

Abstract
Rapid elucidation of neutralizing antibody epitopes on emerging viral pathogens like severe acute respiratory syndrome (SARS) coronavirus (CoV) or highly pathogenic avian influenza H5N1 virus is of great importance for rational design of vaccines against these viruses. Here we combined screening of phage display random peptide libraries with a unique computer algorithm “Mapitope” to identify the discontinuous epitope of 80R, a potent neutralizing human anti-SARS monoclonal antibody against the spike protein. Using two different types of random peptide libraries which display cysteine-constrained loops or linear 13–15-mer peptides, independent panels containing 42 and 18 peptides were isolated, respectively. These peptides, which had no apparent homologous motif within or between the peptide pools and spike protein, were deconvoluted into amino acid pairs (AAPs) by Mapitope and the statistically significant pairs (SSPs) were defined. Mapitope analysis of the peptides was first performed on a theoretical model of the spike and later on the genuine crystal structure. Three clusters (A, B and C) were predicted on both structures with remarkable overlap. Cluster A ranked the highest in the algorithm in both models and coincided well with the sites of spike protein that are in contact with the receptor, consistent with the observation that 80R functions as a potent entry inhibitor. This study demonstrates that by using this novel strategy one can rapidly predict and identify a neutralizing antibody epitope, even in the absence of the crystal structure of its target protein.
Keywords: SARS, antibody, epitope, Mapitope, computational algorithm
Abbreviations used: SARS, severe acute respiratory syndrome; CoV, coronavirus; AAP, amino acid pair; SSA, statistically significant pair; mAb, monoclonal antibody; scFv, single chain variable fragment; ST, statistical threshold; RBD, receptor binding domain
Introduction
With every new and emerging infectious pathogen, particularly those that are capable of causing widespread debilitating illness and death, it is necessary not only to institute local, regional and international public health care measures to prevent and contain the infections, but also to rapidly develop therapeutic strategies to elicit protective host immunity. In the case of respiratory illnesses such as severe acute respiratory syndrome (SARS), highly pathogenic H5N1 avian influenza and West Nile Virus febrile illness/encephalitis, where the importance of neutralizing antibodies in preventing disease onset is clearly established, defining the molecular determinants of the neutralizing epitope(s) is critically important in the development of an efficacious vaccine.1, 2, 3, 4, 5, 6, 7 In particular, recombinant vaccines that are capable of focusing the humoral immune response on neutralizing epitopes can be predicted to be most beneficial and may provide a more rapid way to respond to emerging biothreats than traditional attenuated or inactivated viruses or subunit vaccines.
SARS emerged as a new infectious disease and caused a serious worldwide outbreak in 2002 to 2003 with over 8000 individuals becoming infected. In its most severe form, infection with the novel SARS-coronavirus (SARS-CoV) was associated with progressive pneumonia, respiratory failure, and a fatality rate of ca 10%.8, 9, 10, 11 The receptor for SARS-CoV was shortly thereafter identified as angiotensin-converting enzyme-2 (ACE-2)12, 13 and importance of neutralizing antibodies to the SARS-CoV spike protein in preventing infection in vitro and in vivo was established.1, 2, 3 However, serologic studies from both late outbreak infected humans and with serum from mice immunized with a late outbreak strain demonstrated the presence of antibodies that were able to enhance infection of SARS-like CoV from civet cats in a pseudo-virus reporter assay.14 Since these enhancing mouse antibodies map to the receptor binding domain (RBD) of Spike (S) protein, a region that would obviously be used in a subunit vaccine, it appears that some epitopes contained therein may be detrimental and thus defining the precise nature of the neutralizing epitope(s) is warranted. Therefore, a vaccine should focus on eliciting only neutralizing antibodies and not antibodies that are either non-neutralizing or enhancing in nature.15
We took the first steps toward the goal of identifying the major neutralizing epitope of SARS-CoV as a model of neutralizing epitope identification using a reverse immunological approach. In order to accomplish this task one must backtrack from the antibody of interest to its corresponding neutralizing epitope.16 It is then assumed that, once identified, the epitope can be reconstituted and stabilized with the intent that when administered as a vaccine it will elicit the neutralizing activity characteristic of the original monoclonal antibody (mAb). The human recombinant mAb used in this study, named 80R was isolated from a phage display library after panning against the S1 domain of the SARS-CoV Spike protein.3 80R binds to the RBD, a 193 amino acid fragment (residues 318 to 510) of spike protein with high-affinity (K d=1.7 nM) and is a potent neutralizing mAb in vitro and in vivo.17 It acts as a viral entry inhibitor through blocking the association of S protein to its receptor ACE2. Mutagenesis studies further support this conclusion as Spike determinants involved in the binding of receptor and of 80R are in part overlapping and are likely to result from both common and unique contact residues.17
Results
The principles of the Mapitope algorithm
A unique computer algorithm Mapitope enabled us to map epitopes on spike protein using peptides that bind to 80R. Mapitope is an updated user-friendly version of the algorithm previously published by Enshell-Seijffers et al. 16 The prediction of an epitope is based on the notion that the panel of peptides derived from a random peptide library collectively represents the epitope of the mAb which they bind. The underlying principle of Mapitope is that the simplest meaningful fragment of an epitope is an amino acid pair (AAP) of residues that lie within the footprint of the epitope. These AAPs can be related to one another on the surface of the antigen such that a cluster is defined which constitutes the majority of the epitope footprint, i.e. the epitope is in essence a cluster of connected AAPs. The AAPs of the epitope need not be consecutive tandem residues of the antigen, but often are the result of juxtaposition of distant residues brought together through folding of the polypeptide chain, the distance between their carbon alphas (parameter D), defines what constitutes a legitimate pair. AAPs of the epitope are simulated by tandem residues of the peptides, affinity selected from the random library. Each peptide is assumed to contain one or more epitope relevant AAPs, which is the basis for mAb recognition of that peptide. In order to identify the statistically significant pairs (SSPs) present in the panel of peptides, the peptides are first deconvoluted into AAPs. Thus, for example to deconvolute a peptide into AAPs, a peptide of the sequence ABCDE... would be written as the series of pairs: AB, BC, CD, DE, etc. All the AAPs derived from the panel of peptides are then pooled and the frequency of each type is calculated. It is next determined whether the AAPs representation in the pool is higher than the random expectation and if so, these pairs are considered to be SSPs. A second parameter of the algorithm (the first being D) is the frequency of a specific pair in a given pool of AAPs derived from the panel of peptides. The number of standard deviations above randomness for a given pair is defined as the statistical threshold (ST). Once the most frequent AAPs are identified, the algorithm seeks the pairs for a selected D value on the surface of the antigen and attempts to link them into clusters. A third parameter of the algorithm is E, the surface accessibility threshold. E defines those residues that are sufficiently exposed on the antigen's surface to be included in the predicted epitope. The accessibility of each amino acid is automatically calculated using the software “SurfRace,”18 which has been assimilated in the algorithm software. In this study the SSPs which were mapped on the 3-D structure of the antigen contained residues that are at least 5% exposed (E=5); however, impact of the E parameter was examined as well (see below).
As contacts between the mAb and the antigen are mostly through functional moieties of the R-groups, conserved residues were consolidated into 13 functional subgroups of amino acids and given single-letter notations:
In summary, a mAb is used to screen a random peptide library to generate a panel of peptides recognized by the mAb. These peptides are deconvoluted into AAPs and the SSPs are identified. These are then mapped in the crystal structure of the antigen and the most elaborate and diverse clusters on the surface of the antigen are identified. These are regarded as the predicted epitope candidates.
Phage display peptide panning against 80R scFv
A variety of combinatorial phage display peptide libraries were screened with the 80R single chain variable fragment (scFv) (see Materials and Methods). Two independent panels of peptides were isolated (Table 1 ). The peptides were derived from two different types of random peptide libraries, 42 peptides derived from cysteine constrained-loop libraries were designated as panel 1 and 18 peptides, derived from libraries of random linear peptides, were designated as panel 2. No common homologous motif was observed within the peptides themselves, or between the peptides and the SARS-CoV spike protein. This is not surprising in view of the fact that the epitope of 80R is conformational.3 Each set of peptides was used independently for Mapitope analysis, thus generating two independent predictions of the 80R epitope.
Table 1.
Peptide data sets for Mapitope prediction of the 80R epitope
| Panel 1 (42 peptides) | Panel 2 (18 peptides) |
|---|---|
| RSGGCVGGQYCLTPTH | LDSMHFPFHSRSFWP |
| NDWPCLSHTTVCNGTQ | NLSCTHPLGSPPPAP |
| ATMPCLSHPSVCKHLY | GQICYYGRDAYLCFL |
| PMHECLSAPSVCADNY | CESSLCLMYSLGPPA |
| TELACLSEAYICDRSN | QTPPCPIEHCPSFYQ |
| ETFTCISAPWTCVTWL | QSTCLSHPLLCLSWN |
| EKMACLSTLDVCMENP | PNCWVGLTGAHSCFL |
| NNMSCLSHETICGRNP | THSVPVAYPWPDLNA |
| LPFECISKREVCDTPM | SPLDYECISHATVCF |
| SVDDCRWNLNCEPPP | YSTPSSILDTHPLYK |
| SEVYCPRPDRCLRAP | TLPPPCLSSPSRCVN |
| VQRDCRWTFSCATLI | RTMHPSDEFLPLGMP |
| TPPRCSDQMYCSLSR | GTGLVPLFDPRYRFL |
| THQFCPDPKHCLAQP | SSSRQEPYPLYPLFS |
| RMPPCMNAGECPTIA | HPKVGEGIDFTSIVP |
| DTPDCXGNEKCLEYA | ATDLLAAYPLYSPSL |
| TSNFCPAGGPCSPHG | VVPLGRCVSHPAICA |
| NPRVCMNKWECEQAI | GFPCLSVASACYGIT |
| GPPLGCLSLSCYDVA | |
| WNDYCTMNQCDTHN | |
| KPLHCGDTFCSLNQ | |
| YLEHCTMNECLNAR | |
| NGYHCLSEFCMPHP | |
| SMEECRLWLCPPYE | |
| YKPWCEMNKCKPLA | |
| VMPECLSRLCDFDM | |
| DDMPGCYPMCTLNK | |
| YDSYCIMNFCGHAA | |
| YTAADCPGLLYLCP | |
| NDVRCKLWLCPMPD | |
| NNWPCLNETCPTKG | |
| VQWPCLSKQCNDNI | |
| YQADCLMNRCPTAE | |
| SAPECHLYYCPEQA | |
| ANPVCRLWMCPPIV | |
| RQTEPCNLWFCPQV | |
| REPPCVQVHCSTAK | |
| PKEQPWSEFRPAGM | |
| ADCTLWFCPQTSN | |
| CLSATCDCTLCGP | |
| FPELTCWTCLASS | |
| PPAYSCLCPWAHM |
Panel 1, peptides isolated with the 80R from phage display peptide libraries where cysteine residues are fixed. The pre-fixed cysteine residues are indicated in bold. Panel 2, peptides isolated from linear peptide libraries.
Analyzing the peptides and defining statistically significant pairs (SSPs)
The first step in applying the algorithm is to “translate” the peptides into Mapitope functional notations (see above) and to deconvolute them into AAPs. Deconvolution of peptides into AAPs using the functional notation allows for 13 classes of amino acids and therefore 169 possibilities. However, as 13 pairs are homodimers (e.g. AA, BB, etc.) the total number of different AAPs possible is 156. Deconvolution of the 42 peptides of panel 1 produced a total of 568 AAPs which are represented by 133 different pair types. Taking ST≥3, a total of 11 pair types were found to be statistically significant pairs (SSPs). These 11 pair types (8% of all available 133 pair types) were represented by 108 pairs (19% of all the 568 pairs). Similarly, deconvolution of the 18 peptides of panel 2 produced a total of 252 AAPs represented by 89 different pair types. Taking ST≥3, a total of 12 pair types were found to be SSPs. These 12 pair types (13% of all available 89 pair types) were represented by 60 pairs (24% of all the 252 pairs).
The Mapitope predictions are based on focusing on those pairs that are statistically enriched. Figure 1 (a) gives the 11 SSPs of panel 1 comparing the observed occurrence with the calculated expected occurrence based on total randomness. Note that in Figure 1(b) the highest value for occurrence does not necessarily promise the greatest statistical significance, since the statistical significance depends on the individual expectation of each SSP (for more explanation about random expectation of SSPs and factors that can influence this parameter see Enshell-Seijffers et al. 16). Compare for example, the SSPs CU versus YC; CU appears 26 times in the peptides, which is five standard deviations greater than its expectation in the library (in a panel of 42 totally random peptides, CU is expected to appear 18.1 times). On the other hand, the SSP YC appears only six times, but is two times more abundant than would be expected; consequently its ST value is 4.76. An extreme case is the pair CJ which exists eight times in the peptides; however, its expected occurrence is 9.05 and therefore this pair is actually under-represented (not shown). Similarly, analysis of the 18 peptides of panel 2 is shown in Figure 1(c) and (d). Of the 12 pairs which are defined as SSP (ST≥3) the most significant pairs are PU, CU and PP.
Figure 1.
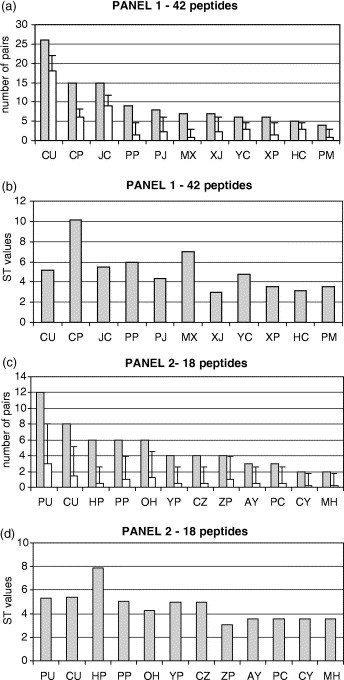
Computation of the SSPs derived from the 80R binding peptides ((a) and (c) for panel 1 and panel 2, respectively) and their comparison between the observed occurrence (gray bars) and calculated expected occurrence (open bars). The error-bars represent statistical threshold (ST) value equals 3. Histograms (b) and (d) show the significance of each pair (ST values) based on the peptides of panel 1 and panel 2, respectively.
Preliminary prediction on the RBD of spike protein
Once the analysis of the peptides was preformed and the most significant amino acid pairs were identified, the next step is to map these pairs on the surface of the SARS-CoV spike protein. The most desirable starting point for this would be to use a solved atomic structure of the antibody's antigen, in this case, the receptor binding domain (RBD), but such a solved structure was not available when this study initiated. Nonetheless, an alternative Mapitope prediction was conducted using a theoretical model of the spike, which was obtained by homology modeling between the SARS-CoV spike and the botulinum neurotoxin B.19 The 3-D structure of botulinum neurotoxin B served as a template for the prediction of the 3-D structure of the SARS-CoV spike.19 As previous studies of 80R have indicated that its epitope is contained within the RBD of the spike, our prediction was focused on this aspect of the modeled spike protein. Application of Mapitope entails a preliminary run of a given data set of peptides using the default parameters (ST=3, D=9 Å, E=5%). Such a procedure generates a first approximation of possible epitope candidates, i.e. “clusters”. The analysis of Table 1 panel 1 gave three possible clusters designated as clusters A, B and C (Table 2 ). The analysis of Table 1 panel 2 gave the same three clusters with an addition of a fourth cluster designated cluster D (Table 2). Therefore, at this point each cluster was analyzed independently.
Table 2.
Amino acids predicted in each cluster A, B and C for panel 1 and panel 2 peptides using the theoretical model
| A |
B |
C |
|||
|---|---|---|---|---|---|
| Panel 1 | Panel 2 | Panel 1 | Panel 2 | Panel 1 | Panel 2 |
| Pro450 | Phe334 | ||||
| Glu452 | Asn318 | Pro335 | |||
| Asp454 | Ile319 | Ile319 | Val337 | ||
| Asn457 | Asn321 | Thr320 | Tyr338 | ||
| Pro459 | Leu322 | Leu322 | Ala339 | ||
| Pro462 | Cys323 | Cys323 | Ala350 | ||
| Asp463 | Pro324 | Pro324 | Tyr352 | ||
| Pro466 | Pro466 | Phe325 | |||
| Cys467 | Cys467 | Phe361 | |||
| Pro469 | Pro469 | Glu327 | Glu341 | Phe364 | |
| Pro470 | Pro470 | Val328 | Val328 | Cys366 | Cys366 |
| Leu472 | Leu472 | Asn330 | Tyr367 | Tyr367 | |
| Asn473 | Thr332 | Val369 | Val369 | ||
| Cys474 | Cys474 | Ala371 | |||
| Tyr475 | Tyr475 | Tyr440 | Tyr440 | ||
| Trp476 | Tyr442 | Tyr442 | Leu374 | Leu374 | |
| Pro477 | Pro477 | Leu443 | Leu443 | Asn375 | |
| Leu478 | His445 | His445 | Asp376 | ||
| Asn479 | Leu377 | Leu377 | |||
| Asp480 | Cys378 | Cys378 | |||
| Tyr481 | Phe379 | ||||
| Asn381 | |||||
| Val382 | Val382 | ||||
| Tyr383 | Tyr383 | ||||
| Ala384 | |||||
| Asp385 | |||||
The prediction for each peptide panel and cluster was made at the respective Q point (see Figure 2(c)) and at ST=3. Amino acids common to both panels are in bold.
Defining the limits of each cluster: modifying the D parameter
The question that arises is how can one rank the clusters and identify which is a better candidate of the epitope as compared to the others? For this, once a set of preliminary clusters is identified, the next step is to evaluate the behavior of each cluster, taking different D values ranging from 4 to 15 (the distance of carbon α to carbon α for tandem residues (n, n+1) is 3–6 Å). Maintaining ST=3, the number of amino acids for each cluster was measured as a function of distance between two amino acids comprising a pair. As an example, Figure 2 illustrates the effect of distance on the four clusters of panel 2. Figure 2(a) shows the change in the number of amino acids in clusters A and C and Figure 2(b) shows the same for clusters B and D. Note that as a function of increasing the D value the number of amino acids increases, as expected. However, beyond a given point this increase gives a “quantum jump” in the number of amino acids associated with a given cluster, this is defined as the “Q point” (indicated by the gray arrows). The significant increase in the number of amino acids beyond the Q point could be the result of merging of adjacent clusters or recruitment of peripheral or underlying irrelevant amino acids thus leading to a sharp increase in the number of amino acids associated with a given D value. For example, for cluster A the jump is at 12 Å, going from 11 amino acid residues to 31, for cluster D the Q point is at 13.5 Å (from 30 to 55 amino acid residues in the cluster; see Figure 2(a)). The Q points for clusters A, B and C in the first panel (42 peptides) and for clusters A–D in the second panel (18 peptides) are shown in Figure 2(c).
Figure 2.
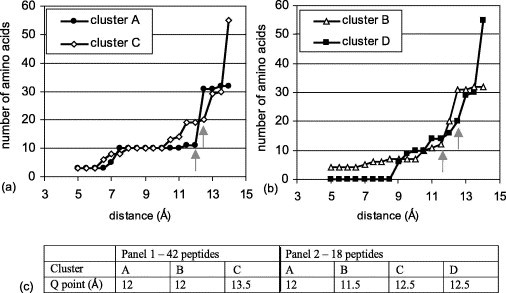
The effect of distance between amino acids comprising a pair on the number of amino acids within a cluster in the analysis of the peptides of panel 2 applied to the theoretical model of the SARS-CoV spike. (a) Clusters A and C; (b) clusters B and D. The arrows indicate the Q points. (c) The table summarizes the Q points for the three clusters of panel 1 (data not shown) and for the four clusters of panel 2. All the predictions were conducted at ST=3.
Cluster D is not predicted in the analysis of panel 1 peptides. Moreover, as can be seen in Figure 2(b), it is based exclusively on pairs which are separated by at least 8.5 Å. This would be an unusual situation as it indicates that none of the pairs in this cluster are tandem in the linear sequence. Therefore, we consider cluster D as least likely to be the epitope of 80R. Figure 3 shows clusters A, B and C as predicted by Mapitope using panel 1 and panel 2 peptides. Table 2 summarizes the amino acids included for the three clusters A, B and C which are predicted at their respective Q points using ST=3 for each panel of peptides. Amino acids common to both panels are in bold.
Figure 3.
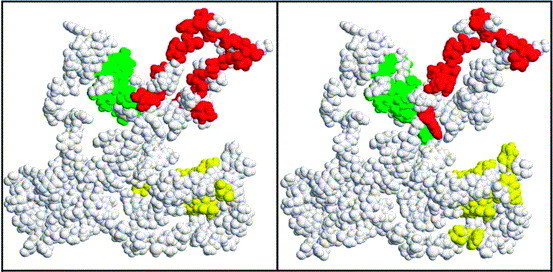
RasTop spacefill presentation of clusters A (red), B (green) and C (yellow) as predicted from Mapitope analyses of panel 1 peptides (left panel) or panel 2 peptides (right panel) using the theoretical model of the spike RBD. Amino acids comprising each cluster are listed in Table 2.
Table 3 shows the SSPs comprising each cluster and their significance according to the calculations that were made in Figure 1. Note that clusters A and B are the most varied as they contain the larger amount of different SSPs and use the SSPs with the highest significance (e.g. the highly significant pair CP in panel 1, or the SSPs HP, PP, OC and PC that are used by clusters A and B but missing from cluster C in panel 2).
Table 3.
The number and the quantity of the SSPs used by each cluster as predicted on the surface of the theoretical model of the 193 amino acid segment of the spike
| Pair |
CU |
CP |
JC |
PP |
PJ |
MX |
JX |
YC |
XP |
HC |
PM |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | ST | 5.15 | 10.15 | 5.50 | 5.95 | 4.34 | 7.00 | 3.00 | 4.76 | 3.55 | 3.155 | 3.55 | |
| A | + | + | + | + | + | + | + | + | |||||
| B | + | + | + | + | + | + | + | + | |||||
| C | + | + | + | + |
| Pair |
PU |
CU |
HP |
PP |
OH |
YP |
CZ |
ZP |
AY |
PC |
CY |
MH |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | ST | 5.29 | 5.38 | 7.85 | 5.06 | 4.29 | 5.00 | 5.00 | 3.04 | 3.57 | 3.57 | 3.53 | 3.53 |
| A | + | + | + | + | + | + | + | + | |||||
| B | + | + | + | + | + | + | + | + | + | + | |||
| C | + | + | + | + | + | + | + |
The table on the top shows panel 1 clusters and the bottom table shows panel 2 clusters. The ST values for each SSP are given (only those SSPs which have ST values greater than 3 are shown).
Mapitope analysis based on the crystal structure of the RBD of spike protein
During the course of this study, Li et al. solved the atomic structure of the RBD of the SARS-CoV with its receptor ACE2.20 This allowed us to repeat the Mapitope analysis; however, this time using the genuine atomic coordinates. Once this was completed, we were able to compare the two sets of predictions, and thereby gain insight as to the utility of Mapitope prediction using theoretical models, for future studies where crystal structures have not been solved. In order to compare the two structures, we employed the FlexProt program,21 which is capable of detecting hinge regions and structurally aligning the rigid subparts of two 3-D structures (pair-wise alignment). In the comparison of the two RBD structures, residues 323–498, we found about 50% correspondence (89 matches out of 174 amino acid residues; RMSD=2.79 Å). This indicates that there is a general similarity between the genuine structure and the theoretical model used above.
As before, we used the SSPs of both peptide panels to perform Mapitope predictions on the crystal structure of the spike using the default parameters. Much to our satisfaction clusters A, B and C described above were partially predicted anew (at least 50% overlap with the clusters predicted using the theoretical model) but this time using the atomic coordinates of the crystal structure (this corresponds well with the FlexProt analysis described above). As is illustrated in Figure 4 the three clusters are easily identified at ST=3. In this case a fourth cluster is also defined (designated as cluster D) as distinct for the panel 1 peptides, which merges with cluster C in the case of panel 2. Increasing the ST value to five eliminates clusters C and D or diminishes cluster C markedly using panel 1 and panel 2, respectively (not shown).
Figure 4.
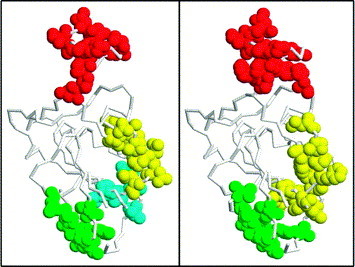
Left and right panels: RasTop representation of clusters A (red), B (green), C (yellow) and D (cyan) on the crystal structure of the SARS CoV S protein RDB in the analysis of panel 1 or panel 2, respectively.
Identification of the Q point for each cluster and its effect on the predictions are shown in Figure 5 . Clusters B and C have a Q point=10.5 Å, above which the two clusters merge into one. In contrast to this, the prediction of cluster A is far more robust and tolerates D values as high as 12.5 before reaching a Q point. This distinguishes this cluster as compared to the other two.
Figure 5.
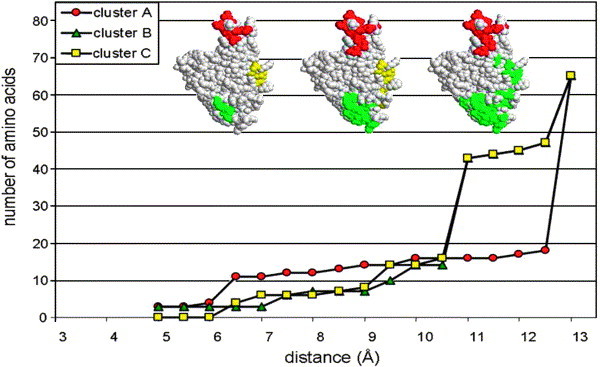
The impact of the distance (parameter D) on the number of amino acids within a cluster. Panel 2 peptides were used for Mapitope prediction on the SARS-CoV S crystalline structure. The images are RasTop space-fill representations of the spike protein RBD indicating the three clusters; A (red), B (green) and C (yellow) at different D values (left image, 6 Å; middle, 10 Å; right, 12 Å). All the predictions were conducted at ST=3.
Considering the usage of SSPs and their ST values, here cluster A ranks the highest as is illustrated in Table 4 . The amino acid residues included in the clusters using the crystal structure are listed in Table 5 . In summary, cluster A stands out as being the most attractive potential candidate for the 80R epitope.
Table 4.
The number and the quantity of the SSPs used by each cluster as predicted on the surface of the crystalline structure of the SARS-CoV RBD spike (D=9 Å)
| Pair |
CU |
CP |
JC |
PP |
PJ |
MX |
JX |
YC |
XP |
HC |
PM |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | ST | 5.156 | 10.15 | 5.50 | 5.95 | 4.34 | 7.00 | 3.00 | 4.76 | 3.55 | 3.155 | 3.55 |
| A | + | + | + | + | + | + | ||||||
| B | + | + | + | + | + | |||||||
| C | + | + | ||||||||||
| D | + | + | + | + |
| Pair |
PU |
CU |
HP |
PP |
OH |
YP |
CZ |
ZP |
AY |
PC |
CY |
MH |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster | ST | 5.29 | 5.38 | 7.85 | 5.06 | 4.29 | 5.00 | 5.00 | 3.04 | 3.57 | 3.57 | 3.53 | 3.53 |
| A | + | + | + | + | + | + | + | + | + | + | |||
| B | + | + | + | + | + | + | + | ||||||
| C | + | + | + | + | + | + | + | + |
The table on the top shows panel 1 clusters and the bottom table shows panel 2 clusters. The ST values for each SSP are given (only those SSPs which have ST values greater than 3 are shown).
Table 5.
Amino acids predicted in each cluster; A, B and C for panel 1 and panel 2 peptides as predicted using the genuine coordinates of the spike RBD
| A |
B |
C |
|||
|---|---|---|---|---|---|
| Panel 1 | Panel 2 | Panel 1 | Panel 2 | Panel 1a | Panel 2 |
| His445 | Cys323 | Cys323 | Phe364 | ||
| Asn457 | Pro324 | Pro324 | Cys366 | Cys366 | |
| Val458 | Val458 | Phe325 | Tyr367 | Tyr367 | |
| Pro459 | Pro459 | Glu327 | Val369 | Val369 | |
| Phe460 | Val328 | Ala398 | |||
| Pro462 | Pro462 | Ile345 | Ile345 | Cys419 | |
| Asp463 | Cys348 | Cys348 | Gln396 | ||
| Pro466 | Pro466 | Val349 | Val349 | Pro399 | Pro399 |
| Cys467 | Cys467 | Ala350 | Gln401 | ||
| Pro469 | Pro469 | Asp351 | Pro413 | Pro413 | |
| Pro470 | Pro470 | Tyr352 | Tyr352 | Asp414 | Phe416 |
| Ala471 | Ala371 | Asp415 | |||
| Leu472 | Leu472 | Met417 | |||
| Asn473 | Cys419 | ||||
| Cys474 | Cys474 | Leu448 | |||
| Tyr475 | Tyr475 | Pro450 | Pro450 | ||
| Trp476 | Phe451 | ||||
| Glu452 | |||||
| Leu499 | Leu499 | ||||
| Phe501 | |||||
Amino acids common to both panels are in bold. Amino acids that were predicted in the analysis of the theoretical model are highlighted in gray. The analysis was conducted when D=9 Å and ST=3.
The amino acids of cluster D are included in this list as well (see the text).
In Figure 6 the cluster A (colored in red) and the common amino acid residues (colored in yellow) predicted by both the theoretical model and genuine structure of the Spike RBD are shown in the crystal structure of the complex of the SARS-CoV S protein RBD and receptor ACE2.20 The compactness of the genuine structure is obvious and here cluster A becomes a tight protrusion comprised of three segments. Residues 455–463 form an ascending strand that then crosses over as a traversing segment (residues 463–472) followed by the descending segment (residues 473–476). The distance maintained by five hydrogen bonds between the ascending and descending segments is about 5 Å, which is shorter than the limits of the traversing segment (13.4 Å). This therefore imposes a force flipping the traversing segment forward (viewing the ascending segment on your right). The orientation and position of this segment is stabilized by the disulfide between Cys467 and Cys474 and a series of nine hydrogen bonds cross-linking the top of the structure within itself and to the ascending and descending segments.
Figure 6.
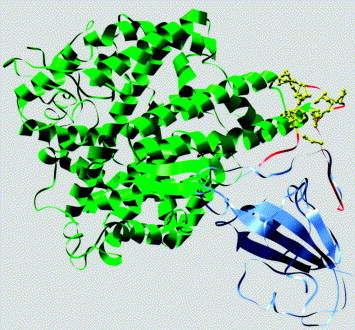
Presentation of the cluster A and the common amino acids predicted by both the theoretical model and genuine structure of the spike RBD in the crystal structure of the complex of the SARS-CoV S protein RBD and receptor ACE2.20 The spike RBD is shown in cornflower blue. The ACE2 is presented in green. Cluster A (residues 450–480 of the spike) are colored red. The predicted common residues (highlighted in Table 5) are colored in yellow.
In view of this compact and stable structure, the Mapitope prediction of cluster A gains a robustness that is lacking for clusters B and C. This is particularly noticeable considering the impact of the D parameter on the predictions (see Figure 5). In the case of the theoretical model, the Q point for cluster A is 12 Å where a sharp increase from 11 to 31 amino acid residues occurs. The Q point for cluster A in the crystal structure shifts to 13.5 Å, where the increase is from 18 amino acid resdues to over 60! This illustrates that the prediction is basically constant and that the structure of cluster A is relatively unchanged throughout the range of D values of 6 Å to 13 Å.
Finally in view of the fact that the mechanism of neutralization by mAb 80R has been proposed to be interference of viral association with its receptor, one cannot escape the fact that in the co-crystal, cluster A overlaps with a critical segment of the Spike:RBD interface. Several amino acids that lie within or juxtaposed to this predicted epitope effect spike protein structure globally (e.g. C464, C474),22 others effect Spike:ACE-2 and Spike:80R specifically (e.g. E452, D454).22 In addition, a critical amino acid in the predicted epitope has been shown to be specifically involved in Spike:80R molecular interactions (D480)17 while another amino acid, L472, had no effect.17 Nevertheless, one can see in Figure 6 how antibodies to the predicted epitope would interfere with Spike:ACE-2 interactions.
Discussion
The Mapitope algorithm was developed for the localization of B-cell epitopes based on the analysis of phage displayed affinity purified peptides.16 Validation of the algorithm has been achieved by first determining the defining parameters using the 17b:HIV gp120 co-crystal as a known control model.23 Subsequently, the algorithm was shown to be efficient in predicting the epitope of the anti- HIVp24 mAb 13b5 also co-crystallized with its antigen (HIVp24).16 In a third co-crystal model, a published panel of 27 phage displayed peptides specific for the Bo2C11 mAb that binds factor VIII24 were used as input with the atomic structure of its antigen (factor VIII) taken from the co-crystal published by Spiegel et al.25 The Mapitope algorithm predicted two clusters, the major one (cluster B) coincided with the genuine epitope (E. Bublil, personal communication). The strategy of using multiple independent peptide data sets has also been tested using the Trastuzumab (Herceptin®) mAb which was co-crystallized with its corresponding antigen (the cellular receptor Her-2/neu). In this case all three segments of the bona fide epitope were correctly predicted when two peptide panels were used for Mapitope data bases (unpublished results). Further validation of Mapitope has been published by Enshell-Seijffers et al. 16 in the analysis of the murine mAb CG10 (an antibody specific for the HIV gp120-CD4 complex) where the prediction was confirmed by functional reconstitution.16 Thus, Mapitope predictions have been validated by four separate mAb:antigen co-crystals and one case of epitope confirmation by physical reconstitution.
Here we apply this system to the analysis of a mAb against the major neutralization epitope in the RBD of spike protein to which 80R and several other human mAbs are directed.3, 26, 27, 28 Our efforts to delineate the structure of the 80R epitope with overlapping peptide ELISA scans were unsuccessful, suggesting along with other published data that the neutralizing epitope(s) are conformational.3, 17, 26 This region of RBD appears to be highly immunogenic and neutralizing human antibodies have been recovered from non-immune phage display libraries, human Ig transgenic mice and EBV-immortalized B cells from convalescent blood of a SARS-CoV infected individual. Other studies have identified two other neutralizing epitopes on spike protein that appear to be mostly linear, one outside the RBD in S1 and a few others to the S2 region; however, the mechanisms by which antibodies to these regions lead to neutralization have not been elucidated.28, 29, 30, 31 Although a number of methods exist to delineate the structure of epitopes (e.g. mutagenesis, docking in silico, neutralization escape studies and others), all ultimately produce a collection of candidate epitopes and there is no current method that provides a single solution with any degree of confidence.32, 33, 34 Thus, the objective of our analysis was to reduce the problem of conformational epitope mapping to a limited number of candidates that can be tested and validated experimentally.
The predictions based on the theoretical model would score clusters A and B as both being the more likely candidates for the 80R epitope as compared to cluster C, when considering the behavior of the clusters as a function of parameter variation. By altering the parameters D and ST, one recognizes that cluster C uses fewer SSPs and of lower ST values (variation in parameter E, surface accessibility, had little bearing on ranking the significance of the clusters). Nonetheless, a dilemma remained; can one discriminate between clusters A and B and identify that cluster which might be the better prediction of the genuine 80R epitope? Here the strength of using a high resolution atomic structure based on empirical X-ray analysis of the antigen's crystal becomes apparent; cluster A, as determined when using the coordinates of the crystal structure of the RBD becomes markedly more significant than cluster B. This provided us a firm basis to focus on cluster A as most likely being the 80R epitope. This furthermore illustrates that whenever possible, one should use the most detailed and highest resolution structure of the antigen as input for Mapitope analysis.
There have been several attempts to map conformational epitopes of antibodies in the absence of solved crystal structures of their corresponding antigens. One approach for this is to use theoretical models of the antigen, based on sequence alignment with an alternative protein-template whose atomic structure has already been worked out.32, 35, 36, 37 Of specific relevance is the study by Myers et al. in which they used a panel of affinity purified phage displayed peptides to assist in the localization of the epitope corresponding to the MICA3 and MICA4 mAbs that bind the major diabetes antigen, glutamic acid decarboxylase (GAD65). Their analyses identified five different prospective solutions which were further studied via mutagenesis. Here we present for the first time a comparative study between predictions based on a theoretical model of the SARS-CoV spike on the one hand and on the recently published crystal structure of the SARS-CoV RBD on the other. As described previously, there is about 50% correspondence between the two structures, nonetheless it appears that this level of similarity is sufficient, as Mapitope analyses of the peptide panels predicted three clusters for each structure that shared 50–70% identity between them (comparing the cluster of the theoretical model with the crystal structure; see Table 2, Table 5). This is an extremely intriguing result as it illustrates the potential of Mapitope analyses in situations where crystal structures are not available. The construction of theoretical models is almost routine where sequence homologies can be identified and as such all that is then necessary is to screen the mAb of interest against phage libraries so to produce a satisfactory peptide database and apply the algorithm for epitope prediction.
Although empirical approaches may lead to successful vaccine development, rational design of epitope-based vaccines using proven neutralizing mAbs as templates for epitope discovery is an important and worthwhile goal that could be applied to other new and emerging infectious diseases. This approach may eliminate the unwanted induction of non-neutralizing and enhancing antibodies that have been documented in SARS, dengue fever38 and respiratory syncytial virus.39 This property may be inherent even in subunit vaccines because of the proximity of these epitopes to the neutralizing epitopes that are sought. For this reverse immunological approach, one must be able to backtrack from the selected mAb to its corresponding epitope and ultimately reconstitute the epitope into a functional immunogen. The current study focuses on the first aspect of this paradigm, i.e. the discovery of a neutralizing epitope of the SARS-CoV protein. The 80R mAb is a very attractive case in point as it has been shown to be extremely potent in virus inactivation in vitro and in vivo. Analysis of the mechanism of action has led to the conclusion that the mAb interferes with virus:receptor binding; however, identifying the specific residues involved in 80R binding, i.e. the precise composition of its epitope, is still a formidable challenge, especially in view of the fact that the epitope has been shown to be conformational.3, 17 While our studies provide a demonstration of a robust computational approach that can be applied to neutralizing epitope discovery and a roadmap of how these advances may be applied in the future, the value of these predictions will ultimately be determined in functional studies where the reconstructed and stabilized neutralizing epitopes based on the cluster predictions are tested in vaccine studies and when the 80R:S1 protein co-crystal is solved.
Materials and Methods
Production of 80R scFv
80R scFv were expressed and purified as described.3 The VH and VL gene of 80R scFv were cloned into prokaryotic expressing vector pSynI for expression. It was expressed in Escherichia coli. XL1-Blue (Stratagene, La Jolla, CA) and purified from the periplasmic fractions by immobilized metal affinity chromatography.
Peptide libraries
The fUSE5/15-mer, F88-4/15-mer, and F88-4/Cys1/13-mer phage display peptide libraries display random linear 13–15-mer peptides. The F88-4/Cys1/13-m23 library is a constrained-loop library containing two cysteine residues within its sequence. The complexity of the libraries is estimated to be 2×108 for fUSE5 and 5.5×107 for F88-4/Cys1. These peptide libraries were selected with the mAb 80R scFv.
Affinity selection with 80R scFv and screening for 80R binding clones
1012 plaque-forming units (pfu) of phage-peptides prepared from each library were screened and introduced individually for panning into Maxisorp immunotubes (Nunc, Naperville, IL) coated with 10 μg of 80R scFv. Non-specifically absorbed phages were removed by intensive washings. Specific bound phages were eluted, neutralized, amplified and used for further selections as described.40, 41 Randomly picked single phage clones were screened for specific binding to 80R scFv by ELISA after three rounds of panning. In brief, 96 well Maxisorp immuno-plates were coated with 0.5 μg/well of 80R scFv or a control scFv, blocked with PBS containing 4% (w/v) non-fat milk. Then, individual phage-peptide clones in phosphate-buffered saline (PBS) containing 2% non-fat milk were added. Specific bound phages were detected by adding HRP-conjugated mouse anti-His6 and the system was developed by adding TMB substrate. Absorbance at 450 nm was measured. Clones that bound to 80R scFv with A 450 values of >1.0 were scored as positive, whereas negative clones gave values of <0.2. Unique positive clones were identified by DNA sequencing and the derived peptide sequences were used for Mapitope analysis.
The Mapitope algorithm
The Mapitope program was implemented in C++ and runs on the order of a minute (on Windows XP, 1 processor, Pentium 4 1.80 GHz, 256 KB cache machine). The output of Mapitope is written as a RasTop script which allows one to easily cut and paste into RasTop in order to easily view the clusters on the surface of the antigen color-coded from the most likely to less likely first five clusters as epitope predictions.
Acknowledgements
This work was supported by AI28785, AI48436, AI061318 and AI053822 (to W.A.M.), by a Center for AIDS Research award AI60654 and by an Israel Science Foundation grant (to J.M.G.). We also thank Dr Wenhui Li, Harvard Medical School for his helpful discussions, Erez Bublil for his assistance in the Mapitope analyses and Dr Tal Pupko and his group for their constructive comments throughout this study.
Edited by I. Wilson
Contributor Information
Jonathan M. Gershoni, Email: gershoni@tauex.tau.ac.il.
Wayne A. Marasco, Email: wayne_marasco@dfci.harvard.edu.
References
- 1.Traggiai E., Becker S., Subbarao K., Kolesnikova L., Uematsu Y., Gismondo M.R. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nature Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ter Meulen J., Bakker A.B., van den Brink E.N., Weverling G.J., Martina B.E., Haagmans B.L. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet. 2004;363:2139–2141. doi: 10.1016/S0140-6736(04)16506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sui J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl Acad. Sci. USA. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smirnov Y.A., Gitelman A.K., Govorkova E.A., Lipatov A.S., Kaverin N.V. Influenza H5 virus escape mutants: immune protection and antibody production in mice. Virus Res. 2004;99:205–208. doi: 10.1016/j.virusres.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Riberdy J.M., Flynn K.J., Stech J., Webster R.G., Altman J.D., Doherty P.C. Protection against a lethal avian influenza A virus in a mammalian system. J. Virol. 1999;73:1453–1459. doi: 10.1128/jvi.73.2.1453-1459.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gould L.H., Sui J., Foellmer H., Oliphant T., Wang T., Ledizet M. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J. Virol. 2005;79:14606–14613. doi: 10.1128/JVI.79.23.14606-14613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliphant T., Engle M., Nybakken G.E., Doane C., Johnson S., Huang L. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nature Med. 2005;11:522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S. A novel coronavirus associated with severe acute respiratory syndrome. New Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 9.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 11.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 12.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimitrov D.S. The secret life of ACE2 as a receptor for the SARS virus. Cell. 2003;115:652–653. doi: 10.1016/S0092-8674(03)00976-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z.Y., Werner H.C., Kong W.P., Leung K., Traggiai E., Lanzavecchia A., Nabel G.J. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl Acad. Sci. USA. 2005;102:797–801. doi: 10.1073/pnas.0409065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sette A., Fikes J. Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Curr. Opin. Immunol. 2003;15:461–470. doi: 10.1016/s0952-7915(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 16.Enshell-Seijffers D., Denisov D., Groisman B., Smelyanski L., Meyuhas R., Gross G. The mapping and reconstitution of a conformational discontinuous B-cell epitope of HIV-1. J. Mol. Biol. 2003;334:87–101. doi: 10.1016/j.jmb.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Sui J., Li W., Roberts A., Matthews L.J., Murakami A., Vogel L. Evaluation of human monoclonal antibody 80R for immunoprophylaxis of severe acute respiratory syndrome by an animal study, epitope mapping, and analysis of spike variants. J. Virol. 2005;79:5900–5906. doi: 10.1128/JVI.79.10.5900-5906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsodikov O.V., Record M.T., Jr, Sergeev Y.V. Novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature. J. Comput. Chem. 2002;23:600–609. doi: 10.1002/jcc.10061. [DOI] [PubMed] [Google Scholar]
- 19.Spiga O., Bernini A., Ciutti A., Chiellini S., Menciassi N., Finetti F. Molecular modelling of S1 and S2 subunits of SARS coronavirus spike glycoprotein. Biochem. Biophys. Res. Commun. 2003;310:78–83. doi: 10.1016/j.bbrc.2003.08.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 21.Shatsky M., Nussinov R., Wolfson H.J. FlexProt: alignment of flexible protein structures without a predefinition of hinge regions. J. Comput. Biol. 2004;11:83–106. doi: 10.1089/106652704773416902. [DOI] [PubMed] [Google Scholar]
- 22.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwong P.D., Wyatt R., Robinson J., Sweet R.W., Sodroski J., Hendrickson W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villard S., Lacroix-Desmazes S., Kieber-Emmons T., Piquer D., Grailly S., Benhida A. Peptide decoys selected by phage display block in vitro and in vivo activity of a human anti-FVIII inhibitor. Blood. 2003;102:949–952. doi: 10.1182/blood-2002-06-1886. [DOI] [PubMed] [Google Scholar]
- 25.Spiegel P.C., Jacquemin M., Jr, Saint-Remy J.M., Stoddard B.L., Pratt K.P. Structure of a factor VIII C2 domain-immunoglobulin G4kappa Fab complex: identification of an inhibitory antibody epitope on the surface of factor VIII. Blood. 2001;98:13–19. doi: 10.1182/blood.v98.1.13. [DOI] [PubMed] [Google Scholar]
- 26.van den Brink E.N., Ter Meulen J., Cox F., Jongeneelen M.A., Thijsse A., Throsby M. Molecular and biological characterization of human monoclonal antibodies binding to the spike and nucleocapsid proteins of severe acute respiratory syndrome coronavirus. J. Virol. 2005;79:1635–1644. doi: 10.1128/JVI.79.3.1635-1644.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traggiai E., Becker S., Subbarao K., Kolesnikova L., Uematsu Y., Gismondo M.R. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nature Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenough T.C., Babcock G.J., Roberts A., Hernandez H.J., Thomas W.D., Jr, Coccia J.A. Development and characterization of a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody that provides effective immunoprophylaxis in mice. J. Infect. Dis. 2005;191:507–514. doi: 10.1086/427242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H., Wang G., Li J., Nie Y., Shi X., Lian G. Identification of an antigenic determinant on the S2 domain of the severe acute respiratory syndrome coronavirus spike glycoprotein capable of inducing neutralizing antibodies. J. Virol. 2004;78:6938–6945. doi: 10.1128/JVI.78.13.6938-6945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S., Chou T.H., Sakhatskyy P.V., Huang S., Lawrence J.M., Cao H. Identification of two neutralizing regions on the severe acute respiratory syndrome coronavirus spike glycoprotein produced from the mammalian expression system. J. Virol. 2005;79:1906–1910. doi: 10.1128/JVI.79.3.1906-1910.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keng C.T., Zhang A., Shen S., Lip K.M., Fielding B.C., Tan T.H. Amino acids 1055 to 1192 in the S2 region of severe acute respiratory syndrome coronavirus S protein induce neutralizing antibodies: implications for the development of vaccines and antiviral agents. J. Virol. 2005;79:3289–3296. doi: 10.1128/JVI.79.6.3289-3296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myers M.A., Davies J.M., Tong J.C., Whisstock J., Scealy M., Mackay I.R., Rowley M.J. Conformational epitopes on the diabetes autoantigen GAD65 identified by peptide phage display and molecular modeling. J. Immunol. 2000;165:3830–3838. doi: 10.4049/jimmunol.165.7.3830. [DOI] [PubMed] [Google Scholar]
- 33.Halperin I., Wolfson H., Nussinov R. SiteLight: binding-site prediction using phage display libraries. Protein Sci. 2003;12:1344–1359. doi: 10.1110/ps.0237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dall'Acqua W., Goldman E.R., Lin W., Teng C., Tsuchiya D., Li H. A mutational analysis of binding interactions in an antigen-antibody protein-protein complex. Biochemistry. 1998;37:7981–7991. doi: 10.1021/bi980148j. [DOI] [PubMed] [Google Scholar]
- 35.Venkatesh N., Krishnaswamy S., Meuris S., Murthy G.S. Epitope analysis and molecular modeling reveal the topography of the C-terminal peptide of the beta-subunit of human chorionic gonadotropin. Eur. J. Biochem. 1999;265:1061–1066. doi: 10.1046/j.1432-1327.1999.00828.x. [DOI] [PubMed] [Google Scholar]
- 36.Kolaskar A.S., Kulkarni-Kale U. Prediction of three-dimensional structure and mapping of conformational epitopes of envelope glycoprotein of Japanese encephalitis virus. Virology. 1999;261:31–42. doi: 10.1006/viro.1999.9859. [DOI] [PubMed] [Google Scholar]
- 37.Marti-Renom M.A., Stuart A.C., Fiser A., Sanchez R., Melo F., Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 38.Halstead S.B. Pathogenesis of dengue: challenges to molecular biology. Science. 1988;239:476–481. doi: 10.1126/science.3277268. [DOI] [PubMed] [Google Scholar]
- 39.Murphy B.R., Prince G.A., Walsh E.E., Kim H.W., Parrott R.H., Hemming V.G. Dissociation between serum neutralizing and glycoprotein antibody responses of infants and children who received inactivated respiratory syncytial virus vaccine. J. Clin. Microbiol. 1986;24:197–202. doi: 10.1128/jcm.24.2.197-202.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu J., Smith G.P. Affinity maturation of phage-displayed peptide ligands. Methods Enzymol. 1996;267:3–27. doi: 10.1016/s0076-6879(96)67003-7. [DOI] [PubMed] [Google Scholar]
- 41.Matthews L.J., Davis R., Smith G.P. Immunogenically fit subunit vaccine components via epitope discovery from natural peptide libraries. J. Immunol. 2002;169:837–846. doi: 10.4049/jimmunol.169.2.837. [DOI] [PubMed] [Google Scholar]