

Highlights
-
•
Benserazide was discovered from a screen of drug-like compounds using a FRET-based enzyme assay of CVB3 3Cpro.
-
•
Enzyme kinetic studies suggest that benserazide is a non-competitive allosteric CVB3 3Cpro inhibitor.
-
•
The 2,3,4-trihydroxybenzyl moiety of benserazide is important for the inhibitory action against CVB 3Cpro.
-
•
The putative allosteric binding site of benserazide in CVB3 3Cpro was predicted using an in silico docking study.
Abbreviations: CVB3, Cosxackievirus B3; CVB3 3Cpro, Cosxackievirus B3 3C protease; FRET, fluorescence resonance energy transfer; l-DOPA, levodopa; Boc, tert-butyloxycarbonyl; TFA, trifluoroacetic acid; DCM, dichloromethane; IC50, the half maximal inhibitory concentration; 1H NMR, proton nuclear magnetic resonance spectroscopy; 13C NMR, carbon nuclear magnetic resonance spectroscopy; ESI, electrospray ionization; TLC, thin-layer chromatography; PCR, polymerase chain reaction; IMPACT-CN system, Intein Mediated Purification with an Affinity Chitin-binding Tag-CN system; DTT, dithiothreitol; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; NaCl, sodium chloride; EDTA, ethylenediaminetetraacetic acid; IGEPAL, octylphenoxypolyethoxyethanol
Keywords: Coxsackievirus B3, 3C protease, Enzyme kinetics, Non-competitive inhibitor, Benserazide, Allosteric binding site
Abstract
Coxsackievirus B3 is the main cause of human viral myocarditis and cardiomyopathy. Virally encoded Coxsackievirus 3C protease (3Cpro) plays an essential role in viral proliferation. Here, benserazide was discovered as a novel inhibitor from a drug library screen targeting Coxsackievirus 3Cpro using a FRET-based enzyme assay. Benserazide, whose chemical structure has no electrophilic functional groups, was characterized as a non-competitive inhibitor by enzyme kinetic studies. A molecular docking study with benserazide and its analogs indicated that a novel putative allosteric binding site was involved. Specifically, a 2,3,4-trihydroxybenzyl moiety was determined to be a key pharmacophore for the enzyme’s inhibitory activity. We suggest that the putative allosteric binding site may be a novel target for future therapeutic strategies.
Interpreting the parameters
- Vmax
the maximum enzyme velocity without inhibitor, expressed in the same units as Y
- Km
the Michaelis–Menten constant, expressed in the same units as X
- Ki
the inhibition constant, expressed in the same units as I
1. Introduction
Human coxsackieviruses (CVs) belong to the picornavirus family [1] of non-enveloped positive-sense single-stranded RNA viruses [2]. Among them, Coxsackievirus B3 (CVB3) is the most common cause of human viral myocarditis [3], [4]. Acute myocarditis typically results in heart failure in children and adolescents and often progresses to chronic myocarditis and dilated cardiomyopathy. Annually, approximately 50% of the symptoms of viral myocarditis after heart transplants are caused by CVB3 infections [4], [5].
Upon viral infection, the positive-strand RNA genome of CVB3 is released to the host cell and translated into a large poly-protein precursor that undergoes a successive proteolytic cleavage process to generate 7 functional viral proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) and 4 structural proteins (VP1–VP4) [6], [7]. The proteolytic processing of the poly-protein is essential for the production of the components of virions, which are primarily generated by two virally encoded proteases, 2A and 3C [8]. The picornavirus 3C protease in particular is an essential multifunctional protein that is required for not only the majority of the proteolytic cleavage events that occur during viral replication [9] but also for targeting several host proteins such as eukaryotic translation initiation factor G and cleavage stimulation factor 64 to halt host protein synthesis and induce apoptosis [10], [11], [12]. The essential roles of this protease in virus replication have drawn attention as attractive targets for the development of antiviral therapeutic agents because effective therapeutic strategies for the prevention or treatment of diseases caused by CVB3 infection are not currently available [13], [14].
There are many picornavirus 3C protease and 3C like protease inhibitors developed by many research groups in the previous studies [13], [14], [15], [16], [17], [18], [19], [20], [23], [24], [25], [26], [27], [28], [29], [30]. Among them, a variety of CVB3 3C protease inhibitors have been recently reported from both the screening of compound libraries and the design of compounds based on the structure of the substrate acting at the active site [15]. Although numerous 3C protease inhibitors have been developed as therapeutic agents for CVB3 infections, no antiviral drugs have been approved to date for the treatment of CVB3 infections in humans [16]. For example, compound 1 (TG-0204998 in Ref. [17]) is a peptidomimetic inhibitor (K i = 0.8 μM) interacting at the active site of CVB 3Cpro, whose binding mode was determined by crystallization [17]. Compound 2 (43146 in Ref. [18]) was discovered from random screening as a non-peptide multifunctional inhibitor (IC50 = 5.4 μM) against CVB3 3Cpro, EV71 3Cpro and SARS-CoV 3CLpro [18]. Our group has reported peptidomimetic inhibitor 3 (9e in Ref. [6]) (IC50 = 130 nM), which is an AG7088 derivative with the lactam ring modified into a glutamine residue and the ethyl ester group modified into an isoquinoline moiety [6]. The α,β-unsaturated carbonyl compounds such as 1 and 3 are known to undergo electrophilic irreversible covalent interaction with the cysteine residue (Cys-147) at the active site (Fig. 1 ) [2], [7], [19]. Although the covalent binding can provide strong irreversible inhibition, the electrophilic nature of CVB3 3Cpro inhibitors increase the off-target safety risks by the possibility of interacting with host cysteine proteases [20]. Therefore, critical unmet needs in the field of the discovery of allosteric reversible inhibitors against CVB3 3Cpro with reduced side effects.
Fig. 1.

The structure and inhibitory effect of various inhibitors against CVB3 3Cpro.
Herein, we report the first discovery of a non-competitive inhibitor of CVB3 3Cpro that interacts with a putative allosteric binding site through the screening of a drug compound library intended to explore a new chemical class of CVB3 3Cpro inhibitors. We also present studies on enzyme kinetics and molecular docking for the characterization of the inhibitor.
2. Materials and methods
2.1. Expression and purification of CVB3 3Cpro
The CVB3 3Cpro gene was transformed into the pTYB12 vector and expressed in Escherichia coli strain BL21 (DE3). The CVB3 3Cpro was purified by chitin column chromatography using the IMPACT system (New England Bio Labs, Beverly, MA) [21]. The identity of CVB3 3Cpro was confirmed by sodium dodecyl sulphate-polyacrlamide gel electrophoresis (SDS-PAGE), based on its size of almost 22kDa. Purified CVB3 3Cpro was maintained in HEPES buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, 1 mM DTT (pH 7.9) and 5% (v/v) glycerol) at −20 °C for an enzyme-based assay.
2.2. FRET-based enzyme assay and screening of compound library
The enzyme assays were performed in 96-well microplates with overall reaction volumes of 100 μL comprising 50 μL 2× reaction buffer (80 mM Tris–HCl, 2 mM DTT, 0.2% IGEPAL), 45 μL water, 1 μL CVB3 3Cpro, 1 μL of 10 mM stock solution in 100% (v/v) dimethyl sulfoxie (DMSO) of various compounds (final concentrations of compounds and DMSO are 10 μM and 1%, respectively) and 3 μL substrate peptide (1 mM concentration). The compounds and enzymes were incubated for 5 min in the reaction solution at 25 °C, and the reactions were initiated by the addition of a fluorogenic substrate (Nma-EALFQGPPVK-Dnp-rrr-NH2/C83H126N28O21, 97% purity) for CVB3 3Cpro, which was purchased from Anygen (Jangseong, Republic of Korea). The fluorescence values were measured at 340 nm (excitation) and 440 nm (emission) and recorded in relative fluorescence units (RFUs). Reactions were incubated at 35 °C and monitored every 10 min by a spectrofluorometer (Bio Tek Instrument Inc., USA) for 2 h.
Two thousand drug-like compounds (provided by LG Life Science Ltd., Daejeon, Republic of Korea) were screened at 10 μM on a FRET-based enzyme assay. The 12 selected hit compounds were further screened at 5 μM, and after measuring the IC50 values which was determined by the series of compound concentrations (0–10 μM final concentration at 3-fold dilution) and calculated by SIGMAPLOT program (SPSS, San Diego, CA, USA). Finally, one compound (Benserazide, 4; IC50 value = 2.4 μM) was selected as a hit compound.
2.3. Enzyme kinetics study and reversibility test
An enzyme kinetics experiment was performed using the following method. CVB3 3Cpro was mixed with different concentrations of Benserazide (4) (0–10 μM) in a final volume of 97 μL for 5 min at 25 °C. Each mixture was placed into a 96-well microplate in triplicate. Three microliters of different concentrations of fluorogenic substrates (0–400 μM) were also added into each respective well. The reactions were incubated at 35 °C, and the enzyme activities were monitored every 10 min by a spectrofluorometer (Bio Teck Instrument Inc., USA) over a period of 2 h. Initial velocities were determined from the linear section of the Michaelis–Menten plot. The data were easily fitted by the SIGMAPLOT program (SPSS, San Diego, CA, USA).
The reversibility of inhibition was determined by measuring the recovery of enzymatic activity after a large and rapid dilution of the enzyme-inhibitor complex. The 50-fold concentration of the IC50 values of inhibitors (Benserazide (4) and compound 3) with the enzyme were pre-incubated for 30 min (equilibration time) at 35 °C. Then, the mixtures were diluted 50-fold with the reaction buffer containing the fluorogenic substrate (50 μM) to initiate the enzyme reaction. The enzyme activities were monitored every 5 min by a spectrofluorometer (Bio Teck Instrument Inc., USA) over a period of 2 h. Reversibility plot was fitted by the SIGMAPLOT program (SPSS, San Diego, CA, USA).
2.4. In silico prediction of binding site and molecular docking study
A molecular docking study of Benserazide (4) was performed based on the X-ray crystal structures of CVB3 3Cpro (PDB code: 2ZU3) and DOPA-decarboxylase (PDB code: 1JS3). An allosteric binding site for the non-competitive inhibitor Benserazide (4) was sought through the detection of cavities using the eraser algorithm method. A possible allosteric binding site was found, and a molecular docking study on this site was performed using CDOKER, a CHARMm forcefield-based docking tool from Discovery Studio 3.5 (Accelrys Inc., San Diego, CA, USA). The process of the CDOKER workflow was followed by using high-temperature molecular dynamics to find possible ligand conformations, random rotation of ligand in the rigid receptors; grid-based simulated annealing; and full energy minimization. The outputs of the refined ligand poses were sorted by CDOKER energy.
2.5. Synthesis and enzyme inhibitory effects of Benserazide (4) derivatives
The 2,3,4-trihydroxybenzyl moiety of Benserazide (4) was replaced by (b–f) di-hydroxy benzyl, mono-hydroxy benzyl (g–i) and benzyl moieties (j) following the synthetic procedure illustrated in Scheme 1 . Those benserazide derivatives were synthesized starting from 3-(tert-butyl) 4-methyl 2,2-dimethyloxazolidine-3,4-dicarboxylate, which was reacted with hydrazine hydrated (excess amount) in methanol to yield compound 5. Conjugation reactions of compound 5 with various hydroxybenzaldehydes (6a–j), removal of the acetonide (7a–j) and subsequent deprotection of the Boc groups afforded hydrazone derivatives, 8a–j. The final compounds (9a–j) were synthesized from 8a–j by hydrogenation. The inhibitory effects of Benserazide derivatives against CVB3 3Cpro are summarized in Table 1 .
Scheme 1.

Reagents and conditions: (a) hydrazine hydrate(excess amount), MeOH, −40 °C, 12 h, 80% yield; (b) EtOH, 40 °C, 8 h, 70–80% yield; (c) 0.5 N TFA in DCM, rt 1 h, 60–70% yield; (d) 20% TFA in DCM, rt, 2 h, 70–75% yield; (e) Pd/C, H2 gas MeOH, rt, 1 h, 40–50% yield.
Table 1.
% Inhibition at 50 μM or IC50 value contained various substituted benzyl moiety compounds (8a–j, 9a–j) tested against CVB3 3Cpro.
| Compd | R |
8 |
9 |
|---|---|---|---|
| % Inhibition 50 μM or (IC50) | % Inhibition 50 μM or (IC50) | ||
| a | 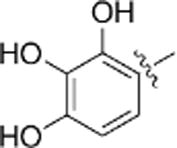 |
(2.72 μM) | (2.40 μM) |
| b | 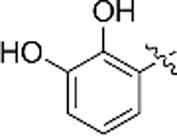 |
N.A. | N.A. |
| c | 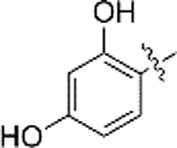 |
N.A. | N.A. |
| d | 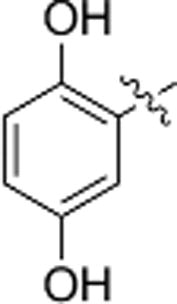 |
N.A. | N.A. |
| e | 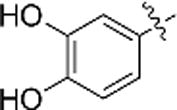 |
55.9% | 56.2% |
| f | 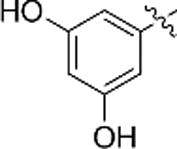 |
N.A. | N.A. |
| g | 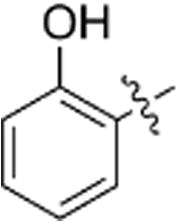 |
N.A. | N.A. |
| h | 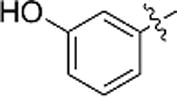 |
N.A. | N.A. |
| i | 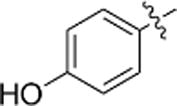 |
N.A. | N.A. |
| j | 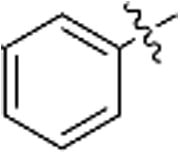 |
N.A. | N.A. |
N.A.: no activity.
3. Results and discussion
3.1. Hit compounds from library screening
A screening of 2000 drug-like compounds in a library using a FRET-based enzyme assay of CVB3 3Cpro identified initial 12 hit compounds, and we selected Benserazide (4) as a final effective compound with an IC50 value of 2.4 μM against CVB3 3Cpro. Benserazide (4) is a commercial drug developed by Roche for the management of Parkinson’s disease in combination with l-DOPA (levodopa) as co-beneldopa (BAN), which is unable to cross the blood-brain barrier. Thus, the drug peripherally inhibits aromatic l-amino acid decarboxylase (AADC) or DOPA decarboxylase [22].
3.2. Enzyme kinetics study and reversibility test
Because the structure of Benserazide (4) is quite different from that of known inhibitors such as 1–3 acting at the active site of CVB3 3Cpro, we performed enzyme kinetics studies to determine the mechanism of the inhibition of Benserazide (4) against CVB3 3Cpro. The effects of Benserazide (4) on the enzyme kinetics of CVB3 3Cpro are illustrated in Fig. 2 . In the Michaelis–Menten plot (Fig. 2a), the V max values are reduced in the presence of Benserazide (4) in a concentration-dependent manner without a change in the K m value (64.5 μM), suggesting that Benserazide (4) should be a non-competitive inhibitor. The Lineweaver–Burk plot (Fig. 2b) of 1/[S] versus 1/V (1/RFU·min−1) further confirmed the characteristics of non-competitive inhibition, showing a series of lines converging at the same point on the x(1/[S]) axis (−1/K m). As shown in Fig. 2c, the Dixon plot of [I] versus 1/V (1/RFU·min−1) also resulted in a family of straight lines with the same x-axis intercept, reflecting non-competitive inhibition [23] against CVB3 3Cpro, and the K i value was calculated as 4.7 μM from this figure [24].
Fig. 2.

Inhibitory mechanism study of 4 (Benserazide) by enzyme kinetic and reversibility studies. (a) Michaelis–Menten plot. In the presence of various concentrations of substrate and inhibitor, CVB3 3Cpro activity was measured by fluorescence. The x-axis is the substrate concentration, and the у-axis is the CVB3 3Cpro activity (RFU min−1). Each plot describes different inhibitor concentrations 0 μM (●), 2.5 μM (△), 5.0 μM (■), 7.5 μM (○), and 10 μM (▾). (b) In the Lineweaver–Burk plot, the Benserazide (4) concentration was set to 0 μM (●), 2.5 μM (△), 5.0 μM (■), 7.5 μM (○), and 10 μM (▾). The x-axis is 1/substrate concentration and the у-axis is the 1/CVB3 3Cpro activity (1/RFU min−1). (c) In the Dixon plot, the substrate concentration was set to 30 μM (●), 150 μM (■), and 300 μM (▽). The x-axis is the inhibitor concentration, and the у-axis is the 1/CVB3 3Cpro activity (1/RFU min−1). (d) The reversibility plot, the x-axis is time and the у-axis is product (fluorescence). Benserazide concentration (△) was set 2.5 μM and compound 3 concentration (●) was set 150 nM.
As explained in the introduction, certain known non-competitive inhibitors act at the active site of CVB3 3Cpro by covalent interactions. However, Benserazide (4) has no electrophilic functional group for covalent interactions; therefore, an allosteric binding site for the Benserazide (4) should exist. To further confirm the reversible allosteric inhibition mechanism of Benserazide (4), a reversibility test was performed with a conventional protocol using large and rapid dilution of inhibitor. As shown in Fig. 2d, the partial inhibition from the condition of full inhibition with high concentration of inhibitors, Benserazide (4) (50-fold concentration of IC50 value) suggested that the mode of enzyme inhibitory activity of benserazide displayed a reversible pattern, while and an irreversible inhibitor, compound 3 showed a typical irreversible pattern of enzyme inhibition.
3.3. In silico search for a putative allosteric binding site and molecular docking study of compound 4 (Benserazide) and its analogs
Therefore, we further pursued the determination of a novel putative allosteric binding site of CVB3 3Cpro using an in silico study. Among a few cavities that were detected using the eraser algorithm method available in commercial in silico software (Discovery Studio 3.5; Accelrys Inc., San Diego, CA), the putative allosteric binding site most fitted to the size of Benserazide (4) was selected, which was positioned opposite the active site (Fig. 3 a) and was composed of Arg13, Ser16, Asn50, Arg79, Glu81 and Arg84 residues (Fig. 3b). The binding mode Benserazide (4) 4 and its derivatives in this binding pocket were investigated by a molecular docking study (Fig. 3c–f). It is speculated that (1) 2-, 3- and 4- hydroxyl groups of compound Benserazide (4) may interact by hydrogen bonding with the side chains of Arg79 and Glu81, the side chain of Asn50, and the carbonyl oxygen of the Ser16 back bone, respectively; (2) the carbonyl oxygen of Benserazide (4) may interact with the guanidine moiety of Arg84; and (3) the -OH, and -NH2 groups of Benserazide (4) may also form hydrogen bonds with the guanidine moiety of Arg13 (Fig. 3c; CDOKER energy (Kcal·mol−1): −34.23). The binding mode of compound 8a, was very similar to that of Benserazide (4) (Fig. 3d; CDOKER energy: −31.95); the compound also exhibits an inhibitory effect (IC50 = 2.72 μM) similar to that of Benserazide (4). In the case of di-hydroxy benzyl (b–f), mono-hydroxy benzyl (g–i) and benzyl (j)-substituted compounds (8b-j, 9b-j), the enzyme inhibitory activity was completely lost, and the binding modes of those analogs (9e and 9l) suggest that decreased hydrogen bonding interactions (Fig. 3e; CDOKER energy: −25.46 and Fig. 3f; CDOKER energy: −24.73) compared to those of compounds 4 or 8a (Fig. 3c and d) may have detrimental effects on the enzyme inhibitory activity. Therefore, the 2,3,4-trihydroxybenzyl moiety may be critically important for the binding to evoke the inhibition of enzyme catalysis.
Fig. 3.

In silico binding site prediction and molecular docking study. (a) Overall structure of CVB3 3Cpro (PDB code: 2ZU3); each known active site and putative allosteric active site is highlighted with red- and blue-colored spheres, respectively. (b) The expanded putative allosteric binding site; it is composed of Arg13, Ser16, Asn50, Arg79, Glu81, and Arg84 residues. (c) and (d) Binding modes of 4 (gray), 8a (green). (e and f) Binding modes of 9e (purple), 9l (yellow). The hydrogen bond interactions are represented by green dotted lines. Important amino acids are described and labeled in green.
3.4. Comparing the binding mode of Benserazide (4) at CVB3 3Cpro and DOPA-decarboxylase using In silico molecular docking study
Since Benserazide (4) is a potent inhibitor of DOPA decarboxylase, we analyzed the possible binding mode of Benserazide in DOPA decarboxylase by docking study. As shown in Fig. 4 b, only one phenolic hydroxyl group of benserazide might be interacted with Lys303, which is interacted with the hydrazine moiety of carbiDOPA [Fig. S1; in Supplementary Information]. In addition, the serine moiety of benserazide was detected to form hydrogen bonding with Ser193 via water molecules as the carboxylate groups of carbiDOPA do the similar interaction. It seems that the important binding moieties of benserazide might be different in between CVB3 3C protease (Fig. 4a) and DOPA decarboxylase (Fig. 4b), which are the tri-hydroxyl groups and serine group, respectively. Therefore, modifications of serine moiety of benserazide such as substitutions of the amine and hydroxyl groups or changing to non-polar groups, may provide selective inhibition of CVB3 3C protease to avoid potential side effects, which may be caused by the inhibition of decarboxylase enzymes.
Fig. 4.
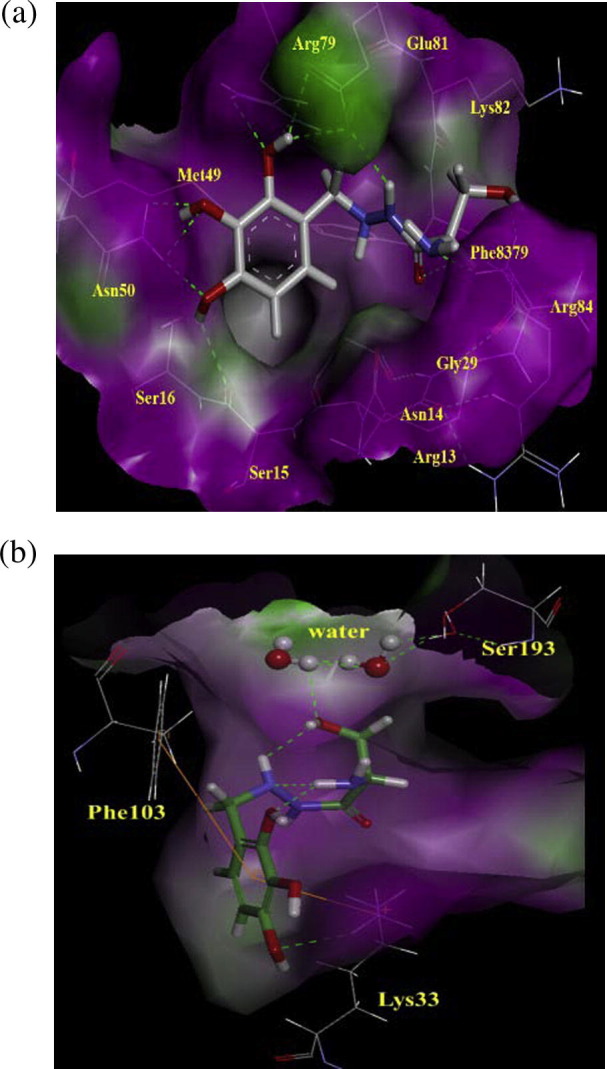
Comparing the binding mode of Benserazide (4) at CVB3 3Cpro and DOPA-decarboxylase. (a) The binding mode of Benserazide (4) at CVB3 3Cpro and (b) DOPA-decarboxylase. The hydrogen bond interactions are represented by green dotted lines. Important amino acids are described and labeled in green. The surface of binding site was highlighted in hydrogen donor (purple) and acceptor (green).
4. Conclusion
Herein, we reported that a novel non-competitive CVB3 3Cpro inhibitor Benserazide (4), a known anti-Parkinson’s disease drug, was discovered through screening using a FRET-based enzyme assay. The mechanism of enzyme inhibition and the binding mode of Benserazide (4) and its derivatives were characterized by enzyme kinetics and in silico molecular docking studies. These studies demonstrate that Benserazide (4) is a non-competitive inhibitor that interacts with a putative allosteric binding site, and the 2,3,4-trihydroxybenzyl moiety of Benserazide (4) is the most important to the inhibitory action against CVB 3Cpro. These results suggest that new therapeutic strategies targeting the newly found putative allosteric binding site of CVB 3Cpro could be applied for the development of non-competitive inhibitors.
Acknowledgements
This research was supported by a Grant of Basic Science Research Program through the National Research Foundation of South Korea (NRF) funded by the Ministry of Education, Science and Technology (Grant No. NRF-2013R1A1A2059917 and NRF-2014M349A9073788).
Footnotes
Author contributions: Bo-Kyoung Kim: Enzyme Assay, Kinetic Study and Chemical Synthesis. Joong-Heui Cho: Expression of Enzyme. Pyeonghwa Jeong: Molecular Docking Study. Youngjin Lee, Jia Jia Lim, Kyoung Ryoung Park, Soo Hyun Eom: Study for X-ray Co-crystal Structure. Yong-Chul Kim: Project Leader and Writing Manuscript.
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.febslet.2015.05.027.
Appendix A. Supplementary data
References
- 1.Yuan J. Inhibition of coxsackievirus B3 in cell cultures and in mice by peptide-conjugated morpholino oligomers targeting the internal ribosome entry site. J. Virol. 2006;80:11510–11519. doi: 10.1128/JVI.00900-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Palma A.M., Vliegen I., De Clercq E., Neyts J. Selective inhibitors of picornavirus replication. Med. Res. Rev. 2008;28:823–884. doi: 10.1002/med.20125. [DOI] [PubMed] [Google Scholar]
- 3.Feldman A.M., McNamara D. Myocarditis. N. Engl. J. Med. 2000;343:1388–1398. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 4.D’Ambrosio A., Patti G., Manzoli A., Sinagra G., Di Lenarda A., Silvestri F., Di Sciascio G. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: a review. Heart. 2001;85:499–504. doi: 10.1136/heart.85.5.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chau D.H., Yuan J., Zhang H., Cheung P., Lim T., Liu Z., Sall A., Yang D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis. 2007;12:513–524. doi: 10.1007/s10495-006-0013-0. [DOI] [PubMed] [Google Scholar]
- 6.Kim B.K., Kim J.H., Kim N.R., Lee W.G., Lee S.D., Yun S.H., Jeon E.S., Kim Y.C. Development of anti-coxsackievirus agents targeting 3C protease. Bioorg. Med. Chem. Lett. 2012;22:6952–6956. doi: 10.1016/j.bmcl.2012.08.120. [DOI] [PubMed] [Google Scholar]
- 7.Lee E.S. Development of potent inhibitors of the coxsackievirus 3C protease. Biochem. Biophys. Res. Commun. 2007;358:7–11. doi: 10.1016/j.bbrc.2007.03.208. [DOI] [PubMed] [Google Scholar]
- 8.Zell R., Markgraf R., Schmidtke M., Gorlach M., Stelzner A., Henke A., Sigusch H.H., Gluck B. Nitric oxide donors inhibit the coxsackievirus B3 proteinases 2A and 3C in vitro, virus production in cells, and signs of myocarditis in virus-infected mice. Med. Microbiol. Immunol. 2004;193:91–100. doi: 10.1007/s00430-003-0198-6. [DOI] [PubMed] [Google Scholar]
- 9.Binford S.L., Maldonado F., Brothers M.A., Weady P.T., Zalman L.S., Meador J.W., 3rd, Matthews D.A., Patick A.K. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 2005;49:619–626. doi: 10.1128/AAC.49.2.619-626.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuo R.L., Kung S.H., Hsu Y.Y., Liu W.T. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 2002;83:1367–1376. doi: 10.1099/0022-1317-83-6-1367. [DOI] [PubMed] [Google Scholar]
- 11.Weng K.F., Li M.L., Hung C.T., Shih S.R. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009;5:e1000593. doi: 10.1371/journal.ppat.1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M.L., Hsu T.A., Chen T.C., Chang S.C., Lee J.C., Chen C.C., Stollar V., Shih S.R. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology. 2002;293:386–395. doi: 10.1006/viro.2001.1310. [DOI] [PubMed] [Google Scholar]
- 13.Wu K.X., Ng M.M., Chu J.J. Developments towards antiviral therapies against enterovirus 71. Drug Discov. Today. 2010;15:1041–1051. doi: 10.1016/j.drudis.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lall M.S., Jain R.P., Vederas J.C. Inhibitors of 3C cysteine proteinases from Picornaviridae. Curr. Top. Med. Chem. 2004;4:1239–1253. doi: 10.2174/1568026043387836. [DOI] [PubMed] [Google Scholar]
- 15.Ramajayam R., Tan K.P., Liu H.G., Liang P.H. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg. Med. Chem. 2010;18:7849–7854. doi: 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnard D.L. Current status of anti-picornavirus therapies. Curr. Pharm. Des. 2006;12:1379–1390. doi: 10.2174/138161206776361129. [DOI] [PubMed] [Google Scholar]
- 17.Lee C.C. Structural basis of inhibition specificities of 3C and 3C-like proteases by zinc-coordinating and peptidomimetic compounds. J. Biol. Chem. 2009;284:7646–7655. doi: 10.1074/jbc.M807947200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo C.J., Liu H.G., Lo Y.K., Seong C.M., Lee K.I., Jung Y.S., Liang P.H. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009;583:549–555. doi: 10.1016/j.febslet.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webber S.E. Design, synthesis, and evaluation of nonpeptidic inhibitors of human rhinovirus 3C protease. J. Med. Chem. 1996;39:5072–5082. doi: 10.1021/jm960603e. [DOI] [PubMed] [Google Scholar]
- 20.Baxter A. Non-covalent inhibitors of rhinovirus 3C protease. Bioorg. Med. Chem. Lett. 2011;21:777–780. doi: 10.1016/j.bmcl.2010.11.110. [DOI] [PubMed] [Google Scholar]
- 21.Cordingley M.G., Register R.B., Callahan P.L., Garsky V.M., Colonno R.J. Cleavage of small peptides in vitro by human rhinovirus 14 3C protease expressed in Escherichia coli. J. Virol. 1989;63:5037–5045. doi: 10.1128/jvi.63.12.5037-5045.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shen H., Kannari K., Yamato H., Arai A., Matsunaga M. Effects of benserazide on l-DOPA-derived extracellular dopamine levels and aromatic l-amino acid decarboxylase activity in the striatum of 6-hydroxydopamine-lesioned rats. Tohoku J. Exp. Med. 2003;199:149–159. doi: 10.1620/tjem.199.149. [DOI] [PubMed] [Google Scholar]
- 23.Ryu Y.B. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg. Med. Chem. 2010;18:7940–7947. doi: 10.1016/j.bmc.2010.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen T.T., Ryu H.J., Lee S.H., Hwang S., Breton V., Rhee J.H., Kim D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. Lett. 2011;21:3088–3091. doi: 10.1016/j.bmcl.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costenaro L. Structural basis for antiviral inhibition of the main protease, 3C, from human enterovirus 93. J. Virol. 2011;85:10764–10773. doi: 10.1128/JVI.05062-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu G. Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. J. Virol. 2011;85:10319–10331. doi: 10.1128/JVI.00787-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan J. 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 2013;87:4339–4351. doi: 10.1128/JVI.01123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mosimann S.C., Cherney M.M., Sia S., Plotch S., James M.N. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 1997;273:1032–1047. doi: 10.1006/jmbi.1997.1306. [DOI] [PubMed] [Google Scholar]
- 29.Matthews D.A. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui S. Crystal structure of human enterovirus 71 3C protease. J. Mol. Biol. 2011;408:449–461. doi: 10.1016/j.jmb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.