

Abstract
The 3C-like proteinase of severe acute respiratory syndrome (SARS) coronavirus has been proposed to be a key target for structural-based drug design against SARS. Accurate determination of the dimer dissociation constant and the role of the N-finger (residues 1–7) will provide more insights into the enzyme catalytic mechanism of SARS 3CL proteinase. The dimer dissociation constant of the wild-type protein was determined to be 14.0 μM by analytical ultracentrifugation method. The N-finger fragment of the enzyme plays an important role in enzyme dimerization as shown in the crystal structure. Key residues in the N-finger have been studied by site-directed mutagenesis, enzyme assay, and analytical ultracentrifugation. A single mutation of M6A was found to be critical to maintain the dimer structure of the enzyme. The N-terminal octapeptide N8 and its mutants were also synthesized and tested for their potency as dimerization inhibitors. Peptide cleavage assay confirms that peptide N8 is a dimerization inhibitor with a Ki of 2.20 mM. The comparison of the inhibitory activities of N8 and its mutants indicates that the hydrophobic interaction of Met-6 and the electrostatic interaction of Arg-4 contribute most for inhibitor binding. This study describes the first example of inhibitors targeting the dimeric interface of SARS 3CL proteinase, providing a novel strategy for drug design against SARS and other coronaviruses.
Keywords: SARS coronavirus, 3C-like proteinase, Mutational study, N-terminal peptide, Dimerization inhibitor, Peptide inhibitor, Zhang–Poorman plot
Human coronaviruses are major causes of upper respiratory tract illness in humans. A novel form of coronavirus has been identified as the major cause of severe acute respiratory syndrome (SARS), a disease that had rapidly spread from southern China to several countries in 2003 [1], [2]. Coronaviruses are members of positive-stranded RNA viruses featuring the largest viral RNA genomes known to date. The SARS coronavirus replicase gene encompasses two overlapping translation products, polyprotein 1a (∼450 kDa) and 1ab (∼750 kDa), which are conserved both in length and amino acid sequence to other coronavirus replicase proteins. Polyprotein 1a and 1ab are cleaved by the internally encoded 3CL proteinase to release functional proteins necessary for virus replication. The SARS 3CL proteinase is fully conserved among all released SARS coronavirus genome sequences and is highly homologous with other coronavirus 3CL proteinases. Due to the functional importance of SARS 3CL proteinase in the viral life cycle, it has been proposed to be a key target for structural-based drug design against SARS [3]. Virtual screening of chemical compound libraries has discovered potential inhibitors as described by various groups [4], [5], [6], [7]. Several bifunctional boronic acid inhibitors were designed to target the serine cluster (Ser-139, Ser-144, and Ser-147) near the catalytic residues [8]. Peptide analogs and organic compounds derived from known rhinovirus 3C protease inhibitors were also synthesized and some of them were found to be active towards SARS 3CL proteinase [9], [10], [11], [12], [13], [14]. The crystal structures of the 3CL proteinases of several coronaviruses have been solved [3], [15], [16], all of which are dimeric. The enzyme was shown to follow a general base catalytic mechanism [17]. As we have reported previously, the peptide cleavage assay shows that the specific activity for proteolysis decreases linearly with the decrease of enzyme concentration, suggesting that the dimer is the major form for biological activity and that the dimeric interface could be targeted for structural-based drug design against SARS 3CL proteinase [18]. The extra helical domain of the SARS 3CL proteinase was also shown to play an important role in controlling the association–dissociation equilibrium and regulating the activity and specificity of the enzyme [19]. However, there is no published report on the inhibitor design for the dimeric interface to date.
The same strategy has been successfully used in the structure-based inhibitor design for human immunodeficiency virus 1 (HIV-1) protease and other viral enzymes [20], [21], [22], [23], [24], [25], [26], [27]. HIV-1 protease is a homodimer of two identical 99-residue subunits in which the active site is generated by the self-assembly of these subunits. The dimeric structure is stabilized by an extended anti-parallel β-sheet formed by the inter-digitations of the N-terminal (residues 1–4) and C-terminal (residues 96–99) β-strands of each monomer [22]. Peptides with the N- and C-terminal amino acid sequences have been designed and confirmed as dimerization inhibitors [21], [27]. A large set of oligopeptides has been derived from the terminal fragments of HIV-1 protease by computer modeling, and their inhibiting activities have been determined experimentally. The best inhibitor has an IC50 value of less than 1 μM [20]. Cross-linked N- and C-terminal peptides also inhibit HIV-1 protease activity and reduce the fraction of dimers in solution as measured by size-exclusion chromatography [25]. Design of dimerization inhibitors has also been reported for many other viral enzymes, such as HIV reverse transcriptase, intergrase, herpes simplex virus ribonucleotide reductase, and DNA polymerase [24].
The crystal structure of SARS 3CL proteinase indicates that the N-finger fragment plays an important role in the dimerization and maintenance of the active form of the enzyme [16]. Peptides derived from this fragment may be used as dimerization inhibitors, as in the case of HIV-1 protease and other viral enzymes. In the present paper, we have studied the key residues in the N-finger of SARS 3CL proteinase by site-directed mutagenesis, activity test, and analytical ultracentrifugation. The N-terminal octapeptide N8 and its nine mutants were also synthesized and tested for their inhibitory activities. The dimer dissociation constants of the SARS 3CL proteinase and N-terminal mutants were determined by sedimentation velocity and equilibrium methods. This study provides important information for the structural-based inhibitor design targeting the dimeric interface of SARS 3CL proteinase, which provides a novel strategy for drug design against SARS and other coronaviruses.
Materials and methods
The expression and purification of SARS 3CL proteinase. The C-terminal His-tagged 3CL proteinase was expressed and purified as described previously [18]. The non-His-tagged 3CL proteinase was expressed and purified as reported [17]. The R4E, K5A, and M6A mutants of SARS-CoV 3CL proteinase were prepared with the QuikChange site-directed mutagenesis kit (Stratagene) using pET 3CLP-21h [17] as a template. The N-terminal deletion mutant, Δ7N, was constructed by inserting the PCR product which carried the NheI/HindIII site fragment with a deletion of the 6 codons coding for Gly-2 to Phe-7 into the NheI/HindIII-digested pET 21a DNA. The mutations were verified by nucleotide sequencing. The purification method used for each mutant was the same as that used for the wild-type proteinase and their purity was verified by SDS–PAGE to be greater than 95%.
Peptide synthesis. The octapeptide inhibitor N8 and its mutants were synthesized by solid-phase peptide synthesis using the standard 9-fluorenylmethoxycarbonyl/tert-butyl chemistry. Cleavage of the peptide from Rink resin and removal of all sidechain-protecting groups were achieved in trifluoroacetic acid solution. The crude peptide was purified by reversed-phase high-performance liquid chromatography (RP-HPLC, Elite P200II, Dalian, China) on a Zorbax C18 semi-preparative column (9.4 by 250 mm, Agilent) using water/acetonitrile gradients containing 0.1% trifluoroacetic acid. The purity of all peptides was measured by analytical RP-HPLC, and the identity was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy.
Colorimetric enzyme assay. The enzyme activity was measured by a colorimetric assay as reported before [17]. In short, 20 μl pNA substrate stock solution (2 mM Thr-Ser-Ala-Val-Leu-Gln-pNA water solution) was added to 180 μl 37 °C preheated reaction buffer (40 mM PBS, 1 mM EDTA, and 3 mM DTT, pH 7.3), which contained 2.8 μM enzyme. Colorimetric measurements of enzymatic activity were performed in 96-well microtiter plates using a multiwell ultraviolet spectrometer (Spectra Max 190, Molecular Device) at 390 nm. Each assay was repeated three times.
Peptide cleavage assay. The proteolytic activity of the His-tagged SARS 3CL proteinase was determined using an HPLC-based peptide cleavage assay as previously reported [18]. The peptide substrates S01: NH2-TSAVLQSGFRK-CONH2 and S12: NH2-SAVLQSGF-CONH2 were synthesized as described previously [18].
Zhang et al. [27] have established the Zhang–Poorman plot to distinguish dimerization inhibitors from competitive inhibitors. The three major hypotheses in their model were: (i) the bioactive dimeric enzyme was in equilibrium with the inactive monomeric enzyme; (ii) the inhibitor only bound with the enzyme monomer; (iii) the substrate concentration in peptide cleavage assay was very low compared with K m of the dimeric enzyme.
For the Zhang–Poorman plot, peptide S12 was used as substrate. Cleavage reaction mixture was incubated at room temperature and contained 400 μM substrate S12, 8.12–43.32 μM His-tagged SARS 3CL proteinase, no inhibitor or 800 μM peptide N8, and 57 μM DTT in 40 mM Tris–HCl buffer, pH 7.3, at room temperature. Reaction was stopped after 5 or 10 min by adding 0.1% trifluoroacetic acid aqueous solution and analyzed as described previously.
The inhibitor peptide was dissolved in DMSO and premixed with the His-tagged proteinase for 1–2 h. Cleavage reaction mixture (400 μM substrate S01, 2.43 μM His-tagged SARS 3CL proteinase, 800 μM inhibitor peptide, and 57 μM DTT in 10% DMSO, 40 mM Tris–HCl buffer, pH 7.3) was incubated at room temperature. Aliquots of reactions were removed every 20 min within 80 min and the reaction was stopped by the addition of 0.1% trifluoroacetic acid aqueous solution, and the reaction mixture was analyzed by RP-HPLC as described above. k cat/K m was determined by plotting the substrate peak area using the equation below:
| (1) |
where PA is the peak area of the substrate peptide, c E is the total concentration of His-tagged 3CL proteinase, and C is an experimental constant. The inhibitory activities of the peptides were estimated using the same dimerization inhibitor model as in the Zhang–Poorman plot [27]. The dimerization inhibition constant K i was thus calculated using the following equation:
| (2) |
where (k cat/K m)app.0 is the apparent k cat/K m without inhibitor and [I] is the concentration of the inhibitor.
Analytical ultracentrifugation analysis. Sedimentation equilibrium and velocity experiments were conducted on a Beckman Optima XLA analytical ultracentrifuge equipped with absorbance optics. An An60Ti rotor and standard six-sector equilibrium centerpieces were used. The freshly prepared wild-type and mutational SARS 3CL proteinase was further purified and buffer-exchanged using a gel filtration column, Superdex 75 10/300 GL (Amersham Bioscience), into sedimentation buffer (40 mM phosphate buffer, 100 mM NaCl, and 0.5 mM EDTA, 0.5 mM DTT, pH 7.3). The molar extinction coefficient at 280 nm (1.04 mg/cm2), density of the sedimentation buffer (1.005 g/ml), partial specific volume (0.723 ml/g), and molecular weight of the monomer (33 914 Da) were calculated based on its amino acid composition using the program SEDNTERP (http://www.bbri.org/rasmb/rasmb.html). For sedimentation velocity experiments, 380 μl samples (concentrations between 0.1 and 4 mg/ml) and 400 μl reference solutions were loaded into cells. The rotor temperature was equilibrated at 20 °C and rotor speeds of 60,000 rpm. Absorbance scans at 230, 280 or 290 nm were collected at a time interval of 4 min. Data were analyzed with the software Sedfit version 8.9 g [28], [29]. For the sedimentation equilibrium experiments, 110 μl samples and 120 μl reference solutions were loaded into nitrogen-flushed cells, followed by degassing and a further nitrogen flush prior to sealing. The protein was equilibrated for data collection at 20 °C and three rotor speeds (15,000, 20,000, and 25,000 rpm). Once equilibrium was reached (typically 24–32 h), absorption data were collected at 280 nm, using a radial step size of 0.001 cm, and recorded as the average of 10 measurements at each radial position. To determine the baseline values in the cell, at the end of the data collection time the rotor speed was increased to 42,000 rpm for 8 h, and the absorbance of the depleted meniscus was measured. Dissociation constants were determined by fitting a monomer–dimer equilibrium model using the Origin-based data analysis software for Beckman XL-A/XL-I (Beckman Instruments, Beckman Coulter, Fullerton, CA). Data from different concentrations and speeds were combined for global fitting.
Results and discussion
Dissociation constant of SARS 3CL proteinase dimer
The sedimentation experiments study the aggregation state of the enzyme in the native solution condition, and no dilution effect exists as compared to gel filtration. AUC is considered more reliable than gel filtration in determining protein aggregation states. Sedimentation velocity provides hydrodynamics information about the molecular size distribution and conformational changes, whereas sedimentation equilibrium can perfectly characterize the thermodynamics of the self-association systems and determine the dissociation constants [30], [31]. Recently, Peter Schuck has developed a method for the analysis of protein self-association by sedimentation velocity experiments [29], [32]. In order to get a reliable result, we performed both sedimentation velocity and equilibrium experiments to determine the dissociation constant of SARS 3CL proteinase. We determined the dissociation constant of the wild-type proteinase with no tags.
Sedimentation velocity experiments on the wild-type enzyme were conducted at nine enzyme concentrations. The resulting c(s) distribution profiles show concentration-dependent peaks at positions between the monomer and dimer (Fig. 1 A). This clearly indicates the fast reversible association of the SARS 3CL proteinase. The isotherm analysis of sw(c) is shown in Fig. 1B based on the integration of the differential sedimentation coefficient c(s) distributions [29]. By fitting to the monomer–dimer equilibrium in the Sedphat program, the dissociation constant was determined to be 22.9 μM, with the monomer sedimentation coefficient of S1 = 2.7 s, the dimer sedimentation coefficient of S2 = 4.0 s (Fig. 1B).
Fig. 1.
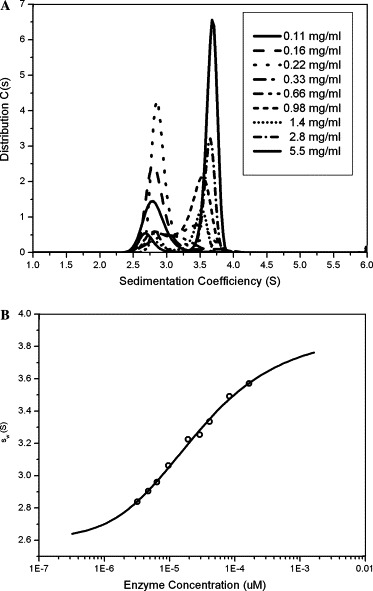
(A) SARS 3CL proteinase sedimentation coefficient distribution c(s) of nine SARS 3CL proteinase concentrations from 0.11 to 5.5 mg/ml. (B) Dimer dissociation constant analyzed by the isotherm analysis of weight-average sedimentation coefficient as a function of concentration. The raw data (circle) were obtained by the integration of the differential sedimentation coefficient c(s) distributions of nine enzyme concentrations. Sedphat program was used to fit the data to the monomer–dimer equilibrium (solid line). The resulted monomer sedimentation coefficient S1 = 2.7 s, dimer sedimentation coefficient S2 = 4.0, and the dissociation constant Kd = 22.9 μM.
The sedimentation equilibrium experiment was performed with three different protein concentrations (0.2, 0.3, and 0.5 mg/ml) and three running speeds (15,000, 20,000, and 25,000 rpm). Fig. 2 shows the result of a global fit of all the data to the monomer–dimer equilibrium with the Beckman Origin-based data analysis software. The dissociation constant for the monomer–dimer self-association model was determined to be K d = 14.0 μM, which is in agreement with the value from the sedimentation velocity experiment. Since the sedimentation velocity method is only a good initial exploration to characterize the self-association of proteins, we take the final K d to be 14.0 μM from the global fit of sedimentation equilibrium data.
Fig. 2.
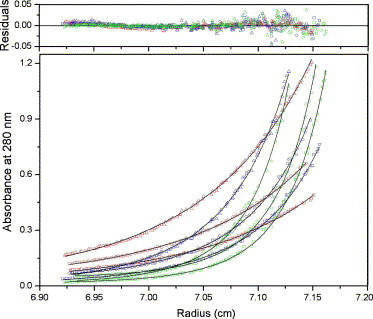
Sedimentation equilibrium data for wild-type SARS 3CL proteinase performed at 20 °C with three different rotor speeds (15,000, 20,000, and 25,000 rpm) and three different enzyme concentrations (0.2, 0.3, and 0.5 mg/ml). The bottom panel is the nine equilibrium data and the best fit (solid line); the upper panel is the residuals’ plot. The dissociation constant (Kd) was determined to be 14.0 μM.
There are several reports on the quantitative measurements of the dimer dissociation constant of the SARS 3CL proteinase by analytical ultracentrifuge or enzyme activity-dependent assays. But the published dissociation constants have large variations from different groups and/or using proteins expressed from different constructs. Chou et al. [33] reported a dissociation constant of 190 ± 14 nM by using the sedimentation velocity method with the C-terminal His-tagged form of SARS 3CL proteinase. Hsu et al. [34] reported a dissociation constant of 0.35 nM by using the sedimentation velocity method on the wild-type protein with no tags. Our sedimentation equilibrium experiments give a dissociation constant of 14.0 μM for the wild-type protein with no tag but one extra alanine residue at the N-terminus. Generally speaking, the sedimentation equilibrium test can provide first-principle analysis of the dissociation constants compared to the sedimentation velocity method [30], [31].
Key residue analysis of the N-finger of SARS 3CL proteinase
The crystal structure of SARS 3CL proteinase suggests that the N-finger fragment contributes significantly to the enzyme dimerization [16]. Our experiments showed that the N-finger deletion mutant Δ7N could not form any dimer (Fig. 3 B), which agrees with the previous report that the N-finger deletion mutant is in monomeric form in solution [35]. However, the seven residues in the N-finger contribute differently to the dimer stability. From Fig. 4 , we can see that the side chains of Phe-3 and Lys-5 point away from the dimer interface to the interior of the same monomer. Ser-1, Gly-2, and Ala-7 do not have remarkable interactions. The two most important residues among the seven are Arg-4 and Met-6. The guanidyl group of Arg-4 in one monomer forms a salt bridge with the side chain of Glu-290 in the other monomer, which is one of the major interactions between the two monomers. Furthermore, the side chain of Met-6 forms hydrophobic interactions with the side chains of the Phe-140 and Tye-126 in its partner. This suggests that residues Arg-4 and Met-6 are the two possible key residues for the N-finger role in dimer stability. However, Chou et al. [33] reported that the mutation of R4A only partly affects the dimerization of SARS 3CL proteinase and keeps most enzymatic activity, which means that Arg-4 might not be the dominating factor to the dimer interface. In this study, R4E and M6A mutants were shown to have very weak dimerization according to sedimentation experiments (Figs. 3C and E) and have no detectable enzyme activity either (Table 1 ). Substituting the positively charged Arg-4 with a negatively charged Glu makes it impossible to form a salt bridge with the Glu-290 in the other monomer and even cause charge repulsion. The mutation of M6A suggests that the hydrophobic interactions between the side chain of Met-6 and Phe-140, Tye-126 in the other monomer contribute significantly to the stability of the dimer.
Fig. 3.
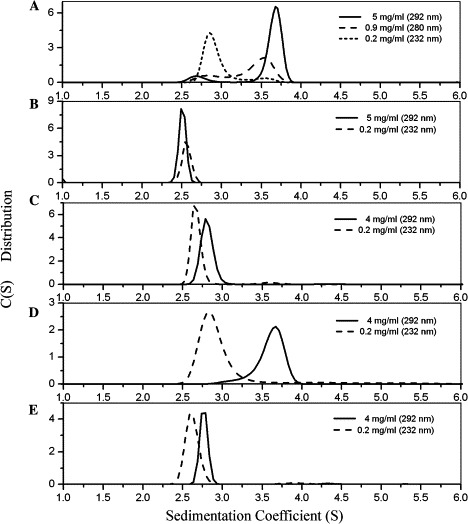
Sedimentation velocity experiments of SARS 3CL proteinase (WT and mutants). Each enzyme solution was run with both high and low concentrations at 60,000 rpm and 20 °C. (A–E) Show the continuous sedimentation coefficient distribution c(s) of wild-type enzyme and mutants. (A) Wild type; (B) Δ7N; (C) R4E; (D) K5A; (E) M6A.
Fig. 4.
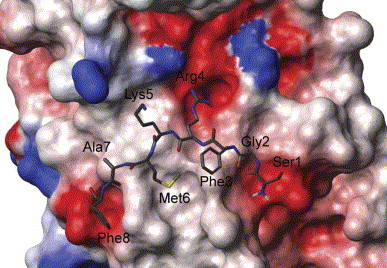
Electrostatic interaction between one monomer of SARS 3CL proteinase and the N-terminal octapeptide of its partner. Figure was drawn by Molmol using its simple charge parameters and the 1UJ1 coordinates from PDB. Met-6 in the N-terminal octapeptide was accommodated in a hydrophobic pocket and Ser-1 and the sidechain of Arg-4 are placed in two negatively charged pockets.
Table 1.
Activity of the mutational SARS-CoV 3CL proteinase
| Enzyme | Relative catalytic activitya (%) |
|---|---|
| WT | 100 |
| Δ7N | <0.01 |
| R4E | 1 |
| K5A | 70 |
| M6A | <1 |
The concentration of the enzyme is 2.8 μM.
Our study confirms that the N-finger plays an important role in stabilizing the dimer of SARS 3CL proteinase. The N-finger of one monomer is important for the enzyme activity of the other monomer [15], [16]. So it is easy to understand the phenomenon of no dimer, no activity. The mutation of R4A kept most of the enzymic activity and did not affect the dimerization severely [33], which means that the electrostatic interaction contributed by the salt bridge between Arg-4 and Glu-290 may not be vital to the dimerization. However, the alanine substitution of Met-6 would be lethal to the dimer. In fact, Arg-4 is not conserved and can be substituted by lysine or valine in several other coronavirus 3CL proteinases, while Met-6 is highly conserved with Leu substitution in very few cases [15]. As a single Met-6 to Ala mutation makes the enzyme to be mostly a monomer with no detectable activity, the hydrophobic effect of Met-6 should play a key role in the dimerization. Since dimerization of SARS 3CL proteinase is necessary for the enzyme activity, the interface of the dimer may be used as a new target for structure-based drug design [18], [19]. From the N-terminal mutational analysis, we propose that a hydrophobic group and an acidic moiety that can form strong hydrogen bonds with ARG-4 are essential to design strong dimer interface binding inhibitors.
The N-finger fragment octapeptide N8 is a dimerization inhibitor of SARS 3CL proteinase
The crystal structure of SARS 3CL proteinase indicates that the N-finger fragment contributes significantly to the enzyme dimerization [16], which may act as a dimerization inhibitor of the enzyme, as in the case of HIV proteinase and other viral enzymes. To determine whether the N-finger fragment is inhibitory towards SARS 3CL proteinase, we have synthesized an octapeptide N8, corresponding to the N-terminal residues 1–8 of the enzyme. The peptide inhibitor (and other peptide mutants) was premixed with the His-tagged enzyme before peptide cleavage assay for 1–2 h. The peptide N8 has obvious inhibitory activity at millimolar level.
As shown in Table 2 , the changes in reaction rate were caused mainly by the changes in apparent k cat. The apparent K m of the His-tagged proteinase changes little at different concentrations of N8, indicating that the N8 peptide works as a non-competitive inhibitor. In other words, the N8 peptide binds to a site distinct from the substrate pocket or to a different enzyme conformation.
Table 2.
The apparent Km and kcat of the SARS 3CL proteinase at different concentrations of inhibitor N8
| Concentration of N8 (mM) | 0 | 0.4 | 0.8 |
|---|---|---|---|
| Apparent Kma (mM) | 1.00 ± 0.03 | 0.96 ± 0.05 | 1.01 ± 0.07 |
| Apparent kcata (min−1) | 18.0 ± 0.4 | 12.7 ± 0.6 | 8.2 ± 0.6 |
The concentration of the enzyme is 1.22 μM.
Zhang et al. [27] have established the Zhang–Poorman analysis to distinguish dimerization inhibitors from competitive inhibitors by plotting vs. in their research about the dimerization inhibitor of HIV protease. In the Zhang–Poorman analysis, a dimerization inhibitor alters the y-intercept value by a factor of [I]/K i, but has no effect on the slope [20], [27]. Dimerization inhibitors are easily identified using the Zhang–Poorman plot as they provide lines that are parallel with that from the protease alone, while competitive and non-competitive inhibitors give non-parallel lines [36]. Zhang–Poorman plot has been successfully applied in many protease dimerization inhibitor studies [20], [23], [25], [27], [36]. Fig. 5 shows that the line in the presence of 0.8 mM peptide N8 is parallel with that in the absence of inhibitor. This strongly suggests that the inhibitor peptide N8 is a dimerization inhibitor of SARS 3CL proteinase, which prevents the enzyme proteolysis activity by disturbing the dimerization of the proteinase.
Fig. 5.
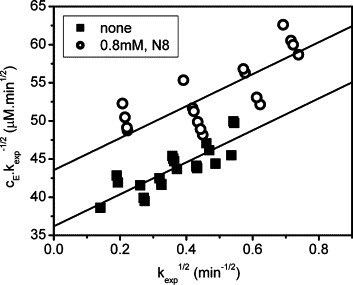
Zhang–Poorman plot of C-terminal His-tagged 3CL proteinase. The lower line (■, without inhibitor) indicates that the enzyme is competitive with Zhang–Poorman’s model, in which only the dimer of the enzyme is biologically active. The upper line (○, with 0.8 mM peptide N8) is parallel with the lower line, indicating that the inhibitor N8 is a dimerization inhibitor of the proteinase, which prevents the enzyme proteolysis activity by disturbing the natural proteinase dimerization.
SARS 3CL proteinase has been proposed to be a key target for structural-based drug design against SARS, because of its functional importance in the viral life cycle. However, all published reports on inhibitor design to target the proteinase are based on the substrate binding pocket [4], [5], [6], [7], [9], [10], [11], [12], [13], [14] and the serine cluster near it [8]. There is no report on the inhibitor design targeting the dimeric interface to date. The octapeptide N8 is the first inhibitor designed to bind to the dimeric interface of coronavirus 3CL proteinase, thus providing a novel strategy for drug design against SARS and other coronaviruses.
The different inhibitory activities of N8 mutants
To further investigate the inhibition mechanism of N8, nine mutants were synthesized and their inhibitory activities were determined (see Table 3 ). According to our mutational study, M6A and R4E mutants disrupted the dimer structure. For the N8 peptide inhibitor, mutation of Arg-4 to Ala (peptide N82) completely abolishes the inhibitory activity. Other single mutations of Arg-4 and Met-6 (peptide N81 and N84) also decrease N8’s inhibition activity significantly. These results correlate well with the mutational study.
Table 3.
The inhibiting activities of N8 and its mutants
| Sequencea | (kcat/Km)appb (mM−1 min−1) | Inhibitory (%) | Experimental pKi | |
|---|---|---|---|---|
| No inhibitor | 23.6 ± 0.9 | 0 | ||
| N8 | SGFRKMAF-NH2 | 12.7 ± 0.2 | 46.2 | 2.66 |
| N81 | SGFEKMAF-NH2 | 17.2 ± 0.8 | 27.1 | 2.33 |
| N82 | SGFAKMAF-NH2 | 22.6 ± 0.6 | 4.2 | 1.44 |
| N83 | SGFRAMAF-NH2 | 14.0 ± 0.3 | 40.7 | 2.57 |
| N84 | SGFRKAAF-NH2 | 19.4 ± 1.7 | 17.8 | 2.11 |
| N85 | SGFEAMAF-NH2 | 18.0 ± 0.8 | 23.7 | 2.26 |
| N86 | SGFAKAAF-NH2 | 18.2 ± 0.8 | 22.9 | 2.24 |
| N87 | SGFEAAAF-NH2 | 21.3 ± 0.2 | 9.7 | 1.82 |
| N88 | Ac-SGFRKMAF-NH2 | 13.6 ± 0.6 | 42.4 | 2.60 |
| N89 | Ac-SGFEAAAF-NH2 | 21.1 ± 1.0 | 10.6 | 1.86 |
The mutant residues are underlined.
The concentration of the enzyme is 2.43 μM.
It is interesting that peptide N86 containing double mutations (Arg-4 to Ala and Met-6 to Ala) shows some inhibitory activity, even though it is predicted to be inactive based on the results from the single mutants. Since the N-finger fragment adopts an extended conformation in the crystal structure, the side chain of Lys-5 points outwards to the dimer interface and does not participate in the enzyme dimerization. The mutation of Lys-5 to Ala (peptide N83) only slightly decreases the inhibiting activity of the N-finger fragment, indicating that Lys-5 contributes little to the inhibitor binding. However, in peptide N86, the peptide backbone may turn around with the Lys-5 taking the place of Arg-4 and interacting with the enzyme, due to the loss of the conformation restriction offered originally by Arg-4 and Met-6. This is confirmed by the triple-mutation peptide N87, which shows little inhibition activity.
In summary, we have carried out a mutational study on the N-finger of SARS 3CL proteinase and determined the dimer dissociation constants for the wild-type protein and the mutants using sedimentation velocity and equilibrium techniques. The Met-6 residue was found to be essential for maintaining the dimer structure, while Arg-4 contributed less. The N-finger octapeptide N8 has been synthesized and confirmed to be a dimerization inhibitor with a K i of 2.20 mM. Several N8 peptide mutants were also synthesized and tested for their inhibition activity. The hydrophobic interaction of Met-6 and the electrostatic interaction of Arg-4 in N8 play important roles in inhibitor binding. Peptide N8 is the first inhibitor designed to target the dimeric interface of coronavirus 3CL proteinase. Our current study provides a novel strategy for drug design against SARS and other coronaviruses.
Acknowledgments
This work was supported by the Ministry of Science and Technology of China and the National Natural Science Foundation of China. The authors thank Prof. Jianguo Chen for providing the clones of SARS 3CL proteinase.
Footnotes
Abbreviations: SARS 3CL, SARS coronavirus 3C-like proteinase; N-finger, N-terminal residues 1-7; DTT, 1,4-dithiothreitol.
References
- 1.Drosten C., Gunther S., Preiser W., van der W.S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLPro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 4.Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y., Yue L.D., Duan W.H., Shan J.K., Qin L., Shi T.L., Li Y.X., Chen K.X., Luo X.M., Shen X., Shan J.H., Jiang H.L. A 3D model of SARS-CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 5.Jenwitheesuk E., Samudrala R. Identifying inhibitor of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003;13:3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toney J.H., Navas-Martin S., Weiss S.R., Koeller A. Sabadinine: a potential non-peptide anti-severe acute respiratory syndrome agent identified using structure-aided design. J. Med. Chem. 2004;47:1079–1080. doi: 10.1021/jm034137m. [DOI] [PubMed] [Google Scholar]
- 7.Lee V.S., Wittayanarakul K., Remsungnen T., Parasuk V., Sompornpisut P., Chantratita W., Sangma C., Vannarat S., Srichaikul P., Hannongbua S., Saparpakorn P., Treesuwan W., Aruksakulwong O., Pasomsub E., Promsri S., Chuakheaw D., Hannongbua S. Structure and dynamics of SARS coronavirus proteinase: the primary key to the designing and screening for anti-SARS drugs. Science Asia. 2003;29:181–188. [Google Scholar]
- 8.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CL Pro. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 9.Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S., Hsu H.H., Huang H.C., Wu D., Brik A., Liang F.S., Liu R.S., Fang J.M., Chen S.T., Liang P.H., Wong C.H. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao R.Y., Tsui W.H.W., Lee T.S.W., Tanner J.A., Watt R.M., Huang J.D., Hu L.H., Chen G.H., Chen Z.W., Zhang L.Q., He T., Chan K.H., Tse H., To A.P.C., Ng L.W.Y., Wong B.C.W., Tsoi H.W., Yang D., Ho D.D., Yuen K.Y. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. Synthesis and evaluation of keto-glutamine analogues as potent inhibitors of severe acute respiratory syndrome 3CLpro. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 12.Chen L.R., Wang Y.C., Lin Y.W., Chou S.Y., Chen S.F., Liu L.T., Wu Y.T., Chih-Jung K.B., Chen T.S.S., Juang S.H. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett. 2005;15:3058–3062. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 14.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., Zhao Q., Zhou Z., Pei D., Ziebuhr J., Hilgenfeld R., Yuen K.Y., Wong L., Gao G., Chen S., Chen Z., Ma D., Bartlam M., Rao Z. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H.T., Yang M.J., Ding Y., Liu Y.W., Lou Z.Y., Zhou Z., Sun L., Mo L.J., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z.H. The crystal structure of severe acute respiratory syndrome virus main proteinase and its complex with an hibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C.K., Wei P., Fan K.Q., Liu Y., Lai L.H. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 18.Fan K.Q., Wei P., Feng Q., Chen S.D., Huang C.K., Ma L., Lai B., Pei J.F., Liu Y., Chen J.G., Lai L.H. Biosynthesis, purification and substrate specificity of SARS coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J.H., Wei Z., Song J.X. Dissection study on the SARS 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: Defining the extra domain as a new target for design of highly-specific protease inhibitors. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schramm H.J., Boetzel J., Buettner J., Fritsche E., Goehring W., Jaeger E., Koeng S., Thumfart O., Wenger T., Nagel N.E., Schramm W. The inhibition of human immunodeficiency virus proteases by “interface peptides”. Antiviral Res. 1996;30:155–170. doi: 10.1016/0166-3542(96)00940-0. [DOI] [PubMed] [Google Scholar]
- 21.Schramm H.J., Nakashima H., Schramm W., Wakayama H., Yamamoto N. HIV-1 reproduction is inhibited by peptides derived from the N- and C-terminal of HIV-1 protease. Biochem. Biophys. Res. Commun. 1991;179:847–851. doi: 10.1016/0006-291x(91)91895-j. [DOI] [PubMed] [Google Scholar]
- 22.Boggetto N., Reboud-Ravaux M. Dimerization inhibitors of HIV-1 protease. Biol. Chem. 2002;383:1321–1324. doi: 10.1515/BC.2002.150. [DOI] [PubMed] [Google Scholar]
- 23.Zutshi R., Chmielewski J. Targeting the dimerization interface for irreversible inhibition of HIV-1 protease. Bioorg. Med. Chem. Lett. 2000;10:1901–1903. doi: 10.1016/s0960-894x(00)00369-3. [DOI] [PubMed] [Google Scholar]
- 24.Zutshi R., Brichner M., Chmielewski J. Inhibiting the assembly of protein-protein interfaces. Curr. Opin. Chem. Biol. 1998;2:62–66. doi: 10.1016/s1367-5931(98)80036-7. [DOI] [PubMed] [Google Scholar]
- 25.Zutshi R., Franciskovich J., Shultz M., Schweitzer B., Bishop P., Wilson M., Chmielewski J. Targeting the dimerization interface of HIV-1 protease: inhibition with cross-linked interfacial peptides. J. Am. Chem. Soc. 1997;119:4841–4845. [Google Scholar]
- 26.Weber I.T. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J. Biol. Chem. 1990;265:10492–10496. [PubMed] [Google Scholar]
- 27.Zhang Z.Y., Poorman R.A., Maggiora L.L., Heinrikson R.L., Kezdy F.J. Dissociative inhibition of dimeric enzymes. J. Biol. Chem. 1991;266:15591–15594. [PubMed] [Google Scholar]
- 28.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 30.Lebowitz J., Lewis M.S., Schuck P. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 2002;11:2067–2079. doi: 10.1110/ps.0207702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laue T.M., Stafford W.F., III Modern applications of analytical ultracentrifugation. Annu. Rev. Biophys. Biomol. Struct. 1999;28:75–100. doi: 10.1146/annurev.biophys.28.1.75. [DOI] [PubMed] [Google Scholar]
- 32.Ali S.A., Iwabuchi N., Matsui T., Hirota K., Kidokoro S., Arai M., Kuwajima K., Schuck P., Arisaka F. Reversible and fast association equilibria of a molecular chaperone, gp57A, of bacteriophage T4. Biophys. J. 2003;85:2606–2618. doi: 10.1016/s0006-3495(03)74683-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou C.Y., Chang H.C., Hsu W.C., Lin T.Z., Lin C.H., Chang G.G. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43:14958–14970. doi: 10.1021/bi0490237. [DOI] [PubMed] [Google Scholar]
- 34.Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P., Shr H.L., Chang G.G., Wang A.H., Liang P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu W.C., Chang H.C., Chou C.Y., Tsai P.J., Lin P.I., Chang G.G. Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J. Biol. Chem. 2005;280:22741–22748. doi: 10.1074/jbc.M502556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shultz M.D., Bowman M.J., Ham Y.W., Zhao X., Tora G., Chmielewski J. Small-molecule inhibitors of HIV-1 protease dimerization derived from cross-linked interfacial peptides. Angew. Chem. Int. Ed Engl. 2000;39:2710–2713. doi: 10.1002/1521-3773(20000804)39:15<2710::aid-anie2710>3.0.co;2-p. [This work was supported by NIH (GM52739) and NSF (9457372-CHE)] [DOI] [PubMed] [Google Scholar]