

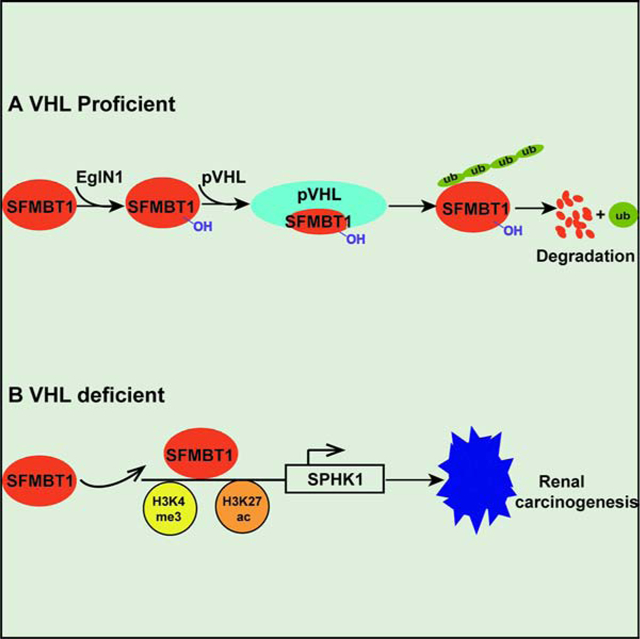
Summary
von Hippel-Lindau (VHL) is a critical tumor suppressor in clear cell renal cell carcinomas (ccRCCs). It is important to identify additional therapeutic targets in ccRCC downstream of VHL loss besides hypoxia inducible factor 2α (HIF2α). By performing a genome-wide screen, we identified Scm-like with four malignant brain tumor domains 1 (SFMBT1) as a candidate pVHL target. SFMBT1 was considered to be a transcriptional repressor but its role in cancer remains unclear. ccRCC patients with VHL loss-of-function mutations displayed elevated SFMBT1 protein levels. SFMBT1 hydroxylation on proline residue 651 by EglN1 mediated its ubiquitination and degradation governed by pVHL. Depletion of SFMBT1 abolished ccRCC cell proliferation in vitro and inhibited orthotopic tumor growth in vivo. Integrated analyses of ChIP-seq, RNA-seq and patient prognosis identified sphingosine kinase 1 (SPHK1) as a key SFMBT1 target gene contributing to its oncogenic phenotype. Therefore, the pVHL-SFMBT1-SPHK1 axis serves as a potential therapeutic avenue for ccRCC.
Keywords: pVHL, SFMBT1, ccRCC, SPHK1
Graphical Abstract
eTOC Blurb
The pVHL-SFMBT1-SPHK1 axis serves as a potential therapeutic avenue for ccRCC.
Introduction
The incidence of renal cancers, including renal cell and renal pelvis types, has been increasing for several decades, but the reasons for this trend are unclear (Godwin et al., 2014). Some patients at risk for clear cell renal cell carcinoma (ccRCC) carry a germline mutation in the von Hippel-Lindau (VHL) tumor suppressor gene, an E3 ubiquitin ligase (Kaelin, 2002). Inactivating VHL mutations play major roles in sporadic RCC (Haase, 2005), and loss of VHL accounts for up to 85% of renal cancers that are classically resistant to cytotoxic chemotherapy (Kaelin, 2002, Cancer Genome Atlas Research, 2013).
pVHL, the protein encoded by VHL, ubiquitinates hypoxia inducible factor α (HIFα, including HIF1α and HIF2α) to mark it for degradation (Ivan et al., 2001, Jaakkola et al., 2001). Loss of pVHL therefore promotes HIFα accumulation, which contributes to the transformation phenotype of renal cancer (Kondo et al., 2003, Kondo et al., 2002). Accumulation and translocation of HIFα factors into the nucleus promotes dimerization of HIFα subunits with a constitutively expressed HIFβ subunit (Semenza, 2012). This dimer transactivates genes containing hypoxia response elements in their promoters or enhancer regions. Recent reports showed that the specific HIF2α inhibitor PT2399 inhibited primary tumor growth and invasion of a subset of kidney cancers (Cho et al., 2016, Chen et al., 2016). However, a significant portion of kidney cancers remained resistant to HIF2α inhibitor treatment (Cho et al., 2016, Chen et al., 2016), highlighting the importance of identifying additional therapeutic vulnerabilities of pVHL-deficient kidney cancer. Evidence suggests that pVHL has substrates other than HIFα (Gamper et al., 2012, Lee et al., 2015). Indeed, non-HIF substrates may account for different subtypes of VHL disease (type 2A and 2B versus 2C) (Clifford et al., 2001, Gordeuk et al., 2004, Hoffman et al., 2001). We recently identified ZHX2 as a new pVHL substrate that contributes to hyper-activation of NF-κB in ccRCC (Zhang et al., 2018).
To systematically identify pVHL substrates, we performed a novel genome-wide in vitro expression strategy coupled with a GST-binding screen for pVHL substrates and identified SFMBT1 as a direct target of pVHL. SFMBT1 was shown to be a histone reader subunit of the LSD1 demethylase complex associated with epipthelial-mesenchymal transition (Tang et al., 2013). The limited understanding of SFMBT1 has primarily focused on binding between SFMBT1 and histone tails (Zhang et al., 2013b). Additional functions for SFMBT1 and its mode of regulation, particularly in cancer, have not been explored. Here we show, for the first time, that SFMBT1 is a target for pVHL and promotes ccRCC cell proliferation, anchorage-independent growth, as well as tumor xenograft growth. Clinically, SFMBT1 is expressed at elevated levels in renal tumors compared to adjacent normal tissue. Thus, our functional characterization of the critical SFMBT1 signaling axis in pVHL-deficient renal cancer may shed light on novel therapeutic modalities.
Results
A Genome-Wide Screen Identifies SFMBT1 as a pVHL Target
Utilizing a genome-wide in vitro expression cloning strategy (Zhang et al., 2018), we were able to identify proteins that bind pVHL and can be competed off by the hydroxylated HIF peptide (Figure 1A). These proteins may represent potential pVHL substrates that bind to the hydrophobic substrate-binding pocket of pVHL. Here, we present evidence that Scm-like with four MBT domains 1 (SFMBT1) is a new pVHL substrate identified in our screen (Figure 1B–C). SFMBT1 was identified as a transcriptional repressor component of the LSD1 demethylase complex that contains multiple MBT domains (Zhang et al., 2013a), and interacts with histone tails (Zhang et al., 2013a, Nady et al., 2012). However, SFMBT1 does not differentiate between unmodified and modified histone peptides (Zhang et al., 2013a). Therefore, there may be additional functions of SFMBT1 that remain uncharacterized. The role of SFMBT1 in cancer, especially in kidney cancer in its relation to VHL, has not been previously explored.
Figure 1. SFMBT1 is a novel pVHL target.
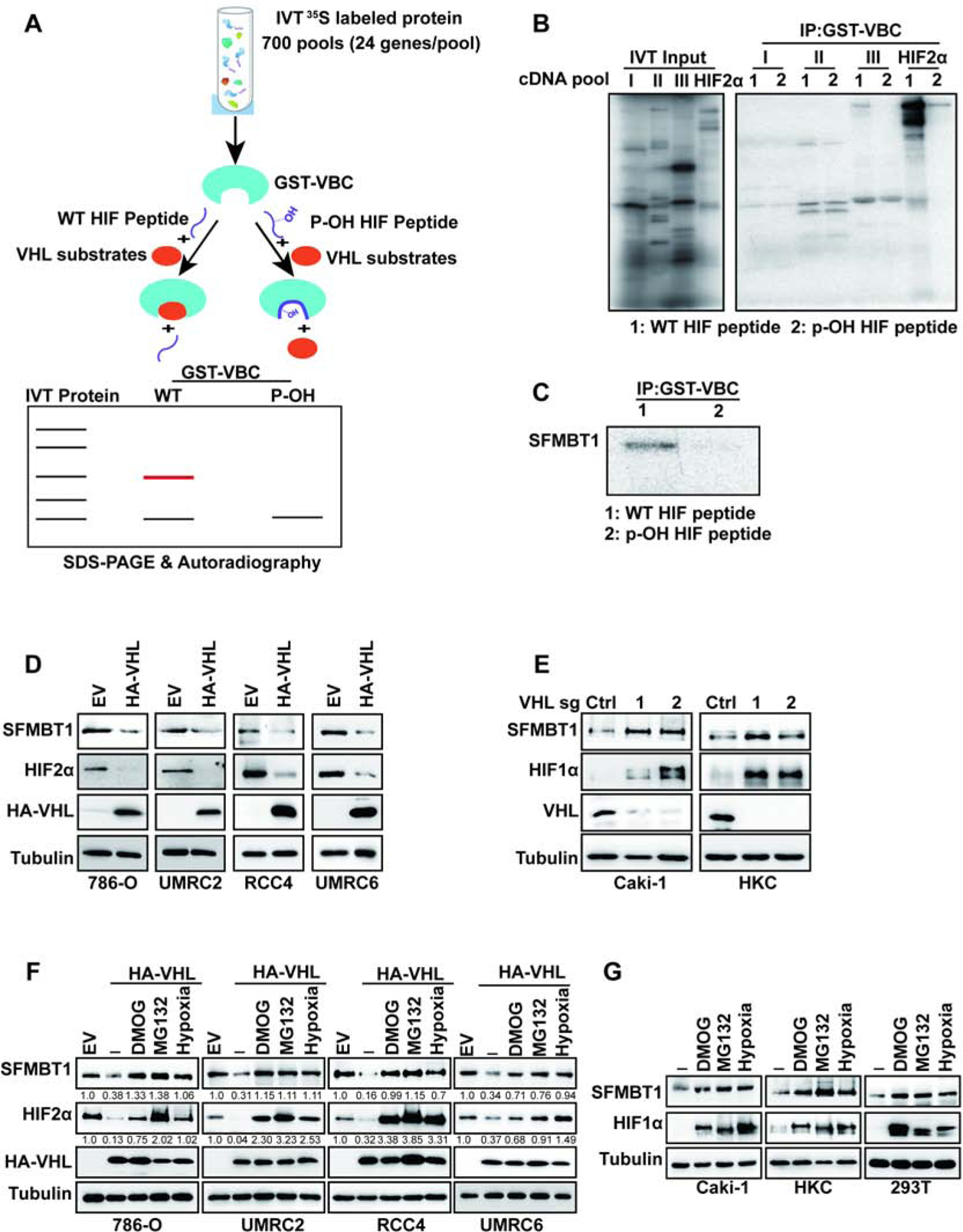
(A) Schematic diagram of pVHL substrate screen.
(B-C) Binding competition assays of 35S-Methionine labelled in vitro translated (IVT) cDNA pools
(B) or IVT SFMBT1(C) and GST-VBC complex in the presence of wildtype (WT) or prolyl hydroxylated (p-OH) HIF peptide followed by SDS-PAGE and autoradiography.
(D-E) Immunoblots for lysates from cells transduced with lentivirus either expressing control (Ctrl) or HA-pVHL (D), control sgRNA (Ctrl) or VHL sgRNAs (1 and 2) (E).
(F) Immunoblots for lysates from cells transduced with lentivirus expressing either control (Ctrl) or HA-pVHL, followed by indicated drugs or hypoxia treatment overnight. The values listed below the blots indicate the relative SFMBT1/HIF2α protein levels with Tubulin normalization.
(G) ) Immunoblots for lysates from cells treated with indicated drugs or hypoxia treatment overnight.
First, we examined SFMBT1 levels in pVHL-null ccRCC cell lines (786-O, UMRC2, RCC4 and UMRC6), with or without pVHL restoration. In all the cell lines examined, pVHL restoration led to the downregulation of SFMBT1, phenocopying HIF2α regulation (Figure 1D). The regulation of SFMBT1 by pVHL may be independent of HIF signaling, since depletion of HIF1 β or HIF2α by two independent CRISPR-Cas9 guide RNAs (sgRNAs) did not affect SFMBT1 protein levels in ccRCC cells (Figure S1A–B). It is also important to note that SFMBT2, a close family member of SFMBT1, was not regulated at the protein level by pVHL restoration in these cells (Figure S1C). We also depleted pVHL expression by using sgRNAs in the pVHL-expressing cell lines Caki-1 and HKC. Two independent sgRNAs against VHL led to SFMBT1 and HIF1α upregulation in these cells (Figure 1E). Canonical regulation of pVHL substrates is through hydroxylation, followed by recognition by the pVHL E3 ligase complex resulting in ubiquitination and proteasomal degradation (Ivan et al., 2001, Jaakkola et al., 2001). To test whether the potential regulation of SFMBT1 by pVHL depends on its hydroxylation, we treated control and HA-pVHL expressing 786-O, UMRC2, RCC4 and UMRC6 cells with dimethyloxalylglycine (DMOG) or hypoxic conditions (1% O2), both able to inhibit hydroxylation. Inhibition of hydroxylation induced SFMBT1 expression to a level comparable to that of cells lacking functional pVHL (Figure 1F). Similar results were observed for the canonical pVHL substrate HIF2α (Figure 1F), indicating that the negative regulation of SFMBT1 by pVHL is hydroxylation-dependent. In addition, treatment of HA-pVHL expressing cells with the proteasome inhibitor MG132 also led to SFMBT1 upregulation (Figure 1F), suggesting that SFMBT1 hydroxylation leads to pVHL-mediated proteasomal degradation. Strengthening the observation from pVHL-null ccRCC cell lines that SFMBT1 regulation is dependent on hydroxylation and proteasomal degradation, treatment of pVHL-expressing renal cells (Caki-1, HKC or 293T) with MG132, DMOG, or hypoxia also resulted in upregulation of SFMBT1 expression (Figure 1G).
Prolyl Hydroxylation Regulates SFMBT1 Binding and Ubiquitination by pVHL
Our results suggest that SFMBT1 may undergo hydroxylation and regulation by pVHL, which is further strengthened by the ability of pan-hydroxyproline antibody to pull-down hydroxylated SFMBT1 in the presence of MG132, an effect abrogated by concurrent MG132 and DMOG treatment (Figure 2A). We further examined the binding between endogenous SFMBT1 and pVHL by reciprocal immunoprecipitations in the presence or absence of prolyl hydroxylase inhibitors FG4592 or DMOG; decreased binding between SFMBT1 and pVHL was observed when prolyl hydroxylation was inhibited (Figure 2B). We observed similar results in 786-O cells with pVHL restoration (Figure S2A). Moreover, GST-pVHL was able to pull-down in vitro translated SFMBT1, indicating that pVHL binds SFMBT1 directly (Figure S2B). GST pull-down of endogenous SFMBT1 from 786-O cells further confirmed the binding between GST-pVHL and SFMBT1, which was diminished when cells were treated with either DMOG or FG4592 (Figure S2C). Next, we aimed to identify the potential SFMBT1 prolyl hydroxylation sites that mediate its binding and potential ubiquitination by pVHL. We overexpressed FLAG-tagged SFMBT1 in 293T cells followed by treatment with either MG132 or MG132 together with DMOG, and performed mass spectrometry (Figure S2D). Specifically, we were interested in those proline sites displaying decreased hydroxylation levels upon DMOG treatment. To increase our confidence, we performed two independent experiments and sent these samples to two different mass spectrometry facilities for the identification of prolyl hydroxylation sites. From both sets of experiments, we identified only two common prolyl hydroxylation sites: Prolines 106 and 651 (Figure S2E–G). Next, we mutated SFMBT1 prolines 106 and 651 to alanines (P106A and P651A). Whereas WT or P106A mutant SFMBT1 bound pVHL efficiently, P651A mutant binding to pVHL was impaired, suggesting that P651 is the major hydroxylation site recognized by pVHL (Figure 2C). Subsequently, we synthesized WT (SFMBT1-WT) or Proline 650/651 hydroxylated (P650-OH and P651-OH) SFMBT1 peptides and performed the binding assay with pVHL (Figure 2D). Only P651-OH peptide was able to bind with pVHL strongly, similar to hydroxylated HIF1α peptide (Figure 2D). We noticed that there was an adjacent Proline (P650) on SFMBT1 but synthesized P650-OH peptide failed to bind with pVHL (Figure 2D), suggesting that proline 651 is the specific site undergoing hydroxylation leading to pVHL binding. Consistently, pan-hydroxylation IP of HA-tagged WT (WT), P106A, or P651A SFMBT1 followed by HA-SFMBT1 immunoblot showed that only P651A mutant abrogated the hydroxylation signal (Figure 2E). We also depleted endogenous SFMBT1 and restored with physiologically relevant levels of SFMBT1 WT or P651A SFMBT1 mutant followed by examination of SFMBT1 hydroxylation. Both SFMBT1 depletion and P651A mutation abrogated the hydroxylation signal (Figure 2F).
Figure 2. SFMBT1 stability is regulated by pVHL through EglN1 hydroxylation.
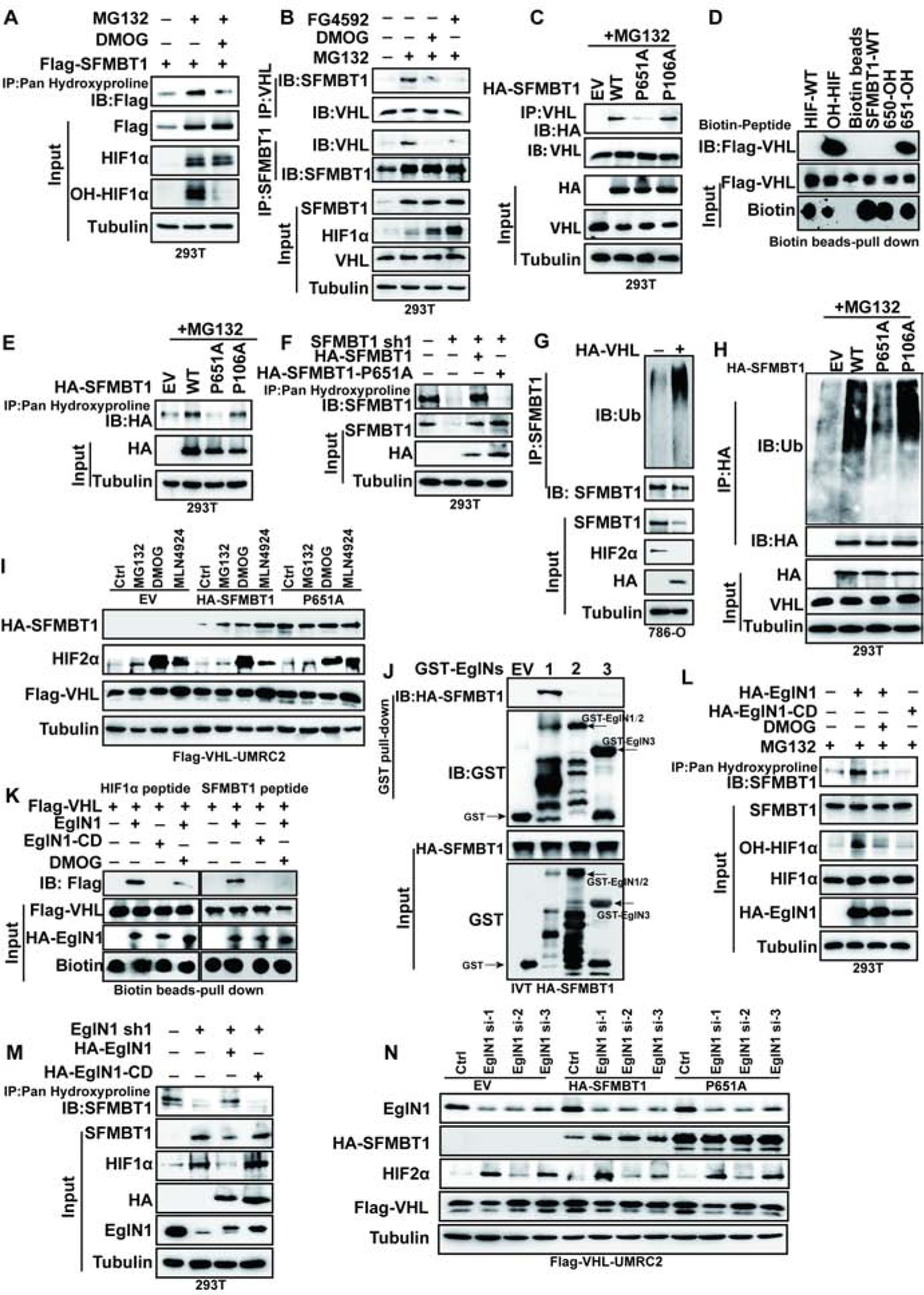
(A) Immunoprecipitaiton of cell lysates for the hydroxylated SFMBT1 from cells treated with MG132 or MG132 plus DMOG treatment.
(B) Immunoprecipitaiton and immunblots of cell lysates treated with of indicated drugs overnight.
(C) Immunoprecipitaiton and immunblots of lysates from cells transfected with indicated plasmids followed by MG132 treatment.
(D) Binding between FLAG-pVHL and HIF1α or SFMBT1 peptides.
(E-F) Immunoprecipitaiton of lysates for the hydroxylated SFMBT1 in cells transfected (E) or transduced with plasmids or lentivirus indicated (F).
(G) Effect of HA-pVHL on ubiquitination of endogenous SFMBT1.
(H) Ubiquitination level of wide type HA-SFMBT1 (WT), P651A and P106A.
(I) Immunoblots for lysates of transduced HA-SFMBT1 and P651A in UMRC2 transduced with lentivirus expressing FLAG-pVHL (FLAG-VHL-UMRC2) followed by treatment with indicated drugs.
(J) GST-pull down assay between GST (EV) or GST-EglNs (EglN1, EglN2 and EglN3) and in vitro translated (IVT) HA-SFMBT1.
(K) Capture of biotinylated HIF1α (left panel) and SFMBT1 (right panel) peptides by FLAG-VHL after in vitro hydroxylation by IVT EglN1 (wide-type) or EglN1 catalytic dead variant (H314VD316/A314VA316, EglN1-CD). HIF1α peptide was used as positive control. Capture of peptides by FLAG-pVHL indicates hydroxylation of peptides by EglN1.
(L) Hydroxylation level of endogenous SFMBT1 in cells transfected with HA-EglN1 or EgLN1-CD, followed by MG132 or MG132 plus DMOG treatment.
(M) Immunoblots of Immunoprecipitated samples to detect endogenous SFMBT1 hydroxylation in cells with EglN1 knock down (sh1) as well as these cells restored with HA-EglN1 or catalytic dead mutant (HA-EglN1-CD).
(N) Immunoblots for lysates of transduced HA-SFMBT1 and P651A protein level in FLAG-VHL-UMRC2 cells transfected with control siRNA (Ctrl) or EglN1 siRNAs (si-1, si-2 and si-3).
To examine whether pVHL regulates SFMBT1 via ubiquitination, we performed an in vivo ubiquitination assay in 293T cells transfected with HA-SFMBT1 in the presence or absence of exogenous FLAG-tagged pVHL. We observed a stronger HA-SFMBT1 ubiquitination with FLAG-pVHL expression, as well as decreased HA-SFMBT1 protein level (Figure S2H). We also confirmed this in vivo ubiquitination assay in pVHL-deficient 786-O renal cancer cells or isogenic cells reconstituted with HA-pVHL, and observed an increase in endogenous SFMBT1 ubiquitination upon HA-pVHL expression (Figure 2G). To investigate whether P651 affects SFMBT1 ubiquitination by pVHL, we performed an in vivo ubiquitination assay in 293T cells. Consistent with Western blot data showing SFMBT1 regulation by pVHL, pVHL promoted ubiquitination of WT or P106A, but not P651A SFMBT1 (Figure 2H). We also transduced FLAG-VHL UMRC2 cells with lentivirus expressing WT or P651A HA-SFMBT1 followed by treatment with MG132, MLN4924 (neddylation inhibitor that inhibits E3 ligase complex formation), or DMOG. WT HA-SFMBT1 was upregulated with these inhibitor treatments compared to control. Conversely, the P651A HA-SFMBT1 mutant was not affected by these inhibitors, and was constantly upregulated compared to its WT counterpart (Figure 2I). Therefore, P651 is the primary site responsible for SFMBT1 hydroxylation and its regulation by the pVHL E3 ligase complex.
EglN1 is the Primary Prolyl Hydroxylase that Regulates SFMBT1 Hydroxylation and Protein Stability in ccRCC
In order to test which prolyl hydroxylase family member may be responsible for SFMBT1 hydroxylation and regulation by pVHL, we first purified GST-EglN1, 2 and 3 recombinant protein followed by GST pull-down from 786-O cell lysates. By implementing an orthogonal strategy, we also performed GST pull-down of in vitro translated SFMBT1 from reticulocyte lysate. In both approaches, only GST-EglN1 could pull-down SFMBT1 (Figure S2I and Figure 2J). Next, we performed an in vitro hydroxylation reaction with SFMBT1 unmodified peptide in the presence of in vitro translated EglN1 enzyme (Figure S2J). Only WT EglN1, but not its catalytic dead variant (EglN1-CD), promoted SFMBT1/HIF1α hydroxylation that was captured by pVHL (Figure 2K). EglN1-triggered hydroxylation of SFMBT1 or HIF1α was inhibited by co-incubation with DMOG (Figure 2K), further confirming that EglN1 promotes SFMBT1 hydroxylation followed by pVHL recognition. We also observed that WT EglN1, but not the EglN1-CD variant, promoted endogenous SFMBT1 hydroxylation that could be inhibited by DMOG treatment, as detected by immunoprecipitation with a pan-hydroxylation antibody (Figure 2L). We then depleted EglN1 and restored with physiologically relevant levels of EglN1 WT or catalytic dead (EglN1 CD) mutant, and examined SFMBT1 hydroxylation. WT EglN1, but not the EglN1-CD variant, could promote SFMBT1 hydroxylation (Figure 2M). Consistently, EglN1 depletion by three different siRNAs led to increased exogenous WT SFMBT1 protein levels (Figure 2N). Mutation of the HA-SFMBT1 prolyl hydroxylation site P651A totally abrogated the regulation of HA-SFMBT1 by EglN1 depletion (Figure 2N). In addition, we also performed an in vitro hydroxylation assay with SFMBT1 peptide and EglN1 enzyme. Our results showed that the Km of SFMBT1 was 3.5 μm (Figure S2K–L). Together, our data suggest that EglN1 is the primary prolyl hydroxylase that hydroxylates SFMBT1 on proline 651, which leads to pVHL binding and proteasomal degradation.
SFMBT1 is upregulated in ccRCC Patient Tumors
Next, to examine the physiological relevance of SFMBT1 in ccRCC, where most of patients carry VHL loss-of-function mutations, we analyzed 11 pairs of tumor and normal tissues from ccRCC patients. Most tumors carrying splice variant, missense, or frame-shift mutations displayed consistent upregulation of SFMBT1 compared to paired normal tissues, which correlated well with HIF2α levels in these patients (Figure 3A). Conversely, ccRCC tumors with wild-type VHL did not display distinctive upregulation of SFMBT1 or HIF2α (Figure 3A). We next examined SFMBT1 mRNA in the 11 pairs of normal and tumor tissues. Among them, 8 showed comparable mRNA levels between normal and tumors, one pair showed decreased SFMBT1 mRNA in the tumor and the other 2 pairs displayed increased SFMBT1 mRNA in tumors (Figure S3A). Therefore, VHL loss induced SFMBT1 protein upregulation is likely due to protein stabilization. We next performed immunohistochemical (IHC) staining for these tumors and found that SFMBT1 expression was increased in the nuclei of tumors with VHL loss-of-function mutations compared to their respective adjacent normal tissues (Figure 3B). We also examined SFMBT1 protein levels in tumors from a previously generated ccRCC mouse model with pVHL loss (Bailey et al., 2017); SFMBT1 was upregulated in multiple ccRCC tumors compared to normal kidney tissues (Figure S3B), suggesting the importance of SFMBT1 in human and mouse ccRCC pathogenesis. To corroborate our IHC staining results for SFMBT1 in ccRCC patient tumors, we also stained two sets of ccRCC tissue microarrays (TMAs) for SFMBT1. Consistent with our previous IHC staining in individual ccRCC tumors, SFMBT1 showed higher nuclear intensity in tumors compared to normal controls in both TMA sets, as reflected by representative IHC staining images as well as quantitative analyses (Figure 3C–D).
Figure 3. SFMBT1 is upregulated in ccRCC patients.
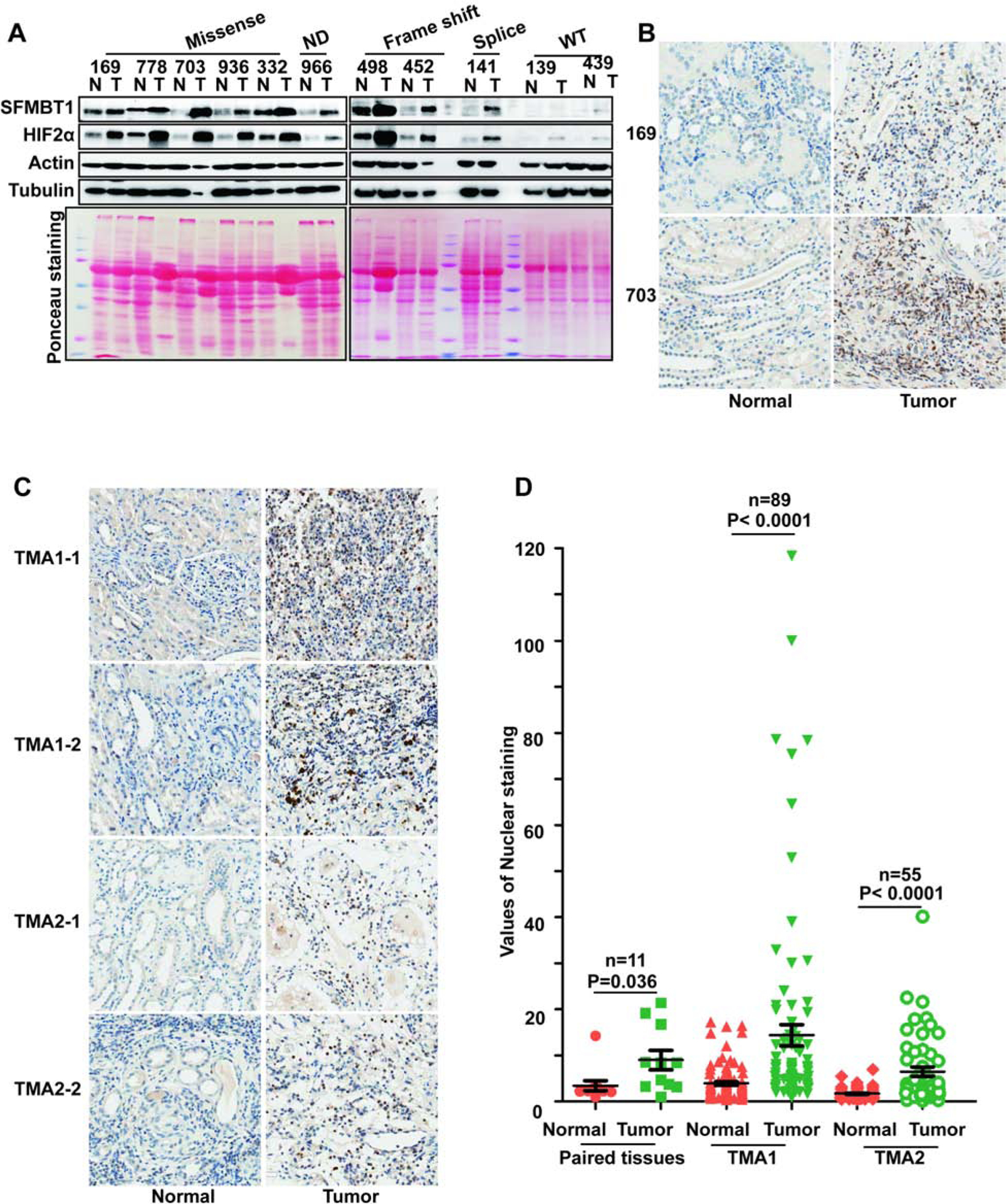
(A) Immunoblots for lysates from indicated ccRCC paired patient non-tumor (N) and tumor (T) tissues.
(B-C) Representative SFMBT1 immunohistochemical (IHC) staining for indicated ccRCC patient paired tissues (B) and tissue microarrays (TMA1 and TMA2)(C).
(D) Quantification of SFMBT1 nuclear staining for B-C.
SFMBT1 Controls ccRCC Cell Proliferation, Anchorage-Independent Growth and Tumorigenesis
SFMBT1 was initially described as a transcriptional repressor (Wu et al., 2007). However, the mechanisms underlying this function, as well as the potential implication of SFMBT1 in cancer, remain largely unexplored. Given the important role of pVHL in ccRCC and the identification of SFMBT1 as a target for pVHL, we aimed to uncover the function of SFMBT1 in ccRCC. To this end, SFMBT1 expression in pVHL-null 786-O renal cancer cells was depleted using three different validated hairpins (1, 2 and 3) in a PLKO-based lentiviral vector. SFMBT1 depletion significantly decreased cell proliferation (Figure 4A–B) and 3-D soft-agar growth (Figure 4C and S4A). We also observed a similar cell proliferation and 3-D soft agar growth defect upon SFMBT1 depletion in another ccRCC cell line, UMRC2 (Figure S4B–E). To further confirm that this phenotype was due to the on-target effects of SFMBT1 hairpins, we depleted SFMBT1 expression and then co-transfected UMRC2 cells with an shRNA-resistant SFMBT1 expression plasmid (Figure 4D). Whereas the SFMBT1 shRNA decreased cell proliferation and colony formation, shRNA-resistant SFMBT1 overexpression efficiently rescued the phenotype (Figure 4E–F and S4F), suggesting that these phenotypes were due to on-target consequences of SFMBT1 depletion.
Figure 4. SFMBT1 regulates ccRCC cell proliferation, anchorage-independent growth and tumorigenesis.
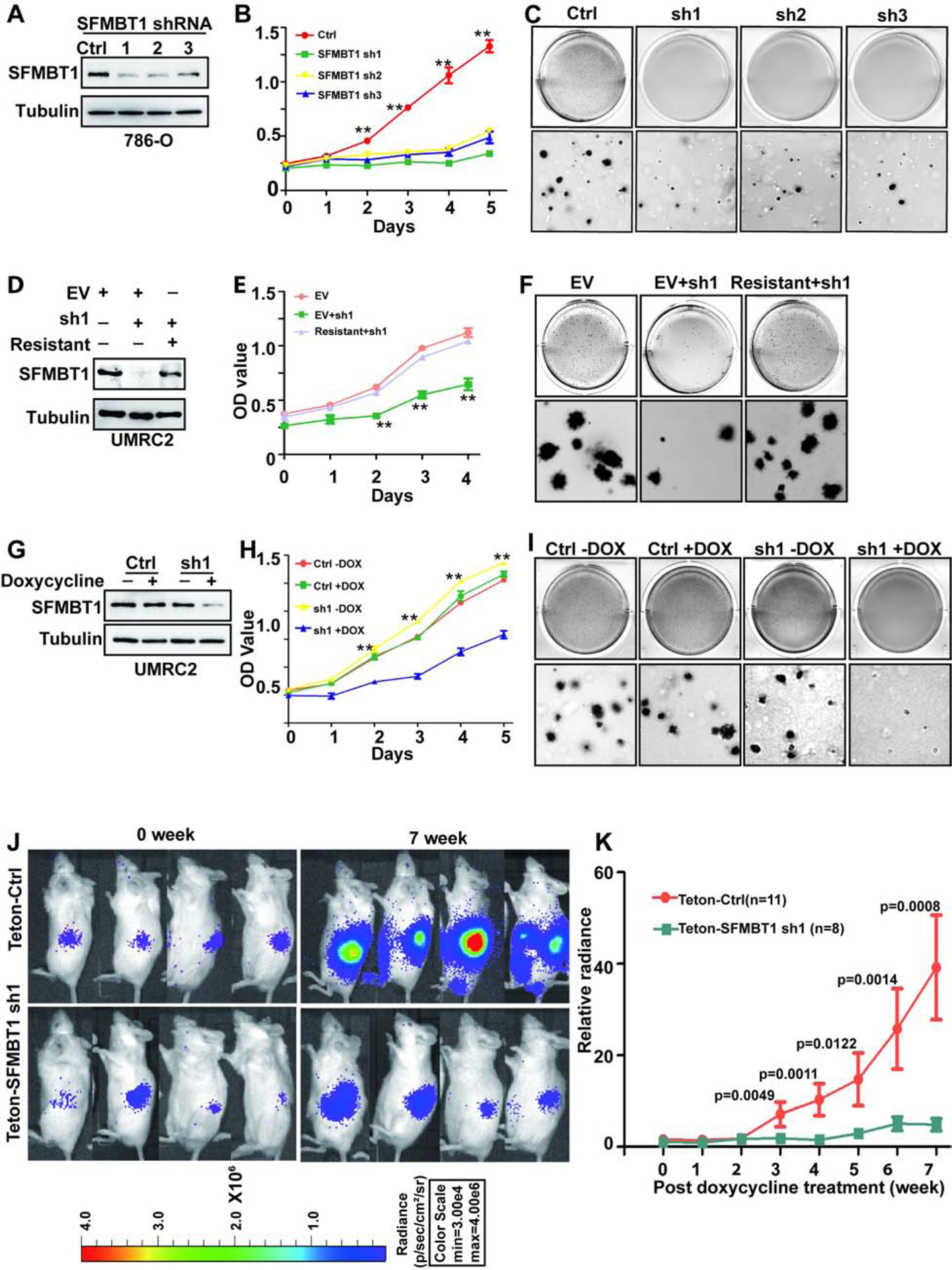
(A-C) Immunoblots for lysates (A), cell proliferation assays (B) and representative anchorage-independent growth assays (C) in cells transduced with lentivirus expressing either control shRNA (Ctrl) or SFMBT1 shRNAs (sh1, sh2 and sh3). * Ctrl vs SFMBT1 sh1/2/3; **, P<0.01.
(D-F) Immunoblots for lysates (D), cell proliferation assays (E) and representative anchorage-independent growth assays (F) in cells transduced with lentivirus expressing either control shRNA (Ctrl) or SFMBT1 sh1, followed by infection with lentivirus encoding empty vector (EV) or HA-SFMBT1 sh1-resistant (resistant). * EV+sh1 vs EV/Resistant+sh1; **, P<0.01; ***, P<0.001.
(G-I) Immunoblots for lysates (G), cell proliferation assays (H) and representative anchorage-independent growth assays (I) in UMRC2 luciferase stable cells tranduced with lentivirus expressing either Teton control shRNA (Ctrl) or Teton-SFMBT1 shRNA1 (sh1) and treated with or without doxycycline as indicated. * sh1+dox vs sh1-dox/Ctrl-dox/Ctrl+dox; **, P<0.01. (J-K) Representative bioluminescence imaging of before (0 week) and 7 weeks post-doxycycline treatment (J) and quantification of post-doxycycline treatment bioluminescence imaging (K) from 786-O luciferase stable cells transduced with lentivirus expressing either Teton control (Teton-Ctrl) or Teton-SFMBT1 sh1 that were injected orthotopically into the renal sub-capsule of NOD SCID gamma (NSG) mice.
To examine whether SFMBT1 was important for ccRCC tumor growth, we constructed 786-O cells expressing an inducible SFMBT1 shRNA, which efficiently depleted SFMBT1 levels upon doxycycline addition in ccRCC cell lines (Figure 4G). SFMBT1 depletion also showed decreased cell proliferation and soft-agar growth upon doxycycline addition in UMRC2 cells (Figure 4H–I and S4G) and 786-O cells (Figure S4H–K). Next, either control or SFMBT1 shRNA (sh1) cells were orthotopically injected into the renal capsules of NOD scid gamma (NSG) mice. Upon confirmation of tumor growth in vivo by consecutive weekly bioluminescence imaging, we administered doxycycline to induce SFMBT1 hairpin expression and monitored live tumor growth. Whereas cells expressing control hairpins grew readily during the 7 weeks after the addition of doxycycline, SFMBT1 hairpin-expressing cells failed to proliferate in vivo (Figure 4J–K). Taken together, our results suggest that SFMBT1 is important for ccRCC both in vitro and in vivo.
SFMBT1 Activates Gene Expression in ccRCC
To investigate how SFMBT1 affects ccRCC tumorigenesis, we performed chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) to assess the genomic localization of SFMBT1 in ccRCC. 3073 binding sites were found, of which 91% overlapped with H3K27ac, 85% with H3K4me3, and only 12% with H3K4me1 (Figure S5A), suggesting that SFMBT1 binds preferentially to active gene promoters (Shlyueva et al., 2014). In addition, there was limited overlap between SFMBT1 and either HIF2α or HIF1β binding sites, indicating that SFMBT1 regulates distinctive downstream events from HIF2α or HIF1β (Figure S5A and 5A). To identify the genes that SFMBT1 may preferentially activate, we performed RNA-seq with two independent SFMBT1 siRNAs and focused on SFMBT1 positively regulated genes. We compared these genes to those activated by HIF2α, as reported in a previous study (Yao et al., 2017). There was very little overlap between SFMBT1 and HIF2α activated genes (Figure S5B), further suggesting that SFMBT1 promotes ccRCC tumorigenesis through HIF2α-independent signaling. It is important to note that these HIF2α activated genes may be an underrepresentation that could result from incomplete depletion of HIF2α by siRNA. However, an analysis of enriched transcription factor motifs within SFMBT1 binding sites did not reveal a HIF motif, and instead showed enrichment for RBPJ1, MYC, AP-1, among others (Figure S5C).
Figure 5. Identification of critical SFMBT1 direct target genes in ccRCC.
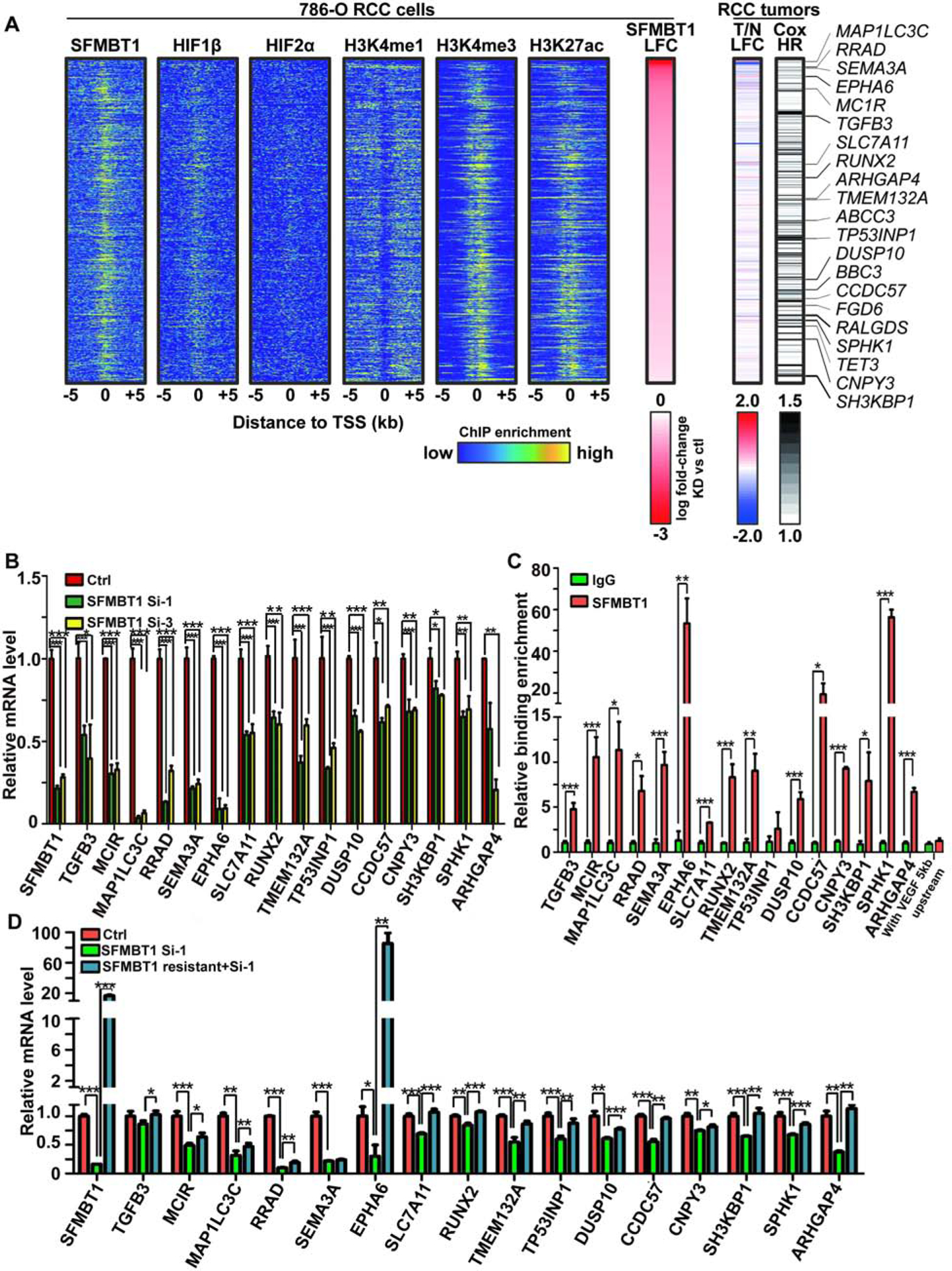
(A) Integrated analyses of ChIP-Seqs (including SFMBT1, HIF1β, HIF2α, H3K4me1, H3K4me3 and H3K27ac), signals expressed as relative to input control when available. Log2 fold change (LFC) for SFMBT1 RNA-seq with two different siRNAs; Log2 fold change for kidney clear cell carcinoma (KIRC) tumor (T) vs normal (N) in TCGA database as well as patient prognosis (Cox HR value). Critical target genes were marked on the right.
(B) qRT-PCR quantification of SFMBT1 target genes from 786-O cells transfected with SFMBT1 control siRNA (Ctrl) or siRNAs (si-1 and si-3)
(C) ChIP-qPCR validation of binding for SFMBT1 at 16 target gene promoters compared to IgG control. VEGF 5kb upstream promoter sequence as a negative locus control for SFMBT1.
(D) qRT-PCR quantification of SFMBT1 target genes from 786-O cells transduced with lentivirus expressing either control or SFMBT1 si1-resistant (SFMBT1 resistant) HA-SFMBT1, followed by transfection with control siRNA (Ctrl) or SFMBT1 siRNA1 (si-1).
For all the panels, *, P<0.05; **, P<0.01; ***, P<0.001.
Next, to identify the critical SFMBT1 target genes involved in ccRCC tumorigenesis, we applied the following stringent criteria for genes: (1) displayed positive regulation by SFMBT1 (downregulated by both SFMBT1 siRNAs) and exhibited SFMBT1 binding in the promoter (±5 kb from transcription start site), resulting in 516 genes (Fig S5D); (2) contained H3K4me3 and H3K27ac ChIP-seq in the promoter (±5 kb from transcription start site) (Figure 5A); (3) showed elevated expression in ccRCC patient tumors compared to normal in TCGA Kidney Clear Cell Carcinoma (KIRC) data set (Cancer Genome Atlas Research, 2013) (Figure 5A); (4) their high expression predicted worse prognosis in ccRCC patients (Figure 5A). By combining all these parameters, we narrowed down the direct SFMBT1 target gene list to 20 genes (Figure 5A and S5E). Among them, sixteen genes displayed reliable gene expression as well as regulation by SFMBT1 (Figure 5B). Next, we further confirmed SFMBT1 binding on these target gene promoters by ChIP-qPCR (Figure 5C). In addition, we performed RT-PCR to examine the potential rescue of gene expression by siRNA resistant SFMBT1 cDNA clones. In nearly all target genes (15 out of 16), expression was completely/partially rescued by the introduction of siRNA-resistant SFMBT1 (Figure 5D), further strengthening our hypothesis that these are direct SFMBT1 target genes contributing to ccRCC pathogenesis.
SPHK1 is the Critical SFMBT1 Target Gene that Contributes to ccRCC Tumorigenesis
We examined the ChIP-seq signals of SFMBT1, HIF2α, HIF1β, H3K4me1, H3K4me3, and H3K27Ac for each of these 16 genes (Figure S5F) and observed particularly robust binding by SFMBT1 to sphingosine kinase 1 (SPHK1), which coincided with strong H3K4me3 and H3K27ac enrichment, but not HIF2α or HIF1β (Figure 6A, 5A, and S5F). Our ChIP-qPCR further confirmed the strong enrichment of SFMBT1 on the SPHK1 gene promoter (Figure 5C).
Figure 6. SPHK1 is an SFMBT1 direct target gene in ccRCC.
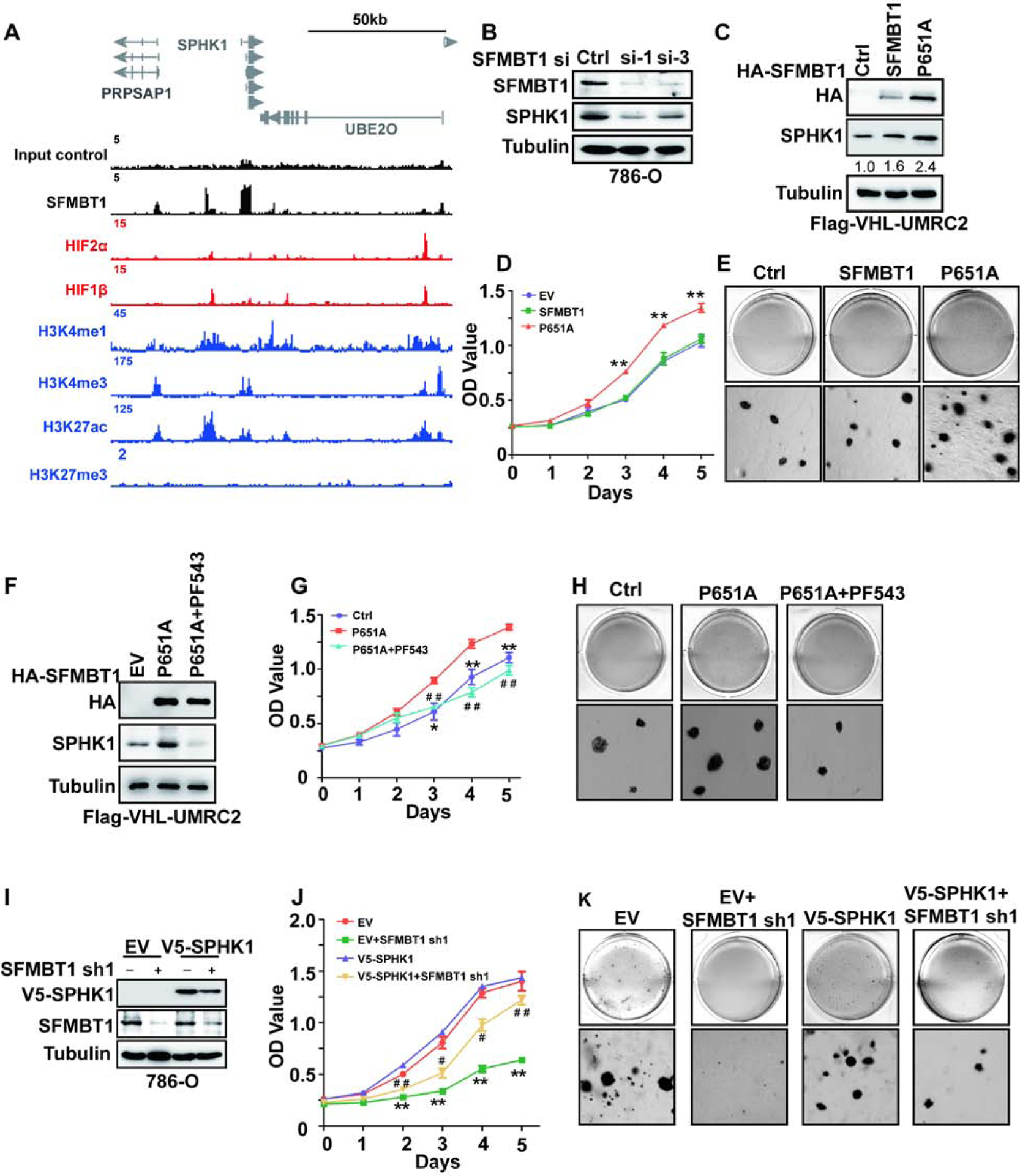
(A) ChIP-seq binding peaks on SPHK1 for SFMBT1, HIF2α, HIF1β, H3K4me1, H3K4me3, H3K27ac and H3K27me3.
(B) SPHK1 protein level in cells transfected with control siRNA (Ctrl) or SFMBT1 si-1 and si-3.
(C-E) Immunoblots for lysates (C), cell proliferation assays (D) and representative anchorage-independent growth assays (E) in cells infected with lentivirus encoding either empty vector (EV), HA-SFMBT1 or P651A. * EV/SFMBT1 vs P651A; **, P<0.01. The values listed below the blots (C) indicate the relative SPHK1 protein levels with Tubulin normalization.
(F-H) Immunoblots for lysates (F), cell proliferation assays (G) and representative anchorage-independent growth assays (H) in cells infected with lentivirus encoding either empty vector (EV), P651A with or without PF543 treatment. * P651A vs Ctrl; # P651A+PF543 vs Ctrl. */#, P<0.05; **/# #, P<0.01.
(I-K) Immunoblots for lysates (I), cell proliferation assays (J) and representative anchorage-independent growth assays (K) in cells transduced with lentivirus expressing either SFMBT1 shRNA control (Ctrl) or SFMBT1 sh1, followed by infection with lentivirus encoding either empty vector (EV) or v5-SPHK1. * EV+shSFMBT1 sh1 vs EV; # v5-SPHK1+SFMBT1 sh1 vs EV. */#, P<0.05; **/# #, P<0.01.
SPHK1 is a kinase that catalyzes the phosphorylation of sphingosine to form sphigosine-1-phosphate (S1P). By analyzing TCGA data, we found that SPHK1 expression was higher in tumors compared to normal tissues, and its high expression predicted worse prognosis in ccRCC (Figure S6A–B). Consistent with this finding, we also found that higher stage (III and IV) tumors expressed higher levels of SPHK1 mRNA compared to lower stage (I and II) tumors and to normal tissue (Figure S6C). Therefore, we decided to focus our efforts on characterizing the role of SPHK1 in SFMBT1 signaling in ccRCC. SFMBT1, but not HIF2α or HIF1β, occupies the proximal promoter region of SPHK1 (Figure 6A). Consistent with the reduction in SFMBT1 following pVHL restoration in pVHL-null ccRCC cell lines (Figure 1D), SPHK1 protein levels were also reduced when pVHL was restored (Figure S6D). SFMBT1 depletion by two independent siRNAs in pVHL-null ccRCC cells led to decreased SPHK1; this effect was rescued by the expression of the siRNA-resistant version of SFMBT1 (Figure 6B and Figure S6E), but was unaffected by HIF2α or HIF1β depletion (Figure S6F–G) in 786-O cells. In addition to SFMBT1 knockdown experiments, we also overexpressed control, WT, or P651A mutant HA-SFMBT1 in UMRC2 cells with FLAG-pVHL restored. As expected, P651A HA-SFMBT1 protein level was higher than that of its wild-type counterpart (Figure 6C), most likely due to the inability of P651A HA-SFMBT1 to be marked for degradation by pVHL. Consistently, ccRCC cells expressing P651A HA-SFMBT1 displayed higher SPHK1 protein levels, which corresponded to increased cell proliferation and soft-agar growth compared to either control vector or WT SFMBT1-infected cells (Figure 6C–E, and Figure S6H). Next, we overexpressed control, WT, or P651A mutant HA-SFMBT1 in the VHL WT cell line HKC, and saw similar effects here (Figure S6I–L) to that of FLAG-pVHL restored UMRC2 cells (Figure 6C). We also cultured these cells under hypoxia; hypoxia led to an increase in protein levels of endogenous SFMBT1 and exogenous WT SFMBT1, but not the P651A SFMBT1 mutant, suggesting that Proline 651 is the major hydroxylation site important for its protein level regulation (Figure S6M). Additionally, we found that EV and WT SFMBT1 expressing HKC cells grew faster under hypoxia compared to normoxia. However, the cell proliferation rate for P651A expressing HKC cells was not affected by hypoxia compared to normoxia (Figure S6N).
Next, we aimed to determine whether SPHK1 mediated the effect of SFMBT1 on ccRCC cell proliferation. For this purpose, we utilized the specific SPHK1 inhibitor PF543 in ccRCC cells (Schnute et al., 2012), which was shown to induce SPHK1 proteasomal degradation (Byun et al., 2013, Baek et al., 2013). PF543 treatment resulted in decreased cell proliferation and 3-D soft-agar growth in 786-O and UMRC2 cells in a dose-dependent manner (Figure S6O–V). Whereas P651A SFMBT1 expression upregulated SPHK1 protein level, this effect was abrogated by PF543 (Figure 6F). ccRCC cells expressing P651A HA-SFMBT1 displayed increased cell proliferation and 3-D soft-agar growth (Figure 6G–H, and S6W); this effect was also disrupted by the co-treatment with PF543. We also depleted SFMBT1 by shRNAs followed by either control or V5-SPHK1 overexpression; V5-SPHK1 overexpression could rescue the effect of SFMBT1 depletion on cell proliferation and soft-agar growth (Figure 6I–K, and Figure S6X). More importantly, PF543 did not grossly affect cell proliferation in HKC cells as measured by either a 2-D MTS assay or 3D soft agar assay (Figure S7A–D). We also found that depleting SPHK1 by two different shRNAs inhibited 786-O and UMRC2 cell proliferation and soft-agar growth (Figure S7E–L). In summary, SPHK1 is a major mediator for the oncogenic phenotype of SFMBT1 on ccRCC cell proliferation.
SPHK1 is upregulated in ccRCC Patient Tumors and Controls Tumorigenesis
Next, to examine the physiological relevance of SPHK1 in ccRCC, we assessed SPHK1 and HIF2α protein levels in the 11 pairs of normal and tumor tissues from ccRCC patients (same as in Figure 4A). Most tumors displayed consistent upregulation of SPHK1 compared to paired normal tissues, which correlated well with HIF2α levels in these patients (Figure 7A). We next performed immunohistochemical (IHC) staining for these tumors and found that SPHK1 expression was increased in the cytoplasm of tumors with VHL loss-of-function mutations compared to their respective adjacent normal tissues (Figure 7B). To corroborate our IHC staining results for SPHK1 in ccRCC patient tumors, we also stained one set of ccRCC tissue microarrays (TMA2) for SPHK1 (same as in Figure 4C–D). Consistent with our previous IHC staining in individual ccRCC tumors, SPHK1 showed higher cytoplasmic staining intensity in tumors compared to normal controls, as reflected by representative IHC staining images as well as quantitative analyses (Figure 7C–D).
Figure 7. SPHK1 is upregulated in ccRCC patients.
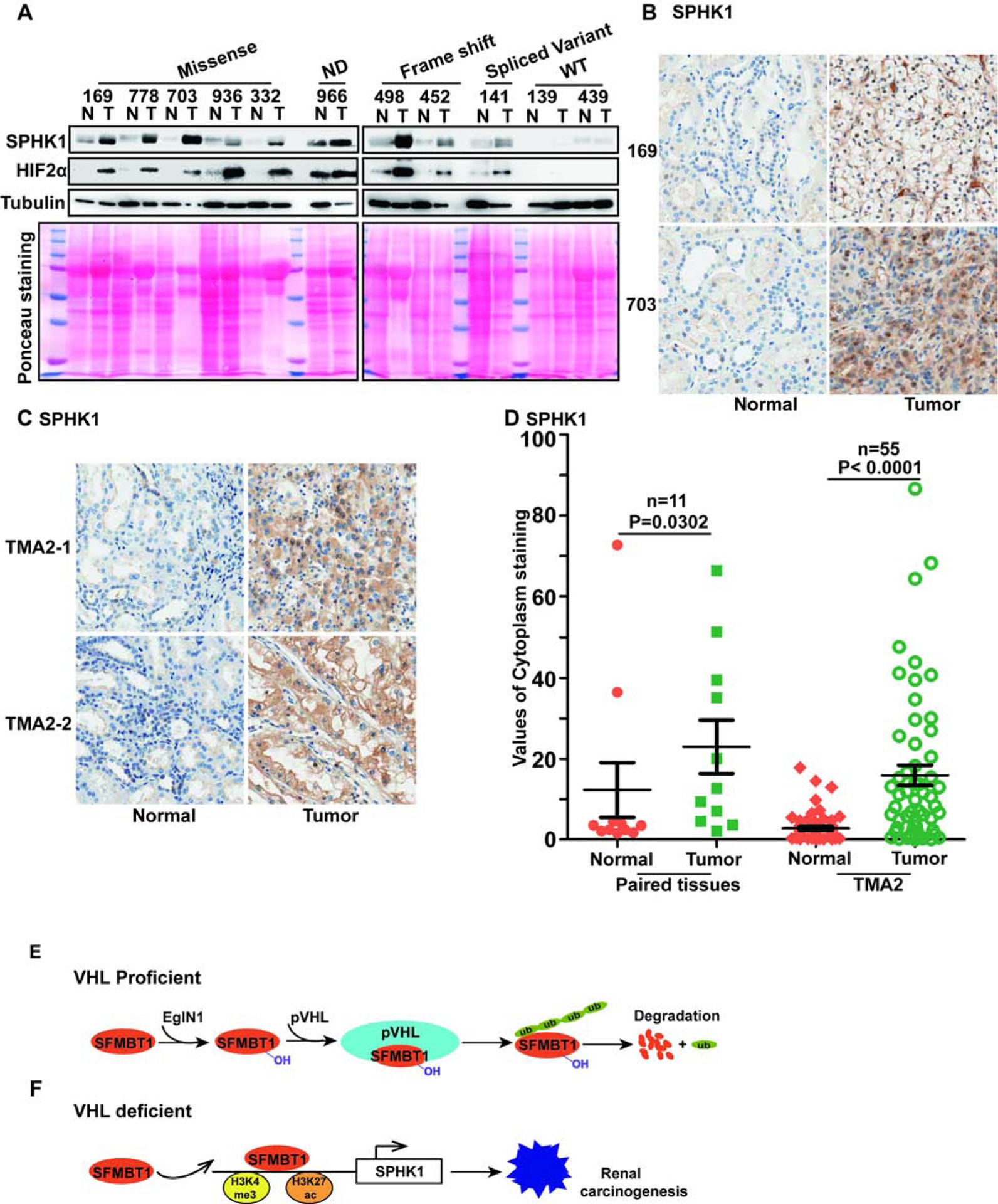
(A) Immunoblots for lysates in indicated ccRCC paired patient non-tumor (N) and tumor (T) tissues.
(B-C) Representative SPHK1 immunohistochemical (IHC) staining for indicated ccRCC patient paired tissues(B) and tissue microarrays (TMA2)(C).
(D) Quantification of SPHK1 cytoplasm staining for B-C.
(E-F) Schematic diagram of relationship of pVHL, SFMBT1 and SPHK1.
We analyzed the correlation of SPHK1 and SFMBT1 staining intensity in the combined dataset of our 11 paired tissues and TMA2. As shown, in normal samples, SPHK1 and SFMBT1 staining showed a significant correlation (Spearman correlation=0.33, p=0.007358) (Figure S7M). In tumor samples, there was a trend that SPHK1 and SFMBT1 correlated but it did not reach statistical significance (Spearman correlation=0.226, p=0.06994) (Figure S7N). Next, we examined whether SPHK1 was important for ccRCC tumor growth by orthotopically injecting 786-O cells into the renal capsules of NSG mice. Upon confirmation of tumor growth in vivo, we treated mice with PF543 to inhibit SPHK1. PF543 did inhibit SPHK1 but not SPHK2 expression level (Figure S7O) in vivo. PF543 also inhibited ccRCC tumor growth in vivo (Figure S7P–Q). Taken together, our results suggest that SPHK1 is important for ccRCC tumor growth in vivo.
Discussion
SFMBT1 is regulated by pVHL through a prolyl hydroxylation- and proteasomal degradation-dependent mechanism (Figure 7E). Loss of pVHL leads to increased SFMBT1 level, which promotes cell proliferation and xenograft tumor growth in ccRCC cells (Figure 7F). Our results provide some new evidence that targeting the SFMBT1-SPHK1 axis decreases kidney cancer fitness in HIF2α-inhibitor-resistant cancer cell lines (such as UMRC2).
VHL is the predominant tumor suppressor gene in renal cancer. The canonical pathway for pVHL E3 ubiquitin ligase complex activity is that VHL loss leads to HIF2α accumulation, which promotes renal cell malignant transformation (Kondo et al., 2003, Kondo et al., 2002). However, recent developments have helped understand the function of pVHL from several different perspectives. Emerging literature suggests the existence of additional pVHL substrates beside HIFs (Li and Kim, 2011), such as NDRG3 (Lee et al., 2015) and ZHX2 (Zhang et al., 2018). Therefore, the function of pVHL appears to be multi-faceted, including both E3 ligase-dependent and -independent functions that reach beyond canonical HIF signaling (Zhang and Zhang, 2018). However, the identification of novel and relevant pVHL E3 ligase complex substrates remains a challenge. One reason is that a unique modification of pVHL substrates (such as prolyl hydroxylation) may be required for pVHL recognition. Therefore, such proteins may not be easily identified by traditional pull-down approaches in the case of low hydroxylated protein levels. Another reason could be the low abundance of these substrates in cell lysates, similar to the case of very low basal levels of HIFα factors. By using an in vitro expression strategy with approximately 17,000 genes divided into 700 pools (24 genes/pool), coupled with a GST-pVHL binding screen, we identified novel pVHL binding partners, as well as potential pVHL substrates. More importantly, our strategy can be broadly applied to search for other E3 ligase substrates.
We have accumulated a substantial amount of evidence showing that SFMBT1 is a potent oncogene and transcriptional regulator in kidney cancer in a HIF-independent manner. However, some questions remain to be answered. SFMBT1 proline 651, the major hydroxylation site that contributes to the protein stability regulated by pVHL, is located outside of its MBT domains. It remains unclear whether the oncogenic phenotype of SFMBT1 is dependent on its MBT domains. SFMBT1 is considered to be a component of polycomb-repressive complex and is largely considered to be a transcription repressor (Sauvageau and Sauvageau, 2010). Specifically, SFMBT1 was shown to form a complex with LSD1 and CoREST in cells (Zhang et al., 2013a, Tang et al., 2013), though our data show that SFMBT1 both activated and repressed gene expression. We have not examined SFMBT1 in other cancer types since VHL loss is primarily restricted to kidney cancer, however this will be explored further in future studies. It is also intriguing that SFMBT1 binding sites were enriched for motifs corresponding to transcription factors RBPJ1, MYC and AP-1. RBPJ1 was reported to be a transcriptional activator for Notch signaling (Nam et al., 2006, Tani et al., 2001). pVHL loss was shown to activate Notch signaling in renal cancer (Sjolund et al., 2008). It is possible that the pVHL-SFMBT1-RBPJ1 axis may be able to modulate Notch activity in renal cancer, though this remains to be determined.
S1P signaling, acting downstream of SPHK1, plays an important role in diseases such as cancer and inflammatory diseases (Kunkel et al., 2013). The anti-S1P antibody was previously identified to be a novel therapeutic option for VEGFR tyrosine kinase inhibitor (TKI)-resistant renal cancer (Zhang et al., 2015). VEGF/VEGFR signaling acts downstream of HIF2α. One of the potential reasons for the VEGFR TKI resistance in renal cancer could be SPHK1 regulation by pVHL loss in an SFMBT1-dependent, but HIF2α-independent manner. It is important to point out that there may be other downstream targets of SFMBT1 that may be functionally important in kidney cancer, which remains to be determined.
During the preparation of our manuscript, a new study suggested a lack of evidence for the hydroxylation of non-HIF prolyl hydroxylase substrates by prolyl hydroxylases in vitro (Cockman et al., 2019). Our current study did perform EglN1-mediated hydroxylation with SFMBT1 peptide in vitro and we found that there was increased hydroxylation signal with various concentrations of SFMBT1 peptides, albeit to much weaker extent compared to HIFα peptide. We also performed various experiments in vivo under physiologically relevant conditions to corroborate our in vitro data supporting the potential for SFMBT1 hydroxylation. However, we acknowledge that our assay did not measure the percentage of SFMBT1 hydroxylation in vitro or in vivo, which remains to be determined in future studies. However, this recently published study suggests that we will need a more rigorous approach to establish that SFMBT1 is indeed a bona fide EglN substrate, which we recognize as a limitation of our current study.
In summary, the most important tumor suppressor in kidney cancer, pVHL, appears to regulate multiple oncogenic signaling pathways in parallel. Whereas HIF2α is a well-established contributor to kidney cancer, SFMBT1 may represent another important oncogenic signaling pathway that could be responsible for a significant portion of renal tumors that do not respond to HIF2α inhibitor or multi-tyrosine kinase inhibitors (including Sorafenib and Axitinib). Since currently no potent SFMBT1 inhibitor is available, targeting its downstream target gene SPHK1 with SPHK1 inhibitors, either alone or in combination with HIF2α pathway inhibitors, may represent a new therapeutic avenue for kidney cancer treatment.
STAR ★ METHODS
LEAD CONTACT AND AVAILABILITY STATEMENT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qing Zhang (qing.zhang@utsouthwestern.edu).
All materials generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Orthotopic Tumor Growth
Six-week old NOD SCID Gamma mice (NSG, Jackson Lab) were used for xenograft studies. About 2×106 viable 786-O kidney cancer cells were resuspended in 20 μL fresh growth medium and injected orthotopically into the left kidney of each mouse as described previously (Li et al., 2013). Bioluminescence imaging was performed as described previously (Zhang et al., 2009), after making sure tumors were successfully implanted in the kidney, mice were fed Purina rodent chow with doxycycline (#5001, Research Diets Inc.). Mice were euthanized 7 weeks after treatment with doxycycline. All animal experiments were in compliance with National Institutes of Health guidelines and were approved by the University of North Carolina at Chapel Hill Animal Care and Use Committee.
METHOD DETAILS
Genome-Wide Screening
A cDNA library containing approximately 17,000 human ORFs was purchased from Mammalian Gene Collection (MGC). These ORF clones were purchased as bacterial stock in a 96-well plate format. To convert the library into plasmid DNA for in vitro protein synthesis, the bacterial stock for each ORF clone was cultured separately in LB media overnight, the density of the cultures was measured the next morning and equal amounts of bacteria was taken from the cultures of 24 different clones and mixed together for miniprep (Qiagen). Eluted plasmid DNA for each pool of 24 clones was arranged in a 96-well plate (Ayad et al., 2005). Through this approach, the whole library was converted to about 700 pools of plasmid DNA with 24 individual clones/pool. For in vitro protein synthesis, 1 μl of each DNA pool was added to 4 μl of in vitro transcription/translation (IVT) master mix (Promega) in the presence of 35S Methionine. The IVT mixtures were incubated at 37C for 90 minutes for the production of in vitro translated proteins. Upon completion of IVT, 2 μl of each IVT mixture was incubated with GST-VHL complex (containing GST-VHL, Elongin B and C mixture) bound to glutathione beads (Yang et al., 2004) with either WT HIF1α peptide or hydroxylated HIF1α peptide as a competitor for 4 hours. Subsequently, the reaction mixtures were washed 4 times with NETN lysis buffer, and proteins bound to the glutathione beads were eluted and resolved on an SDS-PAGE. Finally the protein gels were dried for auto-radiography.
In vitro decarboxylation assay
In brief, peptides were supplemented with 50 mM HEPES (pH 7.4), 1500 U/mL catalase, 100 mM FeSO4, 1 mM ascorbic acid, 0.15 μCi/mL [1-14C] α-KG (Perkin Elmer), and 2 mg of recombinant EglN1 enzyme (Active motif, 81765) in a 100 μl reaction volume. Radiolabeled CO2 was captured with glass fiber filter paper (catalog no. IH-201-A, Inotech Biosystems International).The reaction was quenched by KH2PO4 buffer. The reaction mixture was transferred to a 37°C oven and allowed to incubate for 1 h and then shaken for 30 min at room temperature. The CPM value was determined using a liquid scintillation analyzer (PerkinElmer).
Human ccRCC paired tissues
For human ccRCC paired tissues, fresh-frozen samples of ccRCC and adjacent normal tissues were obtained from the tissue procurement facility at UNC. Tissues were diced, lysed in 8M Urea buffer and followed by sonication. The lysates were subjected to Western blotting as described above. IRB approval (13–1986 from UNC Chapel Hill) was obtained before obtaining these human tumor and normal tissues.
IHC and TMA
For IHC and TMA with SFMBT1 and SPHK1, a rabbit polyclonal antibody against SFMBT1 was purchased from Atlas Antibodies (HPA064564). A rabbit polyclonal antibody against SPHK1 was purchased from Cell Signaling (D1H1). IHC was carried out in the Bond Autostainer (Leica Microsystems Inc.; Norwell MA). Briefly, slides were dewaxed in Bond Dewax solution (AR9222) and hydrated in Bond Wash solution (AR9590). Antigen retrieval was performed for 20 min at 100°C in Bond-Epitope Retrieval solution 1, pH-6.0 (AR9961). Slides were incubated with primary antibody (1:150) for 30min (SFMBT1), and then with second antibody (Leica, RE7261). Antibody detection was performed using the Bond Intense R detection system (DS9263) with ImmPress HRP anti-rabbit IgG (MP-7451; Vector Laboratories; Burlingame, CA). Stained slides were dehydrated and coverslipped. Positive and negative controls (no primary antibody) were included during the run. For digital imaging and image analysis, stained slides were digitally scanned at 20x magnification using Aperio ScanScope-XT (Aperio Technologies, Vista, CA) were uploaded to the Aperio eSlideManager database (Leica Biosystems Inc; eSlideManager version 12.3.3.7075) at the Translational Pathology Laboratory at UNC. TMA images were digitally segmented into cores using TMALab (Aperio). Individual TMA cores were separately analyzed using the Aperio Cytoplasmic v2 algorithm with slight adjustments for cell shape. The number and percentage of cells with light (1+), medium (2+) and strong (3+) nuclei and/or cytoplasmic staining was determined. H Scores were calculated using the following formula: 3 x percentage of strongly staining nuclei + 2 x percentage of moderately staining nuclei + percentage of weakly staining nuclei, giving a range of 0 to 300.
LC-MS/MS analysis
Cell lysate from FLAG-SFMBT1 expressing cells treated with either MG132 or MG132 together with DMOG was immunoprecipitated from 293T cells. Immunoprecipitates were separated by SDS-PAGE and visualized by Coomassie staining. Bands corresponding to SFMBT1 were excised and the proteins were reduced, alkylated and digested with trypsin overnight at 37°C followed by mass spectrometry. Peptides were extracted, desalted with C18 spin columns (Pierce) then dried via vacuum centrifugation.
Transfection, Virus Production and Infection
For immunoprecipitation, 1.5×106 293T cells were treated with drugs for 8 hours or overnight and lysates were harvested 48 hours post transfection (hpt). Lentiviruses were prepared by transfecting 293T cells (2×106 cells in 100-mm dishes) as previously described (Zhang et al., 2009). Following a media change at 12 hpt, the supernatant containing the lentiviruses were harvested at 48 and 72 hpt. After filtering with 0.45-μm filters, viruses were used to infect target cells in the presence of polybrene (8 μg/mL) or stored at −80°C.
Cell Proliferation Assays and Anchorage-Independent Growth assay
For cell proliferation assays, cells were seeded in triplicate in 96-well plates (1000 cells/well) in appropriate growth medium. 10000 cells/mL for 786-O cells and 3000 cells/mL for UMRC2 were used in anchorage-independent growth assay as described previously (Zhang et al., 2018).
Immunoprecipitation and GST pull-down assay
For the protein immunoprecipitation assay (IP), the cell pellets were lysed in EBC buffer for 30 min. After sonication, cells were spun at 12,000 g for 5 min at 4 °C to remove debris. 1 μg protein from whole cell lysate was incubated with indicated antibodies or anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220) / 3F10 HA conjugated beads (Roche Applied Bioscience, 11815016001) overnight or 4h at 4°C. Lysate incubated with antibodies was added to 10 μl of protein G agarose beads (Roche Applied Bioscience, 11243233001) with rotation for an additional 3 hours at 4°C.
For the binding between FLAG-VHL and HIF1α and SFMBT1 peptides, Comparable amounts of Biotin-tagged HIF1α (WT and P564-OH) or SFMBT1 (WT, P650-OH and P651-OH) peptides were incubated with NeutrAvidin Agarose Resin (Thermo, 29200) for 4h at 4°C. Mixtures were centrifuged, washed with EBC buffer and then mixed with in vitro translated (IVT) Flag-VHL overnight at 4°C. The bound complexes were then centrifuged, washed with EBC buffer, subjected to SDS-PAGE, and immunoblotted with FLAG antibody.
For the GST pull-down assay, IVT protein or protein from whole cell lysates were incubated with indicated GST fusion proteins overnight or 4 h at 4°C. Bound complexes were centrifuged, washed with EBC buffer, subjected to SDS-PAGE, and immunoblotted for the indicated targets.
Ubiquitination assay
Cells were harvested and extracted in 100 μL of EBC buffer containing 1% SDS. Cell extracts were heat-denatured for 5 min at 95°C and diluted with 900 μL EBC buffer. After sonication and centrifugation, cell lysates were subjected to immunoprecipitation with ubiquitin antibody (Santa Cruz Biotechnology, sc-8017).
In vitro hydroxylation followed by VHL binding assay
Comparable amounts of Biotin-tagged HIF1α or SFMBT1 peptides were mixed with reaction buffer (50 mM HEPES, 1500 U/mL catalase, 100 μM FeSO4, 5 mM ascorbic acid, 1 mM α-KG) and IVT HA-EglN1, IVT HA-EglN1-CD protein (enzyme) or negative control buffer (150 mM NaCl, 5 M FeSO4, 50 mM Tris-HCl, pH 7.5). The reaction mixture was incubated at 37°C for 1 h.
The reaction mixture was then incubated with NeutrAvidin™ Agrose Resin for 3–5 h, washed by EBC buffer and incubated with IVT FLAG-pVHL overnight at 4°C. Agrose Resin was collected by centrifugation, washed with EBC and subjected to immunoblot with an anti-FLAG antibody.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Seq analysis
RNA was extracted from 786-O cells using RNeasy kit with on-column DNase digestion (Qiagen), and sequenced at BGI as single-end 50 bp reads. Reads were then filtered for adapter contamination using cutadapt (Martin, 2011) and filtered such that at least 90% of bases of each read had a quality score >20. Reads were aligned to the reference genome (hg19) using STAR version 2.5.2b retaining only primary alignments (Dobin et al., 2013). Reads overlapping blacklisted regions of the genome were then removed. Transcript abundance was then estimated using salmon (Patro et al., 2017b), and differential expression was detected using DESeq2 (Love et al., 2014). HIF2α RNA Seq data was obtained from GSE86095 (Yao et al., 2017).
TCGA data
TCGA provides a kidney renal clear cell cancer (KIRC) data set which was previously processed and described (Hoadley et al., 2014). For mRNA expression data, we used RNA-seq by STAR-Salmon (Patro et al., 2017a) to quantify the transcript abundances measured by RNA sequencing and used the log2-transformed up-quantile-normalized (Bullard et al., 2010) STAR-Salmon values of the genes. We compared these gene expression between VHL WT and VHL mutant patients, by two-class unpaired t-test. We compared these gene expression between Stage I, II, III, IV and normal patients by Analysis of Variance (ANOVA).
Chromatin immunoprecipitation and Integrated analysis
ChIP was performed with SFMBT1 antibody (Bethyl, A303–221A) as previously described (Zhang et al., 2018). ChIP-seq libraries (Illumina) were prepared according to manufacturer’s recommendations and sequenced at the UNC High Throughput Genomic Sequencing facility using single-end 50bp reads. Reads were then filtered for adapter contamination using cutadapt (Martin, 2011) and filtered such that at least 90% of bases of each read had a quality score >20. Duplicated sequences were then capped at a maximum of 5 occurrences, and reads were aligned to the reference genome (hg19) using STAR (Dobin et al., 2013) version 2.5.2b retaining only primary alignments. Reads overlapping blacklisted regions of the genome were then removed. Reads were then extended in silico to a fragment size of 250 bp, and regions of significant enrichment relative to input control were identified using MACS2 (Zhang et al., 2008). Motifs were identified by comparing the 200 bp surrounding the peak midpoint to the 200 bp flanking sequence on either side of each identified peak using HOMER (Heinz et al., 2010). HIF ChIP-seq data were obtained from GSE34871, and histone modification ChIP-seq were obtained from GSE86095. Selection of important SFMBT1 direct target genes in ccRCC was based on the following criteria: For TCGA ccRCC data, Cox P-value <0.05, C-index >0.5 and Cox hazard ratio is above 1; Tumor-normal t-test P-value <0.05 and Tumor-normal Log2FC value >0.1. In addition, these genes should have SFMBT1, H3K4me3 and H3K27ac binding peaks in promoter regions.
Statistical analysis
The unpaired two-tail student’s t-test was used for experiments comparing two sets of data, unless otherwise indicated. For quantification of cell proliferation assays, if there were multiple comparisons with multiple time points and comparison groups, we multiplied the raw p-values by the number of comparisons (number of time points x number of experimental groups, in each plot) to get Bonferroni adjusted p-values for each comparison (in each plot). The significance of comparisons were then based in adjusted p-values. Mann-Whitney tests were used for animal experiments. Data represent mean ± SD from at least three independent experiments.
Data and code availability
All data generated or analyzed during this study are included in Figures 1–7 and Figures S1–S7. Additional datasets that support the findings of this study are available from the corresponding author upon reasonable request. ChIP-seq and RNA-seq data have been deposited to GEO under accession number GSE141577.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-SFMBT1 | Bethyl | Cat # A303–221A |
| Anti-HA Tag | Cell Signaling Technology | Cat # 3724S |
| Anti-pVHL | Cell Signaling Technology | Cat # 68547S |
| Anti-HIF1α | Cell Signaling Technology | Cat #14179 |
| Anti-OH-HIF1α | Cell Signaling Technology | Cat # D43B5 |
| Anti-FLAG-tag | Cell Signaling Technology | Cat #14793S |
| Anti-SPHK1 | Cell Signaling Technology | Cat #12071S |
| Anti-Pan hydroxyproline | Abeam | Cat #ab37067 |
| Anti-SFMBT1 | Sigma-Aldrich | Cat #SAB1400557 |
| Anti-HIF2α | Abeam | Cat #ab157249 |
| Anti-HIF1β | BD Biosciences | Cat #611078 |
| Anti-Tubulin | Cell Signaling Technology | Cat #3873S |
| Anti-ubiquitin | Santa Cruz Biotechnology | Cat # sc-8017 |
| Anti-biotin | Cell Signaling Technology | Cat # 5571s |
| Anti-GST | Santa Cruz Biotechnology | Cat # sc-138 |
| Bacterial and Virus Strains | ||
| E. coli BL21 | Homemade | N/A |
| Biological Samples | ||
| Human ccRCC paired tissues | Tissue procurement facility at UNC | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Catalase | Millipore-Sigma | Cat #C-100 |
| MG132 | Peptide international | Cat #IZL-3175-v |
| DFO | Millipore-Sigma | Cat #D9533 |
| FG4592 | Millipore-Sigma | Cat #10338 |
| DMOG | Frontier Scientific | Cat #D1070 |
| Puromycin | Gibco | Cat #A11138–03 |
| Hygromycin | Invitrogen | Cat #10687010 |
| PF543 Citrate | MedChem Express | Cat #HY-15425A |
| Biotin-HIF1 WT Peptide: DLDLEMLAPYIPMDDDFQLR | This paper | N/A |
| Biotin-HIF1 P564-OH: DLDLEMLAP#YIPMDDDFQLR | This paper | N/A |
| Biotin-SFMBT1 WT-OH: GKKKNKRIGRPPGGHSNLACA | This paper | N/A |
| Biotin-SFMBT1 P650-OH: GKKKNKRIGRP#PGGHSNLACA | This paper | N/A |
| Biotin-SFMBT1 P651-OH: GKKKNKRIGRPP#GGHSNLACA | This paper | N/A |
| Critical Commercial Assays | ||
| Quick Change XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat #200516 |
| Lipofectamine 3000 transfection reagent | Thermo Fisher Scientific | Cat #L3000015 |
| TNT® Quick Coupled Transcription/Translation Systems | Promega | Cat #L1170 |
| CellTiter 96 AQueous One Solution Cell Proliferation Assay | Promega | Cat #G3580 |
| Deposited Data | ||
| Mendeley Database | This paper | GSE141577 10.17632/zbx6×92dnv.1 |
| Experimental Models: Cell Lines | ||
| 786-O | American Type Culture Collection | N/A |
| UMRC2 | American Type Culture Collection | N/A |
| UMRC6 | American Type Culture Collection | N/A |
| RCC4 | American Type Culture Collection | N/A |
| HEK293T | American Type Culture Collection | N/A |
| HKC | American Type Culture Collection | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD SCID Gamma mice | UNC Animal Studies Core | N/A |
| Recombinant DNA | ||
| pCDNA-3.1-HA/FLAG-SFMBT1 (WT) | This paper | N/A |
| pGEX-4T1-VHL | This paper | N/A |
| pGEX-4T1-EglN1/2/3 | This paper | N/A |
| pCDNA-3.1-HA-EglN1/2/3 | This paper | N/A |
| pCDNA-3.1-HA-SFMBT1(P106A) | This paper | N/A |
| pCDNA-3.1-HA-SFMBT1(P651A) | This paper | N/A |
| plenti-UBCp-FLAG-HA-SFMBT1-gateway | This paper | N/A |
| plenti-UBCp-FLAG-HA-SFMBT1sh1-resistant-gateway | This paper | N/A |
| pll3.7-UBCP-EF1-SFMBT1 | This paper | N/A |
| pll3.7-UBCP-EF1-SFMBT1(P651A) | This paper | N/A |
| pll3.7-UBCP-EF1-SFMBT1si1/3 | This paper | N/A |
| pLX304-Blast-v5-SPHK1 | UNC Lenti-shRNA Core Facility | N/A |
Highlights:
A genome-wide screen identified SFMBT1 as a pVHL target
Proline hydroxylation of SFMBT1 decreases its protein stability and is regulated by pVHL
SFMBT1 promotes ccRCC tumorigenesis and is upregulated in patients with ccRCC
SFMBT1 increases SPHK1 expression in ccRCC
Acknowledgements
We thank Dr. William G. Kaelin Jr for his support as the initial screen was performed by Q.Z in Dr. Kaelin’s lab in collaboration with Dr. Kirschner’s lab with support from NCI R01 CA068490. We thank UNC LCCC Tissue Procurement Facility, UNC Animal Studies Core and UNC Translational Pathology Laboratory for excellent helps. This work was supported in part by the National Cancer Institute (Q.Z., R01CA211732 and R21CA223675) and Cancer Prevention and Research Institute of Texas (CPRIT, RP190058 to Q.Z). J.M.S. and T.S.P. were supported by The Eunice Kennedy Shriver National Institute of Child Health and Human Development (U54HD079124) and NINDS (P30NS045892). Q.Z is an American Cancer Society Research Scholar, CPRIT Scholar in Cancer Research, V Scholar, Kimmel Scholar, Susan G. Komen Career Catalyst awardee and Mary Kay Foundation awardee. Q.Z is also supported by Kidney Cancer Research Alliance (KCCure). This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- AYAD NG, RANKIN S, OOI D, RAPE M & KIRSCHNER MW 2005. Identification of ubiquitin ligase substrates by in vitro expression cloning. Methods Enzymol, 399, 404–14. [DOI] [PubMed] [Google Scholar]
- BAEK DJ, MACRITCHIE N, PYNE NJ, PYNE S & BITTMAN R 2013. Synthesis of selective inhibitors of sphingosine kinase 1. Chem Commun (Camb), 49, 2136–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY ST, SMITH AM, KARDOS J, WOBKER SE, WILSON HL, KRISHNAN B, SAITO R, LEE HJ, ZHANG J, EATON SC, WILLIAMS LA, MANOCHA U, PETERS DJ, PAN X, CARROLL TJ, FELSHER DW, WALTER V, ZHANG Q, PARKER JS, YEH JJ, MOFFITT RA, LEUNG JY & KIM WY 2017. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat Commun, 8, 15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BULLARD JH, PURDOM E, HANSEN KD & DUDOIT S 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. Bmc Bioinformatics, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BYUN HS, PYNE S, MACRITCHIE N, PYNE NJ & BITTMAN R 2013. Novel sphingosine-containing analogues selectively inhibit sphingosine kinase (SK) isozymes, induce SK1 proteasomal degradation and reduce DNA synthesis in human pulmonary arterial smooth muscle cells. Medchemcomm, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANCER GENOME ATLAS RESEARCH, N. 2013. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 499, 43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN W, HILL H, CHRISTIE A, KIM MS, HOLLOMAN E, PAVIA-JIMENEZ A, HOMAYOUN F, MA Y, PATEL N, YELL P, HAO G, YOUSUF Q, JOYCE A, PEDROSA I, GEIGER H, ZHANG H, CHANG J, GARDNER KH, BRUICK RK, REEVES C, HWANG TH, COURTNEY K, FRENKEL E, SUN X, ZOJWALLA N, WONG T, RIZZI JP, WALLACE EM, JOSEY JA, XIE Y, XIE XJ, KAPUR P, MCKAY RM & BRUGAROLAS J 2016. Targeting Renal Cell Carcinoma with a HIF-2 antagonist. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHO H, DU X, RIZZI JP, LIBERZON E, CHAKRABORTY AA, GAO W, CARVO I, SIGNORETTI S, BRUICK R, JOSEY JA, WALLACE EM & KAELIN WG JR. 2016. On-Target Efficacy of a HIF2alpha Antagonist in Preclinical Kidney Cancer Models. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLIFFORD SC, COCKMAN ME, SMALLWOOD AC, MOLE DR, WOODWARD ER, MAXWELL PH, RATCLIFFE PJ & MAHER ER 2001. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet, 10, 1029–38. [DOI] [PubMed] [Google Scholar]
- COCKMAN ME, LIPPL K, TIAN YM, PEGG HB, FIGG WDJ, ABBOUD MI, HEILIG R, FISCHER R, MYLLYHARJU J, SCHOFIELD CJ & RATCLIFFE PJ 2019. Lack of activity of recombinant HIF prolyl hydroxylases (PHDs) on reported non-HIF substrates. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOBIN A, DAVIS CA, SCHLESINGER F, DRENKOW J, ZALESKI C, JHA S, BATUT P, CHAISSON M & GINGERAS TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAMPER AM, QIAO X, KIM J, ZHANG L, DESIMONE MC, RATHMELL WK & WAN Y 2012. Regulation of KLF4 turnover reveals an unexpected tissue-specific role of pVHL in tumorigenesis. Mol Cell, 45, 233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GODWIN JL, ZIBELMAN M, PLIMACK ER & GEYNISMAN DM 2014. Immune checkpoint blockade as a novel immunotherapeutic strategy for renal cell carcinoma: a review of clinical trials. Discov Med, 18, 341–50. [PubMed] [Google Scholar]
- GORDEUK VR, SERGUEEVA AI, MIASNIKOVA GY, OKHOTIN D, VOLOSHIN Y, CHOYKE PL, BUTMAN JA, JEDLICKOVA K, PRCHAL JT & POLYAKOVA LA 2004. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood, 103, 3924–32. [DOI] [PubMed] [Google Scholar]
- HAASE VH 2005. The VHL tumor suppressor in development and disease: functional studies in mice by conditional gene targeting. Semin Cell Dev Biol, 16, 564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEINZ S, BENNER C, SPANN N, BERTOLINO E, LIN YC, LASLO P, CHENG JX, MURRE C, SINGH H & GLASS CK 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38, 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOADLEY KA, YAU C, WOLF DM, CHERNIACK AD, TAMBORERO D, NG S, LEISERSON MDM, NIU BF, MCLELLAN MD, UZUNANGELOV V, ZHANG JS, KANDOTH C, AKBANI R, SHEN H, OMBERG L, CHU A, MARGOLIN AA, VAN’T VEER LJ, LOPEZ-BIGAS N, LAIRD PW, RAPHAEL BJ, DING L, ROBERTSON AG, BYERS LA, MILLS GB, WEINSTEIN JN, VAN WAES C, CHEN Z, COLLISSON EA, BENZ CC, PEROU CM, STUART JM, ABBOTT R, ABBOTT S, AKSOY BA, ALDAPE K, ALLY A, AMIN S, ANASTASSIOU D, AUMAN JT, BAGGERLY KA, BALASUNDARAM M, BALU S, BAYLIN SB, BENZ SC, BERMAN BP, BERNARD B, BHATT AS, BIROL I, BLACK AD, BODENHEIMER T, BOOTWALLA MS, BOWEN J, BRESSLER R, BRISTOW CA, BROOKS AN, BROOM B, BUDA E, BURTON R, BUTTERFIELD YSN, CARLIN D, CARTER SL, CASASENT TD, CHANG K, CHANOCK S, CHIN L, CHO DY, CHO J, CHUAH E, CHUN HJE, CIBULSKIS K, CIRIELLO G, CLELAND J, CLINE M, CRAFT B, CREIGHTON CJ, DANILOVA L, DAVIDSEN T, DAVIS C, DEES ND, DELEHAUNTY K, DEMCHOK JA, DHALLA N, DICARA D, DINH H, DOBSON JR, DODDA D, DODDAPANENI H, DONEHOWER L, DOOLING DJ, DRESDNER G, DRUMMOND J, EAKIN A, EDGERTON M, ELDRED JM, ELEY G, ELLROTT K, FAN C, FEI S, FELAU I, et al. 2014. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell, 158, 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFFMAN MA, OHH M, YANG H, KLCO JM, IVAN M & KAELIN WG JR. 2001. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet, 10, 1019–27. [DOI] [PubMed] [Google Scholar]
- IVAN M, KONDO K, YANG H, KIM W, VALIANDO J, OHH M, SALIC A, ASARA JM, LANE WS & KAELIN WG JR. 2001. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science, 292, 464–8. [DOI] [PubMed] [Google Scholar]
- JAAKKOLA P, MOLE DR, TIAN YM, WILSON MI, GIELBERT J, GASKELL SJ, VON KRIEGSHEIM A, HEBESTREIT HF, MUKHERJI M, SCHOFIELD CJ, MAXWELL PH, PUGH CW & RATCLIFFE PJ 2001. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science, 292, 468–72. [DOI] [PubMed] [Google Scholar]
- KAELIN WG JR. 2002. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer, 2, 673–82. [DOI] [PubMed] [Google Scholar]
- KONDO K, KIM WY, LECHPAMMER M & KAELIN WG JR. 2003. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol, 1, E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONDO K, KLCO J, NAKAMURA E, LECHPAMMER M & KAELIN WG JR. 2002. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell, 1, 237–46. [DOI] [PubMed] [Google Scholar]
- KUNKEL GT, MACEYKA M, MILSTIEN S & SPIEGEL S 2013. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov, 12, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE DC, SOHN HA, PARK ZY, OH S, KANG YK, LEE KM, KANG M, JANG YJ, YANG SJ, HONG YK, NOH H, KIM JA, KIM DJ, BAE KH, KIM DM, CHUNG SJ, YOO HS, YU DY, PARK KC & YEOM YI 2015. A lactate-induced response to hypoxia. Cell, 161, 595–609. [DOI] [PubMed] [Google Scholar]
- LI LJ, SHEN C, NAKAMURA E, ANDO K, SIGNORETTI S, BEROUKHIM R, COWLEY GS, LIZOTTE P, LIBERZON E, BAIR S, ROOT DE, TAMAYO P, TSHERNIAK A, CHENG SC, TABAK B, JACOBSEN A, HAKIMI AA, SCHULTZ N, CIRIELLO G, SANDER C, HSIEH JJ & KAELIN WG 2013. SQSTM1 Is a Pathogenic Target of 5q Copy Number Gains in Kidney Cancer. Cancer Cell, 24, 738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI M & KIM WY 2011. Two sides to every story: the HIF-dependent and HIF-independent functions of pVHL. J Cell Mol Med, 15, 187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN M 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011, 17, 3. [Google Scholar]
- NADY N, KRICHEVSKY L, ZHONG N, DUAN S, TEMPEL W, AMAYA MF, RAVICHANDRAN M & ARROWSMITH CH 2012. Histone recognition by human malignant brain tumor domains. J Mol Biol, 423, 702–18. [DOI] [PubMed] [Google Scholar]
- NAM Y, SLIZ P, SONG L, ASTER JC & BLACKLOW SC 2006. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell, 124, 973–83. [DOI] [PubMed] [Google Scholar]
- PATRO R, DUGGAL G, LOVE MI, IRIZARRY RA & KINGSFORD C 2017a. Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods, 14, 417–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATRO R, DUGGAL G, LOVE MI, IRIZARRY RA & KINGSFORD C 2017b. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods, 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAUVAGEAU M & SAUVAGEAU G 2010. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell, 7, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHNUTE ME, MCREYNOLDS MD, KASTEN T, YATES M, JEROME G, RAINS JW, HALL T, CHRENCIK J, KRAUS M, CRONIN CN, SAABYE M, HIGHKIN MK, BROADUS R, OGAWA S, CUKYNE K, ZAWADZKE LE, PETERKIN V, IYANAR K, SCHOLTEN JA, WENDLING J, FUJIWARA H, NEMIROVSKIY O, WITTWER AJ & NAGIEC MM 2012. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem J, 444, 79–88. [DOI] [PubMed] [Google Scholar]
- SEMENZA GL 2012. Hypoxia-inducible factors in physiology and medicine. Cell, 148, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHLYUEVA D, STAMPFEL G & STARK A 2014. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet, 15, 272–86. [DOI] [PubMed] [Google Scholar]
- SJOLUND J, JOHANSSON M, MANNA S, NORIN C, PIETRAS A, BECKMAN S, NILSSON E, LJUNGBERG B & AXELSON H 2008. Suppression of renal cell carcinoma growth by inhibition of Notch signaling in vitro and in vivo. J Clin Invest, 118, 217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG M, SHEN H, JIN Y, LIN T, CAI Q, PINARD MA, BISWAS S, TRAN Q, LI G, SHENOY AK, TONGDEE E, LIN S, GU Y, LAW BK, ZHOU L, MCKENNA R, WU L & LU J 2013. The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition. J Biol Chem, 288, 27680–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANI S, KUROOKA H, AOKI T, HASHIMOTO N & HONJO T 2001. The N- and C-terminal regions of RBP-J interact with the ankyrin repeats of Notch1 RAMIC to activate transcription. Nucleic Acids Res, 29, 1373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU S, TRIEVEL RC & RICE JC 2007. Human SFMBT is a transcriptional repressor protein that selectively binds the N-terminal tail of histone H3. FEBS Lett, 581, 3289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG HF, IVAN M, MIN JH, KIM WY & KAELIN WG 2004. Analysis of von Hippel-Lindau hereditary cancer syndrome: Implications of oxygen sensing. Oxygen Sensing, 381, 320–335. [DOI] [PubMed] [Google Scholar]
- YAO X, TAN J, LIM KJ, KOH J, OOI WF, LI Z, HUANG D, XING M, CHAN YS, QU JZ, TAY ST, WIJAYA G, LAM YN, HONG JH, LEE-LIM AP, GUAN P, NG MSW, HE CZ, LIN JS, NANDI T, QAMRA A, XU C, MYINT SS, DAVIES JOJ, GOH JY, LOH G, TAN BC, ROZEN SG, YU Q, TAN IBH, CHENG CWS, LI S, CHANG KTE, TAN PH, SILVER DL, LEZHAVA A, STEGER G, HUGHES JR, TEH BT & TAN P 2017. VHL Deficiency Drives Enhancer Activation of Oncogenes in Clear Cell Renal Cell Carcinoma. Cancer Discov, 7, 1284–1305. [DOI] [PubMed] [Google Scholar]
- ZHANG J, BONASIO R, STRINO F, KLUGER Y, HOLLOWAY JK, MODZELEWSKI AJ, COHEN PE & REINBERG D 2013a. SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes Dev, 27, 749–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG J, BONASIO R, STRINO F, KLUGER Y, HOLLOWAY JK, MODZELEWSKI AJ, COHEN PE & REINBERG D 2013b. SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes & Development, 27, 749–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG J, WU T, SIMON J, TAKADA M, SAITO R, FAN C, LIU XD, JONASCH E, XIE L, CHEN X, YAO X, TEH BT, TAN P, ZHENG X, LI M, LAWRENCE C, FAN J, GENG J, LIU X, HU L, WANG J, LIAO C, HONG K, ZURLO G, PARKER JS, AUMAN JT, PEROU CM, RATHMELL WK, KIM WY, KIRSCHNER MW, KAELIN WG JR., BALDWIN AS & ZHANG Q 2018. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science, 361, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG J & ZHANG Q 2018. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG L, WANG X, BULLOCK AJ, CALLEA M, SHAH H, SONG J, MORENO K, VISENTIN B, DEUTSCHMAN D, ALSOP DC, ATKINS MB, MIER JW, SIGNORETTI S, BHASIN M, SABBADINI RA & BHATT RS 2015. Anti-S1P Antibody as a Novel Therapeutic Strategy for VEGFR TKI-Resistant Renal Cancer. Clin Cancer Res, 21, 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Q, GU J, LI L, LIU J, LUO B, CHEUNG HW, BOEHM JS, NI M, GEISEN C, ROOT DE, POLYAK K, BROWN M, RICHARDSON AL, HAHN WC, KAELIN WG JR. & BOMMI-REDDY A 2009. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell, 16, 413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, LIU T, MEYER CA, EECKHOUTE J, JOHNSON DS, BERNSTEIN BE, NUSBAUM C, MYERS RM, BROWN M, LI W & LIU XS 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol, 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in Figures 1–7 and Figures S1–S7. Additional datasets that support the findings of this study are available from the corresponding author upon reasonable request. ChIP-seq and RNA-seq data have been deposited to GEO under accession number GSE141577.