

Abstract
Picornaviruses (PV) and coronaviruses (CoV) are positive‐stranded RNA viruses which infect millions of people worldwide each year, resulting in a wide range of clinical outcomes. As reported in this study, using high throughput screening against ∼6800 small molecules, we have identified several novel inhibitors of SARS‐CoV 3CLpro with IC50 of low μM. Interestingly, one of them equally inhibited both 3Cpro and 3CLpro from PV and CoV, respectively. Using computer modeling, the structural features of these compounds as individual and common protease inhibitors were elucidated to enhance our knowledge for developing anti‐viral agents against PV and CoV.
Keywords: Coronavirus, Picornavirus, 3C protease, Fluorescence assay, High throughput screening, Computer modeling
1. Introduction
Picornaviruses (PV) are small nonenveloped RNA viruses with a single strand of genomic RNA of 7500–8000 nucleotides [1]. The members of PV include rhinoviruses (RV), enteroviruses (EV), coxsackieviruses (CV), polioviruses, echoviruses, encephalomyocarditis viruses, meningitis virus, foot and mouth viruses, hepatitis A virus, and so on. Among them, RV is the major cause of the common cold, whereas EV and CV infection can cause hand, foot, and mouth diseases in human and animals. In severe cases, EV can damage the central nervous systems leading to viral meningitis, encephalitis, and severe myocarditis, as well as fatal pulmonary edema [2, 3, 4, 5]. CV strain B is a major human pathogen that causes meningitis and mycocarditis leading to heart failure in young adults and congestive heart failure [6]. In these PV, a chymotrypsin‐like protease (named 3Cpro) is required to process polyproteins into mature proteins for viral replication, which represents a promising anti‐viral drug target [7].
On the other hand, coronaviruses (CoV) are the positive‐stranded RNA viruses with larger genome of 27–32 kb, which typically cause respiratory and enteric diseases, pneumonia, exacerbation of asthma, neurological symptoms, and myocarditis in humans and domestic animals. An outbreak of severe acute respiratory syndrome (SARS), caused by a novel human CoV, was spread from China to 29 countries in 2003, infecting a total of ∼8000 people and killing ∼800 patients [8]. SARS‐CoV contains a 3C‐like protease (3CLpro) analogous to the 3Cpro of PV, responsible for processing two overlapping polyproteins, pp1a (486 kDa) and pp1ab (790 kDa). Other members of human CoV including CoV‐229E, CoV‐OC43, CoV‐HKU1, and CoV‐NL63 also require a 3CLpro in the maturation of viral proteins.
Several inhibitors have been developed to inhibit the 3Cpro of RV and EV [9, 10, 11, 12] and 3CLpro of SARS‐CoV [13, 14, 15]. However, their inhibitors can not be mutually used without modification. For example, AG7088, a potent inhibitor of RV and other picornaviral 3Cpro [16], failed to inhibit SARS‐CoV 3CLpro [17]. Unlike the 3CLpro, which is dimeric and in which each subunit is composed of three domains, the 3Cpro is a monomer with only the two catalytic domains. The structure‐based sequence alignment (Fig. 1 ) shows some sequence differences, which may alter inhibitor specificity. In this study, we performed high throughput screening using a library of ∼6800 compounds to find five novel inhibitors of the SARS‐CoV 3CLpro, 4 of which also inhibited another human CoV‐229E 3CLpro, but did not inhibit the 3Cpro from RV14, CVB3, and EV71. But, one compound was found to almost equally inhibit these 3CLpro and 3Cpro. From computer modeling, we rationalized the binding discrepancy of the inhibitors against these proteases. The information is useful to further develop more potent individual or common inhibitors of 3Cpro and 3CLpro of PV and CoV for anti‐viral drug discovery.
Figure 1.

A structure‐based sequence alignment of SARS‐CoV 3CLpro, CoV‐229E 3CLpro, CVB3 3Cpro, EV71 3Cpro, and RV14 3Cpro. The domains according to 3CLpro are shown above the sequence and the secondary elements according to the 3Cpro structure are shown below it. Arrows indicate the essential catalytic amino acids His and Cys for 3CLpro and 3Cpro, and Glu (only for 3Cpro).
2. Methods
2.1. Expression and purification of the proteases
Two types of proteases including 3CLpro from SARS‐CoV and CoV‐229E and 3Cpro from CVB3, EV71, and RV14 were used to assay the inhibitors in this study. The SARS‐CoV 3CLpro and EV71 3Cpro were prepared as reported previously [12, 18]. For expressing CVB3, RV14, and CoV‐229E proteases, the genes were cloned from viral cDNAs by using polymerase chain reaction (PCR) as reported elsewhere.
2.2. Primary screening
For screening, 0.05 μM SARS 3CLpro, 6 μM fluorogenic substrate Dabcyl‐KTSAVLQSGFRKME‐Edans, and 50 μM of approximately 6800 compounds provided by Korea Chemical Bank (Daejeon, Korea) were used. Enhanced fluorescence of the reactions in the buffer of 20 mM Bis‐Tris at pH 7.0 was monitored at 538 nm with excitation at 355 nm using a fluorescence plate reader. The compounds which inhibited more than 50% of the protease activity at 50 μM were selected for the next assay run at 10 μM.
2.3. IC50 determination
The five hits that inhibited SARS‐CoV 3CLpro at 10 μM were also evaluated against CoV‐229E 3CLpro, EV71 3Cpro, CVB3 3Cpro, and RV14 3Cpro. In the assay solution, the activities of these proteases (0.5 μM) with 10 μM fluorogenic substrate in the buffers of 10 mM MES at pH 6.5 and 6.0 (the optimal pH for EV71 and RV14 proteases, respectively) and 10 mM HEPES at pH 7.5 (for CoV‐229E and CVB3 proteases) were measured in the presence of various concentrations of the inhibitors to obtain the IC50 values.
2.4. Computer modeling of the inhibitors binding with the proteases
For the modeling analysis, we used the crystal structure of SARS 3CLpro in complex with a peptide inhibitor (PDB code 1UK4) [19], the structures of CoV‐229E 3CLpro and CVB3 3Cpro solved by us, and the structural model of EV71 3Cpro constructed from the structure of RV 3Cpro (PDB code 1CQQ) [20]. Docking process was performed using an automated ligand‐docking subprogram of the Discovery Studio Modeling 1.2 SBD (Accelrys Inc., San Diego, CA), with a set of parameters chosen to control the precise operation of the genetic algorithm. Docking runs were carried out using standard default settings “grid resolution” of 5 Å, “site opening” of 12 Å, and “binding site” selected for defining the active site cavity.
3. Results
3.1. Screening of the protease inhibitors
We first screened against a library of ∼6800 compounds for inhibiting SARS‐CoV 3CLpro. From the primary screening, there were 66 compounds which showed more than 50% inhibition of the enzyme activity at 50 μM. We further tested their inhibitory activities at 10 μM and five of them (21155, 22723, 27548, 43146, and 48511) showed IC50 values smaller than 10 μM. According to their dose–response curves as shown in Fig. 2A–E , the five hits 21155, 22723, 27548, 43146, and 48511 displayed IC50 values of 7.2 ± 0.7, 10.6 ± 1.3, 7.0 ± 0.8, 3.3 ± 0.2, and 8.1 ± 0.9 μM, respectively, against the SARS 3CLpro. Similar inhibition results were observed for 3CLpro of CoV‐229E (data summarized in Table 1 ), but not for 3Cpro. However, 43146 inhibited both 3Cpro and 3CLpro with IC50 values of 10.3 ± 1.1 μM, 5.4 ± 0.2 μM, 3.3 ± 0.3, and 5.2 ± 0.6 μM, respectively, against CoV‐229E 3CLpro, CVB3 3Cpro, EV71 3Cpro and RV14 3Cpro (Fig. 3A–D and summarized in Table 1). This compound contains a dihydropyrazole ring with three substituents, two phenyl groups and a lengthy N‐butyl‐benzimidazolylamino‐toluene.
Figure 2.
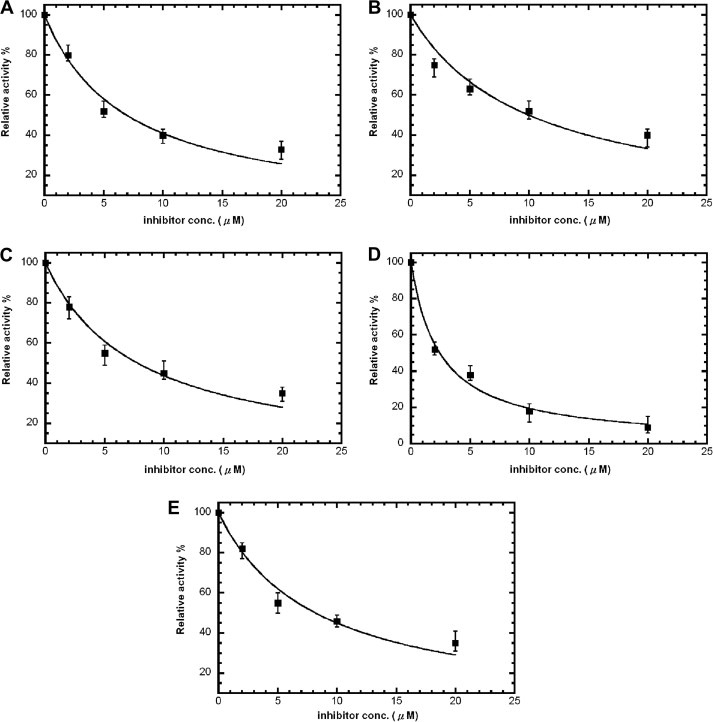
Dose–response curves for the five hits against SARS‐CoV 3CLpro from the screening. IC50 values were determined from the curves using equation 1. These were (A) 7.2 ± 0.7 μM (21155), (B) 10.6 ± 1.3 μM (22723), (C) 7.0 ± 0.8 μM (27548), (D) 3.3 ± 0.2 μM (48511), and (E) 8.1 ± 0.9 μM (43146). The structures and activities of these inhibitors are summarized in Table 1.
Table Table 1.
Summary of IC50 values (μM) of the five hits with SARS‐CoV 3CLpro, and other 3C(L) proteases
| Compound ID | Structure | SARS 3CL | 229E 3CL | CVB3 3C | EV71 3C | RV14 3C |
|---|---|---|---|---|---|---|
| 21155 |
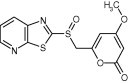
|
7.2 ± 0.7 | 5.6 ± 1.0 | >50 | >50 | >50 |
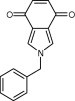
| 22723 |
|
10.6 ± 1.3 | 12.4 ± 0.8 | >50 | >50 | >50 |
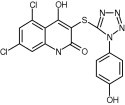
| 27548 |
|
7.0 ± 0.8 | 6.6 ± 0.3 | >50 | >50 | >50 |
| 48511 |
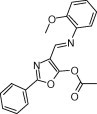
|
3.3 ± 0.2 | 1.8 ± 0.7 | >50 | >50 | >50 |
| 43146 |
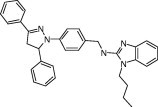
|
8.1 ± 0.9 | 10.3 ± 1.1 | 5.4 ± 0.2 | 3.3 ± 0.3 | 5.2 ± 0.6 |
Figure 3.
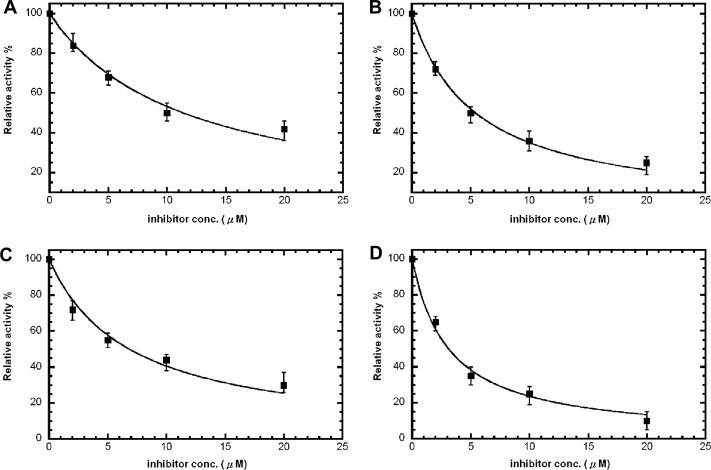
Dose–response curves for 43146 against 229E 3CLpro, CVB3 3Cpro, EV71 3Cpro and RV14 3Cpro. IC50 values were determined from the curves using equation 1. These were (A) 10.3 ± 1.1 μM (229E 3CLpro), (B) 5.4 ± 0.2 μM (CVB3 3Cpro), (C) 3.3 ± 0.3 μM (EV71 3Cpro), and (D) 5.2 ± 0.6 μM (RV14 3Cpro).
3.2. Inhibition potencies of the 43146 analogues
Since 43146 inhibited 3CLpro and 3Cpro, its analogues including 45240, 68638, 55688, and 55585 obtained from another compound library were evaluated. As shown in Table 2 , all of them showed good potencies against the five proteases. The most potent compound was 45240, and its IC50 values in inhibiting the 3C(L) proteases were measured to be 2.5 ± 0.2 μM (SARS‐CoV 3CLpro), 2.6 ± 0.4 μM (CoV‐229E 3CLpro), 1.2 ± 0.3 μM (CVB3 3Cpro), 0.5 ± 0.1 μM (EV71 3Cpro), and 1.7 ± 0.1 μM (RV14 3Cpro) (Table 2). This compound contains four rings, three phenyl groups and one imidazole, surrounding a central dihydropyrazole ring, without the lengthy side chain as seen in 43146. Compound 68638 with benzylcyclohexane ring fused with the dihydropyrazole ring and acetyl and iodobenzyl groups attached to the central ring showed less inhibition against both 3Cpro and 3CLpro (Table 2). The other two compounds, 55688 and 55585, with shorter side chains attached to the benzimidazolyl group showed similar inhibitory activities as compared to 43146 (Table 2).
Table Table 2.
IC50 values (μM) of compound 43146 analogs with SARS‐CoV 3CLpro, and other 3C(L) proteases
| Compound ID | Structure | SARS 3CL | 229E 3CL | CVB3 3C | EV71 3C | RV14 3C |
|---|---|---|---|---|---|---|
| 45240 |
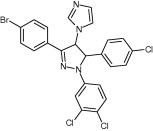
|
2.5 ± 0.2 | 2.6 ± 0.4 | 1.2 ± 0.3 | 0.5 ± 0.1 | 1.7 ± 0.1 |
| 68638 |
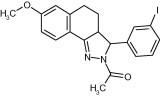
|
9.8 ± 0.8 | 12.4 ± 0.8 | 7.0 ± 0.8 | 10.6 ± 1.3 | 5.3 ± 1.1 |
| 55688 |
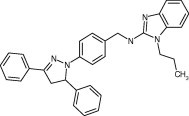
|
8.0 ± 0.5 | 9.6 ± 0.3 | 6.1 ± 0.5 | 8.5 ± 0.6 | 7.7 ± 1.0 |
| 55585 |
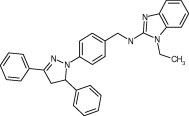
|
8.4 ± 0.2 | 10.2 ± 0.7 | 6.5 ± 0.6 | 4.7 ± 0.2 | 6.4 ± 0.3 |
3.3. Computer modeling of 21155, 22723, 27548, and 48511 binding to the proteases
These inhibitors are competitive inhibitors with respect to the substrate (data not shown), indicating they bind in the active site. To rationalize the binding discrepancy of these inhibitors against these proteases, their binding modes with SARS‐CoV 3CLpro and four other proteases were modeled and some of them are shown in Fig. 4 . The first four inhibitors of SARS‐CoV 3CLpro are more rigid because the thiazolopyridine in 21155, the dichlorobenzoquinolinone in 22723, the isoindoledione in 27548, and the oxazole in 48511 adopt planar structures and the three substituents of the oxazole ring in 48511 are fixed in a conformer, due to the 1,2‐steric interaction between the acetate group and the N‐aryl imino group as well as the biaryl interaction between the phenyl and oxazole to prohibit their free rotation. All these compounds can be considered as two rigid aromatic moieties connected by a small linker. Based on the computer modeling, each of these aromatic moieties is bound to S1 or S2 site of SARS protease by forming H‐bonds and hydrophobic interactions (Fig. 4A–D). As shown in the computer modeling, Glu166 side chain of SARS 3CLpro forms H‐bonds with these four inhibitors. However, the corresponding amino acid residue in 3Cpro is Gly164, which lacks the side chain to form H‐bond with any of these compounds (also see Fig. 4F), leading to loss of inhibition. In addition, the 3Cpro have more open but shallow S2 site (due to its partial blockage by Leu127) than 3CLpro according to the crystal structures of RV 3Cpro [20] and CVB3 3Cpro (Lee et al., unpublished results) compared to 3CLpro (also see Fig. 4F). Thus, 3Cpro can not hold these compounds tightly.
Figure 4.
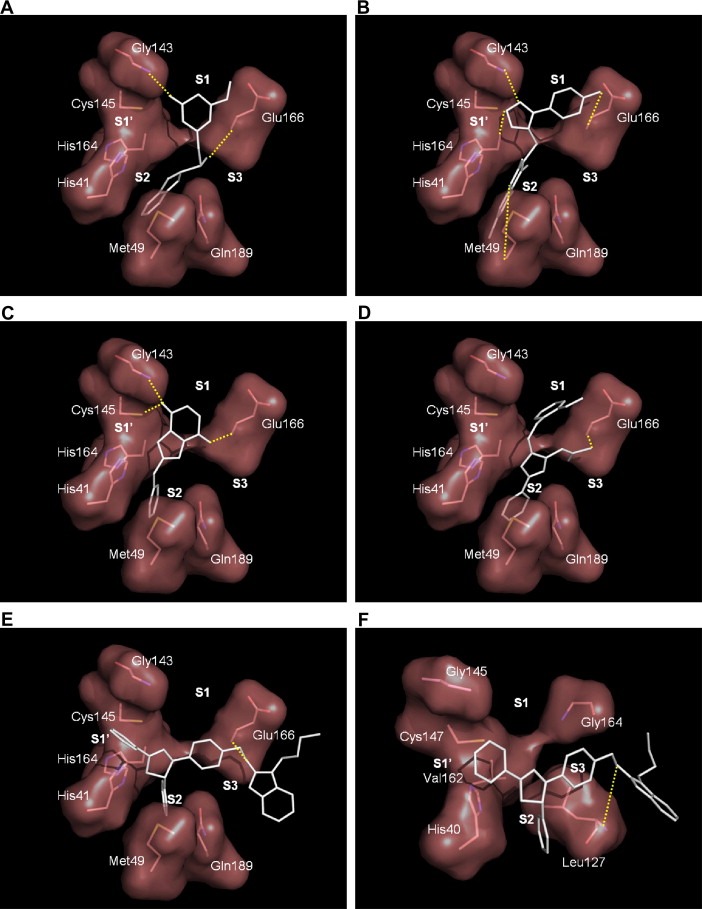
Computer modeling of the binding modes of the inhibitors in the active site of the SARS 3CLpro. The more rigid moieties of the inhibitors 21155, 22723, 27548, 48511 are probably bound to S1 and S2 sites as shown in (A), (B), (C), and (D), respectively. (E) 43146 binds SARS‐CoV 3CLpro differently with the biphenyl 4,5‐dihydro‐1H‐pyrazole moiety anchored at the S1’ and S2 sites and the rest of the molecule at the S3 and the following sites. The hydrogen bond interactions were represented by yellow dotted lines.
3.4. Binding modes of 43146 and its analogues to the proteases
In contrast, the compound 43146 is more flexible, because the dihydropyrazole is not planar, and the phenyl group is linked to the sp3‐hybridized carbon of the dihydropyrazole ring, so it is free for rotation. Different from the binding modes of the other 4 inhibitors, the diphenyl 4,5‐dihydro‐1H‐pyrazole moiety of 43146 fits well at the S1′ and S2 sites in the SARS 3CLpro (Fig. 4E) with the rest of the molecule at the S3 site and beyond. With this binding mode, the compound was predicted to also bind well in the 3Cpro, consistent with the inhibition data. In fact, RV 3Cpro prefers a phenyl group at the S2 site as evidenced by its strong inhibition by AG7088 which has a P2‐fluorophenylalanine. Thus, it could be rationalized by computer modeling that only 43146 among the five hits can inhibit the three 3Cpro in addition to the 3CLpro.
The analogues of 43146, including 45240, 68638, 55688, and 55585, bind in the 3Cpro and 3CLpro active sites with similar modes to that of 43146 (data not shown). Compared to 43146, 55688 and 55585 only have minor structural difference with shorter alkyl groups attached to the benzimidazole ring, so that they showed similar inhibition against the proteases. The fused ring system and the phenyl group in 68638 may also span from S1′ to S2 sites in both kinds of proteases, yielding similar inhibition. However, 45240 showed a significantly better inhibition against the 3Cpro than 43146. Apparently, the lengthy side chain attached to the phenyl group in the compound did not provide additional interaction with the protease, consistent with the binding mode shown in Fig. 4E. However, the additional interaction is provided by the pyridine ring bound near the more open S1′ site in 3Cpro.
4. Discussion
AG7088 is the best inhibitor identified so far for 3Cpro, which not only inhibits the 3Cpro from RV, but also those from CV and EV [16]. However, it did not inhibit 3CLpro from SARS‐CoV [17]. This may be partially due to the blockage of its P1‐lactam ring by the relatively larger Glu166 side chain and also the S2 site of 3CLpro is narrower although it is deeper. Therefore, when the P2‐phenylalanine is changed to non‐planar leucine or cyclohexane without changing the P1‐lactam, they became good inhibitors of SARS 3CLpro [21]. Unlike AG7088, which is a ketomethyl isostere of a tripeptide‐conjugated ester, compound 43146 is not peptide‐like. From the random screening as shown in the study, we have found a starting point toward the development of non‐peptide multiple‐function inhibitors against CoV and PV. With further modification of these individual and common inhibitors of the viral proteases, we hope to find solution for the possible reoccurrence of SARS and other diseases caused by the viruses with the 3Cpro and 3CLpro.
Acknowledgements
This work was supported by a grant from Academia Sinica to PHL, and we thank Korea Chemical Bank for providing their chemical library with which this work was conducted.
Kuo Chih-Jung,Liu Hun-Ge,Lo Yueh-Kuei,Seong Churl-Min,Lee Kee-In,Jung Young-Sik and Liang Po-Huang(2009), Individual and common inhibitors of coronavirus and picornavirus main proteases, FEBS Letters, 583, doi: 10.1016/j.febslet.2008.12.059
Contributor Information
Young-Sik Jung, Email: ysjung@krict.re.kr.
Po-Huang Liang, Email: phliang@gate.sinica.edu.tw.
References
- 1. Melnick, J.L. (1996) Enterovirus: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In Virology, 3rd ed., Lippincott-Raven, Philadephia, PA.
- 2. Blomberg K., New enterovirus type associated with epidemic of aseptic meningitis and-or hand, foot, and mouse disease. Lancet, 2, (1974), 112– [DOI] [PubMed] [Google Scholar]
- 3. Sperber S.J., Hayden F.G., Chemotherapy of rhinovirus colds. Antimicrob. Agents Chemother., 32, (1998), 409– 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lum L.C., Neurogenic pulmonary oedema and enterovirus 71 encephalomyelitis. Lancet, 352, (1998), 1391– [DOI] [PubMed] [Google Scholar]
- 5. Ho M., An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. New Engl. J. Med., 341, (1999), 929– 935. [DOI] [PubMed] [Google Scholar]
- 6. Lee C.K., Characterization of an infectious cDNA copy of the genome of a naturally occurring, avirulent coxsackievirus B3 clinical isolate. J. Gen. Virol., 86, (2005), 197– 210. [DOI] [PubMed] [Google Scholar]
- 7. Krausslich H.G., Wimmer E., Viral proteinases. Annu. Rev. Biochem., 57, (1998), 701– 754. [DOI] [PubMed] [Google Scholar]
- 8. Ksiazek T.G., A novel coronavirus associated with severe acute respiratory syndrome. New Eng. J. Med., 348, (2003), 1953– 1966. [DOI] [PubMed] [Google Scholar]
- 9. Patick A.K., Rhinovirus chemotherapy. Antiviral Res., 71, (2006), 391– 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee E.S., Development of potent inhibitors of the coxsackievirus 3C protease. Biochem. Biophys. Res. Commun., 358, (2007), 7– 11. [DOI] [PubMed] [Google Scholar]
- 11. Maugeri C., New anti-viral drugs for the treatment of the common cold. Bioorg. Med. Chem., 16, (2008), 3091– 3107. [DOI] [PubMed] [Google Scholar]
- 12. Kuo C.J., Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem., 16, (2008), 7388– 7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang P.H., Characterization and inhibition of SARS-coronavirus main protease. Curr. Top Med. Chem., 6, (2006), 361– 376. [DOI] [PubMed] [Google Scholar]
- 14. De Clercq E., Potential antivirals and antiviral strategies against SARS coronavirus infections. Expert Rev. Anti Infect. Ther., 4, (2006), 291– 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhai S., Liu W., Yan B., Recent patents on treatment of severe acute respiratory syndrome (SARS). Recent Patents Anti-Infect Drug Disc., 2, (2007), 1– 10. [DOI] [PubMed] [Google Scholar]
- 16. Binford S.L., Conservation of amino acids in rhinovirus 3C protease correlates with broad-spectrum activity of rupintrivir, a novel rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother., 49, (2005), 619– 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shie J.J., Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α,β-unsaturated esters. Bioorg. Med. Chem., 13, (2005), 5240– 5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuo C.J., Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biophys. Biochem. Res. Commun., 318, (2004), 862– 867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang H., The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA, 100, (2003), 13190– 13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matthews D.A., Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA, 96, (1999), 11000– 11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang S., Synthesis, crystal structure, structure–activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem., 49, (2007), 4971– 4980. [DOI] [PubMed] [Google Scholar]