

Abstract
Modern rational modulator design and structure-function characterization often concentrate on concave regions of biomolecular surfaces, ranging from well-defined small-molecule binding sites to large protein-protein interaction interfaces. Here we introduce beta-cluster as a pseudomolecular representation of fragment-centric pockets detected by AlphaSpace [J. Chem. Inf. Model. 2015, 55, 1585], a recently developed computational analysis tool for topographical mapping of biomolecular concavities. By mimicking the shape as well as atomic details of potential molecular binders, this new beta-cluster representation allows direct pocket-to-ligand shape comparison and can be used to guide ligand optimization. Furthermore, we defined beta-score, the optimal vina score of the beta-cluster, as an indicator of pocket ligandability, and developed an ensemble beta-cluster approach which allows one-to-one pocket mapping and comparison among aligned protein structures. We demonstrated the utility of beta-cluster representation by applying the approach to a wide variety of problems including binding site detection and comparison, characterization of protein-protein interactions and fragment-based ligand optimization. These new beta-cluster functionalities have been implemented in AlphaSpace 2.0, which is freely available on the web at: http://www.nyu.edu/projects/yzhang/AlphaSpace2.
Graphical Abstract
I. INTRODUCTION
Biomolecular recognition usually involves heterogeneous and diverse concave surfaces, ranging from well-defined singular deep pockets for binding native small molecules, which are traditionally considered as prime targets by drug molecules1, 2, to much larger and flatter binding interfaces for protein-protein and protein-peptide interactions, which usually consist of multiple small shallow binding cavities. As better understanding of concave biomolecular surfaces can present many opportunities for modulator discovery including lead optimization3, improved ligand screening4, and even the identification of previously unknown druggable sites5, many computational methods6–16 have been developed for biomolecular surface characterization. With the detected binding cavities, one key element for binding pocket comparison and analysis is pocket representation.
A number of pocket characterization methods use pocket lining atoms to represent the binding site with different levels of abstraction, including graph17–20, feature points21–24, and fingerprint models25–30. Fingerprint methods such as FLAP29 and KRIPO30 abstract the binding site into fingerprints by binning distances between pharmacophore elements within the binding site. Similarly, protein-ligand interactions can be encoded in distance-binned fingerprints as in TIFP28. Graph-based approaches such as ProBIS20 and SOIPPA19 represent the binding site as condensed nodes. Both fingerprint and graph-based methods have the advantage of describing complex 3D binding sites into simpler 2D or 1D representations which are computationally efficient and can be used for rapid binding site comparison31. However, these abstract representations of binding sites would be devoid of detailed 3-D structural information and do not allow for the direct comparison between the binding site and their cognate ligands.
On the other hand, several grid-based computational methods32–37 have been developed to generate 3D representations of binding sites that can be used for direct comparison between binding sites and ligands. For example, Shaper33 fills a cavity with grid points to form an inverse image of the binding site, which can mimic volumetric properties of its potential binding ligands. Another grid-based method termed FLAPpharm uses the molecular interaction fields based on the GRID force field to compare binding sites and ligand ensembles34. Along a similar direction, Sanner et. al. recently developed AutoSite38, in which AutoDock affinity maps are calculated for the entire surface followed by removing low-affinity points and then clustering to generate a pseudomolecular representation of potential binding sites. This AutoSite representation can mimic cognate ligands and can be used for direct comparison of the binding site and the ligand. However, generating pseudomolecules with grid-based methods is sensitive to placement of grid points as well as translation or rotation of a protein structure. Additionally, employment of finer grids over the entire protein surface would significantly increase computational cost.
Recently we developed AlphaSpace 1.039, a computational approach for fragment-centric topographical mapping of concave biomolecular surfaces based on alpha spheres - a geometric feature of a protein’s Voronoi diagram. In comparison with other Voronoi-based methods40–44, one novel feature of AlphaSpace is its introduction of alpha-atom, which has the same center as its associated alpha sphere but with an atomic radius of 1.8 Å. As alpha-atoms are generated from surface alpha spheres and are selected to be within proper interaction distance from their associated pocket lining atoms, they can mimic the overall shape contour of their cognate ligand partner. For the 2P2I data set45, a good volume overlap and volumetric correlation between alpha-atoms and pocket-occupying inhibitors has been observed with an R2 of 0.77. Thus in AlphaSpace1.0, alpha-atoms have been employed to represent fragment-centric concave interaction regions, and the capacity of alpha-atoms to mimic potential binders has been exploited to develop a pocket-centric design strategy for PPI inhibitor optimization46, 47. However, due to the concavity of protein lining atoms, distances between neighboring alpha atoms are often much shorter than typical chemical bonds and thus overlapping alpha atoms cannot properly mimic molecular detail of the partner ligand, as illustrated in Fig. 1A.
Figure 1.
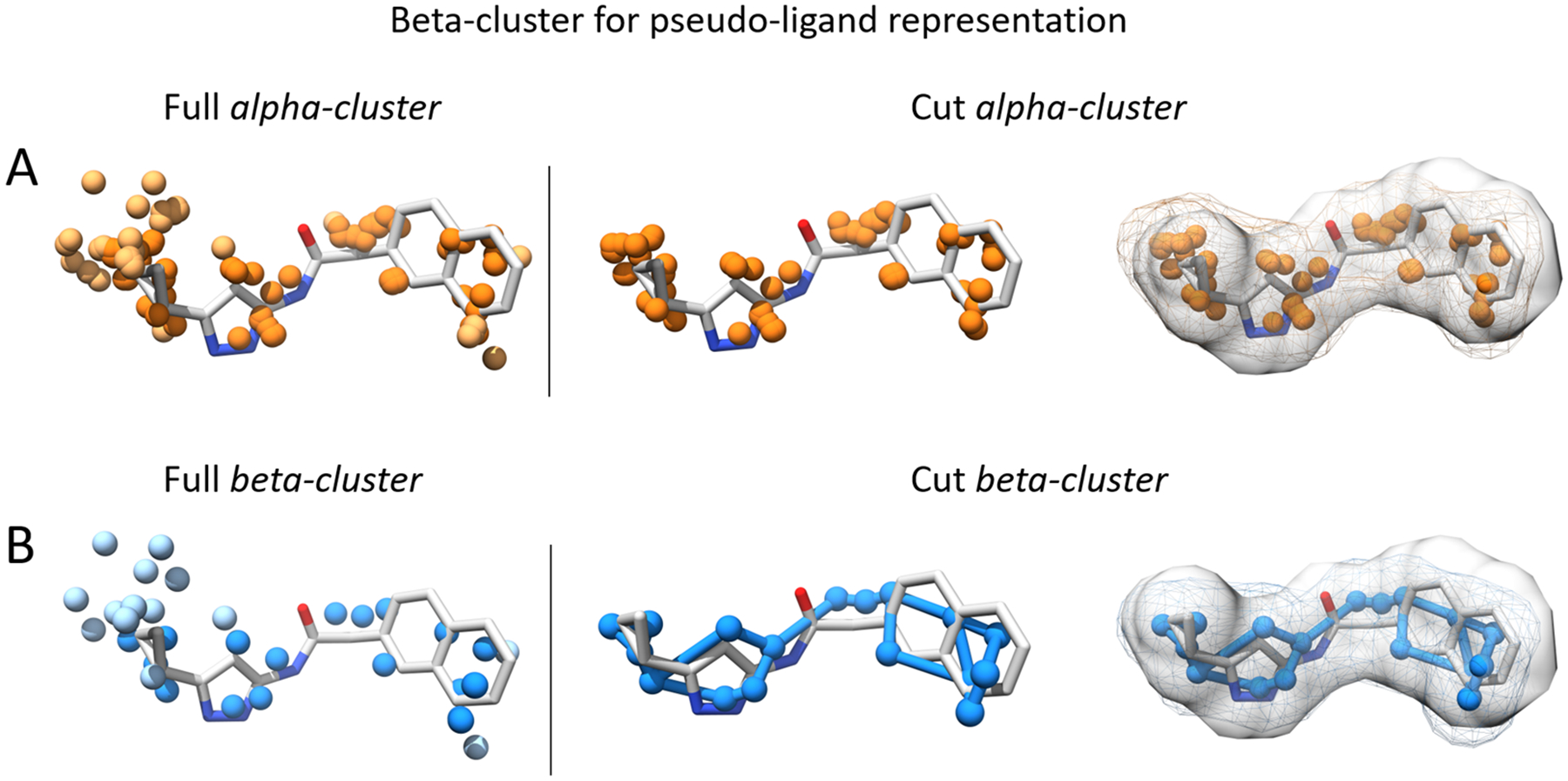
Illustration of how clustering alpha atoms in the alpha cluster leads to a more realistic, pseudomolecular representation of the concave site. (A). The full alpha-cluster is obtained with the average-linkage algorithm as in AlphaSpace1.0. Non-contact alpha atoms, which are more than 1.8 Å away from any ligand atom, are colored in light orange. Removing those non-contact alpha atoms yields the cut alpha-cluster. We can see that the cut alpha-cluster has a shape contour similar to the ligand, but distances between neighboring alpha atoms are often much shorter than typical chemical bonds and thus overlapping alpha atoms can not properly mimic molecular detail of the partner ligand. (B). By clustering alpha atoms into beta atoms, it leads to a more ligand-like pseudomolecule representation, a beta-cluster. The blue edges in the cut beta-cluster are calculated based on a minimum spanning tree algorithm, and are used for illustration.
In this work, we introduce beta-cluster, a pseudomolecular representation that can realistically mimic molecular features and properties of partner ligands that can be accommodated in the cavity. Being a tessellation-based method instead of grid-based, beta-cluster is naturally formed in the concave region and avoids the uncertainty of grid point positioning. By mimicking the shape as well as atomic details of potential molecular binders, this new beta-cluster representation can not only be used for pocket characterization, but also allows for direct pocket-ligand shape comparison and can be used for ligand optimization as well as pocket-guided docking. Furthermore, we defined the BScore, which is the maximum docking score of beta-clusters, as an indicator of pocket ligandability, and developed an ensemble beta-cluster approach which allows one-to-one pocket mapping among aligned protein structures. To illustrate applicabilities of beta-cluster, we apply our new representation of concave surfaces to diverse problems including PPI interfaces, traditional binding sites and whole biomolecular surfaces.
II. METHOD
A. Beta-cluster representation of pockets
In order to generate a pseudomolecular representation of fragment-centric pockets detected by AlphaSpace with better connectivity properties, we cluster alpha-atoms using their pairwise distances and the complete linkage method48 in the hierarchical clustering implementation of Scipy package49 with a distance cut-off of 1.6 Å, a typical aliphatic carbon-carbon bond distance, to eliminate unphysical and extremely close-contact alpha atoms while retaining its overall shape. The complete linkage method generates spherical clusters and each cluster centroid is represented as a beta-atom. The combination of all beta-atoms in a given fragment-centric pocket forms a beta-cluster, which is the pocket’s pseudomolecular representation, as illustrated in Fig 1B. The alpha-space value for each alpha atom in the cluster is summed to obtain the alpha-space of the beta-cluster. Other beta-cluster attributes, including the solvent accessible surface area and the volume, are calculated the same as for a real molecule with a van der Waals radius of 1.6 Å for each beta atom.
B. Beta Cluster Score (BScore).
With beta-cluster mimicking potential molecular binder of a fragment-centric pocket detected by AlphaSpace, we define beta-score, a maximum docking score of the beta-cluster, as an indicator of pocket ligandability. Based on the AutoDock Vina scoring function50, a probe atom corresponding to various Vina atom types (C, N, O, F, P, S, Cl, Br, I) is placed at each beta atom location and AutoDock Vina score is calculated for that probe atom. The beta atom type and score are determined by the probe type with the best vina score (highest affinity). Beta atom types can also be generally classified as polar/non-polar and hydrogen bond donors/acceptors based on the strength of the Vina interaction terms. Finally, the BScore is obtained by summing the best vina score of each beta atom in the beta cluster, which provides an estimate of pocket ligandability.
C. Pocket mapping with aligned protein structures.
Given several aligned protein structures and their determined beta-atoms, consistent pocket mapping among different structures can be achieved by determining ensemble pockets. First, several proteins are aligned together, and each one is processed separately to generate corresponding beta atoms along concave surfaces. Second, all determined beta atoms from different structures are superimposed based on structural alignment and re-clustered altogether by hierarchical clustering using the average linkage method51. This places spatially close beta-atoms from different protein structures into an ensemble beta-cluster. After the clustering step, each individual beta atom in ensemble beta-clusters is re-mapped to its corresponding protein structure. Statistical properties such as the mean, median, or variance of the distribution pocket attributes can be calculated for the ensemble beta-clusters adding more information for the analysis of concave surfaces. A collection of individual pockets coming from the same ensemble beta-cluster are considered to be matched and forms an ensemble pocket. One advantage of ensemble pockets over our previous pocket matching method is the use of structural alignment of the protein to generate consistent pocket definitions that are less sensitive to minor structural changes on the protein surface. Because the alignment is done before the clustering step, it allows pocket-mapping among homologous proteins that share a similar protein fold.
In this work where we apply the ensemble pocket approach to identify the same pockets across different surface states or different homologous proteins, we used the Matchmaker52 program implemented in Chimera53. Matchmaker allows for the superimposition of protein structures using their global sequence alignment or a user-specified alignment based on select residues from the target structures. Restricting the alignment to active site or interface residues allows for consistent matching of surface pockets that are less affected by large conformational changes observed for multidomain proteins. We employed a two-step process to superimpose the protein structures: an initial superimposition was performed using the global sequence alignment, if the global RMSD of the aligned structures fell below 1.5 Å then the structure pairs were considered sufficiently aligned for processing of the ensemble pockets; then for structure pairs that have RMSD >1.5 Å , a second superimposition step was performed using residues selected to be within 5.0 Å from the largest ligand-bound structure in the ensembles to be analyzed. Finally, visual inspection of beta atoms on their respective protein surface was performed to ensure alignment quality.
D. Beta-Cluster and Ligand Features.
Since beta-cluster is sufficiently molecule-like, it allows the direct comparison between beta-cluster and ligand. Meanwhile, a variety of features can be easily calculated for both beta-clusters and ligands, as listed in Table 1, which include geometric features like surface area and volume, as well as chemoinformatic features like those from ChemoPy54. To show the “ligand-like” properties of the beta clusters, we also calculate shape similarity features using the distribution of moments with the USR formulation55–57.
Table 1.
Selected features and their descriptions available for beta-clusters and ligands.
| Feature | Beta -Cluster/Ligand only? | Description | |
|---|---|---|---|
| 1 | Alpha-Space | Beta-Cluster Only | Sum of alpha-space associated with all alpha atoms in the beta-cluster |
| 2 | BScore | Beta-Cluster Only | Sum of the maximum affinity scores for the beta atoms using AutoDock Vina50 atom probes |
| 3 | Occupancy | Beta Cluster Only | Overlapping volume of ligand and beta-cluster divided by total volume of beta-cluster |
| 4 | Coverage | Ligand Only | Overlapping volume of ligand and beta-cluster divided by total volume of ligand |
| 5 | Tanimoto Overlap | Both | Grid-based similarity measure of the overlap between the Beta cluster and Ligands58 |
| 6 | Volume | Both | Grid-based van der Waals volume of the beta clusters with a radius of 1.6 Å for each beta atom |
| 7 | Accessible Surface Area (ASA) | Both | Accessible surface area defined by Grant et al59 with a radius of 1.6 Å for each beta atom |
| 8 | Occluded ASA | Both | Difference in the ASA of covered and non-covered protein lining atoms with pharmacophoric atom types60 |
| 9 | Exposed ASA | Both | Solvent exposed ASA of molecule in complex with the protein |
| 10 | USR | Both | Shape similarity features55–57 based on the statistical moments of pairwise distance distribution |
E. Datasets
The small-molecule dataset was constructed based on the protein-ligand complexes retrieved from the 2013 release of the scPDB data sets61. First we screened the scPDB 2013 release and removed structures that did not have any binding data according to the 2016 release of the PDBbind refined set62, which reduces the number of protein-ligand structures from ~10,000 to 996. The scPDB/PDBbind refined set was further reduced by aggregating all protein family structures using their UNIPROT accession number. The protein-ligand complex structure corresponding to the highest affinity ligand from PDBBind was selected as the representative structure for that UNIPROT group. This non-redundant dataset is called the small-molecule dataset and is composed of 293 structures with properties listed in SI. This data set has been used to determine the clustering parameters for beta-cluster and to investigate correlations between the properties of the ligand and the binding site as represented by the beta-cluster.
To demonstrate applicability of beta-cluster representation, we applied our beta-cluster approach to various data sets that are relevant to fragment-based drug-design (FBDD) and identification of cryptic pockets. The Ichihara Set63 was used to investigate the ability of beta-clusters to make meaningful suggestions for FBDD, which consists of 21 lead/fragment pairs of structures where a lead ligand retains its fragment-binding pose. We applied BScore and pocket matching on a reduced set of the latest release of the 2P2I dataset64 in which, only the structure of the maximum affinity small-molecule PPI inhibitor is analyzed for each PPI interface. The CryptoSite database65 was analyzed to demonstrate the pocket matching capabilities of beta clusters across multiple states of the same protein. The CryptoSite database consists of 79 pairs of unbound (apo) and ligand bound (holo) structures of the same protein selected for their structural and functional diversity. Calmodulin and Ca2+ ATPase 1 were excluded due to the lack of a good structural alignment between the apo and holo pairs while Kynurenine aminotransferase II and Serum transferrin were excluded due to the small size and charged nature of their cognate ligands. This leaves 75 pairs of structures in the CryptoSite dataset for our analysis.
III. RESULTS and DISCUSSION
In this section, we present results to establish three new features of AlphaSpace2.0: beta-cluster as a pseudomolecular representation of detected pockets, beta-score as an indicator of ligandability, and pocket mapping/comparison with aligned biomolecular structures. The applicability of these new features is demonstrated by applying them to a diverse collection of datasets that are closely related to important problems in structure-based inhibitor design. Finally, we use two important classes of PPI related disease targets; MDM2/MDM4 and Bcl-xL/Bcl-2, to retrospectively illustrate how AlphaSpace 2.0 can be used to facilitate the discovery of more potent and specific PPI inhibitors.
Beta-clusters Mimic the Shape of Binding-Site Ligands
To establish the pseudomolecular nature of beta-cluster, we determined shape similarities and property correlations of beta clusters against their cognate ligand partners for the high-quality, small-molecule dataset which consists of 293 protein-ligand complexes. The results in Fig 2. show that there are good correlations between ligand and beta cluster features, including volume, surface area and span. This clearly demonstrates the ability of beta clusters to mimic the shape of potential molecular binders. Meanwhile, one basic assumption in rational inhibitor design is that similar binding sites would interact with similar ligands. Thus we are motivated to directly examine correlations between ligand similarities and beta-cluster similarities using two similarity measures: one is USR similarity which is a shape similarity based on statistical moments of pairwise distance distribution derived from the USR features56, 57; the other is OASA similarity which is based on the total and pharmacophore-atom types of the occluded surface area of the protein surface60 encoded into a feature fingerprint. Both are alignment independent measures, which allow for structural patterns to be compared without the need for computationally exhaustive methods like clique detection or geometric hashing. The results in Figure 3 show the correlation of the similarity features with an R of 0.74 for the USR similarities and 0.88 for the OASA fingerprint. Non-contacting beta atoms and ligand atoms contribute to the noise and reduce the shape similarity for the USR features while the OASA fingerprints suffer from less noise due to common occluded surface area for both beta clusters and ligands. Fig. S1 illustrates two examples how highly similar ligands bind to pockets represented by highly similar beta clusters. Only considering overlapped beta atoms and ligand atoms greatly increases the correlation of USR similarity with an R of 0.94 while the OASA similarity fingerprint also increases to 0.97, which indicates that beta clusters can mimic important pocket-occupying portions of the ligand to a high degree.
Figure 2.

The correlation between various molecular features of cognate ligands against their corresponding beta clusters. (A) Volume R=0.84, (B) Solvent Accessible Surface Area (ASA) R=0.84, (C) Occluded Surface Area (Occl. ASA) R=0.92, and (D) Span R=0.74. For all molecular features, beta clusters tend to be greater in magnitude due to the larger size and extent of the concave surface compared to the cognate ligand. The line of best fit is shown as the black solid line to show the quality of the linear fit for both the ligand and beta cluster features. To capture the variance within the correlation plots, +/− one unit of the standard deviation of the ligand features from the line of best fit is plotted as dashed black lines. Only considering overlapped ligand and beta clusters, R-values can increase to 0.96, 0.98, 0.98 and 0.96 for the volume, ASA, occluded ASA and molecular span respectively.
Figure 3.
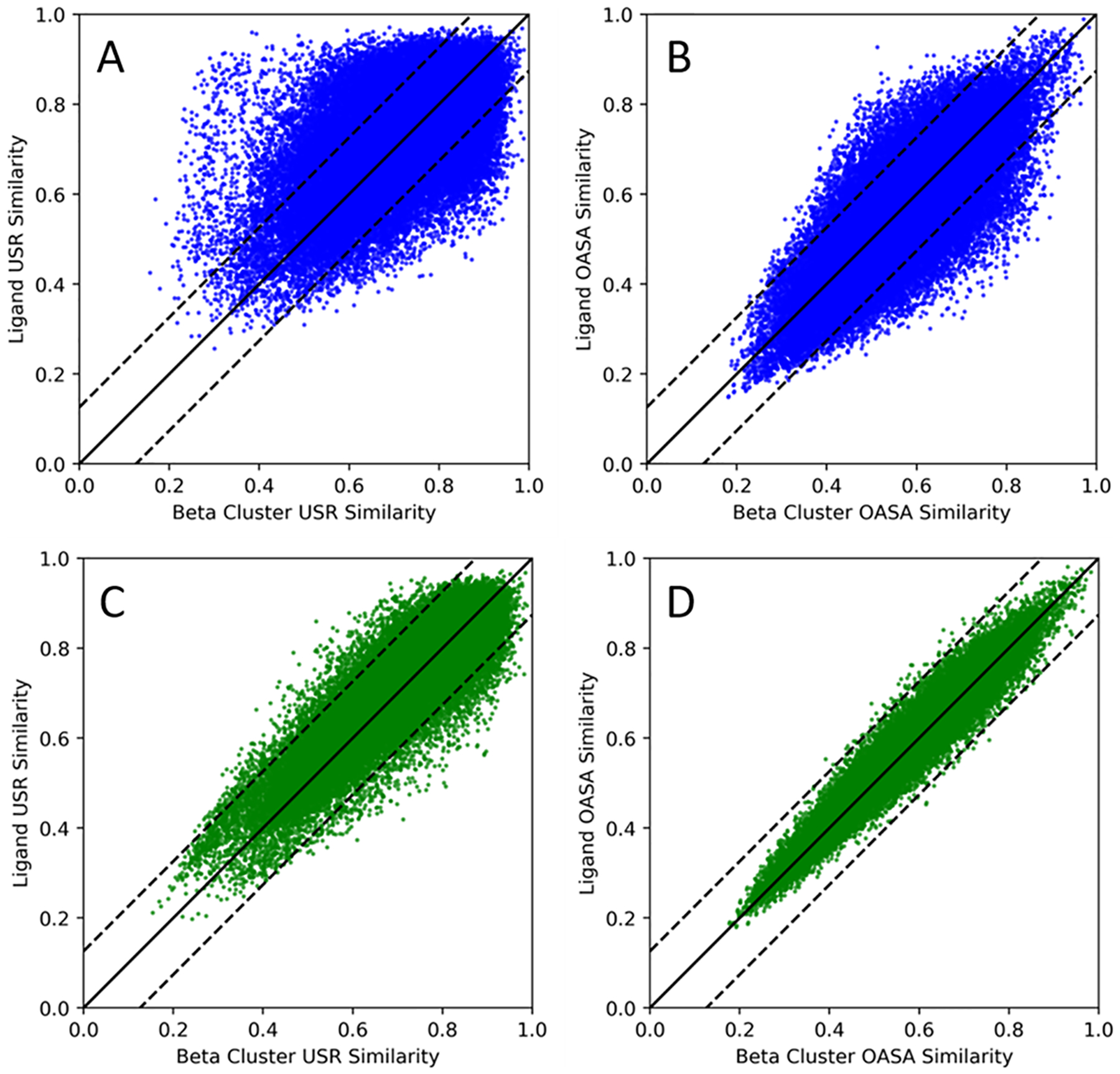
Similarity scores of ligands correlated against the same similarity scores of the pseudomolecular beta clusters. (A) and (B) show the correlation of the USR (R=0.74) and the Occluded ASA pharmacophore fingerprint (R=0.88) similarities respectively for the full beta clusters. (C) and (D) plot the same correlation properties but for overlapped portions of ligands and beta clusters with an improvement in the R-value of 0.94 for the USR and 0.97 for the Occluded ASA similarities. The y=x line is also plotted as the black solid line to show the quality of the linear fit for both the ligand and beta cluster properties. To capture the variance within the y=x plot, +/− one unit of the standard deviation of the ligand similarities from the y=x is plotted as dashed black lines.
Using Beta-cluster Score (BScore) to Estimate Pocket Ligandability
Besides quantifying geometric similarity between beta-clusters and ligands, we have analyzed their energetic similarity using the AutoDock Vina Scoring function by plotting the ligand vina score against the corresponding contact pockets’ BScore. The results in Fig. 4a show that BScore tends to be more negative than the calculated vina score for the ligand among all structures in the small-molecule dataset except for 1QK4. This is not surprising since the contact pockets tend to have unoccupied beta atoms that would otherwise contribute to favorable protein-ligand interactions. Meanwhile, since the overall shape of beta clusters is derived from the Voronoi tessellation of the concave binding site, they naturally represent a maximum possible pseudomolecule packing for that site. Thus, it would be reasonable to consider BScore as an estimate of pocket ligandability. Ligands that have almost complete beta cluster coverage tend to fall closer to the diagonal line indicating that these ligands are well optimized energetically to their binding sites. On the other hand, ligands that have large Vina Score affinity differences with their beta clusters tend to have low ligand coverage of the binding site and can possibly be further optimized by maximizing the overlap of the ligand and the beta cluster as shown in the case of 3ISS in Fig 4b. The individual energetics of the beta atoms also reveals characteristics of localized sites that can facilitate rational ligand design.
Figure 4.

(A) The correlation of the Ligand AutoDock Vina score plotted against the AutoDock Vina based BScore for the beta clusters. The y=x line is plotted as a solid black line. For the majority of the beta cluster/ligand score pairs, the beta clusters have a lower (more favorable) score due to the larger size of the concave surface compared to the ligand. (B) shows two extreme examples where the ligand has a slightly more favorable score compared to its corresponding beta clusters (1QK4) and where the beta cluster is much more favorable than the ligand (3ISS). In both examples, contact beta atoms are colored green while non-contact beta atoms are colored red.
Detected Pocket Communities Represent Potential Binding Sites
We used BScore and alpha space volume to rank detected pocket communities for the small-molecule dataset. Table 2 shows the performance of the community ranking in predicting the small-molecule binding site as highest ranking communities. Top 1 ranking communities based on the alpha space volume and the BScore were predicted to be the small-molecule binding site of the cognate ligand with a success rate of >78%. We observe that all small molecule binding sites can be detected by AlphaSpace as one pocket community. Top 3 ranking communities include the small-molecule binding site for any criteria with a success rate of >95%. The last entry in Table 2 shows the success rate by selecting top ranking communities based on either the alpha-space or the BScore criteria. By using the union of top ranking communities from each criteria , the top 1 success rate improves to 87% while the top 3 success rate improves to 98%. These results further confirm that AlphaSpace can be a useful tool in detecting potential binding sites, which allows the prioritization of high-ranking communities for subsequent studies.
Table 2.
Performance of the beta cluster features of pocket communities in identifying binding sites. The success rate and absolute number of identified sites in parenthesis are shown.
| Success Rate (%) of Identifying Binding Site Community by Ranking | |||
|---|---|---|---|
| N=293 | Top 1 | Top 3 | All Communities |
| alpha-space | 78.8 (231) | 96.2 (282) | 100 (293) |
| Bscore | 79.9 (234) | 95.9 (281) | 100 (293) |
| alpha-space or BScore | 87.0 (255) | 98.3 (288) | 100 (293) |
It should be noted that high or mid ranking communities that do not overlap with the binding site ligands may have other functional roles such as allosteric binding sites66 or protein-protein/peptide interfaces. Cyclin-dependent kinase 2 (CDK2) provides an excellent example to illustrate how AlphaSpace and BScore can be used to identify multiple functionally important binding interfaces for a single protein. Based on the CDK2 monomer structure obtained from the CDK2/Cyclin A complex67, in which CDK2 is bound to ATP and a short substrate peptide, we carried out AlphaSpace analysis and detected 16 different pocket communities, as shown in Fig. 5, and their properties are listed in Table 3. We can see that the ATP binding site associated community (community 1) is ranked at the top in terms of both the BScore and alpha-space, followed by one of the substrate binding communities (community 2). Among 13 remaining detected communities, by analyzing other available CDK2 and related CDK crystal structures, we find that 6 of these are indeed involved in binding other proteins, peptides, and even fragments as shown in Table 3 and Fig 6. For example, important binding residues from the p27 peptide inhibitor68 are found to overlap with the 5th ranking community. Adjacent to this community is a low-ranking community that binds a conserved helix-loop found in all cyclins. The 4th and 9th ranking communities formed the cavity in the C-lobe and A-loop region, which is found to bind loops and helices from Cyclin A67 and Cyclin E69 that may stabilize the open A-loop conformation of the active state of CDK2. Alignment of the CDK1/CKS270 complex to CDK2 shows that the 13th community belongs to the binding site for this regulatory protein. Lastly, a high-scoring fragment-binding site, the 7th community, is found at the solvent exposed region of the C-lobe; a largely uncharacterized region of kinases that may have some functional importance as an allosteric site based on its ability to bind small molecules as shown in multiple crystal structures71–73. In total, 9 out of the 16 detected communities are confirmed to be involved in true binding sites which indicates that the AlphaSpace community definition is sufficiently sensitive for potential binding site identification.
Figure 5.
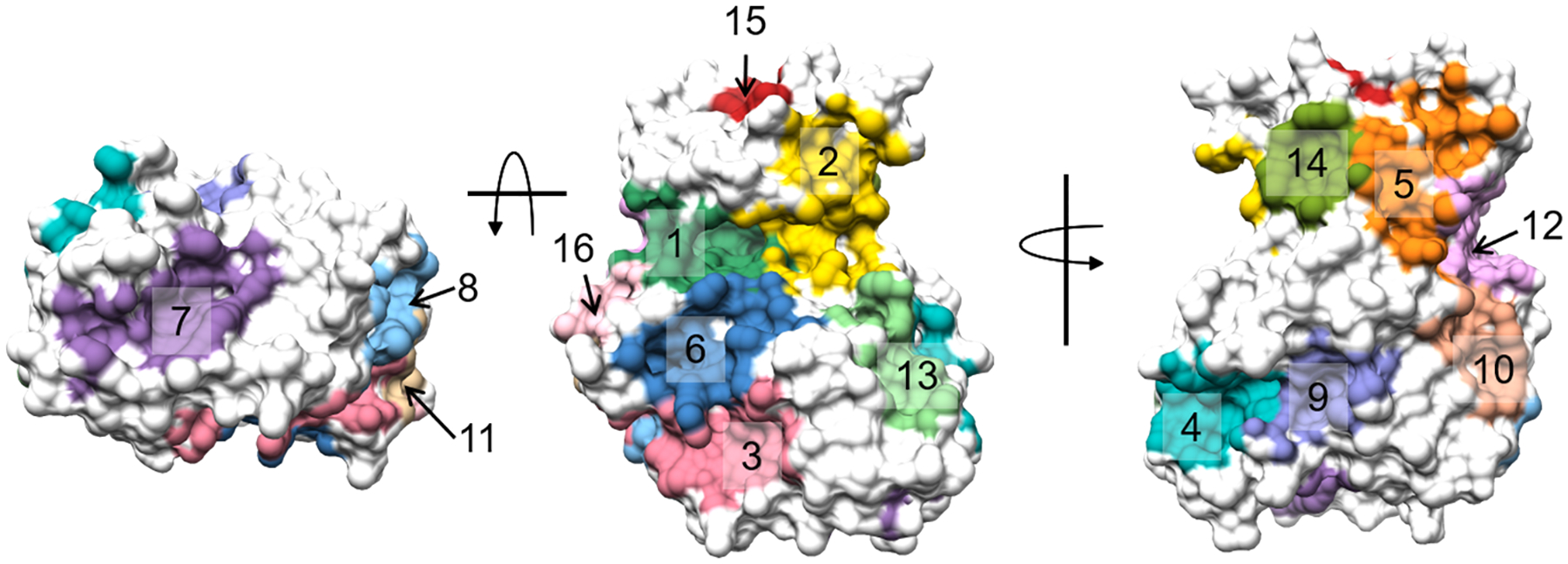
Beta Cluster communities for the active state, CDK2 monomer. Each community is labeled from 1 to 16 colored accordingly. Details for the community features are shown in Table 3.
Table 3.
Detected Communities and their features for AlphaSpace mapping of CDK2 shown in Fig 5. Binding partners detected from other CDK2 or related CDK proteins are listed in the “Binding Partners” column.
| Community Index | Binding Partners | Color | alpha-space (Å3) | BScore (kcal/mol) | Volume (Å3) | % Occupied |
|---|---|---|---|---|---|---|
| 1 | ATP | Green | 1470 | −28.6 | 878 | 30 |
| 2 | Substrate (P,P+1,P+4) | Yellow | 1155 | −18.7 | 757 | 21 |
| 3 | None | Pink | 889 | −15.3 | 622 | - |
| 4 | Cyclin A, Cyclin E | Teal | 886 | −19.8 | 732 | 14 |
| 5 | p27 Inhibitory Protein | Orange | 659 | −13.6 | 547 | - |
| 6 | Substrate (P-2,P-3) | Blue | 647 | −15.5 | 509 | 14 |
| 7 | Fragment binding Site | Purple | 457 | −10.3 | 414 | 30 |
| 8 | None | Lt Blue | 406 | −9.6 | 258 | - |
| 9 | Cyclin A, Cyclin E | Peri | 350 | −5.4 | 219 | 54 |
| 10 | None | Peach | 335 | −6.7 | 247 | - |
| 11 | None | Tan | 286 | −5.5 | 185 | - |
| 12 | None | Plum | 283 | −7.3 | 351 | - |
| 13 | CKS2 Regulatory Protein | Lt Green | 260 | −6.9 | 242 | 37 |
| 14 | Cyclin A, Cyclin E | Olive | 173 | −5.7 | 212 | 64 |
| 15 | None | Red | 131 | −3.4 | 120 | - |
| 16 | None | Lt Pink | 111 | −2.0 | 77 | - |
Figure 6.
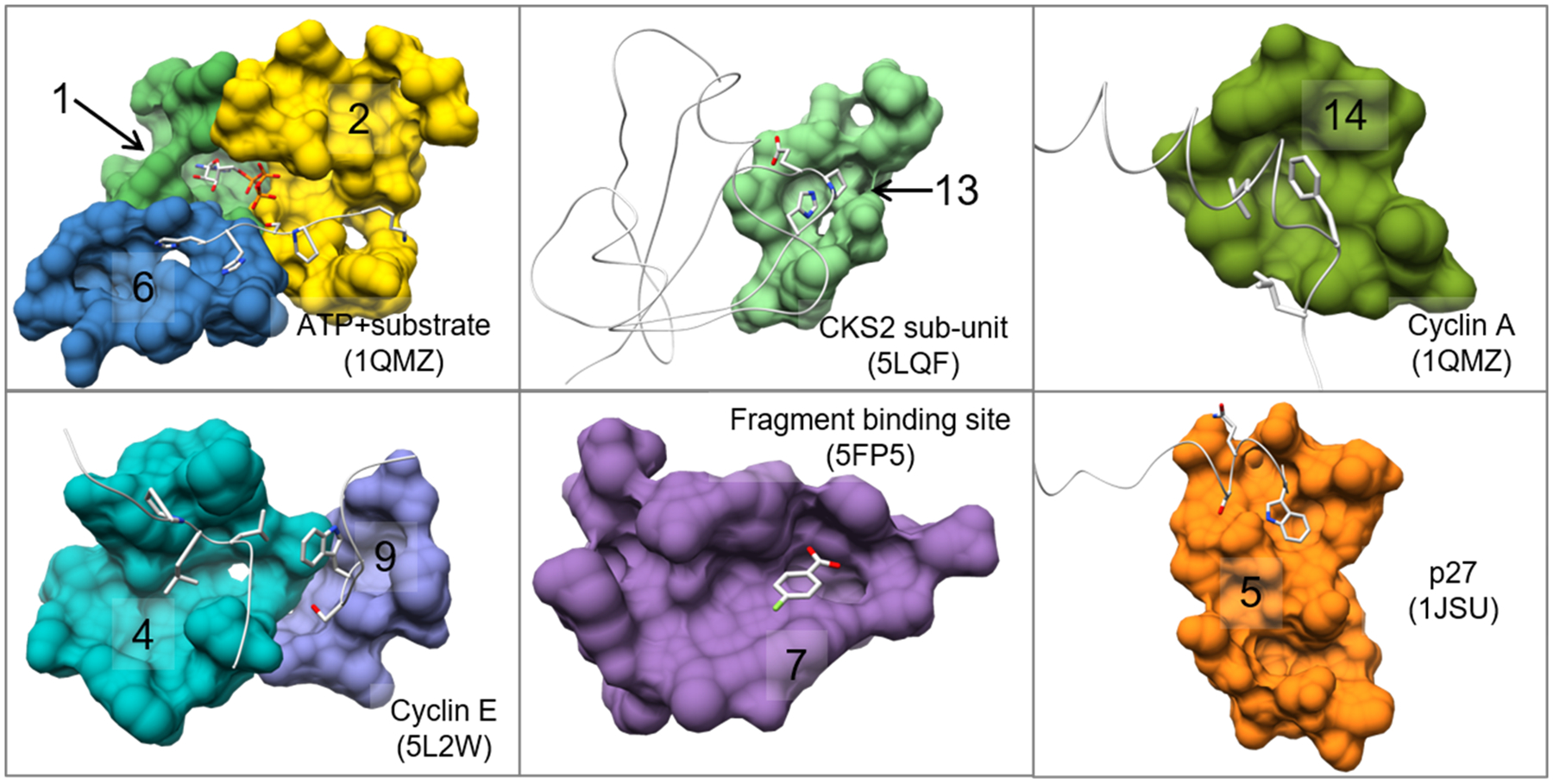
Various communities from the global surface mapping of CDK2 identified as binding sites from the alignment and annotation of similar or homologous proteins. Each community is colored and labeled according to the scheme in Table 3. The aligned binding partners are shown along with their corresponding PDB ID and partner name.
Beta-Cluster can Guide Ligand Optimization
The results for the retrospective analysis of Fragment to Lead pairs from the Ichihara set are listed in Table 4. The Ichihara Set63 is composed of 21 pairs of ligand bound structures covering various important drug targets. For each pair, its fragment member forms the “core” scaffold of the ``lead” and both members have the similar binding mode. Here we employ this dataset to demonstrate how beta-cluster can be used to help guide ligand optimization. For 16 out of 21 systems, the highest-ranking community is the small-molecule binding site with BScore ranging from −15.5 kcal/mol for DPP to −42.8 kcal/mol for PPAR. The large affinity of PPAR is not surprising considering that this target is known to be highly druggable with several contiguous pockets in the binding site that determine the binding mode and activity of PPAR modulators. For 19 out of 21 structures the small-molecule binding site is found among the top 2 ranking communities, thus the community ranking demonstrates the ability of beta clusters to detect and enrich large surface regions of interests which can be useful for focused, high-throughput studies involving only ligandable sites. Within each binding community, the sub-pocket partitioning of AlphaSpace allows for the identification of localized interaction sites that is the preferred binding site for the initial fragment. These core pockets are akin to hot-spots found in PPIs and function as “seeds” for the fragment-based drug design process. Assigning core pockets allows for finding complimentary ligand shapes and pharmacophoric interactions that can maximize initial fragment binding which would carry into an elaborated lead molecule. Out of the 21 systems, only p38 and PKB have core binding community pockets not in the top 1 rank. Nonetheless, all 21 fragment associated core pockets are in the top 2 indicating the ability of BScore to identify the most important pockets for fragment binding. Using BScore ranking information at the global and local level, beta clusters can be used as a prospective tool for the judicious selection of ligandable communities and preferred fragment-targetable pockets within the community.
Table 4.
Ranking and BScores (in kcal/mol) of beta cluster communities of the binding sites of the Fragment structures listed in the Ichihara set. The local rank and BScores of the core pockets are also shown. Core pockets are defined as beta cluster pockets that have extensive contacts with the fragment ligand.
| Ichihara ID | Fragment PDB | Lead PDB | Binding Site Rank (BScore) | Core Pocket Rank (BScore) |
|---|---|---|---|---|
| Aurora | 2W1D | 2W1G | 1 (−26.3) | 1 (−5.9) |
| BACE 1 | 2OHM | 2OHU | 1 (−28.6) | 1 (−4.1) |
| BACE 2 | 2V00 | 2VA7 | 1 (−39.8) | 1 (−5.3) |
| Biotin Cbx | 2W70 | 2W71 | 1 (−40.2) | 1 (−5.2) |
| CDK2 1 | 1VYZ | 1VYW | 1 (−26.5) | 1 (−6.7) |
| CDK2 2 | 2VTA | 2VU3 | 2 (−21.3) | 1 (−6.5) |
| DPP | 3CCB | 3CCC | 10 (−15.5) | 1 (−6.9) |
| HSP90 1 | 2QFO | 2QG0 | 1 (−25.9) | 1 (−5.0) |
| HSP90 2 | 3FT5 | 3FT8 | 1 (−22.6) | 1 (−7.4) |
| HSP90 3 | 2WI2 | 2WI7 | 1 (−19.6) | 1 (−5.9) |
| NS5 prase | 3CIZ | 3CJ5 | 4 (−22.1) | 1 (−4.6) |
| PDE | 1Y2B | 1Y2K | 1 (−33.8) | 1 (−8.0) |
| PKB 1 | 2UW3 | 2UW7 | 1 (−34.6) | 1 (−7.4) |
| PKB 2 | 2UVX | 2VO6 | 1 (−33.4) | 2 (−6.0) |
| PPAR | 3ET0 | 3ET3 | 1 (−42.8) | 1 (−7.3) |
| Jak2 | 3E62 | 3E64 | 1 (−34.7) | 1 (−6.1) |
| Lta4H | 3FU0 | 3FH7 | 1 (−38.9) | 1 (−10.1) |
| p38a 1 | 1W84 | 1WBT | 3 (−26.8) | 2 (−3.9) |
| p38a 2 | 1W7H | 1W83 | 1 (−45.2) | 1 (−7.0) |
| Pantho synthase | 3IMG | 3IUE | 1 (−42.6) | 1 (−7.3) |
| uPA | 2VIN | 2VIW | 2 (−22.1) | 1 (−5.4) |
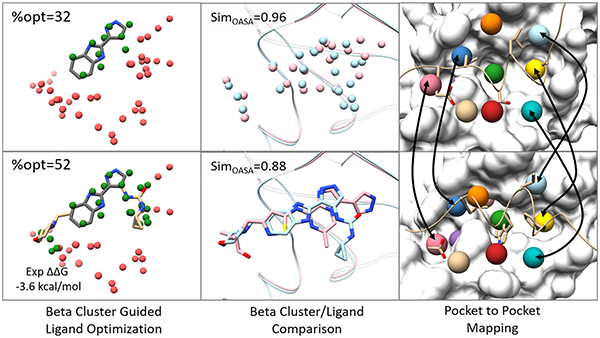
We also apply the concept of optimizability of the binding to the Ichihara dataset, i.e. %optimized, which is calculated as the ratio between the BScore of contact beta atoms within 1.8 Å of any ligand atoms and sum of the BScore of all the beta atoms of the binding site associated community. The higher the value of %optimized indicates the greater overlap between ligand and beta-cluster, i.e., the higher pocket-ligand complementarity. On the other hand, a lower %optimized value suggests that there is more room for further inhibitor optimization. Fig. 7A shows the %optimized for each fragment/lead pair in the dataset while Fig 7B shows the relationship between the change in the %optimized scores with the normalized experimental affinities in going from the fragment to the lead structure. For all systems investigated in the Ichihara set, an improvement in the %optimized score of the fragment to the lead is correlated with an improvement in the affinity as measured by the pIC50 values compiled by Ferenczy and Keseru74.
Figure 7.

Beta cluster complementarity and optimizability for the lead/fragment pairs in the Ichihara set. The %optimized score of the beta clusters improves in going from the fragment to the lead structures for all systems indicating better complementarity of the lead structures for the binding site. (B) Ligand Affinity (pIC50) vs the %optimized Score for all systems in the Ichihara set with lines connecting the fragment (red) to the lead (green) structures.
To illustrate how AlphaSpace2 would be used for ligand optimization, we performed a more detailed retrospective analysis of the PDE and jak2 fragment/lead pairs. Fig 8 shows the overlay of the binding site communities for PDE and jak2 with the fragment and lead pairs shown along with other high-affinity ligands. For both systems, the initial fragment is observed to partially occupy several high-scoring pockets, with neighboring unoccupied pockets highlighted in orange which would be preferable sites for elaboration. For PDE, a nitrophenyl group was used to target this pocket which improves the affinity of the elaborated ligand from 82 μM to 21 nM75. For jak2, the indazole core fragment was grown with a phenyl moiety to target this neighboring pocket, which increased the affinity from 40.9 μM to 1.6 μM76. After this first iteration, another unoccupied pocket is still accessible for further fragment extension, which was subsequently utilized by the final lead compound with a butylsulfonamide moiety. This leads to a potent inhibitor with an affinity of 78 nM. We can see that the fragment-centric pocket representation can clearly indicate those preferred sites for further fragment extension. Considering that there remain unoccupied pockets in PDE and jak2 for the lead member in the Ichihara Set63 , we have searched the PDBBind database and literature for other ligands that can possibly target remaining unoccupied pockets in PDE and jak2 to further improve potency, as shown in Fig. 8. For the PDE lead, the original authors have already performed a QSAR study77 for the initial carboxyl pyrazole scaffold with R1=Phenyl, R2=−4-Chloro-phenyl, R3=-H. This particular substitution extends into a mid-scoring pocket and has the second highest potency amongst the first-round optimizations (150 nM compared to 31 nM) but was unfortunately not further optimized. Comparison with the ligand from PDB ID: 4WCU indicates that moieties from R2 can be extended to interact with this pocket and thus possibly increase the potency for this lead78. Likewise, a phenyl fragment from the ligand in PDB ID: 5K1I extends into this partially occupied pocket, while a benzoic acid like fragment extends into another high-scoring pocket greatly contributing to ligand binding affinity79. Jak2 appears to be further optimizable based on the presence of two nearby high-scoring pockets close to the E1 and gatekeeper regions of the kinase binding site. This can be observed by the overlay with the ligand of PDB ID: 4JI9, in which its methyl-piperazine moiety extends into the unoccupied E1 pocket80. Similarly, a fluoro-hydroxy phenyl fragment of the ligand from PDB ID: 5TQ8 points towards the high-scoring pocket associated with the gatekeeper site81. Both unoccupied pockets can be easily elaborated from the aminoindazole core of the selected Jak2 lead molecule which is a shared scaffold of the ligand in 5TQ8. Finally, we also extended our community based analysis to other systems in the Ichihara set that had an excess of >5 pockets that would be ideal candidates for further elaboration of either the initial fragment or the more potent lead ligands. Fig. S2 shows the overlay of these selected systems with the potency of the leads from the Ichihara set as well as more elaborated ligands from the PDB that co-target nearby unoccupied pockets. We can see that the more advanced ligands not only occupy lead-molecule binding pockets but also display extensive overlaps to nearby, high-scoring, unoccupied pockets suggesting that AlphaSpace and beta clusters not only identify preferred sites of initial fragment binding but can also be used to guide the optimization of lower affinity fragments into more potent lead molecules.
Figure 8.
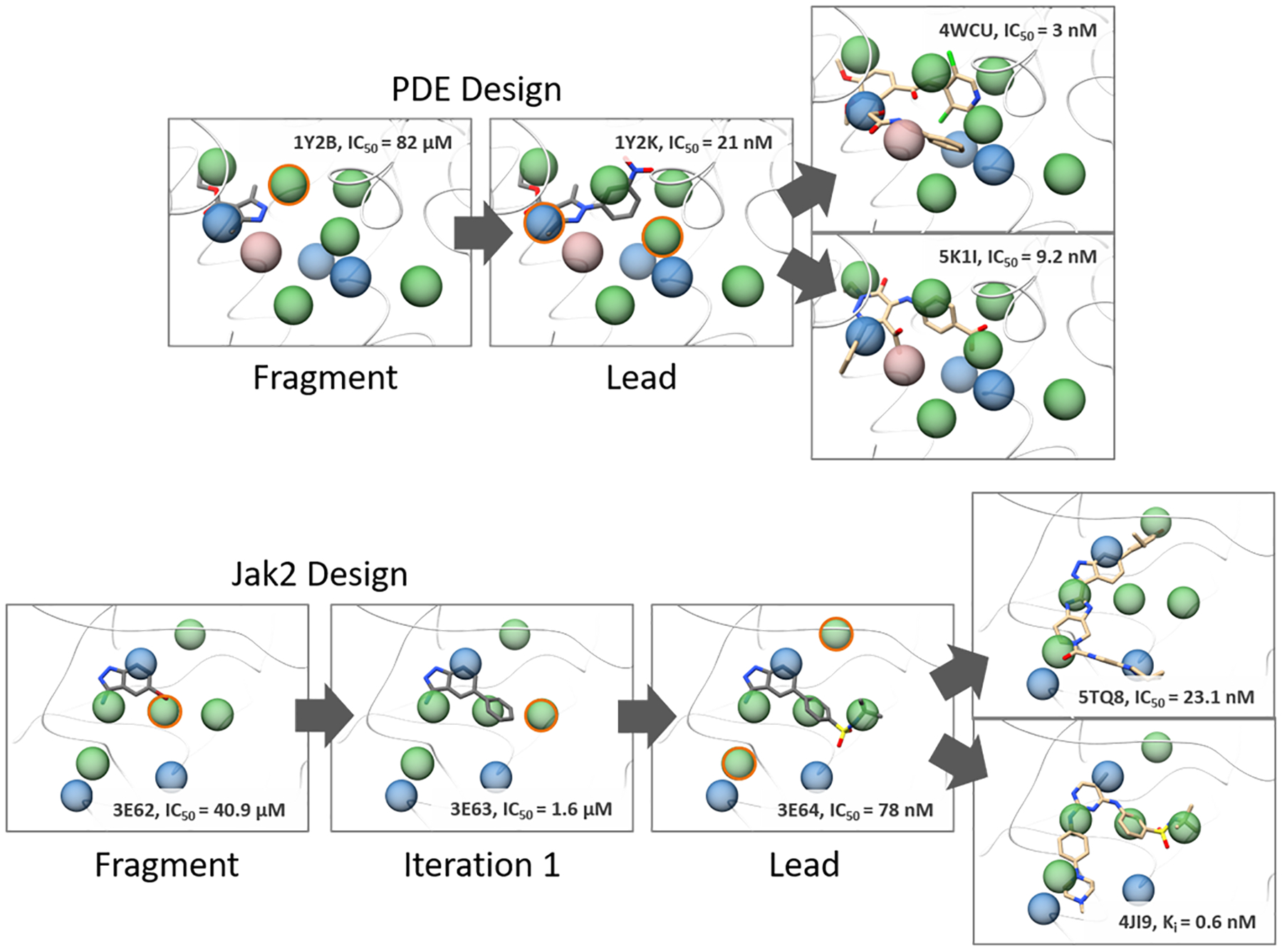
Illustrated pocket-guided optimization for the lead molecules of the PDE and Jak2 systems in the Ichihara set. Multiple, high and mid-scoring pockets were identified for the binding site communities that had limited overlap with the original lead molecule. Lead molecules from the Ichihara set are shown and colored dark grey while more potent binders found in the PDB that target these alternative pockets are shown in tan. High (BScores <−2.5 kcal/mol), mid (−2.5 kcal/mol >= BScores < −1.5 kcal/mol), and low (BScores >= −1.5 kcal/mol) scoring pockets are colored green, blue, and rosy brown respectively. Unoccupied pockets that overlap extensively with the elaborated fragments of the more potent ligands are highlighted in orange.
Analysis of Protein-Protein Interactions using Beta Clusters
Pocket matching was performed for the apo, inhibitor-bound (iPPI), and holo-PPI complexes of the 2P2I set to determine how certain pocket structures are conserved in various states of the PPI. All of the iPPI associated matched ensembled pockets were included in the analysis across the different surface states of the 2P2I set. Fig 9 shows BScores of pockets for various proteins in the 2P2I. Bcl-2 and Bcl-xL have the best BScore for iPPI pockets consistent with the wide availability of high-affinity inhibitors for these systems. On the other hand, TNFa has the worst BScore for its iPPI pockets followed by H-Ras and K-Ras, all of which are targets that have been well known to be challenging for inhibitor design. The variability in BScore among different surface states indicates the effect of protein plasticity on ligandability of iPPI binding sites. For 20 out of 21 systems, BScore difference among apo, iPPI, and PPI beta clusters is less than 8 kcal/mol indicating that “hot spot” regions are conserved for various states of the PPI interface. Only Bcl-xL shows a drastic BScore difference between apo and iPPI/PPI bound states, nonetheless the apo state of Bcl-xL may be still more ligandable than some other PPIs. It should be noted that other authors have commented on the flexibility of Bcl-2 family proteins and how ligands can induce different conformations from the apo state, leading to the formation of more druggable, fragment-centric cavities82, 83.
Figure 9.

Comparisons of the beta cluster ligandabilities as measured by the BScores (kcal/mol) for the interface region of the 2P2I set for different surface states. For most systems, the BScores across different states are within +- 8.0 kcal/mol of each other. Only Bcl-xL, which has a large structural change for the apo state compared to the bound iPPI and PPI states shows a large BScore difference of more than 8.0 kcal/mol.
Meanwhile, we examined complementarity of the ligand partners for the iPPI and PPI associated pockets, as shown in Fig. 10. Only 2 out of 21 systems with %optimized value higher for the PPI structures than for the iPPI structures indicates that, at least for those iPPI associated pockets, small-molecule ligands have been optimized to have higher complementarity for the binding site to some extent. The better complementarity for iPPI structures is not surprising considering that small-molecules take advantage of shape moieties not available to natural amino-acids. On the other hand, shallow binding sites such as in cIAP1, XIAP, and ZIPa can only accommodate similarly flat molecular shapes thus showing similar %optimized values for iPPI and PPI structures.
Figure 10.
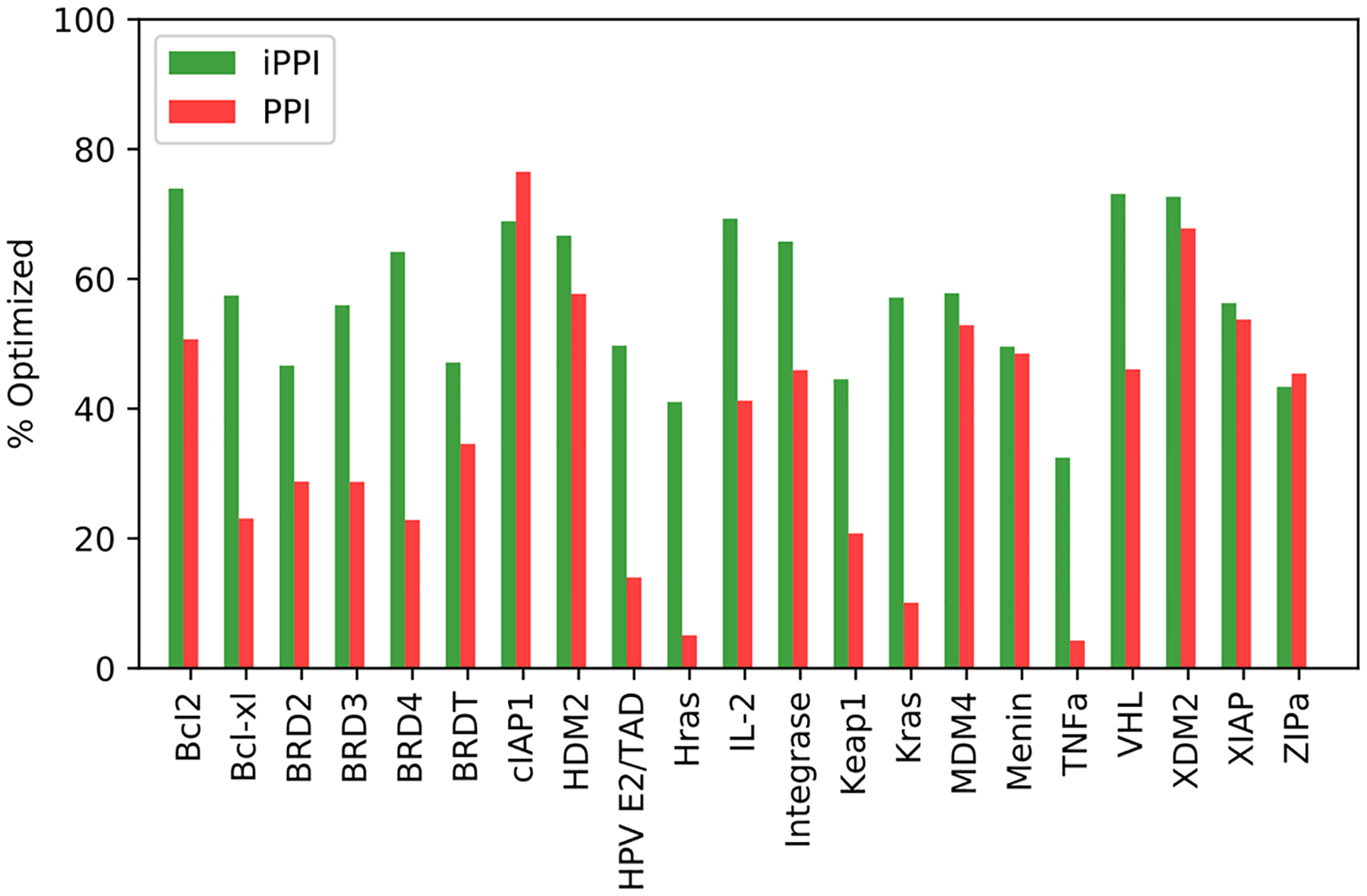
Comparison of the %optimized scores for the iPPI and PPI bound states of the interface regions of the 2P2I. For 19 out of 21 systems, the iPPI bound structures achieve a higher %optimized score due to the ability of non-natural small molecules to interact with concave areas of the interface that are not accessible by the native PPI partner.
Pocket Comparison for the CrypticSite Dataset
One-to-one pocket matching was performed for apo/holo pairs of the CryptoSite dataset and BScores of binding site pockets were calculated and compared as shown in Fig 11A. As expected, the apo state of the proteins tends to be less ligandable than the holo state, with the mean BScore of −12.1 kcal/mol for the apo state versus −18.8 kcal/mol for the holo state. In total, 72 proteins have improved holo state Bscores while only 3 proteins have apo states having better BScores with difference less than 3.0 kcal/mol. On the other hand, there are 17 highly perturbed apo/holo pairs, with the holo state more ligandable by BScore difference larger than 10.0 kcal/mol. To quantify the structural changes between the apo and holo states, we calculate the OASA fingerprint similarity scores between the matched contact pockets of the apo/holo pairs. The distribution of the OASA similarity scores for the apo/holo pairs is shown in Fig. 11B. We observe 21 apo/holo pairs that have high OASA similarity scores >0.80. Visualization of the top 3 most similar apo/holo pairs reveals similar surface states at the binding site with global RMSD differences all less than 0.6 Å. On the other hand, there are 13 apo/holo pairs with OASA similarities less than 0.5. Examination of three least similar apo/holo pairs reveals that the main binding surface difference between apo/holo pairs is the loss of pockets in the apo state due to occlusion by loops or large movements in the secondary structure.
Figure 11.

(A) Correlation plots for the apo and holo state matched beta clusters from the CryptoSite set. The diagonal line which shows perfect correlation between the apo and holo state scores is plotted as a solid black line. Points outside the solid grey lines represent apo/holo pairs with substantial BScore differences of +- 10.0 kcal/mol, while pairs inside the dashed grey lines represent minimally perturbed sites between the apo and holo states with a score difference of only +- 3.0 kcal/mol. (B) the distribution of the Occluded ASA similarities for the Cryptosite apo/holo pairs.
As shown in Fig. 11A, some apo-structures in the CrypticSite dataset consist of ligandable binding sites with BScore more negative than −20.0 kcal/mol, which suggests that these apo-structures may already be targetable in the first place consistent with the extended analysis of the CryptoSite by Vajda et al using their hot-spot mapping method FTMAP84.
Pocket Comparison of NMR Structures for MDM2 and MDM4
The oncogene proteins MDM2 and MDM4 play an important role in many cancers, functioning as a negative regulator of the p53 tumor suppressor protein85–87. Both of them can bind a conserved α-helix segment of p5388. Because of their importance in cancers, dual targeting of MDM2 and MDM4 proteins has been an active area of research, yielding small-molecule PPI disruptors, such as Nutlins89, 90. Here we applied our ensemble pocket approach to help understand similarities and differences between binding interfaces of MDM2 and MDM4 on a total of 40 NMR structures from both MDM2/p7391 (PDB: 2MPS) and MDM4/p5392 (PDB: 2MWY) by using the first structure of MDM2 as the reference for alignment. From the results in Fig 12, we can see that TRP, PHE and LEU pockets corresponding to hot-spot residues of binding partners for MDM2/MDM4 are important local interaction sites for MDM2 and MDM4 based on BScore. Among them, the LEU pocket is slightly more favorable for binding in MDM2 compared to MDM4 which has a BScore difference of ~1.0 kcal/mol. Among the rest of the detected pockets at the interface, pockets 8 and 9 are uniquely associated with MDM4, which have favorable levels of ligandability of −3.1±1.2 and −1.7±0.9 kcal/mol respectively. On the other hand, pocket 7 of the MDM2 interface is more ligandable than its MDM4 counterpart with a score of −2.5±0.6 kcal/mol. Of the remaining pockets, the mid-scoring pocket 1 is optimally positioned near the LEU pocket and may be further explored for the design of dual MDM2/MDM4 PPI disruptors. Throughout this analysis, we leveraged the ability of our ensemble pockets to consistently assign pockets sets for the interface allowing for direct comparison of MDM2 and MDM4 despite that their PPI interface residues are not the same.
Figure 12.
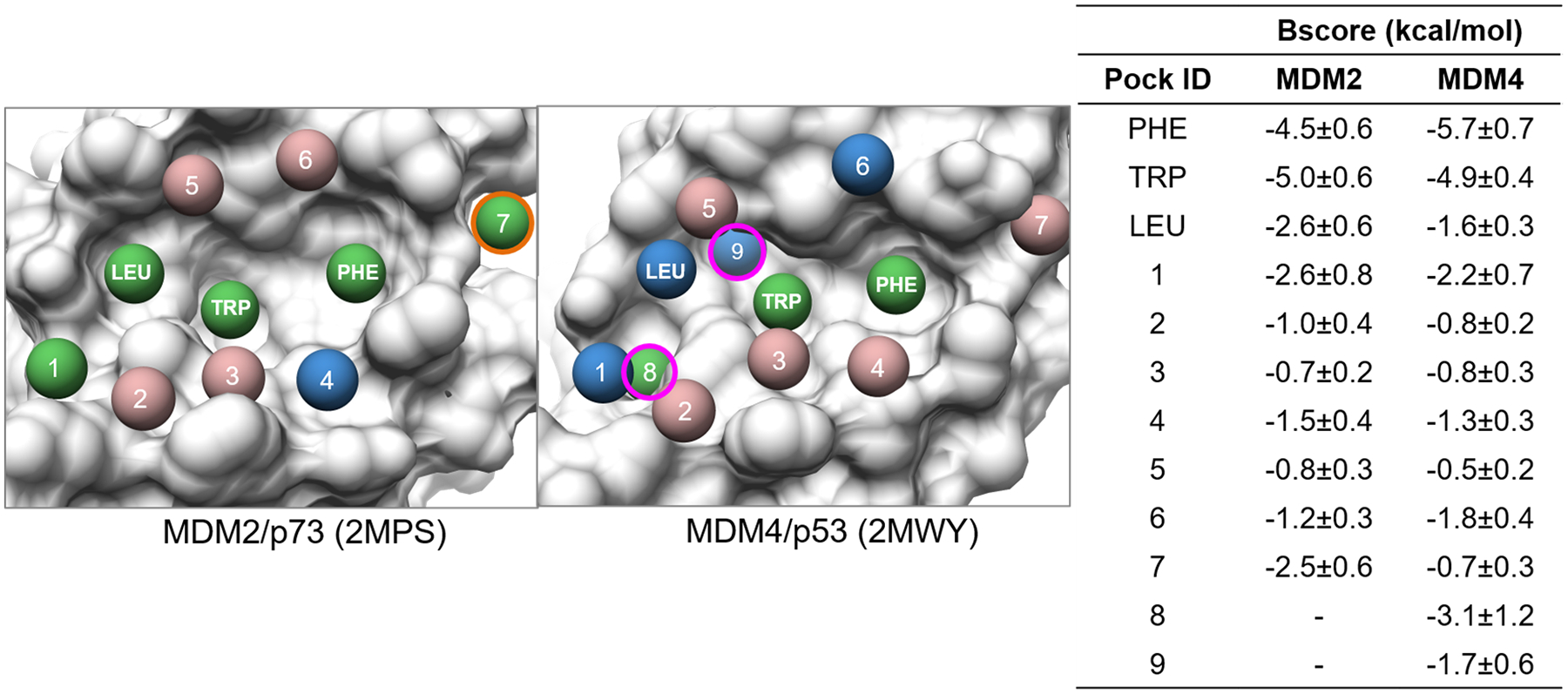
Ensemble matched pockets for 40 NMR structures of MDM2 and MDM4. Pocket ID’s and BScores are listed in companion table. High (BScores <−2.5 kcal/mol), mid (−2.5 kcal/mol >= BScores < −1.5 kcal/mol), and low (BScores >= −1.5 kcal/mol) scoring pockets are colored green, blue, and brosy brown respectively. Orange and magenta highlighted pockets correspond to pockets of interests for the MDM2 and MDM4 interface respectively.
AlphaSpace2 Studies of Bcl-2 family Proteins
Finally, we applied beta AlphaSpace2 to study an important class of PPI related disease targets, Bcl-xL and Bcl-2. Both targets belong to the Bcl-2 family proteins and act as regulators of apoptotic pathways by binding and sequestering pro-apoptotic signaling ligands93. Increase in expression of Bcl-2 or Bcl-xL is associated with resistance to pro-death signals, contributing to multiple diseases such as cancers and tumorigenesis94. By applying AlphaSpace to map the Bcl-xL/BAK (PDB ID: 1BXL) PPI interface, we were able to detect and assign hot-spot associated pockets. As shown in Fig 13, twelve Bcl-xL pockets are detected at the PPI interface and are labeled P1 to P4 corresponding to the hot-spot residue annotation for Bcl-2 family proteins and pockets 1 to 8 corresponding to additional pockets on the interface. We can see that BScores are able to correctly predict and rank hot-spot residue associated pockets as the top 4 ranking pockets with scores more negative than −3.0 kcal/mol, which is very consistent with experimental ΔΔG from alanine scanning mutation experiments of the BAK peptide95. This demonstrates the ability of BScore in correctly identifying important hot-spot interaction sites. The %-optimized score of each pocket also shows the potential to improve ligand/pocket complementarity based on the overlap of ligand and beta atoms. Among hot-spot pockets, P4 has the lowest %optimized score of 20% while other hot-spot pockets have %optimized scores >60%, suggesting a path to improve the affinity of BAK based peptides by designing mutants on the I85 position that maximize the occupation of P4. We also identified mid-scoring, auxiliary pockets 2 and 4 which are adjacent to the hot-spot, core pockets that may further improve the affinity of the BAK peptide. Finally, mid-scoring unoccupied pockets 1, 3, and 5 might be further utilized in the design of non-native peptide-based Bcl-xL PPI disruptors.
Figure 13.
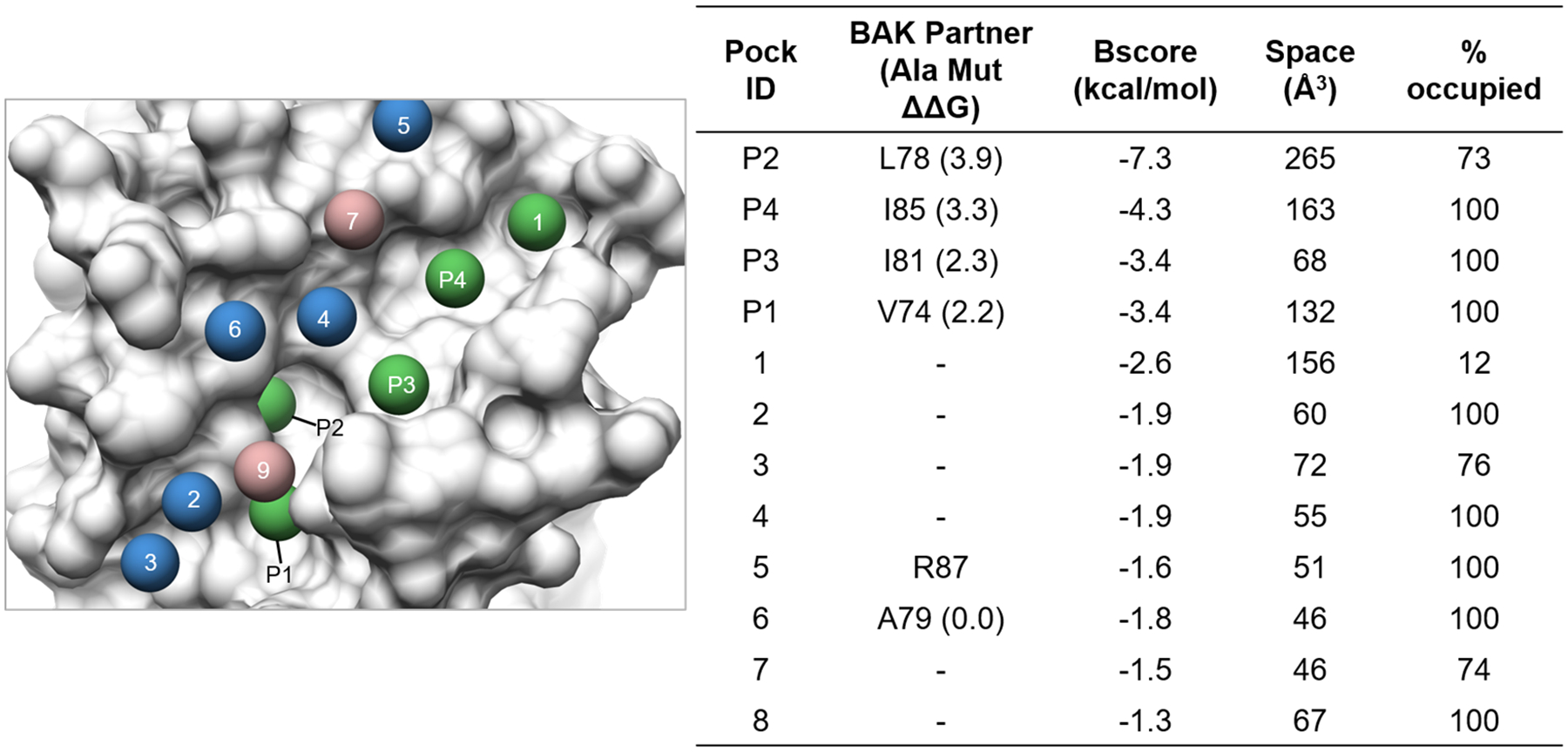
Surface mapping of the Bcl-xL/BAK complex using beta clusters. Corresponding pocket IDs and features are listed in the companion table. High (BScores <−2.5 kcal/mol), mid (−2.5 kcal/mol <= BScores < −1.5 kcal/mol), and low (BScores >= −1.5 kcal/mol) scoring pockets are colored green, blue, and rosy brown respectively.
There has been significant interest to develop Bcl-2 or Bcl-xL specific inhibitors, while the main challenge comes from the close homology and sequence similarity of Bcl-2 family proteins96. Here we applied AlphaSpace to analyze BAX bound complexes of Bcl-297 (PDB ID: 2XA0) and Bcl-xL98 (PDB ID: 3PL7), as shown in Fig 14. The ensemble pocket clustering is able to assign the important P1, P2, P3, and P4 pockets on both Bcl-2 or Bcl-xL surfaces with a score of ~2.5 kcal/mol while also assigning unique pockets specific to either Bcl-2 or Bcl-xL. Among the conserved hot-spot pockets, significant BScore differences observed for P1, P3 and P4 pockets indicate that these pockets may contribute to differential binding profiles between Bcl-2 and Bcl-xL. The more favorable BScore for the P4 pocket of Bcl-xL suggests that this pocket contributes strongly to the affinity of Bcl-xL ligands while this interaction site may be dispensable for Bcl-2. Indeed, Elmore et al99.were able to design Bcl-2 specific inhibitors that intentionally avoided the P4 site causing a drastic drop in affinity for Bcl-xL while only leading to a modest change for Bcl-2, ultimately leading to the design of the first FDA approved Bcl-2 family inhibitor, Venetoclax.
Figure 14.
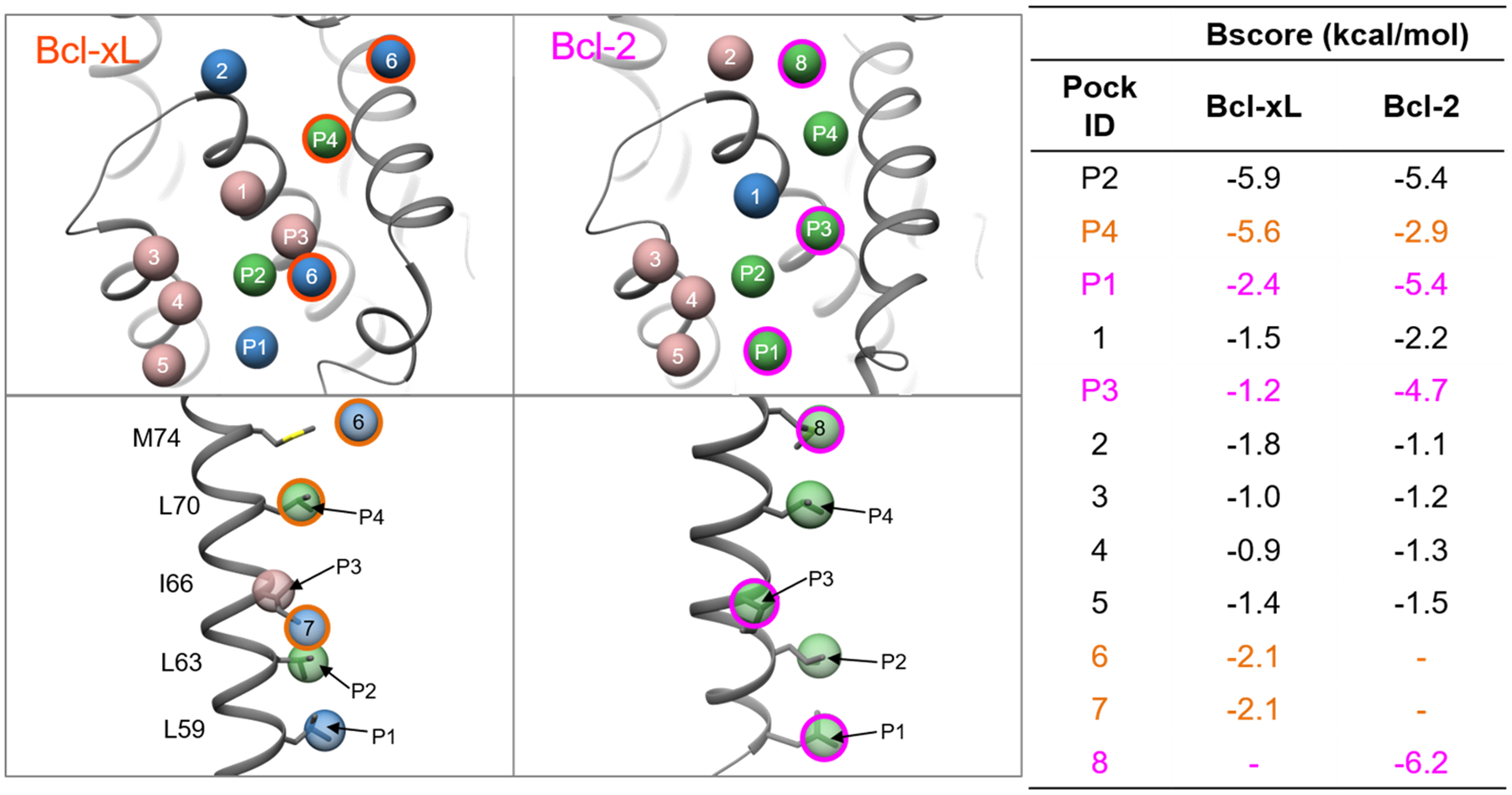
Ensemble pocket matching of Bcl-xL (left) and Bcl-2 (right) complexes bound to BAX peptide. The corresponding pocket ID’s and BScores are listed in the companion table. High (BScores <−2.5 kcal/mol), mid (−2.5 kcal/mol >= BScores < −1.5 kcal/mol), and low (BScores >= −1.5 kcal/mol) scoring pockets are colored green, blue, and rosy brown respectively. Detailed interactions of the ensemble pockets with the side-chains of BAX are shown in the lower images. Bcl-xL and Bcl-2 critical pockets are highlighted in orange and magenta respectively.
CONCLUSION
In this paper, we introduced beta-cluster as a pseudomolecular representation of fragment-centric pockets detected by AlphaSpace, which is demonstrated to mimick the shape as well as atomic details of potential molecular binders. This new beta-cluster representation allows us to develop two new features for pocket characterization and comparison: one is beta cluster score (BScore), the best vina score of the beta-cluster, as an indicator of pocket ligandability; the other is an ensemble beta-cluster approach for one-to-one pocket mapping and comparison among aligned protein surfaces. We demonstrated utility and applicability of these introduced features to a wide variety of problems, including binding site detection and comparison, PPIs and fragment-based ligand optimization.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the U.S. National Institutes of Health (R01-GM079223 and R35-GM127040). We thank NYU-ITS for providing computational resources.
Footnotes
SUPPORTING INFORMATION
Figures S1 and S2. (PDF) Properties of Small Molecule Dataset. (XLS)
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Sheridan RP; Maiorov VN; Holloway MK; Cornell WD; Gao Y-D, Drug-like Density: A Method of Quantifying the “Bindability” of a Protein Target Based on a Very Large Set of Pockets and Drug-like Ligands from the Protein Data Bank. Journal of Chemical Information and Modeling 2010, 50, 2029–2040. [DOI] [PubMed] [Google Scholar]
- 2.Khazanov NA; Carlson HA, Exploring the Composition of Protein-Ligand Binding Sites on a Large Scale. PLOS Computational Biology 2013, 9, e1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruth Brenk GK “Hot Spot” Analysis of Protein-binding Sites as a Prerequisite for Structure-based Virtual Screening and Lead Optimization. In Pharmacophores and Pharmacophore Searches, Thierry Langer RDH, Ed.; 2006; Vol. 32. [Google Scholar]
- 4.Evanthia L; George S; Demetrios KV; Zoe C, Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Current Topics in Medicinal Chemistry 2014, 14, 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ludlow RF; Verdonk ML; Saini HK; Tickle IJ; Jhoti H, Detection of secondary binding sites in proteins using fragment screening. Proceedings of the National Academy of Sciences 2015, 112, 15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters KP; Fauck J; Frömmel C, The Automatic Search for Ligand Binding Sites in Proteins of Known Three-dimensional Structure Using only Geometric Criteria. Journal of Molecular Biology 1996, 256, 201–213. [DOI] [PubMed] [Google Scholar]
- 7.Laskowski RA, SURFNET: A program for visualizing molecular surfaces, cavities, and intermolecular interactions. Journal of Molecular Graphics 1995, 13, 323–330. [DOI] [PubMed] [Google Scholar]
- 8.Laurie ATR; Jackson RM, Q-SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [DOI] [PubMed] [Google Scholar]
- 9.Levitt DG; Banaszak LJ, POCKET: A computer graphies method for identifying and displaying protein cavities and their surrounding amino acids. Journal of Molecular Graphics 1992, 10, 229–234. [DOI] [PubMed] [Google Scholar]
- 10.Hendlich M; Rippmann F; Barnickel G, LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. Journal of Molecular Graphics and Modelling 1997, 15, 359–363. [DOI] [PubMed] [Google Scholar]
- 11.Ngan C-H; Hall DR; Zerbe B; Grove LE; Kozakov D; Vajda S, FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2011, 28, 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang N; Jacobson MP, Binding-Site Assessment by Virtual Fragment Screening. PLOS ONE 2010, 5, e10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durrant JD; de Oliveira CAF; McCammon JA, POVME: An algorithm for measuring binding-pocket volumes. Journal of Molecular Graphics and Modelling 2011, 29, 773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koes DR; Camacho CJ, PocketQuery: protein–protein interaction inhibitor starting points from protein–protein interaction structure. Nucleic Acids Research 2012, 40, W387–W392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brady GP; Stouten PFW, Fast prediction and visualization of protein binding pockets with PASS. Journal of Computer-Aided Molecular Design 2000, 14, 383–401. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez M; Ghersi D; Sanchez R, SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Research 2009, 37, W413–W416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt S; Kuhn D; Klebe G, A New Method to Detect Related Function Among Proteins Independent of Sequence and Fold Homology. Journal of Molecular Biology 2002, 323, 387–406. [DOI] [PubMed] [Google Scholar]
- 18.Najmanovich R; Kurbatova N; Thornton J, Detection of 3D atomic similarities and their use in the discrimination of small molecule protein-binding sites. Bioinformatics 2008, 24, 105–111. [DOI] [PubMed] [Google Scholar]
- 19.Xie L; Bourne PE, Detecting evolutionary relationships across existing fold space, using sequence order-independent profile–profile alignments. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 5441–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konc J; Janežič D, ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics 2010, 26, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powers R; Copeland JC; Germer K; Mercier KA; Ramanathan V; Revesz P, Comparison of protein active site structures for functional annotation of proteins and drug design. Proteins 2006, 65, 124–135. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y; Skolnick J, TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Research 2005, 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao M; Skolnick J, APoc: large-scale identification of similar protein pockets. Bioinformatics (Oxford, England) 2013, 29, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borrel A; Regad L; Xhaard HG; Petitjean M; Camproux A-C, PockDrug: a model for predicting pocket druggability that overcomes pocket estimation uncertainties. Journal of Chemical Information and Modeling 2015, 55, 150402173841007–150402173841007. [DOI] [PubMed] [Google Scholar]
- 25.Liu T; Altman RB, Using multiple microenvironments to find similar ligand-binding sites: application to kinase inhibitor binding. PLoS computational biology 2011, 7, e1002326–e1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weill N; Rognan D, Alignment-free ultra-high-throughput comparison of druggable protein-ligand binding sites. Journal of chemical information and modeling 2010, 50, 123–35. [DOI] [PubMed] [Google Scholar]
- 27.Marcou G; Rognan D, Optimizing Fragment and Scaffold Docking by Use of Molecular Interaction Fingerprints. Journal of Chemical Information and Modeling 2007, 47, 195–207. [DOI] [PubMed] [Google Scholar]
- 28.Desaphy J; Raimbaud E; Ducrot P; Rognan D, Encoding Protein–Ligand Interaction Patterns in Fingerprints and Graphs. Journal of Chemical Information and Modeling 2013, 53, 623–637. [DOI] [PubMed] [Google Scholar]
- 29.Baroni M; Cruciani G; Sciabola S; Perruccio F; Mason JS, A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands And Proteins (FLAP): Theory and Application. Journal of Chemical Information and Modeling 2007, 47, 279–294. [DOI] [PubMed] [Google Scholar]
- 30.Wood DJ; Vlieg J d.; Wagener, M.; Ritschel, T., Pharmacophore Fingerprint-Based Approach to Binding Site Subpocket Similarity and Its Application to Bioisostere Replacement. Journal of Chemical Information and Modeling 2012, 52, 2031–2043. [DOI] [PubMed] [Google Scholar]
- 31.Krotzky T; Klebe G, Acceleration of Binding Site Comparisons by Graph Partitioning. Molecular Informatics 2015, 34, 550–558. [DOI] [PubMed] [Google Scholar]
- 32.Chartier M; Najmanovich R, Detection of Binding Site Molecular Interaction Field Similarities. Journal od Chemical Information and Modeling 2015, 55, 1600–1615. [DOI] [PubMed] [Google Scholar]
- 33.Desaphy J; Azdimousa K; Kellenberger E; Rognan D, Comparison and Druggability Prediction of Protein–Ligand Binding Sites from Pharmacophore-Annotated Cavity Shapes. Journal of Chemical Information and Modeling 2012, 52, 2287–2299. [DOI] [PubMed] [Google Scholar]
- 34.Cross S; Baroni M; Goracci L; Cruciani G, GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. Journal of Chemical Information and Modeling 2012, 52, 2587–2598. [DOI] [PubMed] [Google Scholar]
- 35.Totrov MJBB, Ligand binding site superposition and comparison based on Atomic Property Fields: identification of distant homologues, convergent evolution and PDB-wide clustering of binding sites. 2011, 12, S35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig IR; Pfleger C; Gohlke H; Essex JW; Spiegel K, Pocket-space maps to identify novel binding-site conformations in proteins. Journal of Chemical Information and Modeling 2011, 51, 2666–2679. [DOI] [PubMed] [Google Scholar]
- 37.Kokh DB; Richter S; Henrich S; Czodrowski P; Rippmann F; Wade RC, TRAPP: a tool for analysis of transient binding pockets in proteins. Journal of chemical information and modeling 2013, 53, 1235–52. [DOI] [PubMed] [Google Scholar]
- 38.Ravindranath PA; Sanner MF, AutoSite: an automated approach for pseudo-ligands prediction—from ligand-binding sites identification to predicting key ligand atoms. Bioinformatics 2016, 32, 3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rooklin D; Wang C; Katigbak J; Arora PS; Zhang Y, AlphaSpace: Fragment-Centric Topographical Mapping To Target Protein–Protein Interaction Interfaces. Journal of Chemical Information and Modeling 2015, 55, 1585–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dundas J; Ouyang Z; Tseng J; Binkowski A; Turpaz Y; Liang J, CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Research 2006, 34, W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim D; Cho C-H; Cho Y; Ryu J; Bhak J; Kim D-S, Pocket extraction on proteins via the Voronoi diagram of spheres. Journal of Molecular Graphics and Modelling 2008, 26, 1104–1112. [DOI] [PubMed] [Google Scholar]
- 42.Le Guilloux V; Schmidtke P; Tuffery PJBB, Fpocket: An open source platform for ligand pocket detection. 2009, 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidtke P; Bidon-Chanal A; Luque FJ; Barril X, MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [DOI] [PubMed] [Google Scholar]
- 44.Tian W; Chen C; Lei X; Zhao J; Liang J, CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Research 2018, 46, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basse MJ; Betzi S; Bourgeas R; Bouzidi S; Chetrit B; Hamon V; Morelli X; Roche P, 2P2Idb: a structural database dedicated to orthosteric modulation of protein–protein interactions. Nucleic Acids Research 2012, 41, D824–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rooklin D; Modell AE; Li H; Berdan V; Arora PS; Zhang Y, Targeting Unoccupied Surfaces on Protein–Protein Interfaces. Journal of the American Chemical Society 2017, 139, 15560–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou X; Rooklin D; Yang D; Liang X; Li K; Lu J; Wang C; Xiao P; Zhang Y; Sun J.-p.; Fang H, Computational Strategy for Bound State Structure Prediction in Structure-Based Virtual Screening: A Case Study of Protein Tyrosine Phosphatase Receptor Type O Inhibitors. Journal of Chemical Information and Modeling 2018, 58, 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Defays D, An efficient algorithm for a complete link method. The Computer Journal 1977, 20, 364–366. [Google Scholar]
- 49.Jones E; Oliphant T; Peterson P; others SciPy: Open source scientific tools for Python. http://www.scipy.org/
- 50.Trott O; Olson AJ, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sokal RR; Michener CD; Kansas U. o., A Statistical Method for Evaluating Systematic Relationships. University of Kansas: 1958. [Google Scholar]
- 52.Meng EC; Pettersen EF; Couch GS; Huang CC; Ferrin TE, Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 2006, 7, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 54.Cao D-S; Xu Q-S; Hu Q-N; Liang Y-Z, ChemoPy: freely available python package for computational biology and chemoinformatics. Bioinformatics 2013, 29, 1092–1094. [DOI] [PubMed] [Google Scholar]
- 55.Schreyer AM; Blundell T, USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. Journal of Cheminformatics 2012, 4, 1–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ballester PJ; Richards WG, Ultra Fast Shape Comparison. Journal of computational chemistry 2010, 31, 2967–2970.20928852 [Google Scholar]
- 57.Ballester PJ; Richards WG, Ultrafast shape recognition for similarity search in molecular databases. Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences 2007, 463, 1307–1321. [Google Scholar]
- 58.Koes DR; Camacho CJ, Shape-based virtual screening with volumetric aligned molecular shapes. Journal of Computational Chemistry 2014, 35, 1824–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grant JA; Pickup BT, A Gaussian Description of Molecular Shape. The Journal of Physical Chemistry 1995, 99, 3503–3510. [Google Scholar]
- 60.Fleming PJ; Richards FM, Protein packing: dependence on protein size, secondary structure and amino acid composition11Edited by Cohen FE. Journal of Molecular Biology 2000, 299, 487–498. [DOI] [PubMed] [Google Scholar]
- 61.Desaphy J; Bret G; Rognan D; Kellenberger E, sc-PDB: a 3D-database of ligandable binding sites—10 years on. Nucleic Acids Research 2015, 43, D399–D404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Z; Su M; Han L; Liu J; Yang Q; Li Y; Wang R, Forging the Basis for Developing Protein–Ligand Interaction Scoring Functions. Accounts of Chemical Research 2017, 50, 302–309. [DOI] [PubMed] [Google Scholar]
- 63.Ichihara O; Shimada Y; Yoshidome D, The Importance of Hydration Thermodynamics in Fragment-to-Lead Optimization. ChemMedChem 2014, 9, 2708–2717. [DOI] [PubMed] [Google Scholar]
- 64.Basse MJ; Betzi S; Bourgeas R; Bouzidi S; Chetrit B; Hamon V; Morelli X; Roche P, 2P2Idb: a structural database dedicated to orthosteric modulation of protein–protein interactions. Nucleic Acids Research 2013, 41, D824–D827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cimermancic P; Weinkam P; Rettenmaier TJ; Bichmann L; Keedy DA; Woldeyes RA; Schneidman-Duhovny D; Demerdash ON; Mitchell JC; Wells JA; Fraser JS; Sali A, CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites. Journal of Molecular Biology 2016, 428, 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang Z; Zhu L; Cao Y; Wu G; Liu X; Chen Y; Wang Q; Shi T; Zhao Y; Wang Y; Li W; Li Y; Chen H; Chen G; Zhang J, ASD: a comprehensive database of allosteric proteins and modulators. Nucleic Acids Research 2011, 39, D663–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown NR; Noble MEM; Endicott JA; Johnson LN, The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nature Cell Biology 1999, 1, 438–443. [DOI] [PubMed] [Google Scholar]
- 68.Russo AA; Jeffrey PD; Patten AK; Massagué J; Pavletich NP, Crystal structure of the p27Kip1 cyclin-dependent-kinase inibitor bound to the cyclin A–Cdk2 complex. Nature 1996, 382, 325–331. [DOI] [PubMed] [Google Scholar]
- 69.Chen P; Lee NV; Hu W; Xu M; Ferre RA; Lam H; Bergqvist S; Solowiej J; Diehl W; He Y-A; Yu X; Nagata A; VanArsdale T; Murray BW, Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. 2016, 15, 2273–2281. [DOI] [PubMed] [Google Scholar]
- 70.Coxon CR; Anscombe E; Harnor SJ; Martin MP; Carbain B; Golding BT; Hardcastle IR; Harlow LK; Korolchuk S; Matheson CJ; Newell DR; Noble MEM; Sivaprakasam M; Tudhope SJ; Turner DM; Wang LZ; Wedge SR; Wong C; Griffin RJ; Endicott JA; Cano C, Cyclin-Dependent Kinase (CDK) Inhibitors: Structure–Activity Relationships and Insights into the CDK-2 Selectivity of 6-Substituted 2-Arylaminopurines. Journal of Medicinal Chemistry 2017, 60, 1746–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ludlow RF; Verdonk ML; Saini HK; Tickle IJ; Jhoti H, Detection of secondary binding sites in proteins using fragment screening. 2015, 112, 15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rothweiler U; Eriksson J; Stensen W; Leeson F; Engh RA; Svendsen JS, Luciferin and derivatives as a DYRK selective scaffold for the design of protein kinase inhibitors. European Journal of Medicinal Chemistry 2015, 94, 140–148. [DOI] [PubMed] [Google Scholar]
- 73.Wood DJ; Lopez-Fernandez JD; Knight LE; Al-Khawaldeh I; Gai C; Lin S; Martin MP; Miller DC; Cano C; Endicott JA; Hardcastle IR; Noble MEM; Waring MJ, FragLites—Minimal, Halogenated Fragments Displaying Pharmacophore Doublets. An Efficient Approach to Druggability Assessment and Hit Generation. Journal of Medicinal Chemistry 2019, 62, 3741–3752. [DOI] [PubMed] [Google Scholar]
- 74.Ferenczy GG; Keserű GM, How Are Fragments Optimized? A Retrospective Analysis of 145 Fragment Optimizations. Journal of Medicinal Chemistry 2013, 56, 2478–2486. [DOI] [PubMed] [Google Scholar]
- 75.Card GL; Blasdel L; England BP; Zhang C; Suzuki Y; Gillette S; Fong D; Ibrahim PN; Artis DR; Bollag G; Milburn MV; Kim S-H; Schlessinger J; Zhang KYJ, A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nature Biotechnology 2005, 23, 201–207. [DOI] [PubMed] [Google Scholar]
- 76.Antonysamy S; Hirst G; Park F; Sprengeler P; Stappenbeck F; Steensma R; Wilson M; Wong M, Fragment-based discovery of JAK-2 inhibitors. Bioorganic & Medicinal Chemistry Letters 2009, 19, 279–282. [DOI] [PubMed] [Google Scholar]
- 77.Card GL; Blasdel L; England BP; Zhang C; Suzuki Y; Gillette S; Fong D; Ibrahim PN; Artis DR; Bollag G; Milburn MV; Kim S-H; Schlessinger J; Zhang KYJ, A family of phosphodiesterase inhibitors discovered by cocrystallography and scaffold-based drug design. Nat Biotech 2005, 23, 201–207. [DOI] [PubMed] [Google Scholar]
- 78.Felding J; Sørensen MD; Poulsen TD; Larsen J; Andersson C; Refer P; Engell K; Ladefoged LG; Thormann T; Vinggaard AM; Hegardt P; Søhoel A; Nielsen SF, Discovery and Early Clinical Development of 2-{6-[2-(3,5-Dichloro-4-pyridyl)acetyl]-2,3-dimethoxyphenoxy}-N-propylacetamide (LEO 29102), a Soft-Drug Inhibitor of Phosphodiesterase 4 for Topical Treatment of Atopic Dermatitis. Journal of Medicinal Chemistry 2014, 57, 5893–5903. [DOI] [PubMed] [Google Scholar]
- 79.Gràcia J; Buil MA; Castro J; Eichhorn P; Ferrer M; Gavaldà A; Hernández B; Segarra V; Lehner MD; Moreno I; Pagès L; Roberts RS; Serrat J; Sevilla S; Taltavull J; Andrés M; Cabedo J; Vilella D; Calama E; Carcasona C; Miralpeix M, Biphenyl Pyridazinone Derivatives as Inhaled PDE4 Inhibitors: Structural Biology and Structure–Activity Relationships. Journal of Medicinal Chemistry 2016, 59, 10479–10497. [DOI] [PubMed] [Google Scholar]
- 80.Siu M; Pastor R; Liu W; Barrett K; Berry M; Blair WS; Chang C; Chen JZ; Eigenbrot C; Ghilardi N; Gibbons P; He H; Hurley CA; Kenny JR; Cyrus Khojasteh S; Le H; Lee L; Lyssikatos JP; Magnuson S; Pulk R; Tsui V; Ultsch M; Xiao Y; Zhu B.-y.; Sampath D, 2-Amino-[1,2,4]triazolo[1,5-a]pyridines as JAK2 inhibitors. Bioorganic & Medicinal Chemistry Letters 2013, 23, 5014–5021. [DOI] [PubMed] [Google Scholar]
- 81.Jones P; Storer RI; Sabnis YA; Wakenhut FM; Whitlock GA; England KS; Mukaiyama T; Dehnhardt CM; Coe JW; Kortum SW; Chrencik JE; Brown DG; Jones RM; Murphy JR; Yeoh T; Morgan P; Kilty I, Design and Synthesis of a Pan-Janus Kinase Inhibitor Clinical Candidate (PF-06263276) Suitable for Inhaled and Topical Delivery for the Treatment of Inflammatory Diseases of the Lungs and Skin. Journal of Medicinal Chemistry 2017, 60, 767–786. [DOI] [PubMed] [Google Scholar]
- 82.Lee EF; Czabotar PE; Yang H; Sleebs BE; Lessene G; Colman PM; Smith BJ; Fairlie WD, Conformational Changes in Bcl-2 Pro-survival Proteins Determine Their Capacity to Bind Ligands. Journal of Biological Chemistry 2009, 284, 30508–30517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang C-Y; Wang S, Analysis of Flexibility and Hotspots in Bcl-xL and Mcl-1 Proteins for the Design of Selective Small-Molecule Inhibitors. ACS Medicinal Chemistry Letters 2012, 3, 308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beglov D; Hall DR; Wakefield AE; Luo L; Allen KN; Kozakov D; Whitty A; Vajda S, Exploring the structural origins of cryptic sites on proteins. Proceedings of the National Academy of Sciences 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shadfan M; Lopez-Pajares V; Yuan Z-M, MDM2 and MDMX: alone and together in regulation of p53. J Translational Cancer Research 2012, 1, 88–99. [PMC free article] [PubMed] [Google Scholar]
- 86.Karni-Schmidt O; Lokshin M; Prives C, The Roles of MDM2 and MDMX in Cancer. Annual Review of Pathology: Mechanisms of Disease 2016, 11, 617–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Q; Lozano G, Molecular Pathways: Targeting Mdm2 and Mdm4 in Cancer Therapy. American Association for Cancer Research 2013, 19, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kussie PH; Gorina S; Marechal V; Elenbaas B; Moreau J; Levine AJ; Pavletich NP, Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [DOI] [PubMed] [Google Scholar]
- 89.Vassilev LT; Vu BT; Graves B; Carvajal D; Podlaski F; Filipovic Z; Kong N; Kammlott U; Lukacs C; Klein C; Fotouhi N; Liu EA, In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- 90.Shangary S; Wang S, Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction to Reactivate p53 Function: A Novel Approach for Cancer Therapy. Annual Review of Pharmacology and Toxicology 2009, 49, 223–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shin J-S; Ha J-H; Lee D-H; Ryu K-S; Bae K-H; Park BC; Park SG; Yi G-S; Chi S-W, Structural convergence of unstructured p53 family transactivation domains in MDM2 recognition. Cell Cycle 2015, 14, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grace CR; Ban D; Min J; Mayasundari A; Min L; Finch KE; Griffiths L; Bharatham N; Bashford D; Kiplin Guy R; Dyer MA; Kriwacki RW, Monitoring Ligand-Induced Protein Ordering in Drug Discovery. Journal of Molecular Biology 2016, 428, 1290–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chipuk JE; Moldoveanu T; Llambi F; Parsons MJ; Green DR, The BCL-2 Family Reunion. Molecular Cell 2010, 37, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Letai AG, Diagnosing and exploiting cancer's addiction to blocks in apoptosis. Nature Reviews Cancer 2008, 8, 121. [DOI] [PubMed] [Google Scholar]
- 95.Sattler M; Liang H; Nettesheim D; Meadows RP; Harlan JE; Eberstadt M; Yoon HS; Shuker SB; Chang BS; Minn AJ; Thompson CB; Fesik SW, Structure of Bcl-xL-Bak Peptide Complex: Recognition Between Regulators of Apoptosis. 1997, 275, 983–986. [DOI] [PubMed] [Google Scholar]
- 96.Hennessy EJ, Selective inhibitors of Bcl-2 and Bcl-xL: Balancing antitumor activity with on-target toxicity. Bioorganic & Medicinal Chemistry Letters 2016, 26, 2105–2114. [DOI] [PubMed] [Google Scholar]
- 97.Ku B; Liang C; Jung JU; Oh B-H, Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Research 2010, 21, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Czabotar PE; Lee EF; Thompson GV; Wardak AZ; Fairlie WD; Colman PM, Mutation to Bax beyond the BH3 Domain Disrupts Interactions with Pro-survival Proteins and Promotes Apoptosis. Journal of Biological Chemistry 2011, 286, 7123–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Souers AJ; Leverson JD; Boghaert ER; Ackler SL; Catron ND; Chen J; Dayton BD; Ding H; Enschede SH; Fairbrother WJ; Huang DCS; Hymowitz SG; Jin S; Khaw SL; Kovar PJ; Lam LT; Lee J; Maecker HL; Marsh KC; Mason KD; Mitten MJ; Nimmer PM; Oleksijew A; Park CH; Park C-M; Phillips DC; Roberts AW; Sampath D; Seymour JF; Smith ML; Sullivan GM; Tahir SK; Tse C; Wendt MD; Xiao Y; Xue JC; Zhang H; Humerickhouse RA; Rosenberg SH; Elmore SW, ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Medicine 2013, 19, 202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.