active site. Hydrogen bonds are shown as green dashed lines and van der Waals contacts are shown as bent red combs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this paper.)
Hexachlorophene-like compounds show the anti-SARS-CoV 3CLpro activity
Since hexachlorophene could block the active site of SARS-CoV 3CLpro, further modification of hexachlorophene was carried out to find the necessary side chain from various hexachlorophene derivatives. Nine commercially available compounds whose structures have high similarity with hexachlorophene were investigated (Fig. 4 ). These compounds dose-dependently inhibited the SARS-CoV 3CLpro activity. As shown in Fig. 5 , HL-5 and HL-6 exhibited inhibitory activity higher than those of other compounds and their IC50 values were 9.2 and 7.6 μM, respectively. All of nigh compounds showed inhibitory activity against SARS-CoV 3CLpro, with IC50 values ranging from 7.6 to 84.5 μM.
Fig. 4.
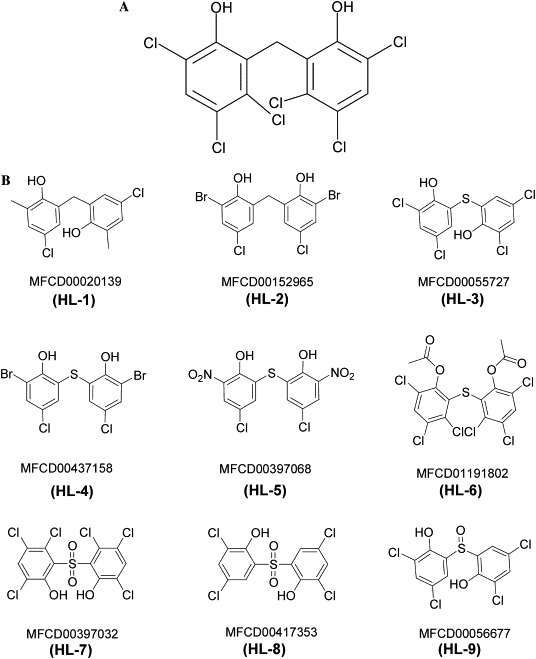
Chemical structures of (A) hexachlorophene (B) hexachlorophene analogues from the MDL’s ACD (Available Chemicals Directory) database.
Fig. 5.
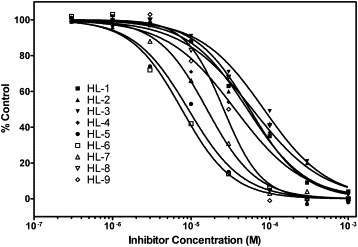
Concentration–response curve for the effect of nine hexachlorophene-like compounds on SARS-CoV 3CLpro activity. Protease (20 nM) was preincubated with varied concentrations of inhibitors for 15 min at 25 °C in buffer B, then the FRET peptide (Abz-SAVLQSGFRK-DNP) was added and the mixture was incubated for a further 15 min at 25 °C, and the results are expressed as a percentage of the digestion in the absence of the inhibitor. The nonlinear regression curves were plotted using GraphPad Prism.
Hexachlorophene has additional hydroxyl groups and chloride atoms, which might make it a better fit for the substrate-binding pocket of SARS-CoV 3CLpro. Therefore, hexachlorophene can be regarded as a lead compound for SARS-CoV 3CLpro inhibitors and the structures of hexachlorophene-like compounds can be used as the basis for further optimization of SARS-CoV 3CLpro inhibitors. Furthermore, our kinetic study showed that hexachlorophene competed with the substrate for the active center. From the docking result, it also implicated that two hydrogen bonds exist between Cys-145 and hexachlorophene. We speculate that hexachlorophene interacts with the active site Cys-145 and hexachlorophene or its analogues may be used to treat SARS disease in humans.
Acknowledgment
This work was supported by Grant NSC-92-2751-B-002-002-Y from the National Science Council of the Republic of China.
References
- 1.Drosten C., Gunther S., Preiser W., van der W.S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 4.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G, Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Ziebuhr J., Siddell S.G. J. Virol. 1999;73:177–185. doi: 10.1128/jvi.73.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziebuhr J., Bayer S., Cowley J.A., Gorbalenya A.E. J. Virol. 2003;77:1415–1426. doi: 10.1128/JVI.77.2.1415-1426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 8.Seybert A., Ziebuhr J., Siddell S.G. J. Gen. Virol. 1997;78(Pt. 1):71–75. doi: 10.1099/0022-1317-78-1-71. [DOI] [PubMed] [Google Scholar]
- 9.Hegyi A., Friebe A., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2002;83:581–593. doi: 10.1099/0022-1317-83-3-581. [DOI] [PubMed] [Google Scholar]
- 10.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hata S., Sato T., Sorimachi H., Ishiura S., Suzuki K. J. Virol. Methods. 2000;84:117–126. doi: 10.1016/s0166-0934(99)00138-x. [DOI] [PubMed] [Google Scholar]
- 12.Koren G., King S., Knowles S., Phillips E. CMAJ. 2003;168:1289–1292. [PMC free article] [PubMed] [Google Scholar]
- 13.Cuendet M., Pezzuto J.M. J. Nat. Prod. 2004;67:269–272. doi: 10.1021/np030304+. [DOI] [PubMed] [Google Scholar]
- 14.Ziebuhr J., Heusipp G., Siddell S.G. J. Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegyi A., Ziebuhr J. J. Gen. Virol. 2002;83:595–599. doi: 10.1099/0022-1317-83-3-595. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J.H., Chung T.D., Oldenburg K.R. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 17.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 18.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Acta Crystallogr. D Biol. Crystallogr. 1998;54(Pt. 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 19.Wallace A.C., Laskowski R.A., Thornton J.M. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 20.Kuo C.J., Chi Y.H., Hsu J.T., Liang P.H. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S., Hsu H.H., Huang H.C., Wu D., Brik A., Liang F.S., Liu R.S., Fang J.M., Chen S.T., Liang P.H., Wong C.H. Proc. Natl. Acad. Sci. USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J., Rabenau H., Doerr H.W., Hunsmann G., Otaka A., Tamamura H., Fujii N., Yamamoto N. Biochem. Biophys. Res. Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]