

Highlights
-
•
TGEV N protein reduces cell viability by inducing cell cycle arrest and apoptosis.
-
•
TGEV N protein induces cell cycle arrest and apoptosis by regulating p53 signaling.
-
•
TGEV N protein plays important roles in TGEV-induced cell cycle arrest and apoptosis.
Keywords: TGEV, N protein, Cell cycle arrest, Apoptosis, p53 signaling
Abstract
Our previous studies showed that TGEV infection could induce cell cycle arrest and apoptosis via activation of p53 signaling in cultured host cells. However, it is unclear which viral gene causes these effects. In this study, we investigated the effects of TGEV nucleocapsid (N) protein on PK-15 cells. We found that TGEV N protein suppressed cell proliferation by causing cell cycle arrest at the S and G2/M phases and apoptosis. Characterization of various cellular proteins that are involved in regulating cell cycle progression demonstrated that the expression of N gene resulted in an accumulation of p53 and p21, which suppressed cyclin B1, cdc2 and cdk2 expression. Moreover, the expression of TGEV N gene promoted translocation of Bax to mitochondria, which in turn caused the release of cytochrome c, followed by activation of caspase-3, resulting in cell apoptosis in the transfected PK-15 cells following cell cycle arrest. Further studies showed that p53 inhibitor attenuated TGEV N protein induced cell cycle arrest at S and G2/M phases and apoptosis through reversing the expression changes of cdc2, cdk2 and cyclin B1 and the translocation changes of Bax and cytochrome c induced by TGEV N protein. Taken together, these results demonstrated that TGEV N protein might play an important role in TGEV infection-induced p53 activation and cell cycle arrest at the S and G2/M phases and apoptosis occurrence.
1. Introduction
Transmissible gastroenteritis virus (TGEV) is an enveloped virus that contains a large, positive-sense, single-stranded RNA genome [1], belonging to family coronaviridae [2]. TGEV, together with human coronavirus 229E (HCoV-229E), porcine epidemic diarrhea virus (PEDV), canine coronaviruses (CCoVs) belongs to alpha coronavirus [2]. TGEV replicates in enterocytes and provokes villous atrophy, resulting in lethal watery diarrhea and dehydration in piglets, which is considered to be a central event in the pathogenesis of TGEV infection [3].
The 5′-two-thirds of the TGEV genome encodes the replicase-transcription complex, Rep1a and Rep1b [4]. During TGEV replication, a 3′-coterminal nested set of subgenomic mRNAs, which encode other viral proteins. TGEV has four major structural proteins: the spike (S), the integral membrane protein (M), the nucleocapsid protein (N) and a small envelope protein (sM) [5]. TGEV N protein, a multifunctional phosphoprotein, plays a primary structural role in packaging the RNA genome into a helical ribonucleoprotein, as well as regulatory roles in viral RNA synthesis (replication and transcription), translation, and modulation of host cell metabolism [5].
TGEV is known to cause cell cycle arrest and apoptosis via activation of p53 signaling in different host cell types including ST and PK-15 cells [6], [7], [15]. The evidence of induction of apoptosis by TGEV implies an involvement of apoptosis in the pathogenesis of TGE [1], [7]. However, it remains to be known which TGEV protein(s) induces cell cycle arrest and apoptosis in cells that support productive viral replication. Current evidence indicates that TGEV N protein might be involved in caspase-mediated proteolysis within the host cell [8]. Further studies revealed that the TGEV N protein could locate nucleolar and might possess the function to disrupt cell division [9]. However, whether TGEV N protein could affect cell division and cell apoptosis and the associated molecular mechanisms are not clear.
In the present study, we demonstrate that TGEV N protein, when expressed within cells, can cause cell cycle arrest and apoptosis in PK-15 cells, in which p53 and p21 activation, cyclin B1 and cdc2 decrease, Bax translocation to mitochondria, cytochrome c release and subsequent activation of caspase-3 are required.
2. Materials and methods
2.1. Antibodies, cells and virus
Monoclonal antibodies against p53, p21, cdc2, cdk2, cyclinB1, Cytochrome c (Cyt c), Bax, actin, Cox4 were purchased from Santa Cruz Biotechnology (Santa Cruz, Inc., CA, USA). PK-15 cells (ATCC, CCL-33) were grown in Dulbecco Minimal Essential Medium (D-MEM) (Gibco BRL, MD, USA) supplemented with 10% heat-inactivated new born bovine serum (Gibco BRL, MD, USA), 100 IU of penicillin and 100 μg of streptomycin per ml, at 37 °C in humidified 5% CO2. The TGEV Shaanxi strain was used as previously described [7], [10].
2.2. Construction of plasmids
Full-length N gene from TGEV was PCR amplified, ligated into the pGEM-T easy vector (Promega, Madison, USA), and then subcloned into between XhoI/EcoRI sites of eukaryotic expression vector pEGFP-N1 (EGFP) (Clontech, Palo Alto, CA), to produce the recombinant His-tagged construct pEGFP-N1-TGEV-N (N-EGFP). This vector contains the cytomegalovirus promoter for the expression of N protein with a six-histidine tag. They were sequenced to confirm that no errors were introduced as a result of PCR amplification. In vitro expression of the N-EGFP was tested in transient expression experiments using PK-15 cells.
PK-15 cells, grown in 60 mm plate, were transfected with EGFP vector only, or N-EGFP. After 24 h, 48 h and 72 h of transfection, the expression of N was directly observed under a fluorescence microscopy and further confirmed by Western blot analysis using His6 antibody (Sigma–Aldrich, USA).
2.3. Cell viability assay
The effects of TGEV N protein on cell viability were determined using the MTT assay. Briefly, PK-15 cells, grown in 96-well culture plates, were transfected with EGFP vector only, or N-EGFP expression vector. After 24 h, 48 h and 72 h of transfection, the cells were treated with 50 μl of 5 mg/ml MTT and the resulting formazan crystals were dissolved in di-methyl sulfoxide (DMSO). The absorbance was measured by microplate spectrophotometer (Bio-tek Instruments, Inc., Winooski, USA) at 570 nm. Results were expressed as percentage of the controls, which were arbitrarily assigned 100% viability.
2.4. Cell cycle analysis
To determine cell cycle status, nuclear DNA content was measured by using propidium iodide staining. Briefly, cells were fixed in 70% ethanol for 30 min at 4 °C. After washing with phosphate-buffered saline (PBS), the cells were stained with 0.1% Triton X-100 (Sigma), 20 μg/ml RNase A (Sigma) and 10 μg/ml propidium iodide (Sigma) for 30 min and analyzed using a FACScan flow cytometer (Beckman Coulter, Inc., Fullerton, CA., USA).
2.5. Apoptotic rate analysis
Annexin V-FITC Apoptosis Kit (BioVision, Inc., CA., USA) was used for apoptosis detection according to the manufacturer’s protocol. Briefly, harvested cells were washed twice with PBS and resuspended in 500 μl binding buffer, followed by adding 5 μl of Annexin V-FITC and 5 μl of PI. After incubation in the dark for 30 min at room temperature, a total of 10,000 cells were acquired and percentage of positive cells were analyzed by flow cytometry (Beckman).
2.6. Western blot analysis
PK-15 cells, grown in 60 mm plate, were transfected with EGFP vector only, or N-EGFP. After transfection, cells were collected and lysed by RIPA. Isolation of mitochondrial and cytosolic proteins was performed using the Mitochondria/cytosol Fractionation Kit (Pierce, Rockford, IL, USA). Protein concentrations were measured using BCA Protein Assay Reagent (Pierce). Equivalent amounts of proteins were loaded and electrophoresed on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Subsequently, proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corp, Atlanta, GA., USA). The membranes were blocked with 5% nonfat dry milk at room temperature for 1 h, and then incubated with indicated primary antibodies over night at 4 °C, followed by HRP-conjugated secondary antibodies at room temperature for 1 h. The signal was detected using ECL reagent (Pierce, Rockford, IL., USA).
2.7. Caspase activity assay
Caspase-3 activity was measured by colorimetric assay kit (BioVision) according to the manufacture’s instructions. Briefly, cell lysates were prepared and protein concentrations were measured using BCA Protein Assay Reagent (Pierce). Then 200 μg of protein in each sample was incubated with caspase-3 substrate at 37 °C for 4 h. Samples were read at 405 nm in microplate spectrophotometer (BioTek Instruments, Inc., Winooski, USA).
3. Results
3.1. TGEV N gene-expressed PK-15 cells show reduced cell viability
The PK-15 cells were transfected with the N-EGFP expression vector, and the expression of N protein was directly examined under the fluorescence microscopy and further confirmed by Western blot using anti-His6 antibody to detect His-tagged N protein. After 24 h of transfection, the N-EGFP protein was dispersed throughout the cytoplasmic region, while the EGFP control showed a diffuse distribution pattern (Fig. 1 A). The expressing N-EGFP proteins were detected as expected, while no signal was detected in the EGFP control when detected using the anti-His6 antibody (Fig. 1B). These results suggested that the N protein of TGEV was expressed in PK-15 cells.
Fig. 1.
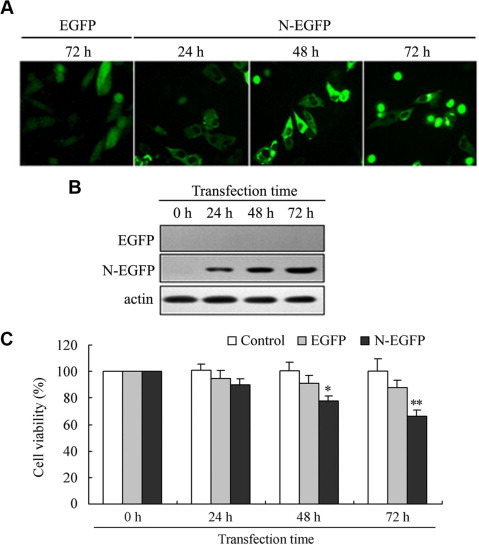
Identification of TGEV N gene expression in transfected PK-15 cells and its effect on cell viability. (A) Fluorescence microscopic observation of PK-15 cells at 24 h, 48 h and 72 h after transfection with the N-EGFP expression vector, at 72 h after transfection with the EGFP empty vector, respectively (Magnification, 400×). (B) Total cell lysates from PK-15 cells transfected with the N-EGFP expression vector and EGFP empty vector were analyzed by Western blot. Anti-His6 antibody was used to detect the His6-N fusion protein. (C) Cell viability of TGEV N gene expressed cells compared to controls at different time points post-transfection. PK-15 cells were transfected in 96-well plates in triplicate with N-EGFP expression vector and EGFP empty vector. At different time points post-transfection, cells viability was measured by MTT assay as described under Section 2. Values are mean ± SD. ∗P < 0.05, ∗∗P < 0.01 versus EGFP empty vector-transfected cells.
To determine the effect of TGEV N protein on expressed PK-15 cells, cell viability was firstly tested by MTT assay. Result showed that the cell viability of PK-15 cells transfected with N-EGFP expression vector had significantly reduced at 48 h of post-transfection (Fig. 1C). The apparent inhibition did not occur in the EGFP empty vector-transfected cells. These data demonstrated that expression of TGEV N gene in PK-15 cells inhibited cell viability.
3.2. Expression of TGEV N gene blocks cell cycle progression at S and G2/M phase
To determine whether the growth inhibition of TGEV N-expressing cells was due to the arrest of cell cycle progression at a certain phase, at different times after transfection, cell cycle profiles were analyzed by flow cytometry (Fig. 2 A, upper panel). The histograms were quantitatively analyzed by use of a curve-fitting program to determine the percentage of cells in each of the G0/G1, S, and G2/M phases (Fig. 2A, lower panel). Results showed that the proportion of cells in S and G2/M phases significantly increased at 24 h of post-transfection, and further increased with transfection time in the cells transfected with N-EGFP expression vector, compared to that in the cells transfected with EGFP control vector (Fig. 2A), which strongly indicated that cell cycle arrest was occurred in TGEV-N-expressing cells.
Fig. 2.
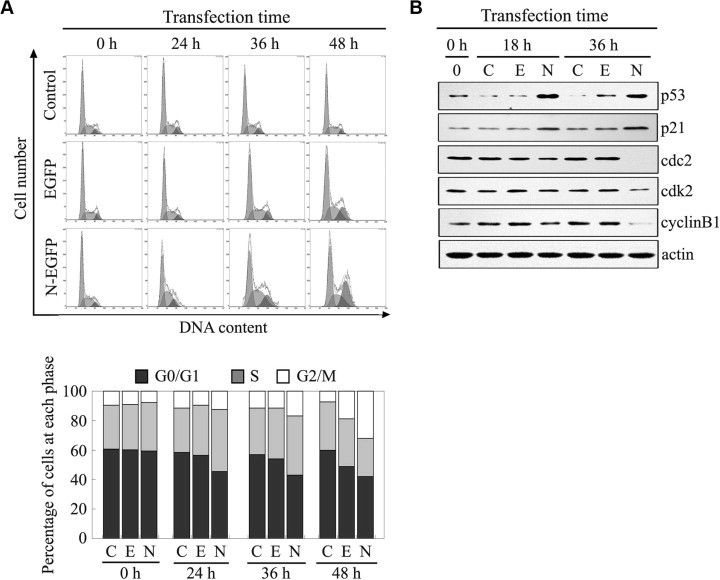
Cell cycle arrest induced by TGEV N protein. (A) PK-15 cells were collected and subjected to cell cycle analysis at 24 h, 36 h and 48 h after transfection with the N-EGFP expression vector and EGFP empty vector, respectively. The data represent results from one of three independent experiments. (B) Effects of TGEV N protein on cell cycle regulating factors. Cell lysates harvested at indicated time points post-transfection with N-EGFP expression vector and EGFP empty vector were used for Western blot analysis. (Note: C, untransfected control cells; E, cells transfected with EGFP empty vector; N, cells transfected with N-EGFP expression vector). The data represent results from one of three independent experiments.
To further determine the effects of TGEV N protein on cell progression through S and G2/M phase, we investigated its effects on protein levels of the p53, p21, cyclin B1, cdc2, and cdk2, which regulate transition through the S and G2/M boundary checkpoint. Fig. 2B showed that the levels of p53 significantly increased at 18 h and 36 h after cells transfected with N-EGFP expression vector compared with EGFP empty vector-transfected cells and non-transfected groups. The amounts of p21 were slightly increased at 18 h, and significantly increased at 36 h in the cells expressed TGEV N gene. Furthermore, the expression of cyclin B1, cdc2 and cdk2 decreased in the cells expressed TGEV N gene compared with the control cells (Fig. 2B), which coincided with the changes of these cycle-related molecules in TGEV-infected cells. These results indicated that TGEV N protein caused S and G2/M-phase arrest through regulation of certain cell cycle factors to modulate the proceeding of S and G2/M phases.
3.3. Expression of TGEV N gene induces apoptosis in PK-15 cells
To examine whether apoptosis was initiated in the cells expressed TGEV N gene, PK-15 cells transfected with N-EGFP expression vector or EGFP empty vector, or untransfected cells were subjected to annexin V and PI staining and flow cytometry analysis. As shown in Fig. 3 A, annexin V positive cells increased about 4.8%, 14% and 21.9% in the cells transfected with N-EGFP expression vector than that in EGFP empty vector-transfected cells at 24 h, 48 h and 72 h of post-transfection, respectively. Whereas, annexin V positive cells increased about 1.6%, 4.3% and 5.9% in the cells transfected with EGFP empty vector than that un-transfected control cells at 24 h, 48 h and 72 h of post-transfection, respectively. Collectively, these data indicated that apoptosis was initiated in the PK-15 cells expressed TGEV N gene.
Fig. 3.
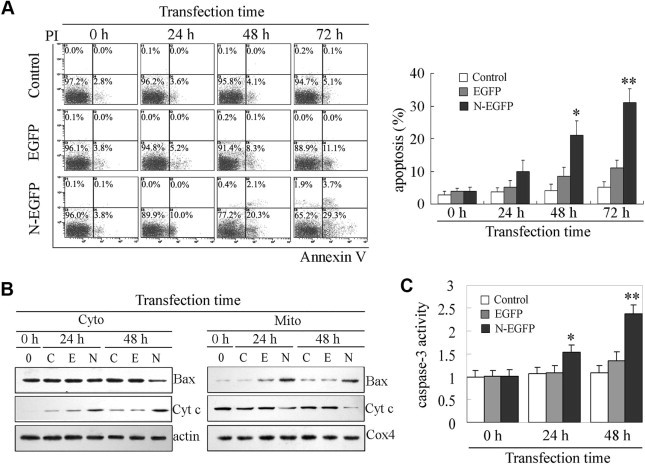
Apoptosis induced by TGEV N protein. (A) Representative dual-labeled quadrants of bivariant fluorescence dot plots showing the induction of apoptosis in the PK-15 cells transfected with N-EGFP expression vector and EGFP empty vector. Percentages shown are proportions of apoptotic cells detected at different time points, as indicated. Results are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01 versus EGFP empty vector-transfected cells. (B) Expression of pro-apoptotic proteins in PK-15 cells after transfection. Cell lysates harvested at indicated time points post-transfection with N-EGFP and EGFP empty vector were used for Western blot analysis. (Note: C, untransfected control cells; E, cells transfected with EGFP empty vector; N, cells transfected with N-EGFP expression vector). (C) Effects of TGEV N protein on the activity of caspase-3 in PK-15 cells. The activity of caspase-3 was measured by colorimetric assay kit. Results are means ± SD. ∗P < 0.05, ∗∗P < 0.01 versus EGFP empty vector-transfected cells.
To further elucidate the mechanism of the TGEV N-induced apoptosis, the translocation of Bax and release of cytochrome c were analyzed by Western blot. As shown in Fig. 3B, Bax was translocated from cytosol to mitochondria at 24 h of post-transfection, and steadily increased in mitochondria with the transfection time. Following Bax translocation to mitochondria, cytochrome c released from mitochondria to cytosol in the cells expressed TGEV N gene. The activity of caspase-3 significantly increased in the cells transfected N-EGFP expression vector compared to un-transfected cell or EGFP empty vector-transfected cells (Fig. 3C). Taken together, the results demonstrate that TGEV N protein triggered the efflux of cytochrome c into the cytosol and the activation of caspase-3, thus leading to apoptosis occurrence in the cells.
3.4. TGEV N protein induces cell cycle arrest and apoptosis by regulation of p53 signaling
In order to investigate whether p53 pathway regulate TGEV N protein-induced cell cycle arrest and apoptosis, we tested cell cycle profiles and apoptosis in TGEV N protein expressed PK-15 cells with specific p53 inhibitor pifithrin-α (PFT-α). Results showed that PFT-α attenuated TGEV N protein-induced cell cycle arrest at S and G2/M phases (Fig. 4 A). In addition, the apoptotic rate induced by TGEV N protein was slightly inhibited by PFT-α (Fig. 4B). To further explore the roles of p53, we investigated the effects of PFT-α on the expression of cell cycle and apoptosis regulating factors in TGEV N protein expressed PK-15 cells. As expected, incubation of PK-15 cells with PFT-α, cdc2, cdk2 and cyclin B1 expression slightly increased in TGEV N protein expressed PK-15 cells with PFT-α treatment compared to that without PFT-α treatment (Fig. 4C). Furthermore, the translocation of Bax and cytochrome c was moderately reversed by PFT-α treatment in TGEV N protein expressed PK-15 cells (Fig. 4C). The activity of caspase-3 was reduced in TGEV N protein expressed cells with PFT-α treatment compared to the cells without PFT-α treatment (Fig. 4D). Taken together, these results suggest that p53 might play key roles in mediation of TGEV N protein induced cell cycle arrest and apoptosis.
Fig. 4.
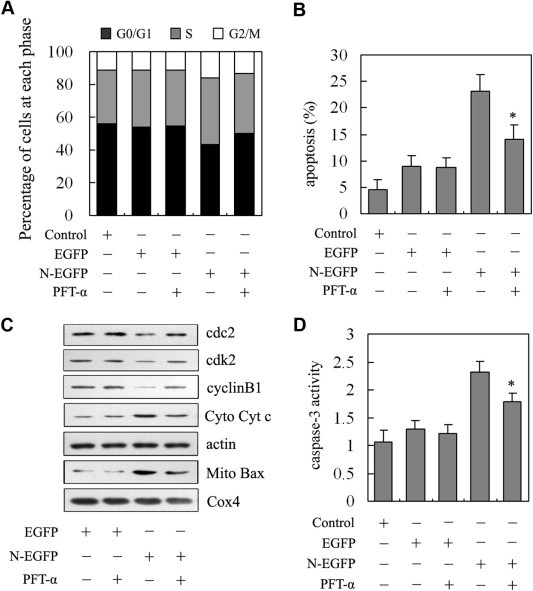
The roles of p53 on regulating TGEV N protein-induced cell cycle arrest and apoptosis. (A) Effect of PFT-α on the changes of cell cycle progression induced by TGEV N protein. PK-15 cells were transfected with N-EGFP expression vector or EGFP empty vector and treated with PFT-α for 36 h. Cells were collected and stained with propidium iodide for FACS analysis. (B) Effect of PFT-α on the apoptosis induced by TGEV N protein. PK-15 cells were transfected with N-EGFP expression vector or EGFP empty vector and treated with PFT-α for 48 h. Cells were collected and stained with annexin V and PI for FACS analysis. Values are mean ± SD. ∗P < 0.05 versus TGEV N gene expressed cells without PFT-α. (C) Effects of PFT-α on the expression of cell cycle and apoptosis regulating factors. PK-15 cells were transfected with N-EGFP expression vector or EGFP empty vector and treated with PFT-α for 24 h. Cells were collected and subjected to Western blot analysis. Cox4 and actin were used as internal controls for the mitochondrial fractions and the cytosolic fractions, respectively. (D) Effects of PFT-α on caspase-3 activity. PK-15 cells were transfected with N-EGFP expression vector or EGFP empty vector and treated with PFT-α for 48 h. Cells were collected and the activity of caspase-3 was measured by colorimetric assay kit. Results are means ± SD. ∗P < 0.05 versus TGEV N gene expressed cells without PFT-α.
4. Discussion
Several viruses-encoded proteins have been shown to activate the cell cycle or apoptotic process by interacting with various cellular components [11], [12], [13]. Virus-induced apoptosis of host cells can either facilitate virus dissemination or be part of the host defense response invoked to counteract viral infection [14]. Several in vitro studies have demonstrated that TGEV infection induced cell cycle arrest and apoptosis through activation of p53 pathway in different host cell lines [6], [15]. However, it is unclear which viral genes play a dominant role in induction of the cell cycle arrest and apoptosis. In this study, we have investigated the effects of TGEV N protein on cell growth, cell cycle progression and cell apoptosis, as well as the mechanism that TGEV N protein causes the cell cycle arrest and apoptosis.
One of the common phenomena of viral infection is to induce the G2/M-phase arrest in host cells. Human immunodeficiency virus type 1 (HIV-1) Vpr activates cell cycle inhibitor p21 to prevent cell proliferation by causing arrest at the G2/M phase of the cell cycle [16]. Human papillomavirus (HPV) type 1 E4 protein induces G2/M arrest through inhibition or delay in the activation of cdk1/cyclin B1 kinase activity [17]. Chicken anemic virus (CAV) apoptin protein and Herpes simplex type 1 (HSV-1) ICP0 protein can interfere with kinetochores to block cell cycle progression at G2/M phase [18], [19]. Previous studies have revealed that the TGEV N could locate nucleolar and might function to disrupt cell division [9]. Here, our results showed that expression of TGEV N protein activated p53 and up-regulated cell cycle inhibitor p21, and then inhibited the activation of cdc2/cyclin B1, leading to G2/M arrest, which was consistent with our previous results found in TGEV-infected cells [6], suggesting that TGEV N protein might play an important role in TGEV-induced cell cycle arrest. In previous studies, we found TGEV infection induces S and G2/M arrest, which is aimed to establish a pseudo-S phase state for viruses to replicate of their genomes [6]. The results presented in here also provided evidences for S phase arrest and following G2/M arrest, suggesting that TGEV N protein might play an important role in establishing a pseudo-S phase state for virus replication. The same phenomena were also found in other viruses [20].
Many viral proteins are involved in induction of apoptosis. Infectious bursal disease virus VP5 [21], Rubella virus capsid protein [22], Hepatitis C virus core protein [23], and Japanese encephalitis virus NS2B-NS3 [24] have been shown to induce apoptosis alone in cell culture and play an important role in viral pathogenesis. Over the past few years, studies report multifaceted roles for the viral N protein in trafficking, translation and in modulation of immune signaling. Previous studies have indicated that TGEV N protein underwent caspase-mediated proteolysis during TGEV-induced apoptosis [8]. In this study, we further investigated its function in modulation of apoptosis and demonstrated that TGEV N protein was capable of inducing apoptosis in the transfected PK-15 cells, which might be important for the pathogenesis of TGEV.
p53 is a key element in the induction of cell cycle arrest or apoptosis in viruses infected cells [25], [26]. Our previous studies have demonstrated that TGEV infection could induce cell cycle arrest and apoptosis via activation of p53 signaling in cultured cells, suggesting that p53 might play important roles in TGEV-induced cell cycle arrest and apoptosis [6]. In the present study, we also found that p53 was activated to induce the change of apoptotic and cell cycle regulating factors such as cdc2, cdk2, cyclinB1, Bax and Cyt c in TGEV N protein expressed PK-15 cells. These studies further suggest p53 might play an important role in the pathogenesis of TGEV.
In conclusion, the present study demonstrates that the expression of TGEV N gene induced S-phase and G2/M-phase arrest, and subsequent apoptosis through the activation of p53 and p21, deduction of cyclin B/cdc2, promoting translocation of Bax to mitochondria, followed by a release of cytochrome c into the cytosol, results in activation of the effector caspase-3, thus leading to cellular destruction.
Contributors and authorship
Li Ding and Yong Huang designed the experiments, interpreted the data and wrote the article. Li Ding performed the experiments with assistance and advice from Qian Du, Feng Dong, Xiaomin Zhao, Wenlong Zhang and Xingang Xu. Yong Huang and Dewen Tong revised the paper. All authors have read the paper and approved to submit it to your journal.
Conflict of interest
There is no conflict of interest of any authors in relation to the submission.
Acknowledgments
This work was supported by grants from the Doctoral Program of Higher Education of China (Grant No. 20110204110014), the Scientific Research Program of Northwest A&F University (Nos. QN2011064, ZD2013009, Z111021103).
References
- 1.Eleouet J.F., Chilmonczyk S., Besnardeau L., Laude H. Transmissible gastroenteritis coronavirus induces programmed cell death in infected cells through a caspase-dependent pathway. J. Virol. 1998;72:4918–4924. doi: 10.1128/jvi.72.6.4918-4924.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carstens E. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses. 2009Arch. Virol. 2010;155:133–146. doi: 10.1007/s00705-009-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enjuanes L., Van der Zeijst B.A.M. Plenum Press; New York, NY: 1995. Molecular Basis of Transmissible Gastroenteritis Virus Epidemiology, The Coronaviridae. 337–376. [Google Scholar]
- 4.Ortego J., Escors D., Laude H., Enjuanes L. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 2002;76:11518–11529. doi: 10.1128/JVI.76.22.11518-11529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yount B., Curtis K.M., Baric R.S. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. J. Virol. 2000;74:10600–10611. doi: 10.1128/jvi.74.22.10600-10611.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L., Huang Y., Dai M., Zhao X., Du Q., Dong F., Wang L., Huo R., Zhang W., Xu X. Transmissible gastroenteritis virus infection induces cell cycle arrest at S and G2/M phases via p53-dependent pathway. Virus Res. 2013;178:241–251. doi: 10.1016/j.virusres.2013.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding L., Xu X., Huang Y., Li Z., Zhang K., Chen G., Yu G., Wang Z., Li W., Tong D. Transmissible gastroenteritis virus infection induces apoptosis through FasL-and mitochondria-mediated pathways. Vet. Microbiol. 2012;158:12–22. doi: 10.1016/j.vetmic.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eléouët J.F., Slee E.A., Saurini F., Castagné N., Poncet D., Garrido C., Solary E., Martin S.J. The viral nucleocapsid protein of transmissible gastroenteritis coronavirus (TGEV) is cleaved by caspase-6 and-7 during TGEV-induced apoptosis. J. Virol. 2000;74:3975–3983. doi: 10.1128/jvi.74.9.3975-3983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wurm T., Chen H., Hodgson T., Britton P., Brooks G., Hiscox J.A. Localization to the nucleolus is a common feature of coronavirus nucleoproteins, and the protein may disrupt host cell division. J. Virol. 2001;75:9345–9356. doi: 10.1128/JVI.75.19.9345-9356.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding L., Zhao X., Huang Y., Du Q., Dong F., Zhang H., Song X., Zhang W., Tong D. Regulation of ROS in transmissible gastroenteritis virus-activated apoptotic signaling. Biochem. Biophys. Res. Commun. 2013;442:33–37. doi: 10.1016/j.bbrc.2013.10.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lam W., Tang J.W., Yeung A., Chiu L., Sung J.J.Y., Chan P.K.S. Avian influenza virus A/HK/483/97 (H5N1) NS1 protein induces apoptosis in human airway epithelial cells. J. Virol. 2008;82:2741–2751. doi: 10.1128/JVI.01712-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnitz R.A., Wan F., Tripuraneni V., Bolton D.L., Lenardo M.J. Protein kinase A phosphorylation activates Vpr-induced cell cycle arrest during human immunodeficiency virus type 1 infection. J. Virol. 2010;84:6410–6424. doi: 10.1128/JVI.02273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davy C., Doorbar J. G2/M cell cycle arrest in the life cycle of viruses. Virology. 2007;368:219–226. doi: 10.1016/j.virol.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poon B., Grovit-Ferbas K., Stewart S.A., Chen I.S.Y. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science. 1998;281:266–269. doi: 10.1126/science.281.5374.266. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y., Ding L., Li Z., Dai M., Zhao X., Li W., Du Q., Xu X., Tong D. Transmissible gastroenteritis virus infection induces cell apoptosis via activation of p53 signaling. J. Gen. Virol. 2013;94:1807–1817. doi: 10.1099/vir.0.051557-0. [DOI] [PubMed] [Google Scholar]
- 16.Chowdhury I.H., Wang X.F., Landau N.R., Robb M.L., Polonis V.R., Birx D.L., Kim J.H. HIV-1 Vpr activates cell cycle inhibitor p21/Waf1/Cip1: a potential mechanism of G2/M cell cycle arrest. Virology. 2003;305:371–377. doi: 10.1006/viro.2002.1777. [DOI] [PubMed] [Google Scholar]
- 17.Knight G.L., Turnell A.S., Roberts S. Role for Wee1 in inhibition of G2-to-M transition through the cooperation of distinct human papillomavirus type 1 E4 proteins. J. Virol. 2006;80:7416–7426. doi: 10.1128/JVI.00196-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teodoro J.G., Heilman D.W., Parker A.E., Green M.R. The viral protein Apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev. 2004;18:1952–1957. doi: 10.1101/gad.1198404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomonte P., Sullivan K.F., Everett R.D. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 2001;276:5829–5835. doi: 10.1074/jbc.M008547200. [DOI] [PubMed] [Google Scholar]
- 20.Belyavskyi M., Braunagel S.C., Summers M.D. The structural protein ODV-EC27 of Autographa californica nucleopolyhedrovirus is a multifunctional viral cyclin. Proc. Natl. Acad. Sci. 1998;95:11205–11210. doi: 10.1073/pnas.95.19.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao K., Vakharia V.N. Induction of apoptosis< i> in vitro</i> by the 17-kDa nonstructural protein of infectious bursal disease virus: possible role in viral pathogenesis. Virology. 2001;285:50–58. doi: 10.1006/viro.2001.0947. [DOI] [PubMed] [Google Scholar]
- 22.Duncan R., Esmaili A., Law L.M.J., Bertholet S., Hough C., Hobman T.C., Nakhasi H.L. Rubella virus capsid protein induces apoptosis in transfected RK13 cells. Virology. 2000;275:20–29. doi: 10.1006/viro.2000.0467. [DOI] [PubMed] [Google Scholar]
- 23.Chou A.H., Tsai H.F., Wu Y.Y., Hu C.Y., Hwang L.H., Hsu P.I., Hsu P.N. Hepatitis C virus core protein modulates TRAIL-mediated apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis signaling pathway. J. Immunol. 2005;174:2160–2166. doi: 10.4049/jimmunol.174.4.2160. [DOI] [PubMed] [Google Scholar]
- 24.Yang T.C., Shiu S.L., Chuang P.H., Lin Y.J., Wan L., Lan Y.C., Lin C.W. Japanese encephalitis virus NS2B-NS3 protease induces caspase 3 activation and mitochondria-mediated apoptosis in human medulloblastoma cells. Virus Res. 2009;143:77–85. doi: 10.1016/j.virusres.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Casavant N., Luo M., Rosenke K., Winegardner T., Zurawska A., Fortunato E. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 2006;80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pampin M., Simonin Y., Blondel B., Percherancier Y., Chelbi-Alix M.K. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 2006;80:8582–8592. doi: 10.1128/JVI.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]