

Abstract
Purpose.
Abnormal glucagon concentrations are a feature of prediabetes but it is uncertain if α-cell dysfunction contributes to a longitudinal decline in β-cell function. We therefore sought to determine if a decline in β-cell function is associated with a higher nadir glucagon in the postprandial period or with higher fasting glucagon.
Methods.
This was a longitudinal study in which 73 non-diabetic subjects were studied on 2 occasions 6.6 ± 0.3 years apart using a 2-hour, 7-sample oral glucose tolerance test. Disposition Index (DI) was calculated using the oral minimal model applied to the measurements of glucose, insulin, C-peptide concentrations during the studies. We subsequently examined the relationship of glucagon concentrations at baseline with change in DI (used as a measure of β-cell function) after adjusting for changes in weight and the baseline value of DI.
Results.
After adjusting for covariates, nadir postprandial glucagon concentrations were not associated with changes in β-cell function as quantified by DI. On the other hand, fasting glucagon concentrations during the baseline study were inversely correlated with longitudinal changes in DI.
Conclusions.
Defects in α-cell function, manifest as elevated fasting glucagon, are associated with a subsequent decline in β-cell function. It remains to be ascertained if abnormal α-cell function contributes directly to loss of β-cell secretory capacity in the pathogenesis of type 2 diabetes.
Keywords: α-cell function, β-cell function, glucagon, insulin action, prediabetes
1.0. Introduction
Type 2 diabetes is characterized by defects in both β-cell and α-cell function. When insulin secretion is decreased and / or delayed, post-prandial glucagon concentrations mimicking those observed in type 2 diabetes raise glucose concentrations by increasing endogenous glucose production [1, 2]. In addition, glucagon expression in the islet is increased in prediabetes and diabetes [3]. Previously, α-cell dysfunction was thought to occur as a secondary consequence of β-cell dysfunction during the development of type 2 diabetes [4]. However, fasting glucagon is elevated in insulin-resistant subjects [5]. More recently, Faerch et al. suggested that elevated glucagon was associated with measures of impaired insulin action but not with impaired insulin secretion [6]. Indeed, in insulin-resistant mice, ceramide accumulation in α-cells impairs insulin-mediated suppression of preproglucagon mRNA [7]. Examination of a large cohort of non-diabetic subjects also demonstrated an inverse relationship of glucagon concentrations with insulin action [8]. However, due to the cross-sectional nature of these observations, firm conclusions as to the role of α-cell dysfunction in prediabetes cannot be made.
In non-diabetic subjects genetically predisposed to type 2 diabetes, glucagon suppression in response to an oral [9] or intravenous [10] challenge is impaired. If defective postprandial suppression of glucagon could drive the development of hyperglycemia, this might suggest a role for glucagon antagonism [11] in preventing the progression of prediabetes. We therefore sought to determine if higher fasting and postprandial glucagon concentrations are associated with longitudinal decline in β-cell function as quantified by the Disposition Index (DI) [12]. To test our hypotheses, we studied non-diabetic subjects using a 75g oral glucose tolerance test (OGTT) administered twice over a ~ 7 year period. We report that fasting, but not nadir, glucagon concentrations during the ‘Baseline’ study are inversely correlated with the change in overall β-cell function (DI) measured during the ‘Follow-Up’ study. These data suggest that fasting glucagon concentrations are associated with a subsequent decline in β-cell function.
2.0. Research Design and Methods
2.1. Subjects, Experimental Design, Calculations and Statistical Analysis.
Participants in previously published studies utilizing a standardized 7-sample, 2 hour, 75g OGTT were invited to participate in the current study. The study was performed after approval by the Mayo Institutional Review Board and informed, written consent was obtained. The OGTT was repeated (and analyzed) as outlined in the Supplementary Data.
Net insulin action (Si) and β-cell responsivity indices calculated using the oral minimal model were used to derive Disposition index (DI) as reviewed previously [12]. Please refer to Supplementary Data for details.
All data are presented as Mean ± SEM unless otherwise noted. Multivariable regression analysis adjusting for the effects of variation in the weight change and the time interval between studies was performed. The target parameter was the fractional change (e.g. in the case of DI: ΔDI = [(DIPresent – DIBaseline)/ DIBaseline]). Please refer to Supplementary Data for details.
3.0. Results
3.1. Subject Characteristics
We studied a total of 73 nondiabetic subjects whose characteristics at the time of the ‘Baseline’ and ‘Follow-Up’ studies are described in Supplementary Table 1.
3.2. Relationship of Fractional Change in Disposition Index (ΔDI) with ‘Baseline’ nadir and fasting glucagon
Nadir glucagon in the Baseline experiments was not associated with ΔDI over the period of observation after adjustment for covariates (Supplementary Figure 1). ‘Baseline’ fasting glucagon was inversely correlated with ΔDI after adjustment for covariates. Spearman Correlation also produced similar results (R = −0.28, p = 0.02 – Figure 1).
Figure 1:
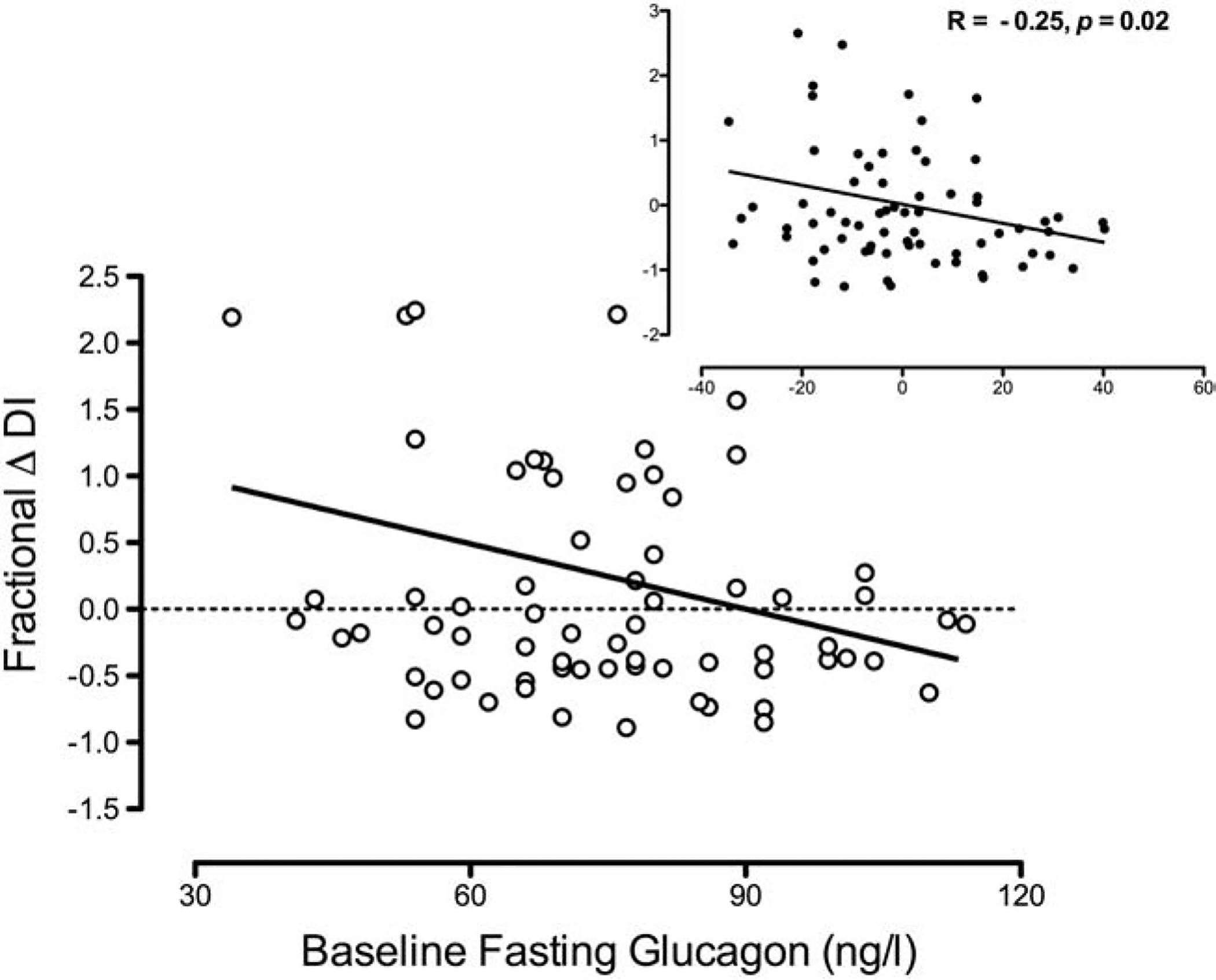
Correlation of fractional change in Disposition Index (DI) in the ‘Follow-Up’ series of experiments with fasting glucagon concentrations in the ‘Baseline’ series of experiments. Inset panels represent residuals after adjusting for the covariates of weight change and time interval between studies.
The fractional change in insulin action (ΔSi) was inversely correlated with ‘Baseline’ fasting glucagon (Supplementary Figure 2 – Panel A) but this was not true for β-cell responsivity (ΔΦ – Panel B).
3.3. Relationship of Δ 120 minute glucose with ‘Baseline’ fasting glucagon
‘Baseline’ fasting glucagon correlated with fractional change in 120 minute glucose after OGTT when adjusted for covariates (Figure 2). Spearman Correlation also produced similar results (R = 0.28, p = 0.02).
Figure 2:
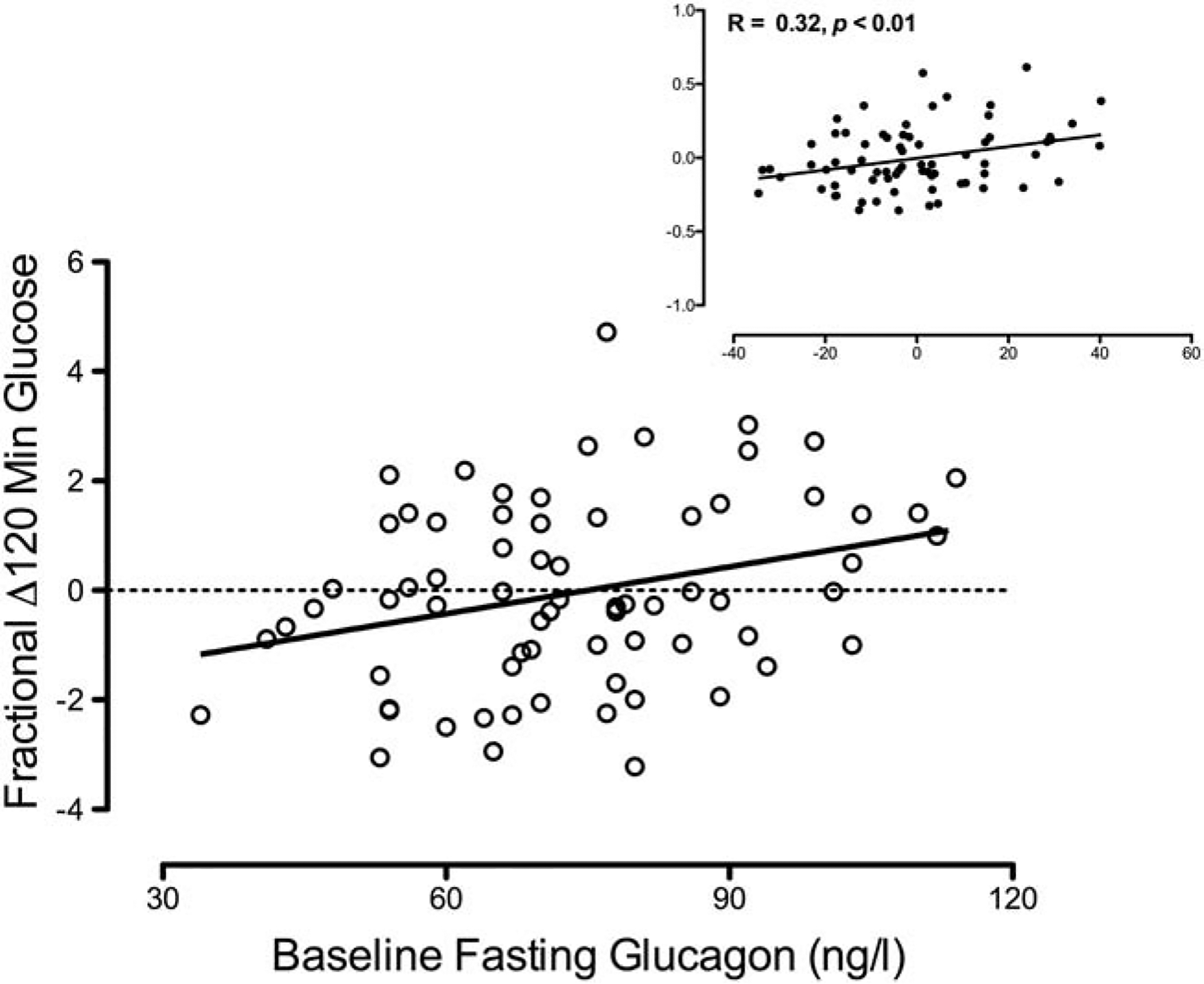
Correlation of fractional change in 120 minute glucose in the ‘Follow-Up’ series of experiments with fasting glucagon concentrations in the ‘Baseline’ series of experiments. Inset panels represent residuals after adjusting for the covariates of weight change and time interval between studies.
Post hoc we divided the cohort into subjects who experienced a >10 (symmetrical) % increase in 120 minute glucose over time versus the remainder. Baseline’ fasting glucagon concentrations were higher in the subjects who had a > 10% increase in 2 hour glucose over time than those who had a ≤ 10% increase in 2 hour glucose (p = 0.02) during the ‘Baseline’ study (Supplementary Figure 3 – Panel A). This pattern was maintained during the ‘Follow-Up’ series of experiments (p = 7 × 10−4 – Panel B). The characteristics of the participants in each group as characterized by changes in their glucose tolerance are described in Supplementary Table 2.
4.0. Discussion
In this small longitudinal study, we demonstrate that fasting glucagon concentrations were inversely correlated with a subsequent decline in β-cell function and glucose tolerance. It is notable that the relationship of ‘Baseline’ fasting glucagon with longitudinal changes in DI is not explained by a relationship with DI during the ‘Baseline’ study. Indeed, initial DI (as well as insulin action and β-cell responsivity) did not differ between subjects categorized by the presence or absence of worsening glucose tolerance (Supplementary Table 2). Taken together, these data suggest that α-cell function is altered early in the evolution of glucose dysregulation, prior to (and independently of [8]), defects in insulin secretion. This α-cell dysfunction is associated with longitudinal decline in β-cell function.
Glucagon secretion and α-cell function have been relatively ignored aspects of islet dysfunction in prediabetes. Indeed our study is relatively unique in the ability to characterize changes in post-(oral) challenge glucagon concentrations over time. However, there are limitations that need to be acknowledged.
The cohort is relatively small and the effect of glucagon on endogenous glucose production was not measured. Glucagon raises endogenous glucose production [13] especially in situations where insulin secretion is impaired [1, 2]. In the absence of tracer-based measures of glucose metabolism, we can only speculate that this is the underlying reason for the longitudinal decline in glucose homeostasis, since glucagon does not impair peripheral glucose uptake [1].
Although the group experiencing decline in glucose tolerance tended to be heavier and older at baseline, these differences were not statistically significant (Supplementary Table 2). In addition, the 120 minute glucose values at baseline were lower in the group who experienced worsening of glucose tolerance. Other factors including age, absolute weight and weight gain did not differ between subjects who did or did not have deterioration in their glucose tolerance as measured by 120 minute glucose.
An important limitation is that the glucagon assay used in this series of experiments cross-reacts with proglucagon-derived peptide fragments [14]. Therefore, it is possible that these, rather than fasting glucagon, are markers of future decline in islet function. However, despite differences in absolute concentrations of fasting and nadir glucagon there is good correlation between these assays and the assay we used in this experiment [14–17]. Nevertheless, future studies using immunoassays with greater specificity for glucagon will be needed to test this possibility.
A final consideration is that in the post hoc analysis grouped by change in glucose tolerance, there was a preponderance of male subjects (in the group experiencing a > 10% rise in 120 minute glucose). Men have consistently been shown to have higher fasting glucagon (and glucose) concentrations compared to women [8, 18, 19], however, baseline DI did not differ between sexes. Further study in larger, better characterized cohorts balanced for sex will be required to confirm this observation and provide mechanistic insights into how elevated fasting glucagon might contribute to a decline in β-cell function.
In conclusion, these data suggest that defects in α-cell function, manifest as elevated fasting glucagon, could drive subsequent decline in insulin action and overall β-cell function. These findings require replication in a larger, independent cohort that addresses the limitations of the current study prior to targeted manipulation of glucagon secretion or signaling in prediabetes.
Supplementary Material
Highlights.
Genetic evidence implicates impaired glucagon suppression in diabetes risk
Could higher glucagon concentrations predict beta-cell dysfunction?
Over a 7-year period higher fasting glucagon predicted a decline in β-cell function
This is independent of weight changes over the period of observation
Acknowledgments:
Funding: This study was funded by funds from the Mayo Clinic General Clinical Research Center (UL1 TR000135) and by the National Institutes of Health (DK78646, DK116231). J.D.A. is supported by training grant 5T32DK007352-37.
Abbreviations
- DI
Disposition Index
- mRNA
messenger Ribonucleic Acid
- OGTT
Oral Glucose Tolerance Test
- SD
Standard Deviation
- SEM
Standard Error of the Mean
- Si
Insulin Action
- Φ
Total β-cell responsivity to glucose
- ϕs
Static β-cell responsivity to glucose
- ϕd
Dynamic β-cell responsivity to glucose
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: Dr. Vella is an investigator in an investigator-initiated study sponsored by Novo Nordisk. He has consulted for XOMA, vTv Therapeutics, Sanofi-Aventis, Novartis and Bayer in the past 5 years. None of the other authors have relevant disclosures.
Data Availability:
Deidentified datasets will be made available on request from the authors after all necessary approvals and agreements are obtained.
References
- [1].Shah P, Basu A, Basu R, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol. 1999;277:E283–90. [DOI] [PubMed] [Google Scholar]
- [2].Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2000;85:4053–9. [DOI] [PubMed] [Google Scholar]
- [3].O’Malley TJ, Fava GE, Zhang Y, Fonseca VA, Wu H. Progressive change of intra-islet GLP-1 production during diabetes development. Diabetes/metabolism research and reviews. 2014;30:661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ahren B, Larsson H. Impaired glucose tolerance (IGT) is associated with reduced insulin-induced suppression of glucagon concentrations. Diabetologia. 2001;44:1998–2003. [DOI] [PubMed] [Google Scholar]
- [5].Ferrannini E, Muscelli E, Natali A, Gabriel R, Mitrakou A, Flyvbjerg A, et al. Association of fasting glucagon and proinsulin concentrations with insulin resistance. Diabetologia. 2007;50:2342–7. [DOI] [PubMed] [Google Scholar]
- [6].Faerch K, Vistisen D, Pacini G, Torekov SS, Johansen NB, Witte DR, et al. Insulin Resistance Is Accompanied by Increased Fasting Glucagon and Delayed Glucagon Suppression in Individuals With Normal and Impaired Glucose Regulation. Diabetes. 2016;65:3473–81. [DOI] [PubMed] [Google Scholar]
- [7].Lee Y, Berglund ED, Yu X, Wang MY, Evans MR, Scherer PE, et al. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc Natl Acad Sci U S A. 2014;111:13217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sharma A, Varghese RT, Shah M, Man CD, Cobelli C, Rizza RA, et al. Impaired Insulin Action is Associated with Increased Glucagon Concentrations in Non-Diabetic Humans. J Clin Endocrinol Metab. 2018;103:314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shah M, Varghese RT, Miles JM, Piccinini F, Dalla Man C, Cobelli C, et al. TCF7L2 Genotype and alpha-Cell Function in Humans Without Diabetes. Diabetes. 2016;65:371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Smushkin G, Sathananthan M, Sathananthan A, Dalla Man C, Micheletto F, Zinsmeister AR, et al. Diabetes-associated common genetic variation and its association with GLP-1 concentrations and response to exogenous GLP-1. Diabetes. 2012;61:1082–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kazda CM, Ding Y, Kelly RP, Garhyan P, Shi C, Lim CN, et al. Evaluation of Efficacy and Safety of the Glucagon Receptor Antagonist LY2409021 in Patients With Type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care. 2016;39:1241–9. [DOI] [PubMed] [Google Scholar]
- [12].Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes. 2014;63:1203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cherrington AD, Stevenson RW, Steiner KE, Davis MA, Myers SR, Adkins BA, et al. Insulin, glucagon, and glucose as regulators of hepatic glucose uptake and production in vivo. Diabetes Metab Rev. 1987;3:307–32. [DOI] [PubMed] [Google Scholar]
- [14].Lund A, Bagger JI, Wewer Albrechtsen NJ, Christensen M, Grondahl M, Hartmann B, et al. Evidence of Extrapancreatic Glucagon Secretion in Man. Diabetes. 2015. [DOI] [PubMed] [Google Scholar]
- [15].Bak MJ, Albrechtsen NW, Pedersen J, Hartmann B, Christensen M, Vilsboll T, et al. Specificity and sensitivity of commercially available assays for glucagon and oxyntomodulin measurement in humans. Eur J Endocrinol. 2014;170:529–38. [DOI] [PubMed] [Google Scholar]
- [16].Wewer Albrechtsen NJ, Hartmann B, Veedfald S, Windelov JA, Plamboeck A, Bojsen-Moller KN, et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels? Diabetologia. 2014;57:1919–26. [DOI] [PubMed] [Google Scholar]
- [17].Wewer Albrechtsen NJ, Veedfald S, Plamboeck A, Deacon CF, Hartmann B, Knop FK, et al. Inability of Some Commercial Assays to Measure Suppression of Glucagon Secretion. J Diabetes Res. 2016;2016:8352957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, DeFronzo RA. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia. 2004;47:31–9. [DOI] [PubMed] [Google Scholar]
- [19].Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. beta-Cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab. 2005;90:493–500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.