

Abstract
Since the emergence of the severe acute respiratory syndrome (SARS) to date, neither an effective antiviral drug nor a vaccine against SARS is available. However, it was found that a mixture of two HIV-1 proteinase inhibitors, lopinavir and ritonavir, exhibited some signs of effectiveness against the SARS virus. To understand the fine details of the molecular interactions between these proteinase inhibitors and the SARS virus via complexation, molecular dynamics simulations were carried out for the SARS-CoV 3CLpro free enzyme (free SARS) and its complexes with lopinavir (SARS–LPV) and ritonavir (SARS–RTV). The results show that flap closing was clearly observed when the inhibitors bind to the active site of SARS-CoV 3CLpro. The binding affinities of LPV and RTV to SARS-CoV 3CLpro do not show any significant difference. In addition, six hydrogen bonds were detected in the SARS–LPV system, while seven hydrogen bonds were found in SARS–RTV complex.
Keywords: SARS, Proteinase, MD simulations, Ritonavir, Lopinavir
1. Introduction
Reported first in the Guangdon province of China, in late 2002, as an atypical pneumonia, severe acute respiratory syndrome (SARS) is of concern, since it subsequently rapidly spread to over 25 countries and displayed an estimated mortality rate of 3–6% in infected humans, leading to more than 700 human deaths (Donnelly et al., 2003). In March 2003, four months after the first case was reported, the causative agent of SARS was identified as a new coronavirus (CoV) (Kuiken et al., 2003; Peiris et al., 2003). The complete genomic sequence (29,751 nucleotides) of the +ssRNA virus was reported a month later (Marra et al., 2003), providing the opportunity for more in-depth studies and the identification of potential targets for drug and vaccine development. The genome of SARS-CoV contains 11–14 major annotated open reading frames including those predicted to encode for known viral proteins, including the replicase polyproteins, S (spike protein), polymerase, M (membrane protein), N (nucleocapsid protein) and E (small envelope protein). The viral proteinase and replicase are the preferred targets for searching and designing of antiviral compounds. Indeed, the SARS-CoV main proteinase, 3CLpro (also called Mpro), exhibits a key role in proteolytic processing of the replicase polyproteins and, as an essential protein for viral replication and function, is viewed as a key target.
Whilst phylogenetic analysis of the SARS genome reveals little resemblance to any of the three known groups of cornoaviruses, several of the specific viral genes, including the main protease gene (SARS-CoV 3CLpro), do show significant homology. Thus, sequence alignment of the SARS-CoV 3CLpro with orthologues from other coronaviruses indicated that the enzyme is highly conserved, with 40% and 44% sequence identity, respectively, to human CoV (HCoV) 229E proteinase and procine transmissible gastroenteritis virus (TGEV) proteinase (Anand et al., 2003). The crystal structure of the 3CLpro protein has been determined (Yang et al., 2003; Hsu et al., 2005; Lee et al., 2005), revealing that it is similar to those of other CoV proteinases. SARS-CoV 3CLpro forms a homodimer, comprised of three domains. Domains I (residues 8–101) and II (residues 102–184) are β-barrels and together resemble the structure of chymotrypsin, respectively, whereas domain III (residues 201–306) consists mainly of α-helices. Domains II and III are connected by a long loop (residues 185–200). The active site of the Mpro is comprised of a catalytic dyad that consists of the conserved residues H41 and C145, which are located at the cleft between domains I and II (Huang et al., 2004). The availability of protein structures and the known biological characteristics of 3CLpro both encourage and help facilitate it to be a key target for searching for drugs directed against SARS.
Although neither an effective antiviral drug nor a vaccine against SARS is currently available, several reports have indicated that HIV-1 protease inhibitors have potential for designing SARS-CoV proteinase inhibitors (Zhang and Yap, 2004; Yamamoto et al., 2004; Jenwitheesuk and Samudrala, 2003). In particular, the proteinase inhibitor Kaletra, which is a mixture of the proteinase inhibitors lopinavir and ritonavir (Fig. 1 ), exhibits encouraging signs of being partially effective against the SARS virus (Vastag, 2003). Therefore, a deeper understanding of known proteinase inhibitors as anti-SARS drugs can be used as a starting point to design and discover better or more specific inhibitors for the treatment of SARS, as well as any mutations by predicting resistance mechanisms and/or new drugs for such mechanisms in advance.
Fig. 1.
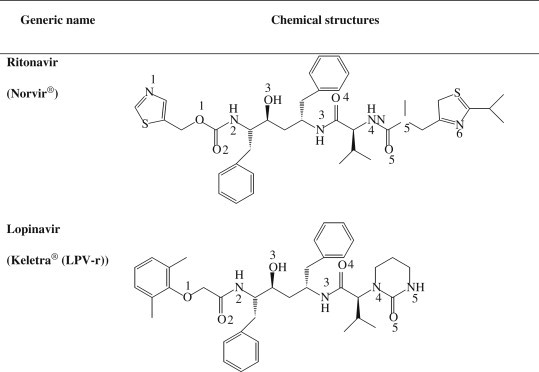
Chemical formula of HIV-1 PR inhibitor, ritonavir and lopinavir, which exhibits signs of effectives against SARS.
In the present research, the evidence for the potential benefits of HIV-1 protease inhibitors against the SARS coronavirus (SARS-CoV) is provided. Molecular dynamic simulations were performed in order to investigate the structure and dynamics behaviours of the free enzyme and its complexes with the lopinavir and ritonavir inhibitors.
2. Methods
2.1. Starting structure and protein preparation
In order to understand ligand–enzyme interactions as well as the effects of inhibitors on the structure and dynamic properties of the free enzyme, three molecular dynamics (MD) simulations were performed: SARS-CoV 3CLpro free enzyme (free SARS) and its two complexes with the lopinavir (SARS–LPV) and ritonavir (SARS–RTV) inhibitors. The X-ray structure of the SARS-CoV 3CLpro (PDB code 1UK3; 2.4 Å resolution) was used as the initial model to construct the SARS–RTV and SARS–LPV complexes (Fig. 2 ). Prior to the simulations, residues SerA1 and GlyA2, which had not been visible in the electron density maps, were modeled using the LEaP module in the AMBER 7 software package (Case et al., 2002). The modeled conformation at the beginning of each simulation was similar to that of residues SerB1 and GlyB2 of the other polypeptide chain in the dimer, which exhibits a well-defined electron density.
Fig. 2.
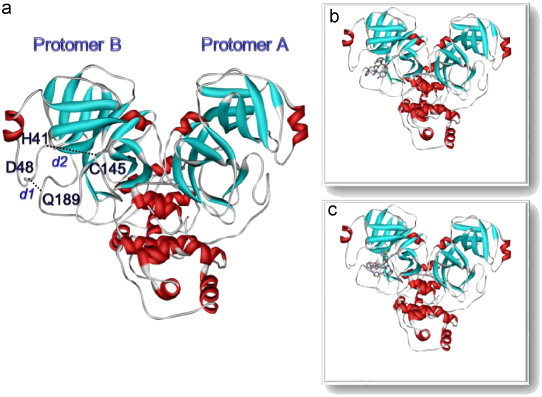
Structures of (a) SARS-free enzyme, (b) SARS–lopinavir complex and (c) SARS–ritronavir complex.
2.2. Flexible docking of SARS and inhibitors
The flexible molecular docking (Muegge and Martin, 1999) with Genetic Algorithm was performed using BioMedCache 2.0 Software to find the most favorable binding interaction. Two drugs, ritonavir and lopinavir, were separately docked into the SARS-CoV 3CLpro binding site. The residues associating in the binding pocket were defined by the conserved amino acid residues, namely H41 and C145, which are also known to be part of the catalytic site of the SARS-CoV proteinase (Anand et al., 2002, Anand et al., 2003; Yang et al., 2003). Additionally, their neighbor residues within a radius of 3 Å of these conserved amino acids were selected and considered as the members of the binding pocket.
The structure of SARS-CoV 3CLpro was obtained from X-ray crystallographic structure (PDB ID:1UK3) (Anand et al., 2003). All missing hydrogen atoms were added into the enzyme using tleap in the AMBER 7 simulation package. The geometries of the inhibitors were generated from X-ray structured and all hydrogen atoms were added using Gaussian03, with the use of antechamber to assign atom types. The geometry of inhibitors was optimized by the semiempirical AM1 method. The PMF scores of the drugs were evaluated by the genetic algorithm with a population size of 50, crossover rate of 0.80, elitism of 5, mutation rate of 0.2, with the maximum generation cycle set to be 40,000. The size of the grid box was set at 25 Å×25 Å×25 Å. Since, as expected, no significant difference was found in terms of their orientations, among the complex structures within the low-energy clusters, the conformations with the lowest PMF energy score were used as the initial model for MD simulations. Finally, the complex structures with the lowest PMF energy score were constructed and used as an initial model for MD simulations.
2.3. Molecular dynamics simulations
Three MD simulations (free SARS, SARS–RTV and SARS–LPV) were carried out using the AMBER 7 simulation package. Energy minimization and MD simulations were achieved using the SANDER module of AMBER 7 with the Cornell force field (Cornell et al., 1995). The parm99 force field, which describes the structure and conformation of each compound, was applied. All MD runs reported here were achieved under an isobaric–isothermal ensemble (NPT) using a constant pressure of 1 atm and a constant temperature of 298 K. The SHAKE algorithm (Ryckaert et al., 1977) was used to constrain all bonds involving hydrogen atoms. Energy minimizations were carried out to relax the system prior to MD runs. To incorporate the solvent and counter ions into the models, each system was neutralized with the counter ions (Na+) and solvated with the TIP3P water model (Jorgensen et al., 1983). The total number of atoms for the three simulated systems, free SARS, SARS–LPV and SARS–RTV, were 77,866, 77,966 and 86,788, respectively. The crystallographic waters were also kept in the simulation box. The simulation time step was set at 2 femtosecond (fs). The simulation steps consist of thermalization, equilibration, and production phases. Initially, the temperature of the system was gradually raised to 298 K for the first 60 picosecond (ps) and then kept constant according to the Berendsen algorithm with a coupling time of 0.2 ps. The systems were maintained at 298 K, until 600 ps. Finally, the production phase started from 600 ps to 2 ns. The 1400 ps trajectories of the production phase were used to calculate the average structure. All MD simulations were carried out for 2 ns. The quality of the geometry and the stereochemistry of the protein structures were validated using PROCHECK. MD trajectories were evaluated in terms of the root-mean-square displacement (RMSD), changes of the distances, torsion angles, H–bond and binding free energies using the CARNAL, Ptraj and MM-PBSA modules of the AMBER7 simulation package.
3. Results and discussion
3.1. Overall enzyme and inhibitor structure
The 2 ns MD trajectories of the three simulated systems were generated. The RMSDs of the overall structure and two inhibitors with respect to the initial configuration were analyzed and plotted (Fig. 3 ). The overall RMSDs for the three systems appeared to have reached equilibrium after 600 ps. Whilst no significant difference was found for the overall (black) and for the back bone (dark grey) RMSDs of all plots, that of the RTV in the SARS–RTV complex (light grey) was found to change substantially. This is due to the rotation of the RTV in the SARS-CoV 3CLpro pocket, i.e., initial (docking) conformation of the RTV is totally different from that after equilibration.
Fig. 3.
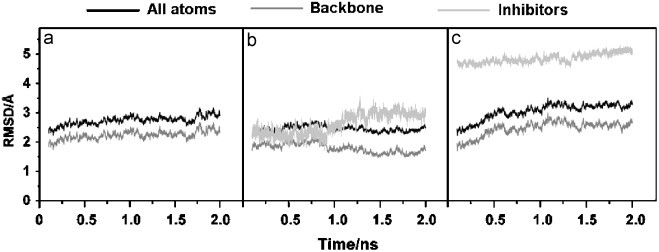
RMSDs of SARS-CoV 3CLpro free enzyme ((a) SARS) and its complexes with lopinavir ((b) SARS–LPV) and ritonavir ((c) SARS–RTV).
3.2. Flexibility of the inhibitor-binding loop regions
Protein flexibility is essential to understand the ligand-binding affinity (Carlson, 2002; Carlson and McCammon, 2000; Lin et al., 2002; Teague, 2003). Conformational changes of the catalytic binding domain play a critical role in ligand binding. Here, the ligand-binding site of the SARS-CoV 3CLpro was evaluated focusing on the amino acid sequence around the active sites (H41 and C145, see Fig. 2) of the CoV proteinase (Anand et al., 2002, Anand et al., 2003; Yang et al., 2003). The binding of the inhibitor is likely to affect the relative motion between the apex of the loop with respect to the D48 and Q189 loops upon the insertion of inhibitor. The probability distributions of the associated interatomic distances from the center of mass of the following residues for the three systems, d1:H41-C145 and d2:D48-Q189, were evaluated and plotted (Fig. 4 ). Clearly, SARS-CoV 3CLpro shows different dynamic flexibilities in the binding region when the SARS-selective inhibitors are bound to their active sites. In the SARS-CoV 3CLpro free enzyme, the most probable d1 distance between H41 and C145 takes place at about 10.0 Å, but this is contracted to ∼7.1–7.3 Å in the enzyme–inhibitor complexes. For the distance between D48 and Q189, d2, in the loop region, no significant differences were observed between free enzyme and two enzyme–inhibitor complexes. Rather the optimal distances, represented by the maximum of the plots, for both SARS-CoV 3CLpro and its two inhibitor complexes are between 8.64 and 10.02 Å (Fig. 4b).
Fig. 4.
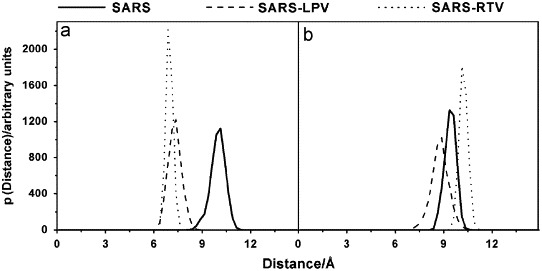
The probability distributions of the associated interatomic distances between the distances (a) d1 and (b) d2 from the center of mass of the following residues, H41 to C145 and D48 to Q189, respectively.
This could be due to the fact that the SARS-CoV 3CLpro free form was initially generated from the complexed SARS-CoV 3CLpro structure, which was identified as having a closed loop conformation. Similarly, this kind of flap (loop) closing was also observed when inhibitors bind in the active site of HIV-1 protease (Hornak et al., 2006a, Hornak et al., 2006b). In addition, the SARS–RTV distribution plots are narrower and higher than those of the SARS–LPV, indicating a lower flexibility of the catalytic pocket and the flap loop of the SARS–RTV complex compared to that of the SARS–LPV complex. This fact relates directly to the greater flexibility of the longer side chain of the RTV than that of the LPV.
3.3. Conformations of the catalytic residues
Another structural feature common to enzyme–inhibitor complexes in general is the conformation of the enzyme-binding site. To investigate the enzyme-binding site conformation of SARS-CoV 3CLpro, we analyzed the changes in the four dihedral angles of the key interactions of the enzyme, involving the amino acids at the catalytic residues and the tip of the loop upon inhibitor insertion. In this study, we present the conformational analysis of the two torsion angles of catalytic residues (Tor1: CA-CB-CG-CD of H41 and Tor2: N-CA-CB-S of C145) and the two amino acids at the tip of the loop (Tor3: CA-CB-CG-OD of D48 and Tor4: CB-CG-CD-CA of Q189), during 600–2000 ps of the simulations (summarized in Fig. 5 and Table 1 ).
Fig. 5.
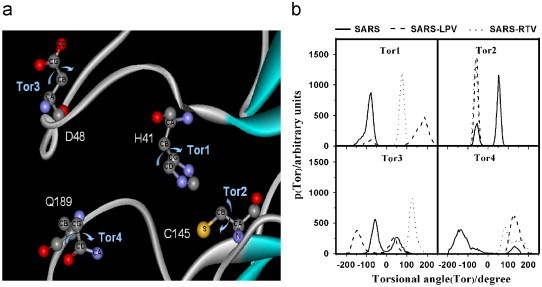
(a) Definitions and (b) probability distributions of the torsion angles of the two catalytic residues (Tor1: CA-CB-CG-CD of H41 and TorR2: N-CA-CB-S of C145) and two amino acids at the tip of the loop (Tor3: CA-CB-CG-OD of D48 and Tor4: CB-CG-CD-CA of Q189) during 600–2000 ps of the simulations.
Table 1.
The four dihedral angles (°), Tor1: CA-CB-CG-CD of H41, Tor2: N-CA-CB-S of C145, TorR3: CA-CB-CG-OD of D48, and TorR4: CB-CG-CD-CA of Q189 taken from the maxima of the distribution plots shown in Fig. 5b
| Tor1 | Tor2 | Tor3 | Tor4 | |
|---|---|---|---|---|
| SARS | −77 | −52, 52 | −57, 52 | −137, 132 (broad) |
| SARS-LPV | −77, 187 | −57 (sharp) | −147, 37 | 132 (broad) |
| SARS-RTV | 77 (sharp) | −62 (sharp) | 127 | 82 |
In free form, Tor1 generates a sharp peak indicating rigidity of the H41 side chain. However, this is not the case for the Q189 side chain, where Tor4 shows a broad peak ranging from −200° to 0°. In contrast, C145 and D48 residues generate two peaks (Tor2 and Tor3), representing two preferential conformations. Changes of their conformations due to complex formations were clearly observed for all four residues. For H41, C145 and Q189, where their orientations were represented by Tor1, Tor2 and Tor4, respectively, their side chains were rotated via complex formations, to the same direction for both inhibitors. This is in contrast to D48, where its side chain was induced by RTV and LPV to turn to the opposite side. Tor3 increases for the RTV complex (dotted line in Fig. 5b), whilst only the first peak of −57° decreases to −147° for the LPV complex (dashed line in Fig. 5b).
3.4. Inhibitor–enzyme interaction
The SARS–LPV and SARS–RTV interactions were analyzed using hydrogen bond interactions and the molecular mechanical energy obtained from the electrostatic and the van der Waals interactions within the systems. The results are summarized in Table 2 .
Table 2.
Percentage occupation of hydrogen bonding between inhibitor and specific residues of SAR-CoV enzyme
| Residue | Type | %Occupation |
|
|---|---|---|---|
| SARS-LPV | SARS-RTV | ||
| T26 | OG1_HG1---O3_LPV | 18.7 | – |
| T26 | LPV_N4_H47---OG1 | 22.8 | – |
| T26 | LPV_N4_H47---O | 36.9 | – |
| H41 | RTV_N2_H5---NE2 | – | 1.7 |
| Y118 | OH_HH---O5_LPV | 2.8 | – |
| N119 | LPV_N4_H47---OD1 | 3.3 | – |
| C145 | RTV_N2_H5---SG | – | 56.3 |
| C145 | SG_HG---O3_RTV | – | 4.4 |
| E166 | N_H---N1_RTV | – | 29.9 |
| Q189 | NE2_HE21---O1_LPV | 9.9 | – |
| Q189 | NE2_HE21---N1_RTV | – | 11.6 |
| Q189 | NE2_HE21---S1_RTV | – | 7.6 |
| Q189 | NE2_HE21---O2_RTV | – | 1.3 |
To analyze the hydrogen bond interactions, the percentage occupancy and number of hydrogen bonds between the inhibitor and binding pocket residues were determined based on the following criteria: (i) a proton donor–acceptor distance of ⩽3.5 Å, and (ii) a donor–H–acceptor bond angle of ⩾120°. It was found that the numbers of hydrogen bonds for the two systems are almost comparable (Table 2). Six hydrogen bonds were detected in the SARS–LPV system, whilst seven were found in the SARS–RTV complex. However, all the hydrogen bonds show a rather low % occupation. Interestingly, excluding Q189, the two inhibitors were found to form hydrogen bonds to different residues, RTV binds to H41, C145 and E166, while LPV binds to T26, Y118 and N119.
In terms of the gas-phase MM interaction energy (ΔEMM), summarized in Table 3 , the molecular mechanical interaction energy for the SARS–LPV complex (−52.9 kcal/mol) is lower than that of the SARS–RTV complex (−48.4 kcal/mol). Taking into account the hydrophobic and electrostatic interactions for the MM interaction energy, a higher contribution from van der Waals energy (−48.1 kcal/mol) and a lower contribution from electrostatic energy (−4.8 kcal/mol) were observed for the SARS–LPV complex than that for the SARS–RTV complex.
Table 3.
Averaged energy contributions (kcal/mol) of the binding free energy for the two simulated systems, SARS-LPV and SARS-RTV
| SARS-LPV | SARS-RTV | |
|---|---|---|
| ΔEelea | −4.8±4.2 | −8.8±3.3 |
| ΔEvdwb | −48.1±3.3 | −39.6±2.9 |
| ΔEMMc | −52.9±5.4 | −48.4±4.6 |
| d | −3.8±0.3 | −2.9±0.2 |
| e | 9.6±1.7 | 6.0±0.8 |
| ΔGsolf | 5.9±1.4 | 3.1±0.8 |
| 4.9±3.4 | −2.8±3.6 | |
| ΔGbindingg | −47.2±5.3 | −45.3±4.3 |
Electrostatic energy.
van der Waals energy.
Electrostatic + van der Waals energy.
Nonpolar contribution to salvation.
Electrostatic contribution to salvation.
Total solvation energy.
Binding free energy in the absence of entropic contribution.
3.5. Inhibitor–enzyme binding free energy
The average binding free energies (ΔGbinding) for the two complexes are shown in Table 3. In this study, the contribution of the entropic term (TΔS) was excluded due to the reasons as in our previous works that the two system models are very similar and, therefore, the contribution of TΔS to ΔGbinding in terms of relative scale is negligible.
From the results, the total electrostatic contribution term, , of SARS–RTV (−2.8 kcal/mol) was found to be more favorable than that of SARS–LPV (4.9 kcal/mol). This can be understood by the higher polarity of the RTV side chain than that of the LPV. However, the van der Waals interactions and nonpolar solvation terms of the SARS–RTV complex are unfavorable by 4.3 kcal/mol relative to those of the SARS–LPV complex. Summing the above contributions, the total binding free energy of the SARS–LPV complex is only slightly more favorable than that of the SARS–RTV complex at −47.2 and −45.3 kcal/mol, respectively. This close similarity, with only −1.9 kcal/mol difference, is due to structural similarity of both complexes. Indeed, the inherent suggestion that the binding affinities of LPV and RTV are not significantly different for the SARS-CoV 3CLpro enzyme is in accordance with the fact that the LPV inhibitor was developed from RTV, so the structures of both inhibitions are quite similar.
3.6. Solvent-accessible region
Ligand solvation, especially in the catalytic pocket, is known to play an essential role in the catalytic mechanism of the enzyme. To ascertain basic information on ligand solvation, the radial distribution function (RDF)—gij(r): the probability of finding a particle of type i in a spherical radii, r, around the particle of type j—centered on the selected atoms of the inhibitors was calculated. The evaluation was done separately for atom sets of both inhibitors, which lay either (i) in equivalent (O2, N2, O3, N3 and N4) or (ii) in different (O1, N4, O5, N5 and N6) environments (see Fig. 1 for atomic label). The results are summarized in Fig. 6 .
Fig. 6.
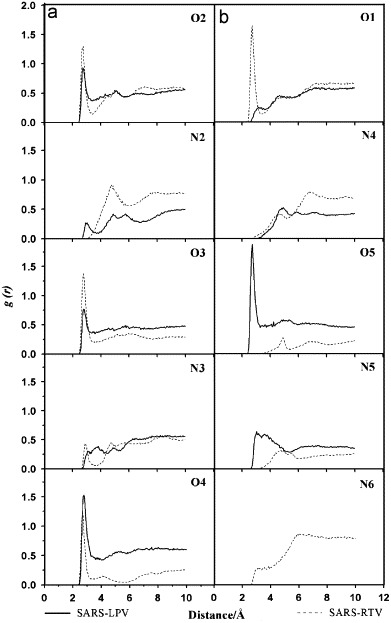
Radial distribution functions, g(r), centered on the inhibitor atoms (see Fig. 1 for definitions) to oxygen atoms of modeled water of the two complexes, SARS–LPV and SARS–RTV. The plots are classified into two categories where the central atom types for the two inhibitors are topologically (a) equivalent and (b) different.
For both of the two complexes, at the central part of the enzyme–inhibitor complex cavity where the atoms of the two inhibitors are topologically equivalent (Fig. 6a), the plots for O2, O3 and O4 show a pronounced first peak at about 3 Å. This indicates that these three atoms were tightly solvated by water molecules. In addition, the three plots for the SARS–RTV complex (dashed line) all show lower minima, indicating that the residence time for the first shell solvation of the SARS–RTV complex is longer than that of the SARS–LPV one. In contrast, the N2 and N3 atoms of both inhibitors are solvated to a significantly lower extent, implying a high hydrophilicity around these regions.
For the outer part (O1, N4, O5, N5 and N6) of the inhibitors (see also Fig. 1), where the RDFs are given in Fig. 6b, O1 of the SARS–RTV complex (dashed line) and O5 of the SARS–LPV complex (solid line) were firmly solvated. Excluding these two regions, the two ends of the two inhibitors are much less solvated, the first peaks are broad and locate far from the referent atoms, indicating a highly hydrophobic characteristic of these two drug molecules.
4. Conclusion
MD simulations and analysis of free SARS, and the SARS–RTV and SARS–LPV complexes give an insight into the dynamic characteristics in terms of the flexibility, conformation, and the inhibitor-enzyme interactions. The analysis of the energetic binding affinity between the HIV-1 proteinase inhibitors has been extensively performed based on free energy calculations. We have found that the interaction of LPV and RTV does not show a significant difference for the SARS-CoV 3CLpro enzyme.
Acknowledgments
This work was jointly supported by the Commission on Higher Education and the Thailand Research Fund (MRG4880041). The authors would like to thank the Computer Center for Advanced Research and the Computational Chemistry Unit Cell, Department of Chemistry, Faculty of Science, Chulalongkorn University, for computing facilities. The authors wish to thank the Publication Counseling Unit, Research Affairs, Faculty of Science, Chulalongkorn University for proofreading the manuscript.
References
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an a-extra helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main protease (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Carlson H.A. Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 2002;6:447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- Carlson H.A., McCammon J.A. Accommodating protein flexibility in computational drug design. Mol. Pharmacol. 2000;57:213–218. [PubMed] [Google Scholar]
- Case D.A., Pearlman J.W., Caldwell T.E., Cheatham J., Jr., Wang W.S., Ross C.L., Simmerling T.A., Darden K.M., Merz R.V., Stanton A.L., Cheng J.J., Vincent M., Crowley V., Tsui H., Gohlke R.J., Radmer Y., Duan J., Pitera I., Massova G.L., Seibel U.C., Singh P.K., Kollman P.A. University of California; San Francisco, CA: 2002. AMBER. (Version 7.0 ed.) [Google Scholar]
- Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. A second generation force-field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- Donnelly C.A., Ghani A.C., Leung G.M., Hedley A.J., Fraser C., Riley S., Abu-Raddad L.J., Ho L.-M., Thach T.-Q., Chau P., Chan K.-P., Lam T.-H., Tse L.-Y., Tsang T., Liu S.-H., Kong J.H.B., Lau E.M.C., Ferguson N.M., Anderson R.M. Epidermiological determinants of spread of causal agent of severe acute respiratory syndrome in Hong Kong. Lancet. 2003;361:1761–1766. doi: 10.1016/S0140-6736(03)13410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V., Okur A., Rizzo R.C., Simmerling C. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2006;103:915–920. doi: 10.1073/pnas.0508452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V., Okur A., Rizzo R.C., Simmerling C. HIV-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. J. Am. Chem. Soc. 2006;128:2812–2813. doi: 10.1021/ja058211x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M.-F., Kuo C.-J., Chang K.-T., Chang H.-C., Chou C.-C., Ko T.-P., Shr H.-L., Chang G.-G., Wang A.H.J., Liang P.-H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wei P., Fan K., Liu Y., Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- Jenwitheesuk E., Samudrala R. Identifying inhibitors of the SARS coronavirus proteinase. Bioorg. Med. Chem. Lett. 2003;13:3989–3992. doi: 10.1016/j.bmcl.2003.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- Kuiken T., Fouchier R.A.M., Schutten M., Rimmelzwaan G.F., van Amerongen G., van Riel D., Laman J.D., de Jong T., van Doornum G., Lim W., Ling A.E., Chan P.K.S., Tam J.S., Zambon M.C., Gopal R., Drosten C., van der Werf S., Escriou N., Manuguerra J.-C., Stöhr K., Peiris J.S.M., Osterhaus A.D.M.E. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.W., Cherney M.M., Huitema C., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N.G. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. J. Mol. Biol. 2005;353:1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.H., Perryman A.L., Schames J.R., McCammon J.A. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J. Am. Chem. Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Muegge I., Martin Y.C. A general and fast scoring function for protein-ligand interactions: a simplified potential approach. J. Med. Chem. 1999;42:791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- Peiris J.S.M., Chu C.M., Cheng V.C.C., Chan K.S., Hung I.F., Poon L.L.M., Law K.I., Tang B.S.F., Hon T.Y.W., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W.M., Guan Y., Yeun K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Caresian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- Teague S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003;2:527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- Vastag B. Old drugs for a new bug: influenza, HIV drugs enlisted to fight SARS. JAMA. 2003;290:1695–1696. doi: 10.1001/jama.290.13.1695. [DOI] [PubMed] [Google Scholar]
- Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J., Rabenau H., Doerr H.W., Hunsmann G., Otaka A., Tamamura H., Fujii N., Yamamoto N. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004;318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.W., Yap Y.L. Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main protease with HIV, psychotic and parasite drugs. Bioorg. Med. Chem. 2004;12:2517–2521. doi: 10.1016/j.bmc.2004.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]