

Abstract
RNA interference (RNAi) plays a pivotal role in the regulation of gene expression to control cell development and differentiation. In plants, insects and nematodes RNAi also functions as an innate defence response against viruses. Similarly, there is accumulating evidence that RNAi functions as an antiviral defence mechanism in mammalian cells. Viruses have evolved highly sophisticated mechanisms for interacting with the host cell machinery, and recent evidence indicates that this also involves RNAi pathways. The cellular RNAi machinery can inhibit virus replication, but viruses may also exploit the RNAi machinery for their own replication. In addition, viruses can encode proteins or RNA molecules that suppress existing RNAi pathways or trigger the silencing of specific host genes. Besides the natural interplay between RNAi and viruses, induced RNAi provides an attractive therapy approach for the fight against human pathogenic viruses. Here, we summarize the latest news on virus–RNAi interactions and RNAi based antiviral therapy.
Keywords: AAV, adeno-associated virus, CMV, cytomegalovirus, DEN, dengue virus, dsRNA, double-stranded RNA, EBV, Epstein Barr virus, HBV, hepatitis B virus, HCV, hepatitis C virus, HDV, hepatitis delta virus, HIV, human immunodeficiency virus, HPV, human papillomavirus, HRV-16, human rhinovirus-16, HSV, herpes simplex virus, IFN dsRNA, interferon-induced dsRNA, IFN-PKR, interferon-induced protein kinase R, JCV, JC virus, KSHV, Kaposi's sarcoma-associated virus, MHV6, murine hepatitis virus 6, miRNA, micro RNA, PFV-1, primate foamy virus type 1, pre-miRNA, precursor miRNA, RISC, RNA-induced silencing complex, RNAi, RNA interference, RSV, respiratory syncytial virus, SARS, severe acute respiratory syndrome, shRNA, short hairpin RNA, SIRCT, siRNA combination therapy, siRNA, small interfering RNA, SV40, simian virus 40, TAR, trans-activating response region, TRBP, TAR RNA binding protein, VA-RNAI, virus-associated RNA I, VA-RNAII, virus-associated RNA II, VSV, vesicular stomatitis virus, RNA interference, Virus infection, Virus–host interaction
1. The RNAi mechanism and viral infection
RNAi is a strongly conserved sequence‐specific gene silencing mechanism in eukaryotic cells that is induced by double‐stranded RNA (dsRNA). Currently, RNAi has been shown to function in two distinct processes. The first is regulation of cellular gene expression via microRNAs (miRNAs). miRNAs represent a family of highly structured small non‐coding RNAs that negatively regulate gene expression at the post‐transcriptional level [4]. They are expressed as primary miRNAs (pri‐miRNA) and processed by the proteins Drosha and Dicer into respectively a ∼70 nucleotide stem‐loop precursor miRNA (pre‐miRNA), and the mature miRNA of 21–25 nts. Only one strand of the mature miRNA, the guide strand, is loaded in the RNA induced silencing complex (RISC). The guide strand targets RISC to the mRNA, where the complex hybridises to (partially) complementary sequences resulting in mRNA cleavage or translational inhibition (Fig. 1 , left panel).
Figure 1.
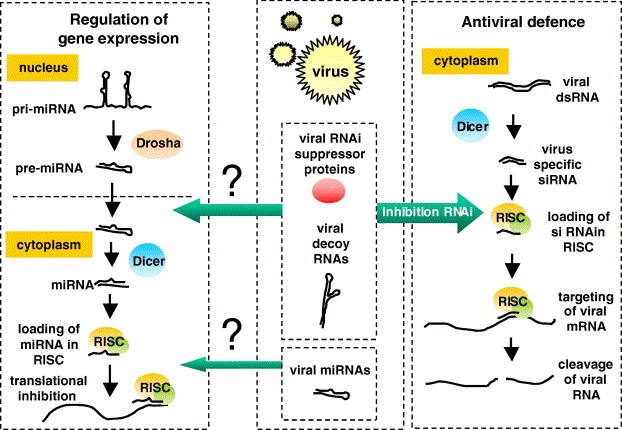
Viral factors affecting cellular RNAi pathways. miRNA regulated gene expression starts with primiRNAs in the nucleus, which are processed into mature miRNAs by Drosha and Dicer (left panel). The antiviral RNAi response is triggered by virus‐derived dsRNAs during infection (right panel). Viral RNAi suppressor factors (proteins or decoy RNAs; middle panel) counter these effects. Viruses can also encode miRNA‐like molecules targeting cellular mRNAs. Both RNAi suppressors and viral miRNAs can potentially affect cellular miRNA processing and function (arrows with question mark).
The second function of RNAi is inhibition of incoming viruses and silencing of transposable elements by generation of small interfering RNAs (siRNAs 21 nts dsRNA, Fig. 1, right panel). siRNAs can originate from extensive secondary RNA structures in the viral RNA or from dsRNA viral replication intermediates and may be fully complementary to viral mRNA. After loading into RISC, siRNAs typically trigger cleavage of the mRNA [2].
Intriguingly, miRNA‐mediated gene regulation and antiviral siRNA activity share Dicer and RISC components, and thus appear to run along similar pathways. Therefore, viruses are likely to affect cellular RNAi processes. Recent data indicate that viruses interact with RNAi mechanisms in various ways, illustrating genetic variation among different virus families. The antiviral RNAi response might represent a general phenomenon. To counter this response, viruses evolved RNAi suppressor proteins or RNA molecules with RNAi‐modulating activity. A more distinct virus–RNAi interaction is the recently reported inhibition of a retrovirus by the cellular miRNA 32 (miR‐32). In contrast, hepatitis C virus (HCV) replicates with the help from the cellular miR‐122, another example of specific virus–RNAi interaction. We will discuss these complex virus–host interactions. In addition, we will address the possibility of RNAi‐based antiviral strategies.
2. Do animals possess an RNAi‐based antiviral mechanism?
RNAi functions as an innate antiviral defence mechanism in plants, insects and nematodes, but the hypothesis that animals possess a similar mechanism remains relatively untested. The fact that several animal viruses encode RNAi suppressor functions (see next chapter) is only indirect evidence for a natural antiviral RNAi mechanism.
Perhaps the best evidence for an RNAi‐like defence against virus infection comes from two recent studies with the nematode Caenorhabditis elegans [46, 55]. Wild type and RNAi‐defective cells of this animal were infected with vesicular stomatitis virus (VSV) engineered to encode a GFP fusion protein. Upon infection, cells lacking components of the RNAi apparatus produce more GFP and infectious particles than the control cells. Furthermore, mutant cells with enhanced RNAi function were shown to produce less GFP. In this way, it was demonstrated that multiple genes required for RNAi are also required for resistance to VSV, suggesting a natural role for RNAi in the resistance to viruses.
Is there any direct evidence for the production of virus‐specific siRNA molecules from dsRNA replication intermediates in infected cells? Recent studies on HIV‐1 probably get the closest to an affirmative answer. Initial attempts to clone virally encoded siRNAs or miRNAs failed [39], but another group reported that an miRNA is encoded by the nef region of the HIV‐1 genome [36, 37]. In addition, Bennasser and colleagues characterized a sequence in the HIV‐1 genome that encodes a rare siRNA precursor: a hairpin structure composed of a 19 base pair perfectly complementary stem and a large loop [5]. This structure is processed by Dicer (or by a Dicer‐like ribonuclease) into functional siRNAs that target the HIV‐1 genome. It was reported the Tat protein functions as an RNAi suppressor protein that blocks the induced antiviral RNAi response by interfering with Dicer activity.
RNAi susceptibility of viruses is largely dictated by their precise replication strategy [23]. For example, RNAi against hepatitis delta virus (HDV) was only successful for the mRNA molecule, and not for the genomic and antigenomic RNA sequences [15]. The latter two RNA molecules may be resistant because their location within the nucleus makes them inaccessible to the cytoplasmic RISC. Similar findings have been reported for influenza virus and respiratory syncytial virus (RSV). The genomic and antigenomic RNA duplexes of HDV also resist Dicer cleavage, which appears to be caused by the extended nature of this imperfect RNA duplex [14]. It is likely that more diverse viral escape mechanisms will be identified in the near future.
3. Viral RNAi suppressors
Antiviral RNAi responses are believed to be triggered by viral dsRNA molecules that are produced during infection. To counter this antiviral effect viruses have evolved strategies to avoid recognition by the RNAi machinery of the host. Many plant viruses encode proteins that interfere with one or more aspects of Dicer action and/or siRNA targeting [31, 42, 51, 52, 53]. These RNAi suppressors were first described as pathogenicity factors, and target the RNAi machinery at various steps along the silencing pathway, enabling the virus to accumulate to higher titers. Recently, RNAi suppressors have also been identified in several important human pathogenic viruses. For example, influenza virus NS1 protein, vaccinia virus E3L protein, HIV‐1 Tat protein and the adenovirus virus‐associated RNAs I and II (VA‐RNAI and II) were shown to exhibit RNAi suppressor activity [2, 5, 30, 32]. This discovery suggests that RNAi functions as an innate antiviral defence mechanism in mammals similar to what has been described in plants and insects.
The mode of action of RNAi suppressors in human viruses is largely unknown. We and others have shown that the adenovirus VA‐RNAs inhibit RNAi by acting as decoy substrates for Exportin 5 (involved in transport of pre‐miRNAs from the nucleus to the cytoplasm), Dicer and RISC [2, 32]. The HIV‐1 Tat protein it thought to block Dicer activity, whereas influenza virus NS1 and vaccinia virus E3L may sequester siRNAs [5, 11, 30]. The presence of a viral suppressor function may have consequences for therapeutic RNAi approaches. For instance, a shRNA therapeutic approach that is dependent on Dicer processing may not be optimal. However, it is clearly too early to toss the towel in the ring, as potent HIV‐1 inhibition can be obtained with such an approach [9, 18, 50, 54].
4. Virus‐encoded miRNAs
Besides suffering from the antiviral RNAi responses, viruses can also exploit RNAi to control the expression of genes of viral or host origin. The first example was recently provided for Epstein Barr virus (EBV), a large DNA virus of the herpes family that preferentially infects human B cells [40]. When the small RNAs from a latently EBV‐infected Burkitt's lymphoma cell line were cloned, 4% of them originated from two regions of the EBV genome. A computational method was used to identify potential targets of these EBV‐encoded miRNAs. Among the predicted targets were regulators of cell proliferation, apoptosis, transcriptional regulators and components of signal transduction pathways. Although these targets should be verified experimentally, it is striking that several of these genes have more than one binding site for a particular EBV‐miRNA. Degradation of a cellular DNA polymerase was demonstrated experimentally [40]. Furthermore, the expression of the EBV‐miRNAs was shown to differ in the lytic versus latent stage, suggesting tight regulation during viral infection. Intriguingly, the viral miRNAs could be involved in tumour formation and may explain how EBV hides so well. Other members of the herpesvirus family and other viruses with a large DNA genome could encode miRNAs in order to exploit RNAi for the regulation of host and viral expression.
Extensive cDNA cloning studies across many families of RNA viruses have failed to identify miRNAs from viruses with RNA genomes [39]. To date, two other members of the γ‐herpes virus subfamily – Kaposi's sarcoma‐associated virus (KSHV) and murine gammaherpesvirus 68 (MHV68) – and one β‐herpes virus – cytomegalovirus (CMV) – have been shown to encode miRNAs [12, 34, 39, 40, 45].
KSHV encodes an array of 11 distinct miRNAs from what appears to be a single genetic locus [12]. All these miRNAs are expressed at readily detectable levels (up to 2200 copies per cell) in latently infected cells. Computer analysis of potential mRNA targets identified a number of interesting candidate genes, including several mRNAs previously shown to be down‐regulated in KSHV‐infected cells. The miRNAs are fully conserved in all KSHV isolates present in the genome database, arguing that they are likely to play a key role in KSHV replication and pathogenesis. The apparent restriction of viral miRNAs to complex DNA viruses may reflect an ability of these viruses to more elegantly manipulate host gene expression relative to the more stripped‐down DNA and RNA viruses.
An exception to this may be the small polyomavirus simian virus 40 (SV40), which encodes a set of miRNAs whose in vivo target has been clearly defined [48]. The pre‐miRNA is part of a viral late premRNA transcript, but cleaved off during polyadenylation and processed further. The SV40 DNA genome is circular, such that the late gene sequences are completely complementary to early mRNAs produced from the opposite strand. The late miRNA were shown to cleave the early miRNAs at the predicted position at late times in the replication cycle, thus reducing early gene expression, and possibly evading immune recognition of the infected cell. Again, conservation of the viral miRNA in other polyomavirus suggests that it is important for the virus.
5. Virus inhibition by a cellular miRNA
Cellular miRNAs are important for the regulation of cellular genes, but recent evidence indicates that cellular miRNAs can also target the genetic material of invading viruses. A comparison of the primate foamy virus type 1 (PFV‐1) genome and human miRNAs revealed several miRNAs that could potentially block gene expression and replication of this retrovirus. When one of these miRNAs, miRNA‐32, was knocked down, the virus nearly doubled its replication rate [28]. Similar to HIV‐1 Tat, the PFV‐1 Tas protein was identified as an RNAi suppressor protein that was required to block the miRNA attack on the virus.
The PFV‐1 Tas suppressor protein was proposed to have evolved in response to miRNA‐32 inhibition. A seemingly more simple evolutionary scenario would provide viral escape by acquisition of one or a few pointmutations within the target sequence in the viral bet gene. Such changes could even be neutral with respect to the encoded Bet protein when silent codon changes are selected. Escape from RNAi via this route has been described for the HIV‐1 retrovirus when inhibited by designed shRNA antivirals [8, 18, 54]. The fact that PFV‐1 did not use this escape route may suggest that, besides the antiviral RNAi pressure of miR‐32, viral sequences themselves also trigger an antiviral RNAi response, similar to antiviral RNAi responses in plants and insects.
6. A cellular miRNA helps HCV
Cellular miRNAs usually act by annealing to the 3′ non‐coding region of an mRNA, thereby repressing mRNA translation. Surprisingly, Jopling et al. identified a liver‐specific miRNA that interacts with the 5′ non‐coding region of the HCV RNA genome and seems to aid virus replication [27]. This is the first time that an miRNA molecule has been found to positively regulate gene expression. The human genome contains at least 800 genes that code for miRNAs. The authors chose to look at miR‐122 because it accounts for 70% of miRNAs found in the liver, the place where HCV replicates, thereby being a major cause of chronic liver disease.
Inhibition of miR‐122 function using an antisense oligonucleotide resulted in a dramatic decrease of HCV RNA by about 80%. With an active miR‐122, a mutant HCV RNA with an altered target sequence in the 5′ non‐coding region failed to accumulate. Most importantly, this defect could be restored by ectopic expression of a mutant miR‐122 with the compensatory mutations, allowing base pairing with the mutant HCV RNA. Thus, the replication defect is due to the lack of miR‐122 interaction, and not due to RNA misfolding because of the 5′ mutation. The miR‐122/HCV RNA interaction does not primarily influence RNA translation or stability, but likely affects viral RNA replication.
The involvement of miR‐122 in HCV replication is reminiscent to the usage of tRNAlys 3 to prime reverse transcription during HIV‐1 replication [1]. These viruses use available cellular resources for their own replication. However, miR‐122 involvement in HCV replication contrasts with the inhibitory effect of miR‐32 on PFV‐1. Apparently viruses can interact with cellular RNAi factors in various ways. Possibly, other viral of host cell mRNAs can also be positively affected by miRNAs. Two plant miRNAs have been demonstrated to interact with the 5′ non‐coding region of their respective targets mRNAs [10], but this has not yet been observed for animal miRNAs [4]. Another intriguing question is about the natural function of miR‐122 in the liver, and whether that function is influenced by HCV infection. These results also have potential therapeutic relevance. First, knocking down miR‐122 in the liver might counteract infection with HCV. Second, antiviral si/shRNA strategies could focus on the miR‐122 target in the 5′ non‐coding region. This target may be highly accessible and is likely to be extremely conserved to maintain miR‐122 interaction. As such, viral escape would be extremely difficult, thus allowing the development of a durable therapy.
7. Viruses that affect the cellular RNAi machinery
Viruses are capable of exploiting cellular resources for their own replication, and they frequently modulate cellular pathways to optimize replication. It is becoming increasingly clear that there is also an intricate relationship between viruses and the cellular RNAi machinery. As we mentioned earlier, miRNA regulated cellular gene expression and the antiviral siRNA activity appear to run along similar pathways (Fig. 1). We discussed examples in which cellular miRNAs inhibit and promote virus replication. Viral RNAi suppressor factors that block Dicer or siRNA function may affect cellular miRNA function in case they cannot discriminate between siRNA and miRNA processing. In that scenario, suppressors will not only inhibit the antiviral RNAi effect, but also interfere with cellular miRNAs function. Such suppressor induced changes in miRNA maturation will indirectly affect cellular gene expression, and may contribute to viral pathogenicity. In plants, the turnip mosaic virus RNAi suppressor protein HC‐Pro blocks miR171 activity, resulting in abnormal plant development [26]. In human cells, adenovirus VA‐RNAI was shown to interfere with miRNA biogenesis [2, 32]. For HIV‐1, the Tat protein was shown to partially repress the processing activity of Dicer [5]. Indeed, the miRNA expression profile was recently demonstrated to differ in cells that express HIV‐1 when compared to control cells [16]. This initial study was performed with cells transfected with an HIV‐1 molecular clone, and should be repeated with virus infected primary human cells. It is conceivable that the downregulation of mature miRNAs may be due to the Dicer suppressive effect exerted by the Tat protein [5].
Another intriguing link between HIV‐1 and the cellular RNAi machinery became apparent with the identification of TRBP as a protein partner of human Dicer. TRBP was previously identified as HIV‐1 TAR RNA‐binding protein [19]. Two recent studies indicate that TRBP is a component that is required for RISC assembly and optimal RNA silencing mediated by siRNAs and endogenous miRNAs [16, 21]. TRBP had previously been assigned several functions, including inhibition of the interferon‐induced (IFN)dsRNA‐regulated protein kinase PKR and modulation of HIV‐1 gene expression by association with TAR. The TRBP–Dicer interaction raises the possibility of crosstalk between RNAi and IFN‐PKR pathways, both in normal and virus‐infected cells. Interestingly, many RNAi suppressors encoded by viruses are also inhibitors of the IFN‐induced PKR, providing another overlap between these two pathways. The finding that TRBP is an essential component of the RISC complex raises several possible scenarios for HIV‐1 interference with the RNAi pathway [20]. For instance, accumulation of HIV‐1 RNA with 5′ and 3′ TAR motifs in virus‐infected cells may trigger the depletion of TRBP, leading to inactivation of RISC and the RNAi machinery. This could facilitate a general suppression of RNAi by HIV‐1, similar to the way in which adenovirus VARNAs block Exportin‐5, Dicer and RISC.
Several alternative scenarios can be proposed. TAR has secondary structure that resembles miRNA precursors, raising the possibility that TAR is a viral pre‐miRNA. Combined with the recent finding that the Tat protein is a suppressor of RNAi, and in particular an inhibitor of Dicer [5], these observations may suggest that TRBP could recruit Dicer to TAR only in the absence of Tat, resulting in functional destruction of HIV‐1 transcripts [43]. Tat may prevent this antiviral action. Any of these speculative models should incorporate the fact that TRBP contributes to HIV‐1 replication [20]. All scenarios obviously require experimental testing. For instance, despite the use of sensitive reporter assays, we never obtained any evidence for functional dicing of TAR in cells (Haasnoot and Berkhout, unpublished results).
8. RNAi based antiviral therapy
The first demonstration of RNAi‐mediated inhibition of a human pathogenic virus was reported by Bitko and Barik in 2001 [7]. These authors reported a 10‐fold inhibition of RSV replication in vitro using nanomolar concentrations of synthetic siRNAs that targeted RNAs encoding the viral polymerase subunit P and the fusion protein F. Currently, many other studies have described RNAi‐mediated inhibition of a large variety of viruses. RNAi‐mediated inhibition of HIV‐1 has received much attention. In addition, 25 different RNA viruses, and 11 different DNA viruses have been efficiently targeted by RNAi [22]. These include important human pathogens such as HCV, dengue (DEN) virus, severe acute respiratory syndrome (SARS) coronavirus, poliovirus, influenza A virus, HDV, human rhinovirus (HRV), hepatitis B virus (HBV), herpes simplex virus type‐1 (HSV‐1), human papillomavirus (HPV), JC virus (JCV), EBV, and CMV.
Initially, the standard method to induce RNAi against viruses in mammalian cells was transfection of synthetic siRNAs shortly before or after viral challenge. Currently, transient transfection of plasmids that express antiviral shRNAs is also commonly used. Both strategies can result in potent, albeit temporary inhibition of virus replication. In order to obtain long‐term virus resistance, researchers have turned to a combined RNAi/gene therapy approach. In this approach, lenti‐, retro‐ or adeno‐associated virus (AAV) vectors are used to stably transduce cells with constructs expressing shRNA, resulting in viral resistance.
9. HIV‐1 inhibition by RNAi, viral escape and human countermeasures
Several studies reported that siRNA can suppress HIV‐1 [13, 17, 24, 25, 29, 33, 35, 38, 41, 49]. There is some evidence suggesting that the genomic RNA present within an infecting virion particle is targeted for destruction, but it appears that new viral transcripts, synthesized from the integrated provirus, are more efficient targets. Most studies used chemically synthesized siRNAs that were transfected into cells either shortly before or after challenge with HIV‐1. Despite the transient nature of such a transfection experiment, a single siRNA application is able to achieve relatively long‐lasting suppression [47]. Other studies used transient transfection of siRNA‐expression vectors. However, the development of efficient vector delivery systems capable of mediating stable siRNA expression in mature T lymphocytes or progenitor stem cells will be a minimal requirement for RNAi to be used as a therapeutic modality against HIV‐1. Lentiviral vectors with a Pol III expression cassette are an efficient means to deliver anti‐HIV siRNAs into haematopoietic precursor cells. In a recent report, the transduced human cells were allowed to differentiate in vivo in the SCID‐hu thymopoiesis mouse model [3], and the mature T lymphocytes derived from this model resisted HIV‐1 infection ex vivo.
Two studies addressed the potency and durability of anti‐HIV RNAi approaches. Boden et al. expressed an shRNA against the tat gene in an AAV vector with an H1‐promoter [8]. Potent inhibition was scored, but an escape virus variant appeared in prolonged cultures. Similar results were described by Das et al. using a lentiviral vector with an H1 unit expressing an siRNA against sequences in the nef gene [18]. The latter study described seven independent HIV‐1 escape variants. The combined results convincingly demonstrate that inhibition was potent and sequence‐specific, but also that HIV‐1 is able to escape from the inhibitory action of a single siRNA. Boden et al. described a single revertant with a point mutation in the target sequence, and Das et al. described a large variety of escape routes (point mutation, double point mutation, partial or complete deletion of the target sequence). A deletion‐based resistance mechanism seems impossible in case essential HIV‐1 genes or critical sequence motifs are targeted. Thus, one should preferentially target essential sequences that are well conserved among HIV‐1 isolates. Interesting targets with relatively little mutational freedom are the multiple overlaps in reading frames within the HIV‐1 genome, including a triple overlap (tat–rev–env). Ideally, one should target more than one of these essential and well‐conserved viral sequences. Such siRNA combination therapy (SIRCT) mimics the successful strategy to combat HIV‐1 with multiple antiviral drugs, and should avoid the evolution of escape variants [6].
We recently discovered an alternative resistance mechanism that is not triggered by mutation of the target sequence. Instead, a mutation in the flanking sequences was selected, which was subsequently shown to induce a conformational change within the target sequence such that it is protected from RISC attack [54]. This finding indicates that it will not be very straightforward to predict viral escape routes. Nevertheless, one could make a first estimation of the chance of viral escape in a therapeutic setting with one or multiple siRNAs [6]. If we assume that an essential viral sequence is targeted, deletion is no option. Therefore, one and more likely two nucleotide substitutions are required per 19‐nucleotide target sequence to obtain a fair level of resistance [18]. Assuming that 2‐point mutations are needed to obtain complete resistance, and further assuming an error rate of the reverse transcriptase polymerase of 2 × 10−5, the chance of viral escape in a single replication cycle is 19 × [(2 × 10−5)]2 = 1.44 × 10−7. Studies in the field of drug‐resistance indicate that an untreated HIV‐infected individual contains an effective viral population size of 104–105 [44], which means that most 1‐nucleotide substitutions will already be present within the viral population. Starting in an untreated patient with a moderate viral load, this means that resistance is likely to occur. Thus, it may indeed be important to consider SIRCT [6]. With four effective siRNAs, the chance of viral escape drops to 2.1 × 10−14. In practice, this means that viral escape is impossible as long as viral suppression is complete. Even if several assumptions are wrong, the prospects are favourable that one can achieve effective and long‐term viral suppression. An alternative strategy is to target unmutable host‐encoded functions that are important for viral replication, but not essential for survival of the host cell.
10. Concluding remarks
Viruses are obligate intracellular parasites whose replication depends on their hosts. This interplay has important consequences, both for the virus and the host. RNAi is a newly discovered mechanism that, through miRNA effector molecules, plays a pivotal role in the regulation of many genes, especially those at nodes of signaling pathways involved in such processes as development, metabolism, apoptosis and aging. It therefore comes as no surprise that viruses also encode miRNAs. In addition, host‐encoded miRNAs have been reported to positively or negatively modulate viral replication. On their part, viruses can encode RNAi suppressors that protect against the cellular antiviral RNAi response, but that may also affect cellular gene expression. Certain viruses even have evolved to utilize that miRNA pathway to their own advantage. Nevertheless, a better understanding of the virus–host interaction at the molecular level should allow us to develop an effective, durable and non‐toxic antiviral therapy. RNAi‐based treatments are quickly moving towards human patients, and the year 2006 should offer the first hints of how this highly publicized technique works in a therapeutic setting. Company‐funded clinical trials in macular degeneration are under way, and targeting of transient viral infections such as pediatric respiratory illness caused by RSV are supposed to launch soon. Chronic viral infections caused by HCV and HIV‐1 will follow.
Acknowledgements
RNA research in the Berkhout lab is sponsored by ZonMw (VICI grant) and NWO‐CW (TOP grant), RNA interference studies are supported by Senter‐NOVEM (TS grant together with Viruvation BV).
Berkhout Ben and Haasnoot Joost(2006), The interplay between virus infection and the cellular RNA interference machinery, FEBS Letters, 580, doi: 10.1016/j.febslet.2006.02.070
References
- 1. Abbink T.E.M., Beerens N., Berkhout B., Forced selection of a human immunodeficiency virus type 1 variant that uses a non-self tRNA primer for reverse transcription: involvement of viral RNA sequences and the reverse transcriptase enzyme. J. Virol., 78, (2004), 10706– 10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andersson M.G., Haasnoot P.C.J., Xu N., Berenjian S., Berkhout B., Akusjarvi G., Suppression of RNA interference by adenovirus virus-associated RNA. J. Virol., 79, (2005), 9556– 9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banerjea A., Li M.J., Bauer G., Remling L., Lee N.S., Rossi J., Akkina R., Inhibition of HIV-1 by lentiviral vector-transduced siRNAs in T lymphocytes differentiated in SCID-hu mice and CD34+ progenitor cell-derived macrophages. Mol. Ther., 8, (2003), 62– 71. [DOI] [PubMed] [Google Scholar]
- 4. Bartel D.P., MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116, (2004), 281– 297. [DOI] [PubMed] [Google Scholar]
- 5. Bennasser Y., Le S.Y., Benkirane M., Jeang K.T., Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity, 22, (2005), 607– 619. [DOI] [PubMed] [Google Scholar]
- 6. Berkhout B., RNA interference as an antiviral approach: targeting HIV-1. Curr. Opin. Mol. Ther., 6, (2004), 141– 145. [PubMed] [Google Scholar]
- 7. Bitko V., Barik S., Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol., 1, (2001), 34– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boden D., Pusch O., Lee F., Tucker L., Ramratnam B., Human immunodeficiency virus type 1 escape from RNA interference. J. Virol., 77, (2003), 11531– 11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boden D., Pusch O., Lee F., Tucker L., Shank P.R., Ramratnam B., Promoter choice affects the potency of HIV-1 specific RNA interference. Nucleic Acids Res., 31, (2003), 5033– 5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brennecke J., Stark A., Russell R.B., Cohen S.M., Principles of microRNA-target recognition. PLoS. Biol., 3, (2005), e85– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bucher E., Hemmes H., Haan P., Goldbach R., Prins M., de The influenza A virus NS1 protein binds small interfering RNAs and suppresses RNA silencing in plants. J. Gen. Virol., 85, (2004), 983– 991. [DOI] [PubMed] [Google Scholar]
- 12. Cai X., Lu S., Zhang Z., Gonzalez C.M., Damania B., Cullen B.R., Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA, 102, (2005), 5570– 5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Capodici J., Kariko K., Weissman D., Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J. Immunol., 169, (2002), 5196– 5201. [DOI] [PubMed] [Google Scholar]
- 14. Chang J., Provost P., Taylor J.M., Resistance of human hepatitis delta virus RNAs to dicer activity. J. Virol., 77, (2003), 11910– 11917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang J., Taylor J.M., Susceptibility of human hepatitis delta virus RNAs to small interfering RNA action. J. Virol., 77, (2003), 9728– 9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chendrimada T.P., Gregory R.I., Kumaraswamy E., Norman J., Cooch N., Nishikura K., Shiekhattar R., TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature, 436, (2005), 740– 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coburn G.A., Cullen B.R., Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol., 76, (2002), 9225– 9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Das A.T., Brummelkamp T.R., Westerhout E.M., Vink M., Madiredjo M., Bernards R., Berkhout B., Human immunodeficiency virus type 1 escapes from RNA interference mediated inhibition. J. Virol., 78, (2004), 2601– 2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gatignol A., Buckler-White A., Berkhout B., Jeang K.T., Characterization of a human TAR RNA-binding protein that activates the HIV-1 LTR. Science, 251, (1991), 1597– 1600. [DOI] [PubMed] [Google Scholar]
- 20. Gatignol A., Laine S., Clerzius G., Dual role of TRBP in HIV replication and RNA interference: viral diversion of a cellular pathway or evasion from antiviral immunity?. Retrovirology, 2, (2005), 65– 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haase A.D., Jaskiewicz L., Zhang H., Laine S., Sack R., Gatignol A., Filipowicz W., TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep., 6, (2005), 961– 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haasnoot P.C.J., Berkhout B., RNA interference: its use as antiviral therapy. Handbook of Experimental Pharmacology Vol. 173, (2006), Springer ; Berlin, Heidelberg: 117– 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haasnoot P.C.J., Cupac D., Berkhout B., Inhibition of virus replication by RNA interference. J. Biomed. Sci., 10, (2003), 607– 616. [DOI] [PubMed] [Google Scholar]
- 24. Hu W.Y., Myers C.P., Kilzer J.M., Pfaff S.L., Bushman F.D., Inhibition of retroviral pathogenesis by RNA interference. Curr. Biol., 12, (2002), 1301– 1311. [DOI] [PubMed] [Google Scholar]
- 25. Jacque J.M., Triques K., Stevenson M., Modulation of HIV-1 replication by RNA interference. Nature, 418, (2002), 435– 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kasschau K.D., Xie Z., Allen E., Llave C., Chapman E.J., Krizan K.A., Carrington J.C., P1/HC-Pro, a viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev. Cell, 4, (2003), 205– 217. [DOI] [PubMed] [Google Scholar]
- 27. Lancaster A.M., Jopling C.L., Yi M., Lemon S.M., Sarnow P., Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science, 309, (2005), 1577– 1581. [DOI] [PubMed] [Google Scholar]
- 28. Lecellier C.H., Dunoyer P., Arar K., Lehmann-Che J., Eyquem S., Himber C., Saib A., Voinnet O., A cellular microRNA mediates antiviral defense in human cells. Science, 308, (2005), 557– 560. [DOI] [PubMed] [Google Scholar]
- 29. Lee N.S., Dohjima T., Bauer G., Li H., Li M.J., Ehsani A., Salvaterra P., Rossi J., Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol., 20, (2002), 500– 505. [DOI] [PubMed] [Google Scholar]
- 30. Li W.X., Li H., Lu R., Li F., Dus M., Atkinson P., Brydon E.W., Johnson K.L., Garcia-Sastre A., Ball L.A., Palese P., Ding S.W., Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. USA, 101, (2004), 1350– 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lichner Z., Silhavy D., Burgyan J., Double-stranded RNA-binding proteins could suppress RNA interference-mediated antiviral defences. J. Gen. Virol., 84, (2003), 975– 980. [DOI] [PubMed] [Google Scholar]
- 32. Lu S., Cullen B.R., Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and microRNA biogenesis. J. Virol., 78, (2004), 12868– 12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez M.A., Gutierrez A., Armand-Ugon M., Blanco J., Parera M., Gomez J., Clotet B., Este J.A., Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS, 16, (2002), 2385– 2390. [DOI] [PubMed] [Google Scholar]
- 34. Miller V., Ait-Khaled M., Stone C., Griffin P., Mesogiti D., Cutrell A., Harrigan R., Staszewski S., Katlama C., Pearce G., Tisdale M., HIV-1 reverse transcriptase (RT) genotype and susceptibility to RT inhibitors during abacavir monotherapy and combination therapy. AIDS, 14, (2000), 163– 171. [DOI] [PubMed] [Google Scholar]
- 35. Novina C.D., Murray M.F., Dykxhoorn D.M., Beresford P.J., Riess J., Lee S.K., Collman R.G., Lieberman J., Shankar P., Sharp P.A., siRNA-directed inhibition of HIV-1 infection. Nat. Med., 8, (2002), 681– 686. [DOI] [PubMed] [Google Scholar]
- 36. Omoto S., Fuji Y.R., Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J. Gen. Virol., 86, (2005), 751– 755. [DOI] [PubMed] [Google Scholar]
- 37. Omoto S., Ito M., Tsutsumi Y., Ichikawa Y., Okuyama H., Andi B.E., Saksena N.K., Fuji Y., HIV-1 nef suppression by virally encoded microRNA. Retrovirology, 1, (2004), 44– 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park W.S., Miyano-Kurosaki N., Hayafune M., Nakajima E., Matsuzaki T., Shimada F., Takaku H., Prevention of HIV-1 infection in human peripheral blood mononuclear cells by specific RNA interference. Nucleic Acids Res., 30, (2002), 4830– 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., Russo J.J., Ju J., Randall G., Lindenbach B.D., Rice C.M., Simon V., Ho D.D., Zavolan M., Tuschl T., Identification of microRNAs of the herpesvirus family. Nat. Methods, 2, (2005), 269– 276. [DOI] [PubMed] [Google Scholar]
- 40. Pfeffer S., Zavolan M., Grasser F.A., Chien M., Russo J.J., Ju J., John B., Enright A.J., Marks D., Sander C., Tuschl T., Identification of virus-encoded microRNAs. Science, 304, (2004), 734– 736. [DOI] [PubMed] [Google Scholar]
- 41. Qin X.F., An D.S., Chen I.S.Y., Baltimore D., Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. USA, 100, (2003), 183– 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qu F., Ren T., Morris T.J., The coat protein of turnip crinkle virus suppresses posttranscriptional gene silencing at an early initiation step. J. Virol., 77, (2003), 511– 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossi J.J., Mammalian Dicer finds a partner. EMBO Rep., 6, (2005), 927– 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rouzine I.M., Coffin J.M., Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. USA, 96, (1999), 10758– 10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samols M.A., Hu J., Skalsky R.L., Renne R., Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol., 9, (2005), 9301– 9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schott D.H., Cureton D.K., Whelan S.P., Hunter C.P., An antiviral role for the RNA interference machinery in Caenorhabditis elegans . Proc. Natl. Acad. Sci. USA, 102, (2005), 18420– 18424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song E., Lee S.K., Dykxhoorn D.M., Novina C., Zhang D., Crawford K., Cerny J., Sharp P.A., Lieberman J., Manjunath N., Shankar P., Sustained small interfering RNA-mediated human immunodeficiency virus type 1 inhibition in primary macrophages. J. Virol., 77, (2003), 7174– 7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sullivan C.S., Grundhoff A.T., Tevethia S., Pipas J.M., Ganem D., SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature, 435, (2005), 682– 686. [DOI] [PubMed] [Google Scholar]
- 49. Surabhi R.M., Gaynor R.B., RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus type 1 replication. J. Virol., 76, (2002), 12963– 12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brake ter O., Berkhout B., A novel approach for inhibition of HIV-1 by RNA interference: counteracting viral escape with a second generation of siRNAs. J. RNAi Gene Silencing, 1, 2 (2005), 56– 65. [PMC free article] [PubMed] [Google Scholar]
- 51. Vance V., Vaucheret H., RNA silencing in plants-defense and counterdefense. Science, 292, (2001), 2277– 2280. [DOI] [PubMed] [Google Scholar]
- 52. Voinnet O., Pinto Y.M., Baulcombe D.C., Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc. Natl. Acad. Sci. USA, 96, (1999), 14147– 14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Waterhouse P.M., Wang M.B., Lough T., Gene silencing as an adaptive defence against viruses. Nature, 411, (2001), 834– 842. [DOI] [PubMed] [Google Scholar]
- 54. Westerhout E.M., Ooms M., Vink M., Das A.T., Berkhout B., HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res., 33, (2005), 796– 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilkins C., Dishongh R., Moore S.C., Whitt M.A., Chow M., Machaca K., RNA interference is an antiviral defence mechanism in Caenorhabditis elegans . Nature, 436, (2005), 1044– 1047. [DOI] [PubMed] [Google Scholar]