

Abstract
Background & Aims
Hartnup amino acid transporter B0AT1 (SLC6A19) is the major luminal sodium-dependent neutral amino acid transporter of small intestine and kidney proximal tubule. The expression of B0AT1 in kidney was recently shown to depend on its association with collectrin (Tmem27), a protein homologous to the membrane-anchoring domain of angiotensin-converting enzyme (ACE) 2.
Methods
Because collectrin is almost absent from small intestine, we tested the hypothesis that it is ACE2 that interacts with B0AT1 in enterocytes. Furthermore, because B0AT1 expression depends on an associated protein, we tested the hypothesis that Hartnup-causing B0AT1 mutations differentially impact on B0AT1 interaction with intestinal and kidney accessory proteins.
Results
Immunofluorescence, coimmunoprecipitation, and functional experiments using wild-type and ace2-null mice showed that expression of B0AT1 in small intestine critically depends on ACE2. Coexpressing new and previously identified Hartnup disorder–causing missense mutations of B0AT1 with either collectrin or ACE2 in Xenopus laevis oocytes showed that the high-frequency D173N and the newly identified P265L mutant B0AT1 transporters can still be activated by ACE2 but not collectrin coexpression. In contrast, the human A69T and R240Q B0AT1 mutants cannot be activated by either of the associated proteins, although they function as wild-type B0AT1 when expressed alone.
Conclusions
We thus show that ACE2 is necessary for the expression of the Hartnup transporter in intestine and suggest that the differential functional association of mutant B0AT1 transporters with ACE2 and collectrin in intestine and kidney, respectively, participates in the phenotypic heterogeneity of human Hartnup disorder.
Abbreviations used in this paper: ACE, angiotensin-converting enzyme; SNP, single nucleotide polymorphism; TEVC, 2-electrode voltage clamp
Hartnup disorder is an autosomal recessive disorder caused by mutations in the SLC6A19 gene, which encodes the main epithelial neutral amino acid transporter B0AT1.1, 2 This disorder is characterized by neutral aminoaciduria due to impaired amino acid transport in kidney proximal tubule epithelial cells, whereas the extent of intestinal transport impairment appears to be less consistent. Various other clinical symptoms such as pellagra-like rash, cerebellar ataxia, or other neurologic dysfunctions may be present in affected individuals, while other subjects remain, besides experiencing aminoaciduria, asymptomatic. Phenotypic variability might be partially explained by the differential impact of various mutations and the frequent compound heterozygosity.
Differential phenotypic effects of mutations could arise from differential interactions of B0AT1 with tissue-specific modulatory and/or associated proteins. We and others have previously shown that B0AT1 requires association with collectrin for luminal surface expression in the kidney proximal tubule, whereas collectrin is nearly absent in the small intestine, the other major site of B0AT1 expression.3, 4 Intriguingly, the closest homologue of collectrin is the angiotensin-converting enzyme (ACE) 2, which functions as a key carboxypeptidase enzyme in the renin-angiotensin system. ACE2 is involved in heart and kidney pathologies5 and has been identified as the severe acute respiratory syndrome receptor in vitro6 and in vivo.7 Importantly, previous studies have reported ACE2 expression and severe acute respiratory syndrome/coronavirus replication in human small intestine.8, 9
We first investigated whether ACE2 associates with B0AT1 in mouse small intestine. Immunofluorescence, coimmunoprecipitation, and functional data presented here clearly show that ACE2 is the specific partner of B0AT1 in the small intestine and thus that the accessory protein of B0AT1 is tissue specific.
We then tested the influence of collectrin and ACE2 on the function of human B0AT1 Hartnup disorder–causing mutations in the Xenopus laevis oocyte expression system. Our results suggest that specific defects in interaction with tissue-specific accessory proteins may lead to differential defects in intestinal versus kidney (re)absorption of neutral amino acids and thereby participate in the variability of phenotypes observed in subjects with Hartnup disorder.
Materials and Methods
Animals
The ace2 and collectrin wild-type and knockout mice were housed in standard conditions and fed a standard diet. Generation of the knockout mice was described elsewhere.3, 5 Animals were either anesthetized and perfused with a fixative solution for localization studies or killed to remove small intestine and kidneys. Scraped small intestine mucosa cells and total kidneys were frozen in liquid nitrogen for subsequent RNA extraction or brush border membrane vesicle preparation. All procedures for mice handling were according to the Swiss Animal Welfare laws and approved by the Kantonales Veterinäramt Zürich.
Organ Fixation
Male mice were anesthetized with ketamine and xylazine (90 mg/kg body wt, Narketan 10; Vétoquinol, Lure, France) and xylazine (10 mg/kg body wt, Xylazin; Streuli, Uznach, Switzerland) intraperitoneally and perfused through the left cardiac ventricle with phosphate-buffered saline (PBS; phosphate buffer, pH 7.4) followed by a buffered paraformaldehyde solution (4%, pH 7) as previously described.10 Small intestine was then harvested, incubated overnight in paraformaldehyde solution, washed several times with PBS, and stored in PBS/0.02% sodium azide at 4°C. Tissues were then mounted with Kryostat OCT (Medite, Nunningen, Switzerland), frozen in liquid propane, and stored at –80°C.
Immunofluorescence
Immunofluorescence was performed as previously described.11 Primary antibodies were diluted (1:200) for rabbit affinity purified anti-mouse B0AT111 and diluted (1:100) for affinity purified goat anti-mouse ACE2 (R&D Systems, Minneapolis, MN). Secondary antibodies were diluted (1:500) for Alexa Fluor 488 donkey anti-goat immunoglobulin (Ig) G and Alexa Fluor 594 donkey anti-rabbit IgG (Molecular Probes/Invitrogen, Carlsbad, CA). Digital images were viewed by using a Nikon Eclipse TE300 epifluorescence microscope (Nikon Instruments Inc, Melville, NY) equipped with a DS-5M Standard charge-coupled device camera (Nikon Instruments Inc) and acquired with NIS-Elements (Nikon Instruments Inc).
Brush Border Membrane Preparations
Brush border membrane vesicles were prepared from small intestine mucosa cells and kidneys using the Mg2+ precipitation technique as described elsewhere.12
Western Blotting
Western blotting was performed as previously described.11 Primary antibodies were diluted to 1:1000 goat affinity purified anti-mouse ACE2 (R&D Systems), 1:1000 goat affinity purified anti-human ACE2 (R&D Systems), 1:2000 or 1:1000 rabbit affinity purified anti-mouse or anti-human B0AT1, respectively11 (Pineda, Berlin, Germany), and 1:10,000 for mouse anti-mouse β-actin (Sigma, St Louis, MO). Secondary antibodies were diluted (1:5000) for enhanced chemiluminescence anti-rabbit or anti-goat IgG horseradish peroxidase linked fragment from donkey or mouse, respectively (Amersham Biosciences, Piscataway, NJ, and Pierce, Rockford, IL) or anti-mouse IgG alkaline phosphatase conjugate from mouse (Promega, Madison, WI). Antibody binding was detected with Immobilon Western Chemiluminescent HRP or AP substrate (Millipore, Billerica, MA) and chemiluminescence visualized with a DIANA III camera (Raytest, Dietikon, Switzerland).
Intestinal Ring Uptake
Uptake of radiolabeled l-isoleucine was performed as previously described13 on ileum segments, with slight modifications. Briefly, everted ileum segments were incubated in bubbling (Oxycarbon) Krebs-Tris buffer (pH 7.4) containing 1 mmol/L l-isoleucine (1 μCi 14C-L-Ile/mL) for 5 minutes at 37°C. Ileum segments were dried at 55°C O/N on cellulose (Sartorius AG, Goettingen, Germany) and weighed. Segments were then lysed in 0.75N NaOH for 6 hours and neutralized with 10N HCl, and the radioactivity was determined by liquid scintillation. Na+ was replaced by N-methyl-d-glucamine in the condition without Na+. Amino acid transport was expressed relative to dry tissue weight.
Identification of New Hartnup Mutations
After informed consent was obtained, genomic DNA was obtained from whole blood of patients with Hartnup disorder as previously described.1 Intronic primers to sequence exons and splice sites of each exon were chosen. Primer sequences are available on request. Polymerase chain reaction products of each exon were separated by electrophoresis on agarose gels and specific bands removed for DNA isolation. This DNA was sequenced in both directions using a Beckman Coulter CEQ8000 (Beckman Coulter, Fullerton, CA) following the manufacturer's protocol. The absence of each recognized missense mutation was confirmed in 100 ethnically matched control alleles.
Isolation and Subcloning of Complementary DNAs and Site-Directed Mutagenesis
Human B0AT1, ACE2, and collectrin complementary DNAs (cDNAs) were amplified from human kidney Marathon-Ready cDNA (Clontech, Mountain View, CA) by polymerase chain reaction using a proofreading polymerase (Pfu; Promega). The primers used are available on request. The amplified ACE2 and collectrin cDNAs were subcloned in pcDNA3 and mouse ACE2 and collectrin in pcDNA 3.1 hygromycin (Invitrogen). The B0AT1 was subcloned into a pBlueScript modified Xenopus laevis expression vector containing both the 5′ and 3′ ends of the β-globin gene (KSM) kindly provided by Dr Leila Virkki. For expression in X laevis oocytes, the plasmids were linearized and used as template for RNA synthesis (mMESSAGEmMACHINE; Ambion, Austin, TX). The mutation in human B0AT1 and ACE2 was performed using the QuikChange Site-Directed Mutagenesis Kit according to the manufacturer (Stratagene, La Jolla, CA). The mutation ACE2-R273Q was shown previously to be catalytically inactive but normally expressed.14 The B0AT1 mutations were performed on the single nucleotide polymorphism (SNP) V252I, which has been shown to behave like the wild type.
Homology Model
The homology model of human B0AT1 transport was based on the crystal structure of LeuTAa (NP_214423) as a template using the I-TASSER server.15, 16 The pdb file was visualized using Pymol (DeLano Scientific LLC, Palo Alto, CA).
Transport Studies in X laevis Oocytes
Expression studies and influx assays using radiolabeled amino acid tracer were performed in X laevis oocytes after 5 to 9 days of expression, as described previously.17 Data are expressed as pmol · h−1 · oocyte−1, and values obtained for noninjected oocytes are subtracted.
Electrophysiology Using 2-Microelectrode Voltage Clamp
The 2-microelectrode voltage clamp (TEVC) technique was used for the recording of whole cell currents from X laevis oocytes. Recordings were performed at room temperature 5 to 9 days after injection with complementary RNA. Recordings were performed as previously described16 at a membrane holding potential of −50 mV. To control for batch variation in transporter expression and measured current, the data from the selectivity experiment were normalized to IL-Ile 10 mmol/L. Pooled data are shown as mean ± SEM where n represents the number of pooled cells. Experimental protocols were repeated at least twice. Nonlinear regression calculations were performed using GraphPad Prism version 4.0 (GraphPad Inc, San Diego, CA).
Labeling of Surface Proteins in Xenopus Oocytes
Surface labeling of oocytes expressing human B0AT1 alone or coexpressed with mouse ACE2 or human collectrin was performed using 2-(aminoethyl)-methanethiosulfonate-biotin (Sigma-Aldrich, Buchs, Switzerland) as previously described.18 Samples were separated on a 10% sodium dodecyl sulfate gel and immunoblotted with affinity purified rabbit anti-human B0AT1 antibody (Pineda). Signal intensity was quantified with the AIDA Image Analyzer (Raytest).
Immunoprecipitations
Mouse brush border membrane proteins
Brush border membrane vesicles were incubated with serum anti-B0AT1 polyclonal antibody11 in EBC solution (20 mmol/L Tris-HCl, pH 8.0, 120 mmol/L NaCl, 0.5% Nonidet P40) at 4°C on a rotator. The immunocomplexes were coupled to Immobilized Protein A/G beads (Pierce) O/N at 4°C on a rotator. The beads were washed with NET-N solution (20 mmol/L Tris-HCl, pH 8.0, 100 mmol/L NaCl, 0.5% Nonidet P40, 1 mmol/L EDTA), and the immunoprecipitate was loaded on a polyacrylamide gel. Western blot was performed as described previously with anti-mouse B0AT1 antibody to check for B0AT1 immunoprecipitation or anti-mouse ACE2 antibody to test ACE2 coimmunoprecipitation. To avoid background staining, protein A/G coupled to alkaline phosphatase was used as a secondary antibody (Pierce).
X laevis oocytes
Immunoprecipitation of human B0AT1, B0AT1 mutants, and human ACE2 was performed as previously described.19 Briefly, oocytes were lysed in EBC buffer (as described for brush border membrane vesicles) and the supernatant was first incubated with serum anti-human B0AT1 and subsequently with Immobilized Protein A/G beads (Pierce) O/N at 4°C on a rotator. Western blot was performed as described previously to test for human ACE2 coimmunoprecipitation.
Statistics
Data are presented as means ± SEM. Analyses were performed by running the GraphPad Prism 4.0 software (GraphPad). Between-group comparisons were performed by Student unpaired t test. Multiple comparisons within groups were performed by repeated-measures one-way analysis of variance, followed by Tukey posttest. Statistical significance was accepted at P < .05.
Results
ACE2 Is the Intestinal Partner of the Amino Acid Transporter B0AT1
We first assessed the potential in vivo role of ACE2 as intestinal B0AT1 associated protein by investigating the B0AT1 expression in ace2-null mice.5 Remarkably, B0AT1 protein was completely absent in small intestine brush border membranes of mice lacking ACE2 (Figure 1 A and B), whereas it was normally expressed in kidney (Figure 1 B). This organ distribution of B0AT1 in ace2-null mice mirrors the situation observed in collectrin-null mice, where B0AT1 is absent in kidney and, as shown in this study, normally expressed in small intestine (Figure 1 B). Furthermore, ACE2 was coimmunoprecipitated with B0AT1 from intestinal brush border membranes of wild-type mice, demonstrating in vivo interaction (Figure 1 C).
Figure 1.

ACE2 and B0AT1 colocalization and functional interaction. (A) Immunofluorescence analysis shows that B0AT1 protein expression is lost in small intestine of ace2-null mice. Small intestine sections from ace2+/y and ace2−/y were colabeled with antibodies against ACE2 (green) and B0AT1 (red) and additionally viewed by phase contrast (PC). (B) Western blot analysis shows that B0AT1 is absent in small intestine but present in kidney brush border membranes of ace2-null mice, whereas B0AT1 is expressed in small intestine and absent from kidney brush border membranes in collectrin-null mice. The loading of brush border membranes (2.5–20 μg) was tested using a β-actin antibody (βA). Note that male ace2+/y or ace2−/y and female ace2+/+ or ace2−/− mice were used with similar results. (C) Coimmunoprecipitation shows interaction of ACE2 with B0AT1 in mouse small intestine. Complexes were immunoprecipitated (IP) from small intestine brush border membrane vesicles (BBMVs) using anti-B0AT1 antibody (AB) and analyzed by Western blot (WB) using anti-mouse ACE2 antibody. (D) Na+-dependent uptake of l-isoleucine (L-Ile) is abolished in ileum segments from ace2−/y mice. The transport was measured in the presence (white bar) and in the absence (black bar) of sodium. Data points represent mean values of 4 intestinal ring uptakes from independent experiments ± SEM, ***P < .001. ns, not significant.
The function of B0AT1 was also shown to depend on ACE2 in mouse intestine. We observed that the lack of B0AT1 protein expression in the intestine of ace2−/y mice abolished the Na+-dependant portion of l-isoleucine transport measured in intestinal rings (Figure 1 D). The total (in the presence of Na+) but not the sodium-independent transport was dramatically reduced when compared with the wild-type littermates. The equivalent effect was previously shown in kidney brush border membrane vesicles of collectrin −/− mice, where the transport was also reduced.3 These data show that ACE2 is essential for expression and function of B0AT1 in intestine, whereas collectrin controls B0AT1 expression in the kidney.
ACE2 and Collectrin Increase the Function of Human Hartnup Transporter B0AT1 In Vitro
The tissue-specific associated proteins collectrin and ACE2 expressed in X laevis oocytes mimicked their effect in vivo on B0AT1 by stimulating its transport function. Expressed alone in oocytes, human B0AT1 induced a low amino acid transport rate as shown by us and others previously.1, 2 We show here that its coexpression with mouse or human ACE2 or human collectrin increases the transport function ∼10-fold (Supplementary Figure 1 A; see supplemental material online at www.gastrojournal.org). The mouse orthologue of collectrin did not activate human B0AT1, although it has been shown to be effective on mouse B0AT1.3 We have as yet no explanation for this differential impact of the collectrin orthologues in the X laevis expression system. Different from collectrin, ACE2 has one catalytic site. It is a carboxypeptidase and converts angiotensin I and II to angiotensin 1–9 and 1–7, respectively,20, 21 functionally antagonizing its homologue ACE. We analyzed the role of the catalytic activity of ACE2 in the functional interaction with B0AT1 (Supplementary Figure 1 B; see supplemental material online at www.gastrojournal.org). The catalytically dead mutant (ACE2 R273Q)14 was as efficient as the wild type in enhancing B0AT1 function, suggesting that it is not necessary for the interaction. This was expected because human collectrin shares 48% identity with ACE2 at the level of its carboxyl-terminal membrane anchor region but entirely lacks the extracellular peptidase domain.22
Supplementary Figure 1.
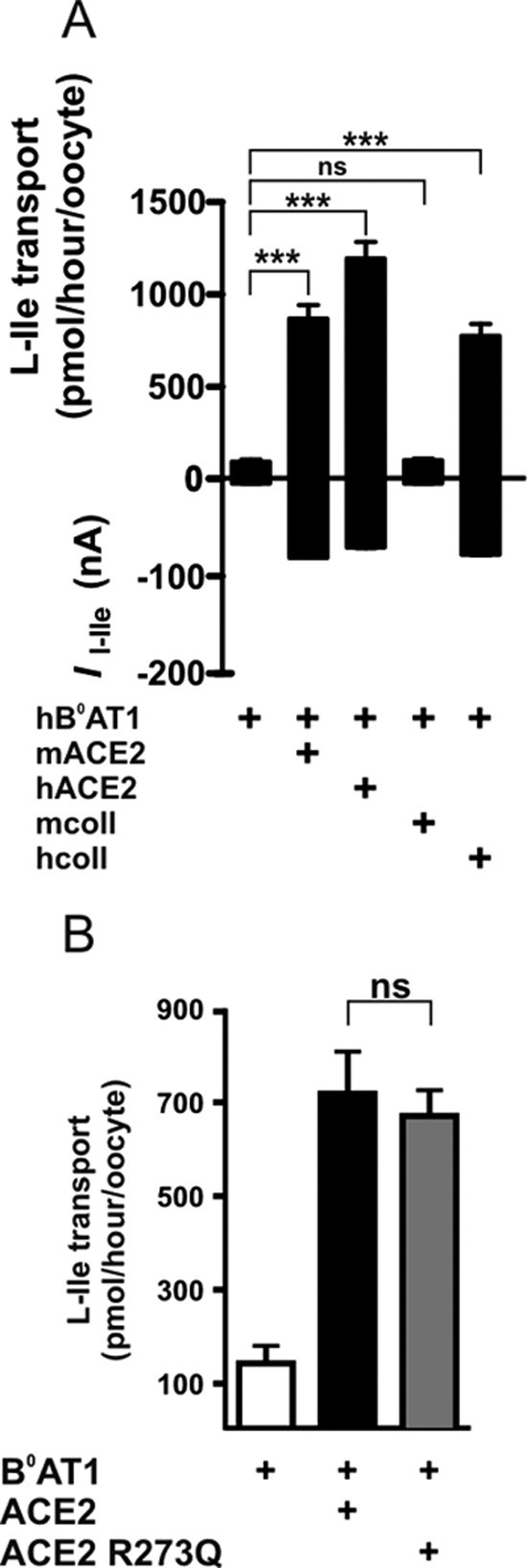
Function of human B0AT1 coexpressed with orthologues of collectrin and ACE2 in X laevis oocytes. (A) Coexpression of ACE2 or human collectrin increases B0AT1 function in X laevis oocytes. Tracer flux experiments (upper graph, n = 16–32) and TEVC (lower graph, n = 11) show that human B0AT1 (hB0AT1) coexpressed with human ACE2 (hACE2), mouse ACE2 (mACE2), or human collectrin (hColl) transports l-isoleucine more efficiently than when expressed alone or in combination with mouse collectrin (mColl). Data points represent means ± SEM. ***P < .001. ns, not significant. (B) Human B0AT1 function is increased by coexpressed human ACE2 independent of its protease activity. The protease-dead human ACE2 mutant (ACE2 R273Q) stimulated the human B0AT1-mediated l-isoleucine influx to the same extent as wild-type human ACE2. Data points represent means ± SEM (n = 15–16). ns, not significant.
The amino acid selectivity, ion dependence, and kinetic characteristics of human B0AT1 coexpressed with ACE2 are, besides a much higher maximal transport rate, very similar to those previously reported for human B0AT1 expressed alone1 (Supplementary Figure 2 A–E; see supplemental material online at www.gastrojournal.org).
Supplementary Figure 2.

Functional characterization of the complex formed by human B0AT1 and ACE2 in X laevis oocytes. (A) The amino acid transport by human B0AT1 coexpressed with mouse ACE2 in X laevis oocytes is Na+ but not Cl− dependent. The ion dependence was tested by substituting Na+ by N-methyl-d-glucamine (NMDG) or Li+ and Cl− by gluconate in the superfusion solution used for TEVC. The values were normalized to the current obtained by superfusion of the oocytes with 10 mmol/L l-isoleucine in solution containing Na+ and Cl− (Inorm). Comparison of means was performed by analysis of variance with Tukey as posttest. Each data point represents the mean ± SEM (n = 16). ***P < .001. (B) Amino acid transport by human B0AT1 coexpressed with mouse ACE2 in X laevis oocytes is maximal at neutral pH. The pH dependence of the transport was measured by TEVC in oocytes clamped at –50 mV and superfused with Na+ solution buffered at pH 5.5, 6.5, 7.4, or 8.0. Values were normalized to the current obtained by superfusion of oocytes with 10 mmol/L l-isoleucine in solution pH 7.4 (Inorm). Comparison of means was performed by analysis of variance with Tukey as posttest. Each data point represents the mean ± SEM (n = 9). *P < .05, **P < .01. (C–E) Human B0AT1 coexpressed with ACE2 transports a broad range of neutral amino acids with low affinity. (C) The substrate selectivity of human B0AT1 coexpressed with mouse ACE2 was measured by TEVC in oocytes clamped at –50 mV and superfused with Na+ solution containing 1 mmol/L (white bars) or 10 mmol/L (black bars) of amino acids. The currents induced by the amino acids were normalized to the currents obtained in oocytes perfused with 10 mmol/L l-isoleucine (Inorm). Data are shown as mean values ± SEM (n = 8–11) and the amino acids are indicated in single letter code. (D and E) The K0.5 and Vmax for l-isoleucine and Na+ of human B0AT1 coexpressed with mouse ACE2 was estimated using TEVC. For the Na+ concentration dependence experiments, the cells were first equilibrated with the indicated Na+ concentration (0, 1, 5, 10, 30, 50, 70, and 100 mmol/L) before the perfusate containing l-isoleucine (10 mmol/L) was applied. For the l-isoleucine concentration dependence experiments, the cells were perfused with increasing concentrations of l-isoleucine (0.1, 0.3, 1, 3, and 10 mmol/L). The continuous line is the nonlinear regression fit of the Michaelis–Menten equation to the data. Each data point represents the mean ± SEM (C, n = 26–27; D, n = 10–22).
Differential Interaction of Accessory Proteins With B0AT1 Hartnup Mutations
To address the question whether Hartnup mutations impact on the interaction of B0AT1 with its accessory proteins ACE2 and/or collectrin, we expressed missense mutations alone or together with either accessory protein in X laevis oocytes and analyzed their transport function and surface expression. Besides 5 previously described missense mutations and one nonsynonymous SNP, we characterized 4 new missense mutations identified in patients with neutral aminoaciduria characteristic of Hartnup disorder belonging to 4 different families (Table 1 and Supplementary Figure 3 A–D; see supplementary material online at www.gastrojournal.org).1, 2 The nonsynonymous SNP V252I essentially behaved as wild-type B0AT1 when coexpressed with collectrin and ACE2 (Figure 2 A, upper and middle panels). Interestingly, the observed 2-fold to 3-fold increase in B0AT1 surface expression measured by surface biotinylation only partially explains the almost 10-fold increase in surface transport function induced by coexpression with collectrin or ACE2 (Figure 2 A, lower panel, and representative Western blot).
Table 1.
Missense Mutations in the SLC6A19/Human B0AT1 Gene Described in Subjects With Hartnup Disorder
| Protein | Wild type | Mutation | Position from the atg | Position in the protein |
|---|---|---|---|---|
| R57C1 | Cgc | Tgc | 169 | 1st TMS |
| A69Ta | Gcc | Acc | 205 | 2nd TMS |
| G93Ra | Ggg | Agg | 277 | 2nd TMS |
| D173N2 | Gac | Aac | 517 | 2nd EL |
| R240Q2 | cGa | cAa | 719 | 3th EL |
| L242P2 | cTg | cCg | 725 | 3th EL |
| P265La | cCg | cTg | 794 | 6th TMS |
| E501K2 | Gag | Aag | 1501 | 10th TMS |
| P579La | Ccg | Tcg | 1735 | 6th EL |
Supplementary Figure 3.
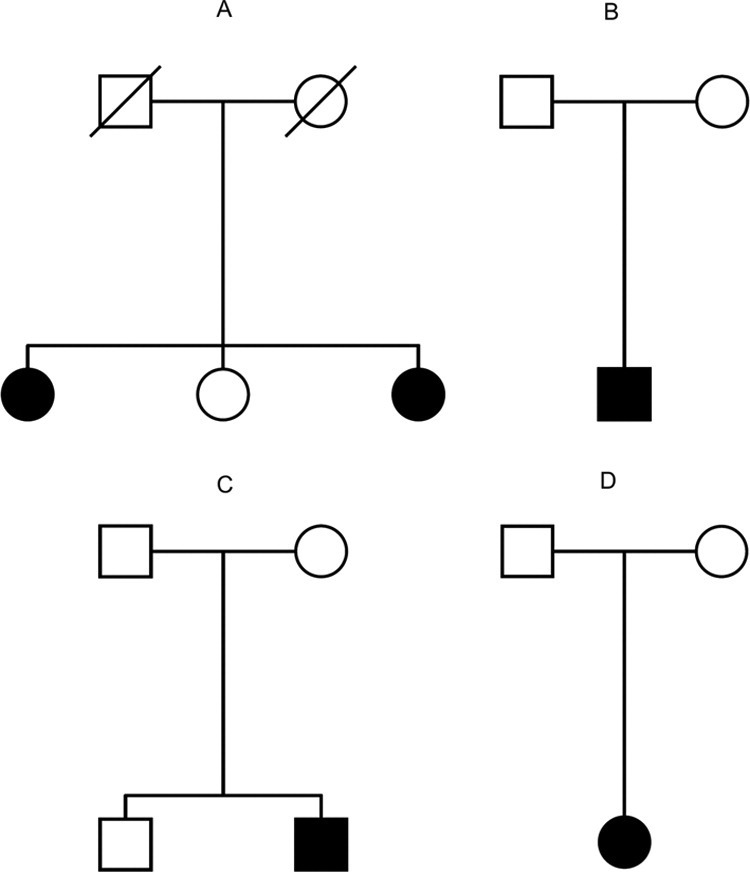
Pedigrees of 4 families with new Hartnup disorder mutations. (A) Danish pedigree: 2 of 3 siblings were compound heterozygous for P265L and L242P. The symptoms observed were quite diverse between the 2 sisters. The younger one was diagnosed with Hartnup disorder in early childhood due to pellagra-like skin rash and an atactic gait that improved with administration of niacin.1 The older affected sibling never showed skin pathologies or ataxia. (B) Another Danish pedigree, where the only child affected was a compound heterozygous for A69T and the original Hartnup mutation IVS8+2T→G. This patient was the first child of unrelated parents and was diagnosed at 1 year of age after a suspected seizure prompted a metabolic screening of urine. Plasma amino acids and electroencephalography were normal. The patient subsequently had normal growth and development without epilepsy, ataxia, or pellagra. (C) The other Danish patient was a compound heterozygous for P579L and the original Hartnup mutation IVS8+2T→G. This patient was the second child of unrelated parents and was diagnosed at 2 years of age after onset of partial complex epilepsy with great difficulty to control seizures. The patient had normal plasma amino acids and never had ataxia or pellagra. (D) Another patient from a German family was diagnosed as homozygous for the new missense mutation G93R. This patient was diagnosed at 6 years of age during a migraine workup and also never had ataxia or pellagra. The affected subjects are depicted as filled symbols.
Figure 2.

Differential function of Hartnup mutations coexpressed with collectrin or ACE2 in X laevis oocytes. Wild-type and mutant human B0AT1 are shown in this figure as 3 groups with different functional characteristics and labeled A–C. (A) The transport function and the surface expression of both wild-type and nonsynonymous SNP V252I of B0AT1 are increased by coexpression with human collectrin or mouse ACE2. (B) The relatively low transport activity of B0AT1 D173N and P265L mutants expressed alone is increased by coexpression with ACE2 but not with collectrin. The surface expression of the D173N mutant is also significantly increased by coexpression with ACE2. (C) The wild-type–like function of the A69T or R240Q mutants expressed alone is not increased in the presence of collectrin or ACE2, although in the case of A69T its surface expression is increased. The upper panels of A–C show the transport function measured as radiolabeled l-isoleucine (L-Ile) uptake (means ± SEM of 13–31 oocytes from 3 independent experiments), and the middle panel shows the transport function as current measured by TEVC (9–11 oocytes from 3 batches). *P < .05, ***P < .001. The lower panels show the surface expression assayed by Western blot using surface-biotinylated proteins and a representative Western blot. The data correspond to the means ± SEM (n = 3–5 independent experiments) of the results normalized to the value obtained for B0AT1 expressed alone. For the transport experiments, the significance was evaluated using analysis of variance with Tukey posttest and for the surface biotinylation the comparison of means was performed by unpaired t test. (D) B0AT1 and the mutants D173N, P265L, R240Q, and A69T interact with ACE2 in X laevis oocytes. The transporters expressed alone or together with human ACE2 were immunoprecipitated (IP) from oocyte lysates using anti-human B0AT1 antibody and complexes were analyzed by Western blot (WB) using anti-human ACE2 antibody. NI, noninjected.
Surprisingly, collectrin and ACE2 coexpression differentially impacted on the surface expression and function of the 2 B0AT1 mutants D173N and P265L (Figure 2 B). In both cases, coexpression with ACE2 increased the transport rate whereas coexpression with collectrin either had the opposite effect or no effect (Figure 2 B, upper panel). The transport function of these mutants was shown to be Na+ dependent as for wild-type B0AT1 (Figure 3, upper and middle panels). Surface biotinylation experiments suggested that the differential function of these B0AT1 mutants with the 2 associated proteins is due, in the case of D173N, to a difference in surface expression, whereas the surface expression appeared in the case of P265L to be similar to both associated proteins (Figure 2 B, lower panel, and representative Western blot). Finally, the mutants D173N and P265L coprecipitated with human ACE2, confirming that they interact with ACE2 (Figure 2 D).
Figure 3.

The ion dependency of D173N, P265L, A69T, and R240Q coexpressed with ACE2 or collectrin is not altered; transport is Na+ but not Cl− dependent. Wild-type human B0AT1 (WT) and the mutants D173N, P265L, A69T, and R240Q were expressed alone or with human collectrin or mouse ACE2. The ion dependence was tested by substituting Na+ with N-methyl-d-glucamine (NMDG) and substituting Cl− with gluconate. Function was assayed by influx of radiolabeled l-isoleucine (L-Ile). For each mutation, 3 independent experiments (15–24 oocytes) were pooled. Comparison of means was performed by analysis of variance with Tukey as posttest. Data represent means ± SEM. *P < .05, **P < .01, ***P < .001. ns, not significant.
We next analyzed the 2 B0AT1 mutations A69T and R240Q that, when expressed alone, display the same l-isoleucine transport function and ion dependence as wild-type B0AT1, whereas their transport function was not activated in the presence of either associated protein (Figure 2 C, upper and middle panels, and Figure 3, lower panel). In the case of the newly described A69T, this was particularly surprising, because its surface expression was increased 2-fold to 3-fold by coexpression with both accessory proteins (Figure 2 C, lower panel, and representative Western blot) and its interaction with ACE2 confirmed by coimmunoprecipitation (Figure 2 D). In contrast, the surface expression of R240Q did not appear to be increased by the presence of the accessory proteins (Figure 2 C, lower panel, and representative Western blot), although the mutant transporter was coimmunoprecipitated with human ACE2 to some extent (Figure 2 D).
The mutations R57C, L242P, and E501K have previously been tested by expression in X laevis oocytes and showed no transport capacity.1, 2 Coexpression of these mutants with collectrin and ACE2 did not change their impaired function and did not affect their cell surface expression (Figure 4). Similarly, the newly described mutations G93R and P579L lack function also in the presence of associated proteins. However, the surface expression of G93R was selectively increased by ACE2 (Figure 4, lower panel, and representative Western blot).
Figure 4.

The B0AT1 mutants R57C, G93R, L242P, E501K, and P579L exhibit no transport activity when expressed alone or coexpressed with collectrin or ACE2. Function was assayed by influx of radiolabeled l-isoleucine (L-Ile) (upper panels) or TEVC (middle panels). For each mutant, 3 independent experiments (13–31 oocytes for influx and 9–11 for TEVC) were pooled. Comparison of means was performed by analysis of variance with Tukey as posttest. Data represent means ± SEM. ns, not significant. Cell surface expression of labeled proteins (lower panels) was analyzed by Western blot. The bands were quantified and data normalized to the values obtained from oocytes expressing B0AT1 proteins alone. For each mutation, 3 to 4 independent experiments were pooled. Comparison of means was performed by unpaired t test. Data represent means ± SEM. *P < .05. ns, not significant.
Discussion
We recently reported that collectrin (Tmem27) associates with and controls the expression of B0AT1 in the kidney proximal tubule.3, 4 In this study, we showed a novel and unexpected function of the important renin-angiotensin system enzyme ACE2 that associates with the luminal amino acid transporter B0AT1 in small intestine and thereby controls its surface expression and function. B0AT1 associates in a tissue-specific manner with ACE2 in small intestine and with collectrin in the kidney. Thus, ace2- and collectrin-null mice can be used to study the role of the neutral amino acid transporter B0AT1 in kidney and intestine independently.
The same effect was observed on human B0AT1 orthologue expressed in the heterologous expression systems, X laevis oocytes. The coexpression of the 2 accessory proteins, ACE2 or collectrin, increased the transport rate and the cell surface expression of the transporter. The analysis of the function and surface expression of B0AT1/Hartnup disorder–causing mutations expressed alone or together with the 2 partner proteins allowed us to discriminate classes of mutants that might differentially impact on the phenotype of the patients.
The Hartnup disorder–causing mutations in B0AT1 are localized throughout the protein, as shown on the homology model depicted in Figure 5. This is also the case for the largest class of mutations mentioned here, the “dead” mutants, 3 of which were reported earlier (R57C, L242P, and E501K1, 2) and 2 of which are newly described here (G93R and P579L) (Supplementary Figure 3 C and D; see supplemental material online at www.gastrojournal.org). These mutants do not display any measurable function, which is consistent with the fact that, with one exception, they do not reach the plasma membrane neither when expressed alone nor upon coexpression with associated proteins.
Figure 5.

Missense mutations in the SLC6A19 gene causing Hartnup disorder. The model of B0AT1 is based on the Slc6 bacterial homologue from Aquifex aeolicus LeuTAa16 and the transmembrane segments are numbered. The localization of mutations causing a loss of surface expression (R57, G93, L242, E501, P579) is depicted in the left panel. The sites of mutations leading to a differential interaction with the 2 accessory proteins (D173 and P265) are depicted as black spheres on the middle panel, and the sites of the mutations preventing both accessory proteins of stimulating the transport function are depicted on the right panel (A69 and R240).
The second class of mutants corresponds to those that, expressed alone, function as wild-type B0AT1 but are not stimulated by either associated protein. In the case of R240Q, its surface expression is not increased upon coexpression with ACE2 or collectrin, but coimmunoprecipitation demonstrates that its interaction with ACE2 is not abolished. Assuming a qualitative effect of the R240Q mutation on this interaction, our observation is nonetheless potentially compatible with the hypothesis recently put forward by the group of Broer, namely that the localization of this mutation at the surface of B0AT1 might impact on the interaction with accessory proteins.23 Further studies are necessary to clarify the mechanism by which this mutation, initially classified as an SNP, prevents the normal activation of B0AT1 by the accessory proteins. In contrast, in the case of the other functional mutant of this class A69T, an increase in cell surface expression of this mutant B0AT1 was demonstrated upon interaction with the associated proteins (Figure 2 C and D). One possible explanation of why the increase in surface expression did not lead to an increase in transport rate is that ACE2/A69T-B0AT1 heterodimers reaching the cell surface remain inactive, meaning that the associated protein inhibits the function of A69T-B0AT1. The fact that the residue mutated in A69T is part of a highly conserved motif (NGGGAF) shown to undergo conformational changes during the transport cycle is compatible with this hypothesis.24 Additionally, that the coexpression of ACE2 or collectrin apparently activates the transport rate of wild-type B0AT1 several-fold more than it increases its surface expression (∼10-fold vs ∼2.5-fold) also suggests that associated proteins may impact on transporter function, although normally by increasing the cycling rate.
The most intriguing class of mutants in regard to its potential phenotypic impact is represented by D173N and the newly described P265L. These mutants differentially interact with the 2 tissue-specific accessory proteins such that their function is stimulated only by the intestine-specific associated protein ACE2. Interestingly, the D173N allele is relatively frequent in unrelated Hartnup pedigrees and can also be observed in healthy white patients with a high heterozygote frequency (1:122 healthy individuals).2, 25 We know of no potential selective advantage for heterozygous carriers that explains the persistence and geographic spread of this mutant allele.26, 27 The selective functional interaction of D173N with the intestinal accessory protein ACE2 might prevent deleterious effects due to a lack of amino acid absorption and thereby allowing the “survival” of this frequent allele. Although as yet no evidence of genotype-phenotype relationship performed in patients were reported, studies performed long before the identification of the Hartnup transporter have shown that intestinal amino acid absorption differs between subjects with Hartnup disorder.28, 29
Taken together, our data show that the expression and function of the main epithelial amino acid transporter are modulated by tissue-specific accessory proteins. Moreover, the intriguing fact that these accessory proteins can interact differentially with Hartnup mutations and thereby differentially affect kidney and intestinal amino acid transport function suggests that collectrin and ACE2 may also contribute significantly to the phenotypic heterogeneity among individuals with Hartnup disorder.
Acknowledgments
S.M.R.C. and D.S. contributed equally to this work.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding Supported by Swiss NF grant 31-108021/1 (to F.V.), the EUGINDAT (The European FP6 Project) (to F.V.), a fellowship from the University Research Priority Program “Integrative Human Physiology” at the University of Zurich (to D.S.), a grant from the Institute of Molecular Biotechnology of the Austrian Academy of Sciences (to J.M.P.), a grant from the Austrian Ministry of Sciences (to J.M.P.), and the EU network grant EuGeneHeart (to J.M.P.).
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi:10.1053/j.gastro.2008.10.055.
Supplementary data
References
- 1.Kleta R., Romeo E., Ristic Z. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet. 2004;36:999–1002. doi: 10.1038/ng1405. [DOI] [PubMed] [Google Scholar]
- 2.Seow H.F., Broer S., Broer A. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet. 2004;36:1003–1007. doi: 10.1038/ng1406. [DOI] [PubMed] [Google Scholar]
- 3.Danilczyk U., Sarao R., Remy C. Essential role for collectrin in renal amino acid transport. Nature. 2006;444:1088–1091. doi: 10.1038/nature05475. [DOI] [PubMed] [Google Scholar]
- 4.Malakauskas S.M., Quan H., Fields T.A. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am J Physiol Renal Physiol. 2007;292:F533–F544. doi: 10.1152/ajprenal.00325.2006. [DOI] [PubMed] [Google Scholar]
- 5.Crackower M.A., Sarao R., Oudit G.Y. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 6.Li W., Moore M.J., Vasilieva N. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuba K., Imai Y., Rao S. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamming I., Timens W., Bulthuis M.L. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.To K.F., Lo A.W. Exploring the pathogenesis of severe acute respiratory syndrome (SARS): the tissue distribution of the coronavirus (SARS-CoV) and its putative receptor, angiotensin-converting enzyme 2 (ACE2) J Pathol. 2004;203:740–743. doi: 10.1002/path.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paunescu T.G., Da Silva N., Marshansky V. Expression of the 56-kDa B2 subunit isoform of the vacuolar H(+)-ATPase in proton-secreting cells of the kidney and epididymis. Am J Physiol Cell Physiol. 2004;287:C149–C162. doi: 10.1152/ajpcell.00464.2003. [DOI] [PubMed] [Google Scholar]
- 11.Romeo E., Dave M.H., Bacic D. Luminal kidney and intestine SLC6 amino acid transporters of B0AT-cluster and their tissue distribution in Mus musculus. Am J Physiol Renal Physiol. 2006;290:F376–F383. doi: 10.1152/ajprenal.00286.2005. [DOI] [PubMed] [Google Scholar]
- 12.Biber J., Stieger B., Haase W. A high yield preparation for rat kidney brush border membranes: Different behaviour of lysosomal markers. Biochim Biophys Acta. 1981;647:169–176. doi: 10.1016/0005-2736(81)90243-1. [DOI] [PubMed] [Google Scholar]
- 13.Inigo C., Barber A., Lostao M.P. Na+ and pH dependence of proline and beta-alanine absorption in rat small intestine. Acta Physiol (Oxf) 2006;186:271–278. doi: 10.1111/j.1748-1716.2006.01538.x. [DOI] [PubMed] [Google Scholar]
- 14.Guy J.L., Jackson R.M., Jensen H.A. Identification of critical active-site residues in angiotensin-converting enzyme-2 (ACE2) by site-directed mutagenesis. FEBS J. 2005;272:3512–3520. doi: 10.1111/j.1742-4658.2005.04756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita A., Singh S.K., Kawate T. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 17.Camargo S.M., Makrides V., Virkki L.V. Steady-state kinetic characterization of the mouse B(0)AT1 sodium-dependent neutral amino acid transporter. Pflugers Arch. 2005;451:338–348. doi: 10.1007/s00424-005-1455-x. [DOI] [PubMed] [Google Scholar]
- 18.Ehnes C., Forster I.C., Kohler K. Structure-function relations of the first and fourth predicted extracellular linkers of the type IIa Na+/Pi cotransporter: I: Cysteine scanning mutagenesis. J Gen Physiol. 2004;124:475–488. doi: 10.1085/jgp.200409060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franca R., Veljkovic E., Walter S. Heterodimeric amino acid transporter glycoprotein domains determining functional subunit association. Biochem J. 2005;388:435–443. doi: 10.1042/BJ20050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donoghue M., Hsieh F., Baronas E. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 21.Tipnis S.R., Hooper N.M., Hyde R. A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H., Wada J., Hida K. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem. 2001;276:17132–17139. doi: 10.1074/jbc.M006723200. [DOI] [PubMed] [Google Scholar]
- 23.Kowalczuk S., Broer A., Tietze N. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880–2887. doi: 10.1096/fj.08-107300. [DOI] [PubMed] [Google Scholar]
- 24.Sato Y., Zhang Y.W., Androutsellis-Theotokis A. Analysis of transmembrane domain 2 of rat serotonin transporter by cysteine scanning mutagenesis. J Biol Chem. 2004;279:22926–22933. doi: 10.1074/jbc.M312194200. [DOI] [PubMed] [Google Scholar]
- 25.Azmanov D.N., Rodgers H., Auray-Blais C. Persistence of the common Hartnup disease D173N allele in populations of European origin. Ann Hum Genet. 2007;71:755–761. doi: 10.1111/j.1469-1809.2007.00375.x. [DOI] [PubMed] [Google Scholar]
- 26.Moalem S., Percy M.E., Kruck T.P. Epidemic pathogenic selection: an explanation for hereditary hemochromatosis? Med Hypotheses. 2002;59:325–329. doi: 10.1016/s0306-9877(02)00179-2. [DOI] [PubMed] [Google Scholar]
- 27.Schroeder S.A., Gaughan D.M., Swift M. Protection against bronchial asthma by CFTR delta F508 mutation: a heterozygote advantage in cystic fibrosis. Nat Med. 1995;1:703–705. doi: 10.1038/nm0795-703. [DOI] [PubMed] [Google Scholar]
- 28.Shih V.E., Bixby E.M., Alpers D.H. Studies of intestinal transport defect in Hartnup disease. Gastroenterology. 1971;61:445–453. [PubMed] [Google Scholar]
- 29.Tarlow M.J., Seakins J.W., Lloyd J.K. Absorption of amino acids and peptides in a child with a variant of Hartnup disease and coexistent coeliac disease. Arch Dis Child. 1972;47:798–803. doi: 10.1136/adc.47.255.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reference
- 1.Nielsen E.G., Vedso S., Zimmermann-Nielsen C. Hartnup disease in three siblings. Dan Med Bull. 1966;13:155–161. [PubMed] [Google Scholar]