

Abstract
This review presents a brief outline and novel developments of electrophoretic separation in microfluidic chips. Distinct characteristics of microchip electrophoresis (MCE) are discussed first, in which sample injection plug, joule heat, channel turn, surface adsorption and modification are introduced, and some successful strategies and recognized conclusions are also included. Important achievements of microfluidic electrophoresis separation in small molecules, DNA and protein are then summarized. This review is aimed at researchers, who are interested in MCE and want to adopt MCE as a functional unit in their integrated microsystems.
Keywords: Microchip electrophoresis, Injection, Turn, Joule heating, Surface modification, Concentration, DNA, Protein, Amino acids
1. Introduction
Microfluidics refers to the manipulation of fluids in a microchannel with a dimension of tens to hundreds micrometers and related technology and science [1]. Capillary electrophoresis (CE) has also been performed in the capillary format with similar dimensions all along. Therefore, microchip-based capillary electrophoresis (MCE) was the earliest format of microfluidics when it was first introduced by Harrison et al. [2].
Electrophoresis itself can be performed much more efficiently on microfluidic devices because the heat dissipation is much better in chip format than that in a capillary format of the same material, and the injection plug is significantly shorter via more flexible control. Thus, higher electric field can be applied across a much shorter separation channel, so MCE can be performed much faster. When integrated with serial sample preparation steps on a chip, MCE will have much low sample and reagent consumption.
Until now, electrophoresis has remained the primary source of separation in microfluidic chip. Most of the papers published on separation in microfluidics are electrophoresis-based, rather than chromatography. Electrophoresis can be performed more directly and conveniently in a microfluidic chip. First, the driving force of electrophoresis is the electric field, which can be applied via the contact of electrodes with the running buffers, and without high pressure needed in chromatography. Second, in some electrophoresis modes, such as capillary zone electrophoresis (CZE), micelle electrokinetic chromatography (MEKC) and isoelectric focusing (IEF), the separation efficiency is mostly determined by the amount of the voltage applied. Therefore, in the microchip format, electrophoresis can still be performed with high efficiency under a higher electric field in a much shorter channel, while the efficiency of chromatography increases with column length. Third, buffers only or buffers with specially designed additives are enough for electrophoretic separation instead of the stationary phase of beads or monolithic material in chromatography. Now there are various additives in running buffers to meet different requirements in various separation modes, such as, water soluble polymers in capillary gel electrophoresis (CGE), ampholytes in IEF, and surfactants in MEKC. Generally, these all simplify the operation of MCE, lower the cost of each chip, and contribute to its miniaturization and portability. While chromatography is generally not performed because (1) glass and plastics cannot withstand the pressures that would be needed, (2) it is difficult to pack the channels without voids, (3) there are mass transfer problems in the corners of the channels, and (4) voids cause unacceptable band broadening.
Photolithography and related MEMS technologies used for chip fabrication provide many novel elements to MCE. More complex fluid networks can be implemented, which would be nearly impossible in capillary formats. Therefore, samples can be collected, pretreated, introduced, injected and separated continuously under the complete control in a microchip. Buffers can be added and mixed simultaneously or one by one at any scheduled time and location. Other techniques developed in microfluidic chip also benefit electrophoresis separation at nearly every step. Microheaters and sensors are useful for sample reaction and separation. Pneumatic valves allow the flexible segregation and collection of analytes interested, and pumps can drive and mix samples through different regions in microchip [3], [4]. Fluorescence detection with optofluidic techniques and the wave guiding fiber [5], [6], chip-based electrochemical detection [7], and the miniaturized chip–MS interface [8] can facilitate chip integration.
The basic physical nature of electrophoresis itself is still the same in both capillary and microfluidic formats, which has been well studied in all modes and most applications. With these ideas in mind, some distinct characteristics of MCE, especially as one key unit in integrated microdevices, are discussed, and its typical applications in small molecules, DNA and protein are presented here.
2. Basic considerations on microchip electrophoresis
Through a complete cycle of electrophoresis, the sample is first introduced and injected, then separated into individual analyte zones by the electric field, which are then pulled through a detection point, and detected one by one. Analyte will diffuse spontaneously under the concentration gradient along the axial direction during separation, and this is accelerated by joule heat. Sometimes, the analyte zone will be twisted and stretched by turns in the folded channel, and tailed by surface adsorption. At least these six factors, sample injection , chip detection , molecular diffusion joule heating , turn geometry and surface adsorption , contribute to total peak broadening (σ 2) in MCE.
Fortunately, these all have occurred in CE. Therefore, the theory and experience of CE can be used as a reference for MCE in most cases. Typically, the contributions from variances of sample injection, chip detection and molecular diffusion to plate height are still calculated as those in CE.
where l inj and l det are the lengths of initial sample plug and chip detection window, D is the molecular diffusion coefficient, and t is the separation time.
However, some adjustments must be made in the transfer from capillary to microchip. New injection strategies are adopted for integrated microdevices, since complex fluid network allows much easier control of the sample plug profile. Various chip materials and channel structures used by MCE require different surface coating methods and heat dissipation controls. Turns of small curvature radii within the microchip can generate more significant peak broadening than that in a bended capillary. Some related theories and control methods for these special factors in MCE relative to conventional CE are discussed in detail below.
2.1. Sample injection
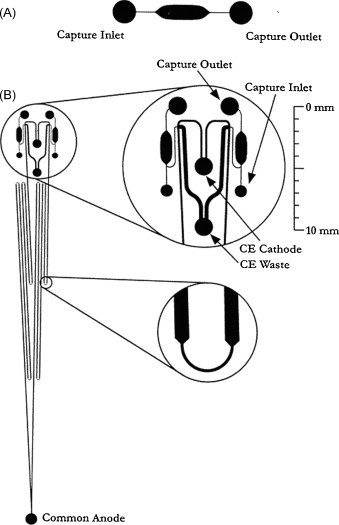
Sample injection is the first step of MCE. Electrokinetic injection is the most preferred choice [9], [10], which is completed in two steps, loading and dispensing. In a typical cross-type microchip, the sample flow is first pulled into injection channel by an electric field, then the sample plug in the cross zone is brought to separation channel when voltages are switched, and separation starts immediately (Fig. 1 ).
Fig. 1.
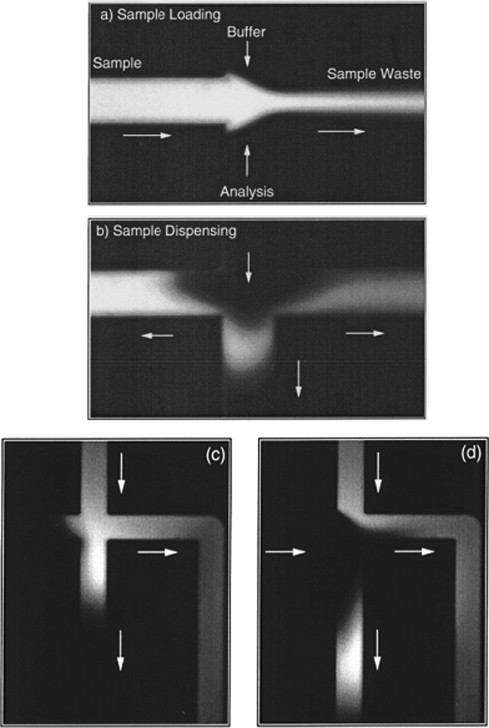
Pinched (top) and gated (down) electrokinetic injection for MCE. From Refs. [10], [14] with permission.
Two impediments in electrokinetic injection have to be dealt with carefully. One is the sample plug dispersion into separation channel during sample loading, which increases the sample plug length; the other is continuous sample leakage from loading channel into separation channel during sample dispensing and the subsequent separation, which results into a tailing peak and gradually elevated background noise. Both will impair separation efficiency significantly.
Ramsey and co-workers clearly introduced electric pinched injection and inhibition leakage strategy to deal with these problems earlier [9]. The electric fields from separation channel to sample waste channel effectuate a virtual valve, which allows the volume of the sample plug to be accurately controlled and is time independent, enabling a constant volume to be injected. The inhibited electric valve can also completely avoid electric leakage during electrophoresis. Electric field distribution on microchip valve and concentration focusing from sample stacking were also studied. In order to increase the sample injection frequency, two tee intersections were added in close proximity of cross point. The tee intersection in the sample channel preserved the sample near to cross point under “pullback” conditions, therefore, allowing faster loading next time. Tee intersection in separation channel allowed unidirectional transport enabling loading of subsequent injections during an analysis. Injection at 5 Hz was demonstrated successfully [11]. In order to finely tune the sample plug in pinched injection, double-cross injector was fabricated, which employed electrokinetic focusing on deliver sample plugs of variable volume [12]. Electrokinetic double-focusing injection technique for microchip electrophoresis was also presented [13].
Gated injection is another form of electrokinetic injection [10]. Instead of “pull” and “push” cycles in pinched injection mode, steady sample and analysis streams are kept all along except instant electric injection.
Both gated and pinched injection methods suffered from electrokinetic injection bias [14], [15], [16]. In pinched format, a larger volume is loaded and dispensed for neutral species than anionic species, and up to 27% difference in injected volumes was observed. The overall sample bias in gated injection was shown to be time-dependent and resulted in a larger sample bias against components of negative electrophoretic mobility [15].
Other novel injection strategies were also presented to alleviate sample leakage and simplify voltage control for sample injection. Zhang and Manz presented cross and tee injectors having narrow sampling channels to accomplish “simple injection” with no pinched or inhibited electric field needed [17]. With tee design, the number of reservoirs in multichannel systems can be reduced to the theoretical limit. Unfortunately, it is hard to produce very thin sampling channels in the glass and silica microchip. Recent, Fu and co-workers found that the injector with 30° included angle also reduced sample leakage successfully [18]. It has great potential in high-throughput microdevice for its simplicity and practicability. Pressure instead of electrokinetic dispensing also could reduce sample leakage effectively [19]. Baba and co-workers demonstrated it in the high-speed microchip CGE of protein and DNA [20], [21].
Pressure-based injection has got attention for its simplicity. Hydrostatic pressure injection method was introduced in Lin's group [22], [23]. This method was first proved effective in leading cells to a separation channel for single-cell analysis. More recently, an improved double-cross hydrostatic pressure sample injection for MCE was reported. It allowed adjustment of the sample volume, could reduce the number of electrodes, and had been used in multichannel systems for fluorescent dye free electrophoresis and chiral selector screening. Negative pressure pinched sample injection was developed by Fang and co-workers, and adopted in a multichannel system [24].
Pneumatic pump and valve were also useful for sample introduction and injection. Early in 2003, Zare and co-workers designed a microfluidic flow injection analysis system [25], which was a two-layer polydimethylsiloxane (PDMS) monolith multiple pneumatic injection system that mimicked the operation of a standard sixport, two-way valve used in liquid chromatography. The hydrolysis of fluorescein diphosphate by alkaline phosphatase was monitored in time [26]. In 2005, utilizing an integrated diaphragm pump on a hybrid PDMS-glass microchip, Landers and co-workers developed a pressure injection for electrophoretic analysis of submicroliter samples [27], in which, as little as 500 nL sample could be injected correctly. This injection system had been integrated into a microchip associated with chip-DNA extraction and chip-PCR for rapid detection of gene of bacteria, virus and human beings [28]. Martin and co-workers incorporated a reduced-volume pneumatic valve that actuated to allow analyte from a continuously flowing sampling channel to be injected into a separation channel for electrophoresis, and a pushback channel was added to flush away stagnant sample associated with the injector dead volume [29]. After evacuating air dissolved in PDMS, loading of a sieving polymer solution and injection of a sample solution could be carried out autonomously [30], [31].
Sample introduction and injection is one important section to “world-to-chip” interface [32]. Smarter methods are still needed in highly integrated microsystems.
2.2. Joule heating
Separation efficiencies in some elelctrophoresis modes are mainly dependent on the voltages applied. If a similar resolution is needed in MCE as that in CE, a higher electric field will be applied in the shorter channel. However, the higher electric field (E) also induces a higher electric current within the separation microchannel, which produces joule heating, and its power (Q) is calculated as Q = ρE 2, where ρ is the buffer's electric conductivity.
There are at least three aspects that should be considered about the effect of joule heating on the performance of MCE, first, an overall temperature increase of the buffer solution, ΔT; second, the establishment of a radial temperature gradient within microchannel, ΔT x; and third, the inhomogeneous temperature distribution along the axial direction.
Microfluidic chip can be simplified as a thick wall capillary without polyimide coating outside [33], and then the overall temperature in the center of the microchannel (T) can be deduced as
where T 0 is the environment temperature, r 1 is the radius of microchannel lumen, r 2 is the radius of chip wall, k is the thermal conductivity of chip wall, and h is the heat transfer coefficient to the surroundings (power radiated per unit area per unit temperature difference between the outer wall and the environment). For simplicity, the heat generation rate is considered as a constant. In fact it is a function of temperature, and the temperature distribution within the buffer itself also has a parabolic profile.
Under the same electric field, running buffer and microchannel structure, T is only dependent on k and h of the chip material, so in glass, silica chip of higher k and h, electrophoresis only leads to little increased temperature, while for plastics, there is much higher temperature increase for their lower k and h. Kutter and co-workers presented a serial data for microfluidic chip of different materials in different dimensions and filled with buffers of different electric conductivities [34], in which the top limits of electric field (before boiling of the buffer)were presented clearly. They concluded that about 3 times electric field can be applied in glass and silica microchip than that in a silica capillary of the same dimension, and leads to the same temperature increase in buffers. As to plastic and PDMS microchip, a much low electric field could be subjected [35].
As to the temperature gradient in buffer along radial direction (ΔT x) [36], [37], which can be described as
where x is the radius distance from the center of microchannel, k b is the thermal conductivity of the buffer. The temperature drop from the channel center (x = 0) to liquid/solid interface (x = r 1) is determined as , so the temperature gradient across channel radius is very similar in microchips of various materials. Properties of the chip itself, such as the external dimensions, thermal conductivity and heat transfer to the surroundings have no direct effect on the temperature profile in the channel lumen. It has been widely assumed that a radial temperature gradient lower than 1.5 K would not degrade the efficiency obviously. It can be deduced that when joule heat sets a limit to the efficiency, one can lower the electric conductivity of the buffer, reduce the channel radius, and extend separation channels to decrease the electric field and the corresponding Q.
Another important phenomenon from joule heating is the temperature jump along the axial direction [38]. Due to the thermal end effect from buffer and sample reservoirs in two ends, there are a serial of things that happened, first, sharp temperature drops appear, second, the local electric field rises to keep the current continuously, third, pressure gradients have to be induced to hold mass continuity, which results into curved fluid velocity profile and increased molecular diffusion, and both contribute to the increased dispersion of samples. In microfluidic formats, reservoirs also exist and complex fluid network cross to separation channel also bring to inhomogeneous temperature profile along axial direction. Yang and co-workers discussed about joule heating effect on electroosmotic flow (EOF) and electrophoretic transportation electrokinetic flow focusing, and found that it caused the diffusion coefficient of the sample to increase, the potential distribution to change, and the flow velocity field to adopt a nonuniform profile [39].
In fact, most microfluidic channels are rectangular, and the steady-state heat transfer problem is governed by a two-dimensional equation.
where x and y are the height and width of microchannel. The effect of joule heating on electroosmotic flow and electrophoretic transportation in the rectangular channel is also reported [33], [40]. Then, with computational fluid dynamics (CFD), the convection–diffusion equation can be formulated and solved in terms of spatial moments of the analyte concentration. When the microchip is of the same material, the square shape allows the most efficient heat dissipation and minimized joule heating effects.
2.3. Turn optimization
The highest electric field strength is ultimately limited for all chip materials due to their inherent heat dissipation coefficient. Furthermore, some electrophoresis modes cannot be performed under a very high electric field level, such as DNA and protein sizing within polymer solution. They will migrate with biased reptation at much high electric fields, so the dependence of molecule mobility on fragment length (molecular weight) becomes marginalized. To achieve adequate resolution and separation efficiency, folded channel is necessary sometimes for a longer separation distance, and turn geometry is unavoidable in these cases.
However, turn will introduce additional geometrical contributions to analyte dispersion via lateral variations in both migration distance and field strength. If sample streams on both the inside of the turn and the outside of the turn are assumed traveling at the same velocity, and only geometry variation is considered, length variance of (Δl) can be calculated as [9], where θ is the turn arc, is the width of separation channel. If the turn induced electric field difference is also included, and as is large, so E c ≈ E av is assumed, then Δl can be calculated as [41], where r c is the radius of curvature along the turn center, E c is electric flied along the turn center, and E av is the average electric field within a turn. Turn transit time (t t) can be calculated as t t = θr c/ν c, where ν c is the velocity along turn center.
If the molecule transverse diffusion time across the channel is also included in turn process, there are two cases that can be classified briefly according to the ratio of t D/t t. First, t D/t t ≤ 1, which corresponds to turns of relatively large radius of curvature or large diffusion coefficients. In this case, cross-stream is dominant, so that the concentration is almost constant across the radius. Molecular diffusion dominates turn variance, , which means that peak dispersion purely from turn geometry is nearly negligible compared to other peak variances, especially molecular diffusion dispersion. Second, t D/t t ≥ 1, which corresponds to small radius of curvature and low molecular diffusion coefficients. In this case, molecules in an analyte band follow single axial streamlines around a turn, and geometrical dispersion dominates turn variance, , this means that peak dispersion purely from turn geometry is significant.
For MCE, the latter has happened in most cases. For shorter separation channel and time saving, chip CZE, chip CEC and chip MEKC are commonly performed at the super-high speed, molecular diffusion coefficients of small molecules are large, but t t is also very small, so t D/t t is usually larger than 1. For example, t D/t t is 3.2, if , r c =250 μm θ = π, ν av = 1 mm/s, D = 5 × 10−10 m2/s. As to chip CGE of protein and DNA in the moderate electric field, D is very small, so its t D/t t is also larger than 1. For example, t D/t t is 16, if , θ = π, ν av = 0.1 mm/s, D = 10−11 m2/s. Larger is t D/t t, more significant is the effect of pure turn geometry on peak dispersion.
Decreasing channel width is the most effective method to control turn dispersion as . Mathies and co-workers created tapered turns by narrowing the separation channel width before the start of a turn and widening the channel after the turn was complete [42]. Examining the separation of DNA sizing ladder, they carefully explored the radius of curvature of the turn, the length over which the channel is tapered, and the degree of tapering. They found that the case of the smallest radius of curvature (250 μm), the shortest tapering length (55 μm), and the largest tapering ratio (4:1) generated the highest separation resolution, which was >95% of that from a straight separation channel. They thought that smallest radius of curvature and shortest tapering length could decrease negative effect of DNA biased reptation in taped channel to an upmost extent, and the largest tapering ratio could control the pure turn geometry on peak dispersion. Taped turn and another similar turn geometry of constrained turn [43] have become two popular schemes in the folded channel for MCE (Fig. 2 ).
Fig. 2.
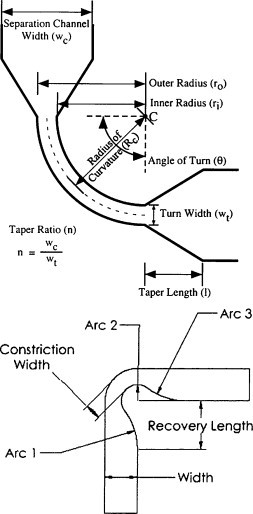
Tapered turn (top) and constrained turn (down) in folded microchannel. From Refs. [42], [43] with permission.
There are also some alternative methods to deal with turn dispersion. Griffiths and Nilson have proposed a comprehensive formula for the turn induced axial variance, where convective mass transfer was also included [44]. Increasing the effective radius of curvature was presented by separation channel of spiral geometry; in order to compensate both the higher electric field and the ‘race track’ effect, optimal designs of channel shapes were also presented by waved turn and polygonal channel; EOF tailoring of side channel of turn was also introduced with laser ablation [45] and microelectrodes [46].
2.4. Surface adsorption and surface modification
Surface property is one of key factors to the success of MCE. Chip surface is getting much more complex than conventional capillary as various materials have been adopted in chip fabrication. Except glass and silica, qualities of other plastics and rubber are unfamiliar to most analysts, so many experiences and skills from CE are out of effect, sometimes, even lead to mistakes. As surface coating is dependent on its substrate properties, surface modification for MCE is discussed on three common used chip materials, glass/silica, plastics and PDMS, individually. Surface modification is usually a critical step for reaction and immunoassay in microfluidic chip, but their related methods are not covered in this paper.
2.4.1. Surface modification of glass/silica microchip
In CE, surface properties and coating methods have been studied in particular. They also contributed a lot to the successes of genome sequencing projects with capillary array electrophoresis. Most of these well-developed experiences have also been directly used on glass and silica microchip. Self-assembly of silane is a common approach of surface modification of glass and silica. Vinyl silane is usually used for the subsequent radical initiated polymerization, in which poly(acrylamide) (PAAm) has been extensively studied, which has excellent compatibility with DNA, protein and other biomolecules, and becomes a “gold standard” for surface coating in electrophoresis since it was first introduced by Hjerten and Zhu in 1985 [47].
Recently, some improvements of PAAm coating have been reported. Zare and co-workers presented photo induced polymerization of PAAm coating within glass microchip [48]. After surface treatment with vinyl silane, the microchannel was filled with the aqueous solution of 5% acrylamide, 0.2% photoinitiator and 250 ppm hydroquinone, which was illuminated for 15 s with a 40 μJ/μm2 diode laser. This method could permit surface pattern within microchannel rapidly. Gao and Liu demonstrated enhanced stability of PAAm coating with bis-acrylamide added [49]. It was thought that dimer could cross-link PAAm molecules nearby, which would cover some surface defects and blank zones, where there was no silane or PAAm coupled. With this coating, chip CZE of basic proteins and IEF of protein standards could be performed for several hundred times without any obvious decrease of separation efficiencies.
Poly(ethylene glycol) (PEG) silane could be used as surface coating reagent directly, and longer chain PEG could lead to lower surface electrosmotic flow [50]. Heat-immobilized poly(vinylalcohol) (PVA), a typical adsorbed coating in CE, was also adopted in glass microchip by Belder et al. [51]. Multilayer assembly coating of positive charged polyelectrolyte and gold nanobeads could improve the electrophoresis separation and electrochemical detection of mixtures of o-, m- and p-aminophenols [52]. A method to rejuvenate surface coating in glass chip was reported. It was found that surface activation by hot H2SO4 and 1 M HCl was necessary before next poly(dimethylacrylamide) (PDMA) coating, which allowed high-resolution separation of single-stranded DNA (ssDNA) standards [53].
2.4.2. Surface modification of plastic microchip
Various thermoplastics, such as poly(methyl methacrylate) (PMMA), poly(carbonate) (PC), polyethylene terephthalate (PET), etc. are commonly used for chip fabrication via mold injection in large amounts. Some special characteristics of plastic chips must be noted before surface modification for electrophoretic separation. First, compared with glass and silica, plastics usually show much poorer resistance to higher temperature and low-polar organic solvents. Second, the properties of one kind plastic often vary with producers and batches, as there are usually some additives in plastic resins, such as antioxidant, fire resistance, and toughness reinforce. Third, polymer molecular weight and distribution often differ between brands and batches. Last, fabrication parameters such as temperature and pressure between mold injections also changed sometimes. Therefore, surface smoothness, hydrophilicity and UV–vis absorption of microchannel are not consistent all the time, and surface modification is usually unavoidable. Now, additives in bulk resins, surface adsorbed coating and surface covalent coating are three primary approaches used for plastic microchip surface modification.
Additives in bulk resins are the simplest, as no additional surface treatment or reaction is needed after chip fabrication. Lin's lab added 1–20% acrylonitrile–butadiene–styrene (ABS) in PMMA resins for chip molding, and found that with the increase of ABS amount, chip surface hydrophilicity was improved, but light permeability was decreased and the fluorescent background was increased slightly [54]. Lee and co-workers fabricated plastic chip via UV induced copolymerization of MMA, PEG monomer and dimer, which allowed the high-efficient electrophoresis separation of peptides and proteins [55].
Adsorbed coating is a convenient and flexible method to control surface quality. Baba and co-workers reported hybrid dynamic coating using n-dodecyl β-d-maltoside (DDM) and methylcellulose (MC) for suppression of EOF and surface adsorption of labeled sugars in PMMA microchip [56]. This coating allowed high-speed and high-throughput profiling of the N-linked glycans In order to get the high-resolution separation of ssDNA ladders within PMMA microchip, different static adsorbed coatings were studied, respectively [57]. Finally, the microchips irradiated with UV for 10 min and coated with PVA as well as the microchips treated with HNO3 and coated with hydroxypropylmethylcellulose (HPMC) were found to have the best performance. The copolymer of butyl methacrylate (BMA) and PEG monomer could introduce a brush-like PEG coating on the PMMA surfaces via the anchoring effect of the hydrophobic BMA units, which allowed bovine serum albumin (BSA) and lysozyme separated with modest efficiencies [58].
Covalent coating is a complex but robust method to tailor surface qualities. Oxygen plasma could activate PMMA surface and introduce some hydroxyls for coupling 2-bromoisobutyryl bromide, which induced atom transfer radical polymerization (ATRP), and PEG was grafted [59]. On this chip, peptides and proteins were separated with very high efficiencies (Fig. 3 ). Soper and co-workers reported the chemical modification of PMMA surfaces by the reaction with the monoanion of diaminoalkanes to yield amine-terminated PMMA surfaces, and it was found that the amine was quite uniform and generated a reversed EOF within pH 3–10 [60].
Fig. 3.
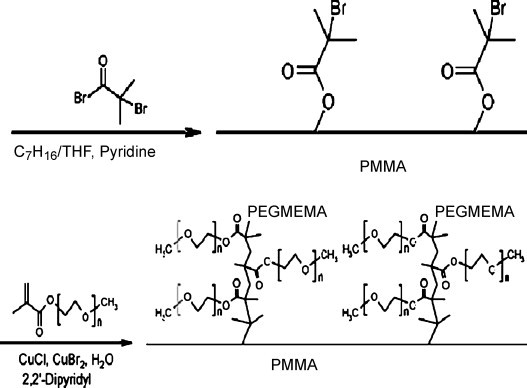
Immobilization of ATRP initiator and grafting of PEG on the PMMA surface. From Ref. [59] with permission.
2.4.3. Surface modification of polydimethylsiloxane microchip
PDMS has become one of the most widely used materials in microfluidics. Rapid prototyping of PDMS microchip via soft photolithography was an important milestone for microchip fabrication [61], which allowed more researchers to testify their ideas quickly. Moreover, pneumatic pump and valve, and chip cell culture on PDMS microchip largely prompted the integration and application of microfluidic chip [3], [4]. Unfortunately, native PDMS can adsorb most biopolymers and even absorb small hydrophobic molecules. By far, many PDMS coating strategies have been proposed based on the distinct chemical and physical properties of PDMS itself.
Native PDMS is hydrophobic and negatively charged, so physically adsorbed coating, such as polyelectrolytes based on electrostatic interaction, neutral chemicals based on hydrophobic interaction, and both of them were also reported. Nearly all surfactants and hydrophobic polymers can be adsorbed on PDMS to control its EOF and protein adsorption, such as neutral surfactants n-dodecyl β-d-maltoside [62], tween-20 and brij-35, cationic surfactants Cetyl Trimethyl Ammonium Bromide (CTAB) and Didodecyl Dimethyl Ammonium Bromide (DDAB) [63], anionic surfactant [64], and hydrophobic copolymer poly(styrene-alt-maleic anhydride) [65]. With sodium dodecyl sulfate (SDS) dynamic coating, amino acid and proteins were separated successfully [66]. Positive charged polyelectrolyte aminodextran [67] also could be adsorbed on PDMS surface to generate a stable EOF and inhibit protein adsorption.
Methyl groups are the main chemical groups on the native PDMS surface, which can be utilized for surface grafting via hydrogen abstraction induced by photo, ions or plasma. Regnier and co-workers reported cerium catalyzed polymerization [68]. Combined with monolith micropillars, peptides and proteins were separated successfully. Allbritton and co-workers demonstrated UV induced surface grafting on PDMS (Fig. 4 ) [69], [70]. The photoinitiator could be preadsorbed on PDMS surface, and then induced polymerization only on PDMS surface. Peptides were separated successfully. Surface pattern also could be produced easily for spatially selective immobilization of cells and biomolecules.
Fig. 4.
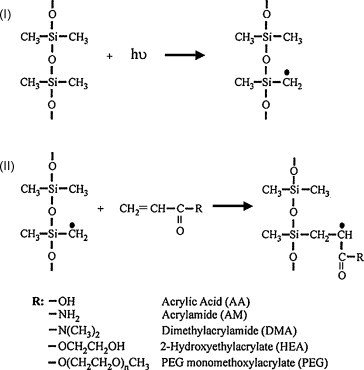
UV graft polymerization on PDMS surface. From Ref. [70] with permission.
Additives in PDMS prepolymer mixture, such as acrylic acid, could gradually migrate out to tailor EOF [71]. Moreover, PDMS is porous, so both small hydrophobic organic compounds immersion and solvent-assisted polymers interpenetration with PDMS surface molecules could tailor PDMS surface charge and hydrophilicity. For example, tetraethyl orthosilicate (TEOS) immersion followed with hydrolysis and cross-linking could generate ∼1 nm silica particles within PDMS [72], which induced an immense EOF, and reduced rodamine B permeability into PDMS bulk significantly.
Silanols and other active groups generated by UV irradiation, HCl–H2O2 immersion and plasma can be used for covalent monolayer self-assembly of silane compounds [73], [74], and hydrophilic polymer adsorption [75]. Recently, Lin and co-workers found that epoxy-modified hydrophilic polymers could be adsorbed on oxidized PDMS surface much more strongly than these polymers themselves, and hard baking of PDMS before surface coating could further improve the coating stability. PDMS chip with epoxy-modified polymer coating could be used for electrophoresis separation of DNA and protein with very high efficiencies [76], [77].
Surface modification techniques have not yet fully developed to face various challenges in different applications. Simple and high-efficient surface coatings are still desperately expected especially to protein electrophoresis in plastic and rubber microchip.
Some distinct issues about MCE over CE have been discussed, which include sample injection, turn in channel, joule heating and surface modification. Moreover, some recognized theories and well-developed methods have also been introduced. Related experimental details can be found in the original papers, and some comprehensive discussions on special subjects have also been presented in some excellent reviews.
3. Applications
In the earliest period of microfluidic chip, MCE was performed in most cases. Up to now, MCE has become one highly developed functional unit in microfluidic chip, and found a lot of application fields in the pure separation research and served as a key part in integrated microdevices. In the following, the applications of MCE in small molecules, DNA and proteins were discussed, individually. MCE of glycan [78], cell and cell contents [79], [80], [81], [82] are also hot points now, but these will not be included this time.
3.1. Small molecules
As well-defined sample plug of several to decades of micrometers long can be injected, super-fast electrophoresis can be achieved easily within microfluidic chip. Moreover, the separation behavior and mechanism of small molecules will not be destroyed under high electric field with chip CZE, CEC and MEKC in glass/silica microchip for their fast heat dissipation rate. Therefore, the high-speed separation of small molecules has been demonstrated very early, and is still one important subject in microfluidic chip.
3.1.1. Various microchip electrophoresis modes
In the early 1990s, Ramsey group studied the characteristics of a series of MCE modes for small molecules. First, in chip CZE, rhodamine B and fluorescein were separated with baseline resolution within 150 ms with a 1.5 kV/cm electric field and 0.9 mm separation length in 1994 [83]. The plate height decreased with increasing electric field, and finally a constant value was obtained, but no optimal electric field could be found. With a re-designed microchip and highly pinched injection plug, rhodamine B and dichlorofluorescein were separated within 0.8 ms with a 0.2 mm effective separation distance, when the electric field was increased to 53 kV/cm [84]. Here, an optimal electric field ∼30 kV/cm was confirmed.
CEC and MEKC are two common electrophoresis modes for MCE of neutral analytes. Earliest chip CEC was also presented in 1994 [85]. The channel surface modified with octadecylsilane was functioned as the stationary phase in open tubular CEC. Using an isocratic running buffer in non-optimized turn channel, a low efficiency was obtained. Later, when the gradient solvent and shallow channel were used, separation efficiency was improved sharply [86]. If the channel depth was only 2.9 μm, the separation efficiency could be increased all along with electric field within the experiment range, while an optimal electric field was occurred for the deeper channel. Chip MEKC was introduced in 1995 [87]. At first, the most optimal electric field was found as 400 V/cm, and about 3.0 μm plate height was obtained. After turn optimization and solvent programming, much better results were obtained, and the effects between methanol and acetonitrile on gradient elution were also discussed [88].
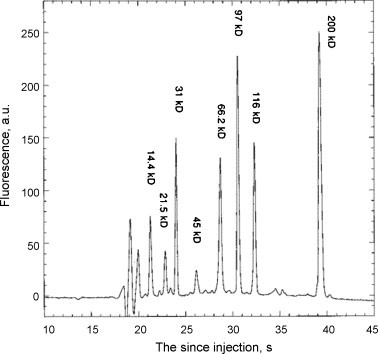
Chip MEKC has been widely used for amino acid separation. A spiral-shaped separation channel on a glass microchip was fabricated by Ramsey and co-workers for high-efficient separation of 19 amino acids [89], which was accomplished within 165 s, using sodium tetraborate–SDS–methanol/isopropanol buffer with 770 V/cm electric flied and an 11.87 cm separation distance (Fig. 5 ). Similar results were also obtained from PDMS and PDMS/glass microchip after surface modification [66]. In 1999, Mathies group reported a chip amino acid chirality analyzer for the extraterrestrial exploration [90]. After extraction from the meteorite and labeling with FITC out of chip, enantiomeric ratios of amino acids were determined within the folded electrophoresis channel of 19.0 cm long, using a SDS-γ-CD-carbonate buffer. The effects of temperature and γ-CD concentration were carefully characterized. In 2005, Mars organic analyzer (MOA) for amino acid biomarker detection and analysis was introduced [91], which integrated high voltage CE power supplies, pneumatic controls, and fluorescence detection optics for field operation. Amino acids extracted from various soils and minerals were analyzed successfully after sample collection, preparation, and derivation with fluorescamine out of chip. The results were comparable and even superior to those from commercial HPLC. Subsecond chiral separation of amino acids was also achieved with highly sulfated cyclodextrins (HS-γ-CD) as chiral selectors [92].
Fig. 5.
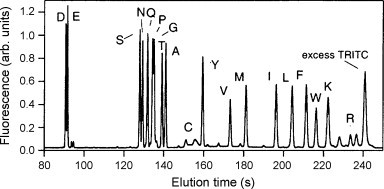
Chip MEKC separation of 19 amino acids. From Ref. [89] with permission.
3.1.2. On-chip concentration-microchip electrophoresis
Low detection limit for small molecules is a problem for MCE. Thus, on-line concentration before MCE is an important step of sample preparation, and of great significance to chip integration. Electric focusing is an effective tool beside conventional solid-phase extraction (SPE). Santiago and co-workers demonstrated the super-high-efficient coupling of transient isotachophoresis (ITP) and CE in a simple cross channel microchip [93]. When 1 M NaCl and 5 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) were used as leading electrolyte (LE) and tailing electrolyte (TE), respectively, Alexa Fluor 488 and bodipy could be stacked more than a million fold, and only a very short injection/ITP time (40 s) was needed. With an upgraded optical system, 100 aM fluorophores could be analyzed without manual buffer exchange steps [94]. This was the highest sensitivity obtained with electrophoretic separation ever reported. Unfortunately, no real sample testing with this method was reported until now. Trace phenolic compounds were analyzed by a chip MEKC technique coupled with field-amplified sample stacking (FASS) and field amplified sample injection (FASI) [95]. FASS, FASI and separation were carried out continuously in three connected channel segments. Sample was injected and stacked with amplified electric field in the first channel, then was transferred to second channel filled with one plug of water, and stacked for the second time under amplified electric field, at the end, focusing sample plug was injected into the third channel, separated and detected by an amperometric detector of a cellulose-dsDNA-modified, screen-printed carbon electrode. With this method, about 5200-fold concentration was achieved, and trace phenolic compounds in tap water and surface water were directly analyzed.
3.1.3. On-chip labeling-microchip electrophoresis
Laser induced fluorescence (LIF) is the primary detection tool for MCE. Therefore, labeling on chip before CE is necessary as most analytes are non-fluorescent themselves. Various on-chip labeling strategies were also presented, such as precolumn and postcolumn reactions, which were first demonstrated by Ramsey and co-workers [96], [97]. However, precolumn labeling was preferred, as it could be performed for a longer time, with no interruption of electrophoresis separation and sampling frequency. Fluorescent covalent labeling and affinity labeling are two most common methods. Generally covalent coupling of fluorescent tag was usually applicable to amino acid [98], bioamines [99] and other active groups-contained molecules, as high reaction rate was needed for a short reaction time in microchip. As to affinity labeling, both direct binding to labeled antibody and competitive binding with labeled standard molecules to antibody were reported [7], [100].
Bioamines and their contents in drinking water had been labeled and separated within a microchip by Hahn and co-workers and deMello and co-workers [99], [101]. Kennedy and co-workers demonstrated an integrated microchip coupled to microdialysis for in vivo monitoring of amino acid neurotransmitters in rat brain [98], which contained sample introduction channel for dialysate, precolumn reactor for derivatization with ophthalaldehyde (OPA), gated injector, and separation channel. The dialysate stream could be analyzed at 130-s intervals, in which the separation time was 20 s with an electric field of 1320 V/cm. In 2006, they presented a capillary–PDMS hybrid chip for MCE-based sensing of neurotransmitters, in which a pneumatic pump and valve system were integrated to accomplish low-flow push–pull perfusion sampling, on-line derivatization, and flow-gated injection onto an embedded capillary for high-speed separation [102]. In this system, in vivo perfusion sample could be sampled every 30 s for over 4 h.
3.1.4. Ultraviolet absorption and other detectors
Labeling small molecules for LIF detection is not well developed as that for DNA and protein, and many small molecules cannot be labeled easily for LIF detection [103], so other strategies have to be introduced. UV absorption and deep UV induced fluorescence were suitable to aromatic compounds. UV detection was reported for the high-speed chiral separations on silica microchip [104] and Chinese traditional drug analysis on hybrid silica/PDMS microchip [105]. In order to decrease the UV absorption background of chip bulk, PDMS was sharply thinned at the detection window zone. UV detection has a wide application range over LIF indeed, but it is usually limited for its poor sensitivity. Thus, the coupling of chip concentration and UV detection can significantly extend the application field of MCE for small molecules [106]. A quartz microchip for UV detection was fabricated by using multi-point pressure method in normal temperature and lower pressure during bonding process in Lin's group (Fig. 6 ), in which ITP and CZE was integrated, and two flavonoids were concentrated up to 32-fold, compared to the CZE mode only. Deep UV fluorescence detection at 266-nm excitation wavelength has been realized for sensitive detection in MCE, and low-micromolar concentrations sensitivity could be achieved in fused silica microchip [107]. Enantioselective catalysis and analysis also were monitored with the same method on an integrated microchip [108].
Fig. 6.

Chip platform for CE and UV detection in B.C. Lin's Lab.
Electrochemical (EC) detection was also widely used to detect small molecules in MCE, in which inorganic ions were specifically reviewed by Haddad and co-workers [109], and environment pollutants by Wang and co-workers [110].
3.2. DNA
Rapid evolution in DNA electrophoretic separation has important impacts on biological and biomedical sciences. Technological innovations in microfluidic chip promise to provide other far reaching benefits. In the early days, most of works are dedicated to transfer capillary electrophoresis of DNA into chip format, as chip array electrophoresis system (chip CAE) was highly expected to grow up quickly and alleviate the huge burden of great amount of various genome sequencing. Fast PCR products analysis was another interest from biological laboratories. Recently, integrated and portable DNA analysis microdevices have become another powerful tool to take over challenges from anti-terrorism, food and environment safety, in which sample pretreatment, DNA/RNA extraction and amplification, electrophoresis separation and detection are all included in a microchip. This time, DNA electrophoresis in sieving media, especially the high-resolution DNA sizing is the main subject.
3.2.1. Microchip gel electrophoresis
Ultra-high-speed DNA separations were reported in the beginning of the concept of μTAS. Manz and co-workers accomplished the separation of antisense oligonucleotides on a micromachined capillary electrophoresis device in 1994 [111]. The ssDNA ranging from 10 to 25 bases could be separated within 45 s under the electric field of 2300 V/cm with a 3.8 cm distance. In the following years, high-resolution electrophoretic separations of DNA restriction fragments [112], single-color sequencing to ∼433 bases in 10 min, and four-color DNA sequencing to −150 bases with 97% accuracy in only 540 s were also reported by Mathies group [113]. Four-loci STR analysis were performed in less than 2 min, and even achieved baseline resolved elelctrophoretic separations of single-locus STR samples in 30 s [114]. However, lots of problems appeared when more applicable DNA electrophoresis microchip was pursued after these original successes. First of all, the unavoidable turn geometry in chip CAE sharply decreased the efficiencies. Moreover, injector size, electric field, separation matrix and temperature did matter when super-high resolution was required to the long-range DNA sequencing and valid STR genotyping.
Therefore, systematic studies and optimization of chip-DNA electrophoresis were started. In 1998, the performance of a single-channel single-color ssDNA sequencing with PAAm was quantitatively studied as a model [115]. Optimized parameters were presented and demonstrated experimentally. On a straight channel of 11.5 cm long with an injection width of <200 μm, 400 bases were sequenced in ∼14 min. These were close to the predicted performance limits of linear 4% PAAm in microchip. An improved result was reported next year, with a 250 μm twin-T injector, 7.5-cm-long separation channel at 35 °C, four-color ssDNA sequencing to 500 bases were appeared in 9.2 min with a resolution of >0.5, and the separation could be extended to 700 bases for single-color sequencing [116]. Extend separation channel length to 40 cm, then an average read length of 800 bases in 80 min (98% accuracy) were also obtained [117]. These all made sure clearly that the high-speed and high-efficient DNA resolution within microfabricated devices was definitely feasible.
As discussed in peak broadening above, the solution to ameliorate turn induced peak dispersion was found by incorporating tapered turn or constrained geometries. Therefore, the 96 16-cm long channels, suitable for sequencing on a 150 mm diameter wafer were produced in a radial format around a central common anode reservoir instead of original chips relied on a rectilinear channel [118]. These serpentine tapered-turn microchannels functioned at ∼95% the efficiency of a straight channel of the equivalent length [42]. At the same period, other technologies about chip fabrication, signal detection, data management, and some auxiliary equipments also were developed, [118], [119]. Therefore, the high-throughput DNA electrophoretic analysis microfluidic systems gradually came to maximum excellence, and started its state-of-art molecule biology applications.
The high-resolution size-based electrophoretic separation of DNA is the most critical prerequisite for all genetic analyses, in which SNP and STR genotyping and DNA sequencing are the widest applications. Multiplex SNP genotyping analysis was demonstrated within 96 chip CAE [121]. Three HHC variants and PCR controls were amplified using energy-transfer allele-specific primers. The PCR products together with MapMarker standard were simultaneously separated with single-base-pair resolution within 10 min, in comparison to 75 min by MegaBACE. DNA sequencing with an average read length of 430 bp within 95 lanes out of 96 lanes completed in 24 min after injection, using 4.5% (w/v) linear PAAm, 15.9 cm effective distance, under ∼200 V/cm electric field and 50 °C, with the sample of energy-transfer labeled M13mp18 forward sequencing products [122]. The chip CAE system could produce sequencing data at a rate of 1.7 kbp/min, a 5-fold increase over current commercial CAE technology (Fig. 7 ).
Fig. 7.
Chip CGE with integrated nanovolume sample purification for DNA sequencing. From Ref. [120] with permission.
High-speed analysis of multiplexed STRs was demonstrated by Ehrlich and co-workers within a chip-based microdevice (Fig. 8 ). The microchip had the effective separation length of 11.5 cm and a double T injector with 200 μm offset. Separation was performed with 4% linear PAAm in 1× TTE with 7 M urea after surface PAAm coating and several minutes of preelectrophoresis and 2 min sample loading. Analyses of the eight combined DNA index system (CODIS) STR loci were performed in 20 min with single-base resolution ranging from 0.75 to 1 [123]. A simultaneous analysis of fifteen loci ladders and a gender marker Amelogenin based on the PowerPlex 16 System was achieved in less than 35 min. A 16 channel microdevice designed for DNA forensics analysis was reported in 2004. With a 20 cm separation distance, peak accuracy of 0.4–0.9 base was achieved for the CODIS 13-locus multiplex with a sample consumption of 0.5 μL [124]. This instrument was coupled with a high-speed galvanometer-scanned four-color detector, and could promise temperature-dependent recalibration and environmental sensitivity in a wild field.
Fig. 8.
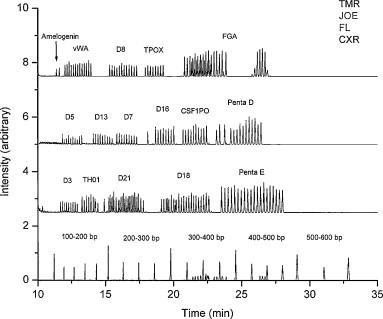
Electropherogram for Promega PowerPlex 16 allelic ladder and internal lane standard. From Ref. [124] with permission.
Fast chip CGE of PCR products has also shown great promises in the molecular biology research and clinical diagnosis. Lin's group reported lots of applications with clinical samples. As to severe acute respiratory syndrome (SARS) early diagnosis, 17 positive results were obtained out of 18 clinically diagnosed SARS patients with samples of nasopharyngeal swabs, while only 12 positive results were confirmed by the conventional RT-PCR with platform gel electrophoresis [125]. Using a modified semi-nested methylation-specific PCR combining PMMA chip CGE, this group scanned tumor-associated aberrant hypermethylation of the p16 gene in plasma and tissue DNA from 153 specimens [126]. They also assessed the –6A/G polymorphism in the core promoter region of angiotensinogen gene in 123 patients with essential hypertension and 103 healthy controls [127]. Besides, microchip CGE was also developed for rapid authentication of ginseng species. The results showed that at two microsatellite loci, American ginseng and Oriental ginseng had distinct allele patterns themselves [128]. Moreover, cultivated and wild American ginseng could be distinguished with similar methods. Recently, integrated chip ITP-CGE was demonstrated for highly sensitive and rapid genotyping of the hepatitis B virus, with a LOD of 0.0021 ng/mL, and 200 clinical cases were studied with 93% classification rate, which was 6% higher than that obtained with the conventional method [129].
3.2.2. Novel sieving matrix
The sieving matrix of low viscosity and high resolution is still pursued in chip-DNA electrophoresis, as gel loading is a hard task in the thin microchannel, and the total efficiency is limited to a shorter channel [130]. Up to now, lots of novel separation matrixes have been developed. Barron and co-workers reported two new polymer matrixes. One was sparsely cross-linked “nanogel” matrixes [131], which was synthesized by inverse emulsion copolymerization of acrylamide and ∼10−4 mol% bisacrylamide. These “nonogel” had typically a radius of ∼230 nm, and was ∼75% cross-linked. Because of the physical network stability of nanogel, longer average read lengths were obtained under standard conditions compared to the conventional PAAm matrix. The other was thermoresponsive polymers [132], which decoupled matrix loading and sieving properties. Baba and co-workers demonstrated the feasibility of nanospheres for DNA separation [20]. The nanosphere had a hydrophobic core of cross-linked poly(lactic acid) and a hydrophilic shell of PEG with a narrow size distribution and low viscosity in aqueous media (0.94 centipoise at 1% (w/v)). DNA fragments up to 15 kbp were successfully separated with 100 s without any saturation in migration rates. Intrachain segregated structure of DNA was observed during DNA migration under an electric field. Monolithic quartz nanopillar array was also used as “sieving media” for long dsDNA fragments, which was about 100–500 nm diameter and 500–5000 nm tall [133]. Lin and co-workers reported an ultra-low viscosity matrix composed of low molecular weight cellulose derivative and glucose [130]. Recently, self-assembled colloidal arrays [134] and magnetic matrixes [135] as 3D nanofluidic sieves for DNA separation were demonstrate in microchip for large dsDNA fragments.
End labeled free solution electrophoresis (FLFSE) is a choice for DNA electrophoresis separation without polymer or gel matrix. A newly synthesis strategy for DNA-tag was presented by Barron and co-workers recently [136]. Primers labeled with a series of monodisperse polyamide “drag-tags” were produced using both chemical and biological synthesis, and they were used to perform highly multiplexed single-base extension (SBE) assays of point mutants in exons 5–9 of the p53 gene, which allowed to separate genotyping reaction products by microchannel electrophoresis within 80 s (Fig. 9 ).
Fig. 9.
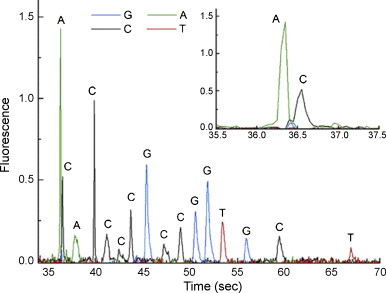
Chip-FLFSE of multiplexed p53 mutation with polyamide drag-tags. From Ref. [136] with permission.
3.2.3. Integrated DNA analyzer
Since the introduction of pneumatic pump and valve technologies, the development of chip PCR with the integrated microheater and microsensor, and the maturation of high-efficient chip CGE and chip CAE, the ability to integrate DNA extraction, DNA amplification and electrophoretic separation within a microchip was strengthened significantly. Although early in 1998, Manz and co-workers reported fast flow chip PCR [137], integrated PCR-CE DNA analysis mostly was relied on static chip PCR, which was introduced by Mathies group, Ramsey lab and many other groups. Chip PCR-CE has been used to carry out genetic analysis from animal, human, virus and bacteria. Two most prominent achievements have been presented recently. One was nanoliter-scale Sanger DNA sequencing with reaction, sample purification and chip sequencing in a microchip [138]. The other was a fully integrated microfluidic genetic analysis from whole blood as a crude biological sample to specific PCR electrophoregram integrated with DNA extraction, amplification and electrophoresis separation [28].
3.3. Proteins
MCE is a promising technique for protein analysis in the future. As the integrated sample preparation and short analysis cycle in microchip allow in situ inspection and monitoring possible. Moreover, the high-throughput potential can also offer as a powerful tool in the proteomics research. This part is dedicated to a brief introduction of the principles of protein electrophoresis in microfluidic chip with the emphasis on approaches to improve peak capacity and detection limit.
3.3.1. Microchip gel electrophoresis
The rapid sizing of proteins in microchip is highly powerful and desirable. Caliper, Agilent and Bio-Rad have commercialized it successfully [139], [140]. Protein sample denatured out of chip was sized by polymer solution, and at the same time, protein–SDS complex was dynamic labeled by the dyes in polymer solution. Then, an intersection fluid flow was introduced to dilute SDS below its critical micelle concentration before detection, which sharply reduced the fluorescent background. Both on-chip staining and SDS dilution steps occurred within 100 ms, which was about 104 times faster than that in SDS-PAGE. Here, 11 model proteins were sequentially separated, with a sizing accuracy better than 5% and high sensitivity (30 nM for carbonic anhydrase) (Fig. 10 ). By omitting the protein denaturing process, Baba and co-workers achieved protein analysis in a shorter total time, with dramatically improved sensitivity [141]. The fact that chip CGE required only a small amount of proteins probably helped to remove the need for the denaturing procedure. About 600 pg BSA was detectable, which equaled levels detectable by silver stain. They also applied this method to analysis of protein expression of Jurkat cells during heat Shock. Instead of polymer solution, Sandia National Laboratories separated proteins with in situ UV initiated cross-linked PAAm gel, and covalent protein labeling protocol instead of dynamic labeling [142].
Fig. 10.
Chip CGE for protein sizing. From Ref. [139] with permission.
3.3.2. Microchip isoelectric focusing
Capillary IEF shows a very high resolution and low detection limit, and is a good qualitative tool for proteins identification. Therefore, its corresponding part in microchip is also a strong tool for protein analysis. Fan and co-workers demonstrated that IEF resolution was essentially independent of focusing length when the voltage was kept and within a range that it did not cause a significant joule heat, which suggested that miniaturization offered IEF great benefits in addition to typical advantages associated with microchip [143]. Moreover, a mobile LIF detection system was also presented. The efficiency of 1.5 × 105 plates for lysozyme was achieved within a LabCard microchip, and a focusing distance of 4.7 cm. The protein–protein interaction was also detected within chip IEF with a detection limit of 50 fM. Later, a whole-channel imaging detection system was developed to monitor proteins while they were moving under an electric field [144]. The phenomenon about the compression of pH gradient to the middle of a channel and the relative amount of the gradient compression decreased with the increase of channel length was confirmed [145].
Three-stage IEF in a PDMS microchip was demonstrated by Ivory and co-workers [146]. Narrow pH range ampholytes as anode and cathode solutions in the second and third IEF stages allowed ready control of pH range. All focusing was completed within 25 min under the electric field of 50–214 V/cm. Recently, a two-dimensional (2D) finite-volume model was developed to simulate nonlinear IEF in complex microgeometries and predicted that IEF resolution could be improved by mobilizing them through a contraction zone. To further increase sample loading and therefore the concentrations of focused analytes, a dynamic approach, which was based on electrokinetic injection of proteins/peptides from solution reservoirs, was demonstrated by Lee and co-workers [147]. The sample was continuously injected and encountered the pH gradient established by carrier ampholytes originally present in the channel. The sample loading was enhanced by approximately 10–100-fold in comparison with conventional IEF after 30 min electrokinetic injection with a 500 V/cm electric field.
Only IEF along fluid flow direction has been discussed in this part. In fact, in microfluidic chip, another kind of protein focusing across the direction of fluid flow was also studied substantially, which was called as free flow isofocusing, or transverse IEF sometimes, in different chip formats. This IEF was commonly carried out for sample preparation and fractionation collection. Related papers could be found from Manz, Han, and Yager et al.
3.3.3. Microchip zone electrophoresis
Chip CZE is still one simple and fast electrophoresis mode for protein mixtures, as chip IEF, chip CGE and chip CEC will run improperly under super-high electric field. Lin's group developed a method for characterizing chicken and turkey ovalbumin by chip CZE combined with exoglycosidase digestion [148]. By using a porous monolith column fabricated by UV-initiated polymerization, lectin affinity chromatography was miniaturized into a microfluidic format [149]. The glycoproteins could be separated into several fractions with different affinities after chip CZE. More important, running buffers for chip CZE are quite close to physiological condition, with no complex additives added. This will facilitate the exploration of protein structure and its function. Some interaction-based MCE works have been reported [150]. Lin and co-workers developed affinity MCE methods based on indirect LIF detection [151] and frontal analysis [152] to study the interaction between heparin and serum albumin. With indirect LIF detection, the binding constant could be determined more precisely without possible negative influence from the fluorescent tag on protein. With two injectors, when sample mixture was injected, another separate sample plug was introduced into separation channel simultaneously, and used as the internal standard during chip CZE, so the potential interference from EOF markers was completely avoided.
Chip CZE–MS is also one ideal tool to identify target protein and to discover interesting biomarkers from complex samples, which exceeds the approximate molecular weight and isoelectric point of a target protein from chip CGE and chip IEF. In the early days, various chip CZE–MS interfaces were demonstrated on glass, PDMS and plastic microchips, mostly from Karger and co-workers [8], [153] and Ramsey and co-workers [154]. Recently, Lin and co-workers improved the sheath-flow nanoelectrospray interface of chip CZE–MS for glycoprotein and glycopeptide analysis [155], in which femtomole detection limit for peptides was achieved, all the glycoforms and glycopeptides of ribonuclease B were identified, and some detailed structures of the glycopeptides were even further characterized with MS/MS, a quadrupole IT-MS. An integrated nanospray emitter on glass microchip for coupling to a mass spectrometer was also reported [156].
3.3.4. Two-dimensional microchip electrophoresis
Two-dimensional gel electrophoresis is still the main workhorse now in proteomics. However, it is suffered from manual operation, long cycle term, and low sample throughput. Therefore, many efforts have been carried out to achieve chip 2D protein analysis. Mimicking 2D gel, chip 2D can be established by focusing samples in one channel, and then sizing IEF sections in parallel in cross channels [157]. The prototype 2D chip electrophoresis was demonstrated in PDMS microchip in 2002 [158]. Here, IEF was performed out, and focused samples were orthogonally placed on the microchannels in parallel and then started chip CGE. Chang et al. improved this system. IEF and μ-CGE were incorporated in a plastic chip [159], in which focused proteins were electrokinetically transferred into orthogonal microchannels directly for subsequent chip CGE. All analysis was completed within 10 min with an overall peak capacity of ∼1700. Han and co-workers coupled IEF with μ-CGE by the active microvalve control, in which protein plug after IEF could be isolated with pneumatic valve, and subjected chip CGE. Thus, the interruption between running buffers of CGE and IEF could be inhibited to a high extent [160].
Some other promising chip 2D systems were also accomplished. Mostly, the sample plugs after the first separation were transferred into the second sequentially at regular intervals. In 2000, Ramsey and co-workers first coupled MEKC (the first dimension) with CZE (the second dimension) on a glass microchip [161]. Gated injection was implemented for CZE injection, and the electric valve was switched every 3–4 s for 0.3 s to inject samples of effluent from the first dimension into the second dimension, with a peak capacity of 500–1000. In 2001, CEC was used instead of MEKC as the first dimension and coupled with CZE [162]. A 25-cm spiral separation channel modified with octadecylsilane for CEC and 1.2 cm straight separation channel for CZE. The sample plug from CEC was loaded into CZE channel for 0.2 s with a cycle of 3.2 s. The peak capacity was estimated as 150. In 2003, an optimized chip MEKC-CZE was introduced with 1 Hz sampling frequency for CZE [163]. BSA tryptic digest was separated within 10 min with a peak capacity of 4200 (Fig. 11 ). In 2006, Soper group presented another tandem PMMA chip 2D [164], in which CGE and MEKC were used as the first and second dimension, respectively. The sample plug from μ-CGE was loaded into MEKC for 0.5 s with a cycle of 10.5 s. This system was demonstrated with 10 model proteins, and provided a theoretical peak capacity of ∼1000.
Fig. 11.
Chip 2D (MEKC-CZE) for separations of BSA digests. From Ref. [163] with permission.
3.3.5. On-chip concentration-microchip electrophoresis
Real protein samples are usually very dilute, and the amount of sample injection in microfluidic chip is also limited, so on-line protein concentration before electrophoresis separation in microchip is highly desired in most cases. Up to now, there are two primary methods have been usually adopted in microfluidic format associated with electrophoresis, one is filtering, the other is electric field gradient focusing (EFGF).
Electric field driving force is obviously more convenient and feasible than pressure or centrifugal force in microfluidic chip. Ramsey and co-workers reported one convenient filter method for sample concentration [165]. Silicate solution was spin-coated on glass substrate, and then the chip was bonded at lower temperature. Proteins were concentrated electrokinetically before porous silica layer during sample loading. This system functioned well in both coated and uncoated open channels, and about 600-fold enhancement was achieved before chip CGE. The nanofluidic filter of the interface between native PDMS and glass, and a nanogap generated by junction gap breakdown could work similarly [166].
Organic membrane was produced by laser induced polymerization at the junction of a cross channel in a microchip [167]. Averaged concentration was increased by 4 orders of magnitude with proteins (>5.7 kDa), and the degree of concentration was limited only by the solubility limit of the proteins. Another organic membrane was fabricated in situ using photopolymerization of cross-linked PAAm (Fig. 12 ). Proteins (>10 kDa) were concentrated rapidly (<5 min) over 1000-fold before μ-CGE [168], [169]. Lin and co-workers produced multilayer devices consisting of 10 nm pore diameter membranes sandwiched between two layers of PDMS substrates with embedded microchannels, which could be used for effective deproteinization, desalt or concentration on-line [170].
Fig. 12.
Chip concentration of protein with nanofluidic filter and photopatterned cross-linked polyacrylamide gels. From Ref. [168], [175] with permission.
ITP is a well-established method in CE. It is also useful for protein concentration in MCE. Lin and co-workers coupled ITP with μ-CGE. Instead of frequent substitution of buffers vials in capillary format, automatically switching electric fields between running buffers in glass microchip was performed. A 5 mm SDS–protein sample plug in double T injection was focusing with a leading electrolyte of 50 mM Tris, 0.5% SDS, 2% dithiothreitol (DTT), pH 6.8, and a tailing electrolyte of 192 mM glycine, 25 mM Tris, pH 8.3, and then sizing in polymer solution of 100 mM Tris–NaH2PO4, 0.1% SDS, 10% glycerol, 10% dextran, pH 8.3, all of which could be completed within 300 s with an enhancement factor of ∼40 [171]. Recently, a simple transient ITP was used to concentrate HSA and its immunocomplex just before electrophoretic separation on a standard cross PMMA microchip. With 5 or 10 mM NaCl added in the leading electrolyte, an 800-fold signal enhancement was achieved [172].
EFGF was a powerful tool to achieve both concentration and separation of protein sample simultaneously. Instead of hollow fiber column in conventional EFGF instruments, the conductive membrane was fabricated in microchip in situ to block the protein molecules while keeping electric field connection [173]. Woolley and co-workers produced this membrane just on the fluid microchannel using phase-changing sacrificial layers (PCSL), which had also been used for PMMA chip bonding. With this method, about 3-fold resolution increase and an enrichment factor of 10,000 within 40 min were obtained. An alternative method was presented by Lee and co-workers [174]. A weir structure was fabricated between main separation channel and electric field gradient channel on one side on their own developed plastic chip. Ion-permeable membrane was polymerized on the weir by surface modification, prepolymer loading, nitrogen gas purging and UV radiation induced polymerization. With this EFGF device, GFP was concentrated up to 4000-fold.
4. Outlook and conclusion
MCE can be performed at higher speed with superior efficiency, for its stable laminar flow, plug flow profile, short initial sample length, diffusion-dominant molecule dispersion and rapid heat dissipation rate. At the same time, well-established microfabrication technologies facilitate MCE with more freedom and compactness. Other related functional units such as microheater, pneumatic pump and valve developed in microfluidic chip can be recruited as required. Therefore, MCE can be carried out in an integrated and controllable way. Generally, MCE has demonstrated the ability for the fast and efficient separation of various analytes, from small ions to large biomolecules, provided a feasible method for conducting numerous experiments in parallel, while consuming little reagent and achieving even better results.
Some factors that determined MCE efficiencies have been studied extensively, and lots of successful experiences and procedures can be used as reference right now, especially for sample injection, turn optimization and joule heating control. Surface modification methods are specially required upon chip material and electrophoresis system, so its complexity and diversity have to be enriched gradually.
Lots of achievements have been obtained in MCE, even chip sizing of DNA and protein has been commercialized, but portable and integrated microdevices based on MCE are still in research by laboratories and companies. MCE is not self-contained in most out-of-lab cases, and it alone dose not fulfill the full potential of miniaturization. On-line sample preparation is highly desirable for most MCE-based applications. DNA extraction and amplification from a crude sample is a typical and successful pioneer technology, and its integration with MCE has also been demonstrated recently. A sample volume of several microliters was needed, and the analysis cycle was within 1 h [28]. Perfusion sampling of proteins and bioamines in vivo, followed with derivation and high-speed MCE were integrated within a microchip [176]. The sampling interval was less than 1 min, and could last several hours. However, more challenges still exist in sample preparation, such as the extraction of a target fraction from solid samples, or from fragile and complex biological samples.
Development of compact detection setups is also an unavoidable task for the portability of MCE. At present, optical accessories are still too large relative to microchip itself, and many limitations in other detection methods also need to be overcome. Recently, optiofluidic, novel light sources and lens may provide new ways to accomplish the next generation detection system. Furthermore, new sensor techniques will probably present other better choices.
It can be expected that MCE technologies will continue to grow stronger, and its integration with sample preparation and facile detection system will lead to a wider range of important applications.
Acknowledgements
This work was funded by NNSF of China (Nos. 20635030, 20575067 and Y2005006), and 863 Program of China (No. 2006AA02Z305), and 973 Programs of China (Nos.2007CB714505 and 2007CB714507).
Contributor Information
Jianhua Qin, Email: jhqin@dicp.ac.cn.
Bingcheng Lin, Email: bclin@dicp.ac.cn.
References
- 1.Whitesides G.M. Nature. 2006;442:368. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 2.Harrison D.J., Manz A., Fan Z.H., Ludi H., Widmer H.M. Anal. Chem. 1992;64:1926. [Google Scholar]
- 3.Unger M.A., Chou H.P., Thorsen T., Scherer A., Quake S.R. Science. 2000;288:113. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 4.Grover W.H., Skelley A.M., Liu C.N., Lagally E.T., Mathies R.A. Sens. Actuators, B. 2003;89:315. [Google Scholar]
- 5.Ng J.M.K., Gitlin I., Stroock A.D., Whitesides G.M. Electrophoresis. 2002;23:3461. doi: 10.1002/1522-2683(200210)23:20<3461::AID-ELPS3461>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Mayers B.T., Vezenov D.V., Vullev V.I., Whitesides G.M. Anal. Chem. 2005;77:1310. doi: 10.1021/ac048692n. [DOI] [PubMed] [Google Scholar]
- 7.Wang J., Ibanez A., Chatrathi M.P., Escarpa A. Anal. Chem. 2001;73:5323. doi: 10.1021/ac010808h. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B., Liu H., Karger B.L., Foret F. Anal. Chem. 1999;71:3258. doi: 10.1021/ac990090u. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson S.C., Hergenroder R., Koutny L.B., Warmack R.J., Ramsey J.M. Anal. Chem. 1994;66:1107. [Google Scholar]
- 10.Jacobson S.C., Ermakov S.V., Ramsey J.M. Anal. Chem. 1999;71:3273. doi: 10.1021/ac990059s. [DOI] [PubMed] [Google Scholar]
- 11.Thomas C.D., Jacobson S.C., Ramsey J.M. Anal. Chem. 2004;76:6053. doi: 10.1021/ac035475y. [DOI] [PubMed] [Google Scholar]
- 12.Effenhauser C.S., Manz A., Widmer H.M. Anal. Chem. 1993;65:2637. [Google Scholar]
- 13.Fu L.M., Yang R.J., Lee G.B. Anal. Chem. 2003;75:1905. doi: 10.1021/ac020741d. [DOI] [PubMed] [Google Scholar]
- 14.Alarie J.P., Jacobson S.C., Ramsey J.M. Electrophoresis. 2001;22:312. doi: 10.1002/1522-2683(200101)22:2<312::AID-ELPS312>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 15.Slentz B.E., Penner N.A., Regnier F. Anal. Chem. 2002;74:4835. doi: 10.1021/ac020301m. [DOI] [PubMed] [Google Scholar]
- 16.Blas M., Delaunay N., Ferrigno R., Rocca J.L. Electrophoresis. 2007;28:2961. doi: 10.1002/elps.200600692. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C.X., Manz A. Anal. Chem. 2001;73:2656. doi: 10.1021/ac010138f. [DOI] [PubMed] [Google Scholar]
- 18.Tsai C.H., Wang Y.N., Lin C.F., Yang R.J., Fu L.M. Electrophoresis. 2006;27:4991. doi: 10.1002/elps.200500913. [DOI] [PubMed] [Google Scholar]
- 19.Lacharme F., Gijs M.A.M. Electrophoresis. 2006;27:2924. doi: 10.1002/elps.200500723. [DOI] [PubMed] [Google Scholar]
- 20.Tabuchi M., Ueda M., Kaji N., Yamasaki Y., Nagasaki Y., Yoshikawa K., Kataoka K., Baba Y. Nat. Biotechnol. 2004;22:337. doi: 10.1038/nbt939. [DOI] [PubMed] [Google Scholar]
- 21.Tabuchi M., Kuramitsu Y., Nakamura K., Baba Y. Anal. Chem. 2003;75:3799. doi: 10.1021/ac030051p. [DOI] [PubMed] [Google Scholar]
- 22.Gai H.W., Yu L.F., Dai Z.P., Ma Y.F., Lin B.C. Electrophoresis. 2004;25:1888. doi: 10.1002/elps.200305835. [DOI] [PubMed] [Google Scholar]
- 23.Luo Y., Wu D.P., Zeng S.J., Gai H.W., Long Z.C., Shen Z., Dai Z.P., Qin J.H., Lin B.C. Anal. Chem. 2006;78:6074. doi: 10.1021/ac0605142. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L., Yin X.F., Fang Z.L. Lab Chip. 2006;6:258. doi: 10.1039/b511924c. [DOI] [PubMed] [Google Scholar]
- 25.Leach A.M., Wheeler A.R., Zare R.N. Anal. Chem. 2003;75:967. doi: 10.1021/ac026112l. [DOI] [PubMed] [Google Scholar]
- 26.Wu H.K., Wheeler A., Zare R.N. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12809. doi: 10.1073/pnas.0405299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlinsey J.M., Monahan J., Marchiarullo D.J., Ferrance J.P., Landers J.P. Anal. Chem. 2005;77:3637. doi: 10.1021/ac048115z. [DOI] [PubMed] [Google Scholar]
- 28.Easley C.J., Karlinsey J.M., Bienvenue J.M., Legendre L.A., Roper M.G., Feldman S.H., Hughes M.A., Hewlett E.L., Merkel T.J., Ferrance J.P., Landers J.P. Proc. Natl. Acad. Sci. U.S.A. 2006;103:19272. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li M.W., Huynh B.H., Hulvey M.K., Lunte S.M., Martin R.S. Anal. Chem. 2006;78:1042. doi: 10.1021/ac051592c. [DOI] [PubMed] [Google Scholar]
- 30.Inoue A., Ito T., Makino K., Hosokawa K., Maeda M. Anal. Chem. 2007;79:2168. doi: 10.1021/ac0616097. [DOI] [PubMed] [Google Scholar]
- 31.Ito T., Inoue A., Sato K., Hosokawa K., Maeda M. Anal. Chem. 2005;77:4759. doi: 10.1021/ac050122f. [DOI] [PubMed] [Google Scholar]
- 32.Liu J., Hansen C., Quake S.R. Anal. Chem. 2003;75:4718. doi: 10.1021/ac0346407. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y., Lin Q., Mukherjee T. Lab Chip. 2004;4:625. doi: 10.1039/b406752e. [DOI] [PubMed] [Google Scholar]
- 34.Petersen N.J., Nikolajsen R.P.H., Mogensen K.B., Kutter J.P. Electrophoresis. 2004;25:253. doi: 10.1002/elps.200305747. [DOI] [PubMed] [Google Scholar]
- 35.Erickson D., Sinton D., Li D.Q. Lab Chip. 2003;3:141. doi: 10.1039/b306158b. [DOI] [PubMed] [Google Scholar]
- 36.Xuan X.C., Li D.Q. Electrophoresis. 2005;26:166. doi: 10.1002/elps.200406141. [DOI] [PubMed] [Google Scholar]
- 37.Huang K.D., Yang R.J. Electrophoresis. 2006;27:1957. doi: 10.1002/elps.200500721. [DOI] [PubMed] [Google Scholar]
- 38.Xuan X.C., Li D.Q. J. Chromatogr. A. 2005;1064:227. doi: 10.1016/j.chroma.2004.12.033. [DOI] [PubMed] [Google Scholar]
- 39.Tang G.Y., Yan D.G., Yang C., Gong H.Q., Chai J.C., Lam Y.C. Electrophoresis. 2006;27:628. doi: 10.1002/elps.200500681. [DOI] [PubMed] [Google Scholar]
- 40.Tang G.Y., Yan D.G., Yang C., Gong H.Q., Chai C.K., Lam Y.C. Sens. Actuators, A. 2007;139:221. [Google Scholar]
- 41.Culbertson C.T., Jacobson S.C., Ramsey J.M. Anal. Chem. 1998;70:3781. [Google Scholar]
- 42.Paegel B.M., Hutt L.D., Simpson P.C., Mathies R.A. Anal. Chem. 2000;72:3030. doi: 10.1021/ac000054r. [DOI] [PubMed] [Google Scholar]
- 43.Molho J.I., Herr A.E., Mosier B.P., Santiago J.G., Kenny T.W., Brennen R.A., Gordon G.B., Mohammadi B. Anal. Chem. 2001;73:1350. [Google Scholar]
- 44.Griffiths S.K., Nilson R.H. Anal. Chem. 2002;74:2960. doi: 10.1021/ac011218m. [DOI] [PubMed] [Google Scholar]
- 45.Johnson T.J., Ross D., Gaitan M., Locascio L.E. Anal. Chem. 2001;73:3656. doi: 10.1021/ac010269g. [DOI] [PubMed] [Google Scholar]
- 46.Lee G.B., Fu L.M., Lin C.H., Lee C.Y., Yang R.J. Electrophoresis. 2004;25:1879. doi: 10.1002/elps.200305880. [DOI] [PubMed] [Google Scholar]
- 47.Hjerten S., Zhu M.D. J. Chromatogr. 1985;346:265. [Google Scholar]
- 48.Kirby B.J., Wheeler A.R., Zare R.N., Fruetel J.A., Shepodd T.J. Lab Chip. 2003;3:5. doi: 10.1039/b210621n. [DOI] [PubMed] [Google Scholar]
- 49.Gao L., Liu S.R. Anal. Chem. 2004;76:7179. doi: 10.1021/ac049353x. [DOI] [PubMed] [Google Scholar]
- 50.Razunguzwa T.T., Warrier M., Timperman A.T. Anal. Chem. 2006;78:4326. doi: 10.1021/ac052121t. [DOI] [PubMed] [Google Scholar]
- 51.Belder D., Deege A., Kohler F., Ludwig M. Electrophoresis. 2002;23:3567. doi: 10.1002/1522-2683(200210)23:20<3567::AID-ELPS3567>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 52.Pumera M., Wang J., Grushka E., Polsky R. Anal. Chem. 2001;73:5625. doi: 10.1021/ac015589e. [DOI] [PubMed] [Google Scholar]
- 53.Ma R.B., Crabtree H.J., Backhouse C.J. Electrophoresis. 2005;26:2692. doi: 10.1002/elps.200410418. [DOI] [PubMed] [Google Scholar]
- 54.Zhou X.M., Dai Z.P., Liu X., Luo Y., Wang H., Lin B.C. J. Sep. Sci. 2005;28:225. doi: 10.1002/jssc.200401920. [DOI] [PubMed] [Google Scholar]
- 55.Liu J.K., Sun X.F., Lee M.L. Anal. Chem. 2007;79:1926. doi: 10.1021/ac0617621. [DOI] [PubMed] [Google Scholar]
- 56.Dang F.Q., Kakehi K., Cheng J.J., Tabata O., Kurokawa M., Nakajima K., Ishikawa M., Baba Y. Anal. Chem. 2006;78:1452. doi: 10.1021/ac051702f. [DOI] [PubMed] [Google Scholar]
- 57.Shah J.J., Geist J., Locascio L.E., Gaitan M., Rao M.V., Vreeland W.N. Electrophoresis. 2006;27:3788. doi: 10.1002/elps.200600118. [DOI] [PubMed] [Google Scholar]
- 58.Bi H.Y., Meng S., Li Y., Guo K., Chen Y.P., Kong J.L., Yang P.Y., Zhong W., Liu B.H. Lab Chip. 2006;6:769. doi: 10.1039/b600326e. [DOI] [PubMed] [Google Scholar]
- 59.Liu J.K., Pan T., Woolley A.T., Lee M.L. Anal. Chem. 2004;76:6948. doi: 10.1021/ac040094l. [DOI] [PubMed] [Google Scholar]
- 60.Henry A.C., Tutt T.J., Galloway M., Davidson Y.Y., McWhorter C.S., Soper S.A., McCarley R.L. Anal. Chem. 2000;72:5331. doi: 10.1021/ac000685l. [DOI] [PubMed] [Google Scholar]
- 61.Duffy D.C., McDonald J.C., Schueller O.J.A., Whitesides G.M. Anal. Chem. 1998;70:4974. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 62.Huang B., Wu H.K., Kim S., Zare R.N. Lab Chip. 2005;5:1005. doi: 10.1039/b509251e. [DOI] [PubMed] [Google Scholar]
- 63.Liu B.F., Ozaki M., Hisamoto H., Luo Q.M., Utsumi Y., Hattori T., Terabe S. Anal. Chem. 2005;77:573. doi: 10.1021/ac0490447. [DOI] [PubMed] [Google Scholar]
- 64.Mora M.F., Giacomelli C.E., Garcia C.D. Anal. Chem. 2007;79:6675. doi: 10.1021/ac070953g. [DOI] [PubMed] [Google Scholar]
- 65.Makamba H., Hsieh Y.Y., Sung W.C., Chen S.H. Anal. Chem. 2005;77:3971. doi: 10.1021/ac0502706. [DOI] [PubMed] [Google Scholar]
- 66.Roman G.T., McDaniel K., Culbertson C.T. Analyst. 2006;131:194. doi: 10.1039/b510765b. [DOI] [PubMed] [Google Scholar]
- 67.Bergstrom S.K., Edenwall N., Laven M., Velikyan I., Langstrom B., Markides K.E. Anal. Chem. 2005;77:938. doi: 10.1021/ac0492618. [DOI] [PubMed] [Google Scholar]
- 68.Slentz B.E., Penner N.A., Regnier F.E. J. Chromatogr. A. 2002;948:225. doi: 10.1016/s0021-9673(01)01319-x. [DOI] [PubMed] [Google Scholar]
- 69.Hu S.W., Ren X.Q., Bachman M., Sims C.E., Li G.P., Allbritton N.L. Anal. Chem. 2004;76:1865. doi: 10.1021/ac049937z. [DOI] [PubMed] [Google Scholar]
- 70.Hu S.W., Ren X.Q., Bachman M., Sims C.E., Li G.P., Allbritton N.L. Anal. Chem. 2002;74:4117. doi: 10.1021/ac025700w. [DOI] [PubMed] [Google Scholar]
- 71.Luo Y.Q., Huang B., Wu H., Zare R.N. Anal. Chem. 2006;78:4588. doi: 10.1021/ac052274g. [DOI] [PubMed] [Google Scholar]
- 72.Roman G.T., Hlaus T., Bass K.J., Seelhammer T.G., Culbertson C.T. Anal. Chem. 2005;77:1414. doi: 10.1021/ac048811z. [DOI] [PubMed] [Google Scholar]
- 73.Jon S.Y., Seong J.H., Khademhosseini A., Tran T.N.T., Laibinis P.E., Langer R. Langmuir. 2003;19:9989. [Google Scholar]
- 74.Xiao D.Q., Van Le T., Wirth M.J. Anal. Chem. 2004;76:2055. doi: 10.1021/ac035254s. [DOI] [PubMed] [Google Scholar]
- 75.Wu D.P., Luo Y., Zhou X.M., Dai Z.P., Lin B.C. Electrophoresis. 2005;26:211. doi: 10.1002/elps.200406157. [DOI] [PubMed] [Google Scholar]
- 76.Wu D.P., Zhao B.X., Dai Z.P., Qin J.H., Lin B.C. Lab Chip. 2006;6:942. doi: 10.1039/b600765a. [DOI] [PubMed] [Google Scholar]
- 77.Wu D.P., Qin J.H., Lin B.C. Lab Chip. 2007;7:1490. doi: 10.1039/b708877a. [DOI] [PubMed] [Google Scholar]
- 78.Zhuang Z., Starkey J.A., Mechref Y., Novotny M.V., Jacobson S.C. Anal. Chem. 2007;79:7170. doi: 10.1021/ac071261v. [DOI] [PubMed] [Google Scholar]
- 79.Ye N.N., Qin J.H., Shi W.W., Lin B.C. Electrophoresis. 2007;28:1146. doi: 10.1002/elps.200600450. [DOI] [PubMed] [Google Scholar]
- 80.Qin J.H., Ye N.N., Yu L.F., Liu D.Y., Fung Y.S., Wang W., Ma X.J., Lin B.C. Electrophoresis. 2005;26:1155. doi: 10.1002/elps.200410136. [DOI] [PubMed] [Google Scholar]
- 81.Qin J.H., Ye N.N., Liu X., Lin B.C. Electrophoresis. 2005;26:3780. doi: 10.1002/elps.200500113. [DOI] [PubMed] [Google Scholar]
- 82.El-Ali J., Sorger P.K., Jensen K.F. Nature. 2006;442:403. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 83.Jacobson S.C., Hergenroder R., Koutny L.B., Ramsey J.M. Anal. Chem. 1994;66:1114. [Google Scholar]
- 84.Jacobson S.C., Culbertson C.T., Daler J.E., Ramsey J.M. Anal. Chem. 1998;70:3476. [Google Scholar]
- 85.Jacobson S.C., Hergenroder R., Koutny L.B., Ramsey J.M. Anal. Chem. 1994;66:2369. [Google Scholar]
- 86.Kutter J.P., Jacobson S.C., Matsubara N., Ramsey J.M. Anal. Chem. 1998;70:3291. doi: 10.1021/ac971367y. [DOI] [PubMed] [Google Scholar]
- 87.Moore A.W., Jacobson S.C., Ramsey J.M. Anal. Chem. 1995;67:4184. [Google Scholar]
- 88.Kutter J.P., Jacobson S.C., Ramsey J.M. Anal. Chem. 1997;69:5165. [Google Scholar]
- 89.Culbertson C.T., Jacobson S.C., Ramsey J.M. Anal. Chem. 2000;72:5814. doi: 10.1021/ac0006268. [DOI] [PubMed] [Google Scholar]
- 90.Hutt L.D., Glavin D.P., Bada J.L., Mathies R.A. Anal. Chem. 1999;71:4000. doi: 10.1021/ac9903959. [DOI] [PubMed] [Google Scholar]
- 91.Skelley A.M., Scherer J.R., Aubrey A.D., Grover W.H., Ivester R.H.C., Ehrenfreund P., Grunthaner F.J., Bada J.L., Mathies R.A. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1041. doi: 10.1073/pnas.0406798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piehl N., Ludwig M., Belder D. Electrophoresis. 2004;25:3848. doi: 10.1002/elps.200406028. [DOI] [PubMed] [Google Scholar]
- 93.Jung B., Bharadwaj R., Santiago J.G. Anal. Chem. 2006;78:2319. doi: 10.1021/ac051659w. [DOI] [PubMed] [Google Scholar]
- 94.Jung B.G., Zhu Y.G., Santiago J.G. Anal. Chem. 2007;79:345. doi: 10.1021/ac060949p. [DOI] [PubMed] [Google Scholar]
- 95.Shiddiky M.J.A., Park H., Shim Y.B. Anal. Chem. 2006;78:6809. doi: 10.1021/ac0606002. [DOI] [PubMed] [Google Scholar]
- 96.Jacobson S.C., Hergenroder R., Moore A.W., Ramsey J.M. Anal. Chem. 1994;66:4127. [Google Scholar]
- 97.Jacobson S.C., Koutny L.B., Hergenroder R., Moore A.W., Ramsey J.M. Anal. Chem. 1994;66:3472. [Google Scholar]
- 98.Sandlin Z.D., Shou M.S., Shackman J.G., Kennedy R.T. Anal. Chem. 2005;77:7702. doi: 10.1021/ac051044z. [DOI] [PubMed] [Google Scholar]
- 99.Ro K.W., Lim K., Kim H., Hahn J.H. Electrophoresis. 2002;23:1129. doi: 10.1002/1522-2683(200204)23:7/8<1129::AID-ELPS1129>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 100.Bromberg A., Mathies R.A. Anal. Chem. 2003;75:1188. doi: 10.1021/ac020599g. [DOI] [PubMed] [Google Scholar]
- 101.Beard N.P., Edel J.B., deMello A.J. Electrophoresis. 2004;25:2363. doi: 10.1002/elps.200305919. [DOI] [PubMed] [Google Scholar]
- 102.Shackman J.G., Dahlgren G.M., Peters J.L., Kennedy R.T. Lab Chip. 2005;5:56. doi: 10.1039/b404974h. [DOI] [PubMed] [Google Scholar]
- 103.Qin J.H., Fung Y.S., Zhu D.R., Lin B.C. J. Chromatogr. A. 2004;1027:223. doi: 10.1016/j.chroma.2003.10.055. [DOI] [PubMed] [Google Scholar]
- 104.Ludwig M., Kohler F., Belder D. Electrophoresis. 2003;24:3233. doi: 10.1002/elps.200305427. [DOI] [PubMed] [Google Scholar]
- 105.Ma B., Zhou X.M., Wang G., Huang H.Q., Dai Z.P., Qin J.H., Lin B.C. Electrophoresis. 2006;27:4904. doi: 10.1002/elps.200600392. [DOI] [PubMed] [Google Scholar]
- 106.Ma B., Zhou X.M., wang G., Dai Z.P., Qin J.H., Lin B.C. Electrophoresis. 2007;28:2474. doi: 10.1002/elps.200600619. [DOI] [PubMed] [Google Scholar]
- 107.Schulze P., Ludwig M., Kohler F., Belder D. Anal. Chem. 2005;77:1325. doi: 10.1021/ac048596m. [DOI] [PubMed] [Google Scholar]
- 108.Belder D., Ludwig M., Wang L.W., Reetz M.T. Angew. Chem., Int. Ed. 2006;45:2463. doi: 10.1002/anie.200504205. [DOI] [PubMed] [Google Scholar]
- 109.Evenhuis C.J., Guijt R.M., Macka M., Haddad P.R. Electrophoresis. 2004;25:3602. doi: 10.1002/elps.200406120. [DOI] [PubMed] [Google Scholar]
- 110.Chen G., Lin Y.H., Wang J. Talanta. 2006;68:497. doi: 10.1016/j.talanta.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 111.Effenhauser C.S., Paulus A., Manz A., Widmer H.M. Anal. Chem. 1994;66:2949. [Google Scholar]
- 112.Woolley A.T., Mathies R.A. Proc. Natl. Acad. Sci. U.S.A. 1994;91:11348. doi: 10.1073/pnas.91.24.11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Woolley A.T., Mathies R.A. Anal. Chem. 1995;67:3676. doi: 10.1021/ac00116a010. [DOI] [PubMed] [Google Scholar]
- 114.Schmalzing D., Koutny L., Adourian A., Belgrader P., Matsudaira P., Ehrlich D. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10273. doi: 10.1073/pnas.94.19.10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schmalzing D., Adourian A., Koutny L., Ziaugra L., Matsudaira P., Ehrlich D. Anal. Chem. 1998;70:2303. doi: 10.1021/ac971381a. [DOI] [PubMed] [Google Scholar]
- 116.Liu S.R., Shi Y.N., Ja W.W., Mathies R.A. Anal. Chem. 1999;71:566. doi: 10.1021/ac980783v. [DOI] [PubMed] [Google Scholar]
- 117.Koutny L., Schmalzing D., Salas-Solano O., El-Difrawy S., Adourian A., Buonocore S., Abbey K., McEwan P., Matsudaira P., Ehrlich D. Anal. Chem. 2000;72:3388. doi: 10.1021/ac9913614. [DOI] [PubMed] [Google Scholar]
- 118.Medintz I.L., Berti L., Emrich C.A., Tom J., Scherer J.R., Mathies R.A. Clin. Chem. 2001;47:1614. [PubMed] [Google Scholar]
- 119.Shen Z., Liu X.J., Long Z.C., Liu D.Y., Ye N.N., Qin J.H., Dai Z.P., Lin B.C. Electrophoresis. 2006;27:1084. doi: 10.1002/elps.200500689. [DOI] [PubMed] [Google Scholar]
- 120.Paegel B.M., Yeung S.H.I., Mathies R.A. Anal. Chem. 2002;74:5092. doi: 10.1021/ac0203645. [DOI] [PubMed] [Google Scholar]
- 121.Medintz I., Wong W.W., Berti L., Shiow L., Tom J., Scherer J., Sensabaugh G., Mathies R.A. Genome Res. 2001;11:413. doi: 10.1101/gr.164701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paegel B.M., Emrich C.A., Weyemayer G.J., Scherer J.R., Mathies R.A. Proc. Natl. Acad. Sci. U.S.A. 2002;99:574. doi: 10.1073/pnas.012608699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mitnik L., Carey L., Burger R., Desmarais S., Koutny L., Wernet O., Matsudaira P., Ehrlich D. Electrophoresis. 2002;23:719. doi: 10.1002/1522-2683(200203)23:5<719::AID-ELPS719>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 124.Goedecke N., McKenna B., El-Difrawy S., Carey L., Matsudaira P., Ehrlich D. Electrophoresis. 2004;25:1678. doi: 10.1002/elps.200305897. [DOI] [PubMed] [Google Scholar]
- 125.Zhou Z.M., Liu D.Y., Zhong R.T., Dai Z.P., Wu D.P., Wang H., Du Y.G., Xia Z.N., Zhang L.P., Mei X.D., Lin B.C. Electrophoresis. 2004;25:3032. doi: 10.1002/elps.200305966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou X.M., Shao S.J., Xu G.D., Zhong R.T., Liu D.Y., Tang J.W., Gao Y.N., Cheng S.J., Lin B.C. J. Chromatogr. B. 2005;816:145. doi: 10.1016/j.jchromb.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 127.Qin J.H., Liu Z.Y., Wu D.P., Zhu N., Zhou X.M., Fung Y.S., Lin B.C. Electrophoresis. 2005;26:219. doi: 10.1002/elps.200406158. [DOI] [PubMed] [Google Scholar]
- 128.Qin J.H., Leung F.C., Fung Y.S., Zhu D.R., Lin B.C. Anal. Bioanal. Chem. 2005;381:812. doi: 10.1007/s00216-004-2889-2. [DOI] [PubMed] [Google Scholar]
- 129.Liu D.Y., Shi M., Huang H.Q., Long Z.C., Zhou X.M., Qin J.H., Lin B.C. J. Chromatogr. B. 2006;844:32. doi: 10.1016/j.jchromb.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 130.Wang H., Qin J.H., Dai Z.P., Wang L., Bai J.L., Lin B.C. J. Sep. Sci. 2003;26:869. [Google Scholar]
- 131.Doherty E.A.S., Kan C.W., Paegel B.M., Yeung S.H.I., Cao S.T., Mathies R.A., Barron A.E. Anal. Chem. 2004;76:5249. doi: 10.1021/ac049721x. [DOI] [PubMed] [Google Scholar]
- 132.Buchholz B.A., Doherty E.A.S., Albarghouthi M.N., Bogdan F.M., Zahn J.M., Barron A.E. Anal. Chem. 2001;73:157. doi: 10.1021/ac001023z. [DOI] [PubMed] [Google Scholar]
- 133.Kaji N., Tezuka Y., Takamura Y., Ueda M., Nishimoto T., Nakanishi H., Horiike Y., Baba Y. Anal. Chem. 2004;76:15. doi: 10.1021/ac030303m. [DOI] [PubMed] [Google Scholar]
- 134.Zeng Y., Harrison D.J. Anal. Chem. 2007;79:2289. doi: 10.1021/ac061931h. [DOI] [PubMed] [Google Scholar]
- 135.Minc N., Futterer C., Dorfman K., Bancaud A., Gosse C., Goubault C., Viovy J.L. Anal. Chem. 2004;76:3770. doi: 10.1021/ac035246b. [DOI] [PubMed] [Google Scholar]
- 136.Meagher R.J., Coyne J.A., Hestekin C.N., Chiesl T.N., Haynes R.D., Won J.I., Barron A.E. Anal. Chem. 2007;79:1848. doi: 10.1021/ac061903z. [DOI] [PubMed] [Google Scholar]
- 137.Kopp M.U., de Mello A.J, Manz A. Science. 1998;280:1046. doi: 10.1126/science.280.5366.1046. [DOI] [PubMed] [Google Scholar]
- 138.Blazej R.G., Kumaresan P., Mathies R.A. Proc. Natl. Acad. Sci. U.S.A. 2006;103:7240. doi: 10.1073/pnas.0602476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bousse L., Mouradian S., Minalla A., Yee H., Williams K., Dubrow R. Anal. Chem. 2001;73:1207. doi: 10.1021/ac0012492. [DOI] [PubMed] [Google Scholar]
- 140.Thongboonkerd V., Songtawee N., Sritippayawan S. J. Proteome Res. 2007;6:2011. doi: 10.1021/pr060586+. [DOI] [PubMed] [Google Scholar]
- 141.Tabuchi M., Kuramitsu Y., Nakamura K., Baba Y. J. Proteome Res. 2003;2:431. doi: 10.1021/pr034009m. [DOI] [PubMed] [Google Scholar]
- 142.Renzi R.F., Stamps J., Horn B.A., Ferko S., VanderNoot V.A., West J.A.A., Crocker R., Wiedenman B., Yee D., Fruetel J.A. Anal. Chem. 2005;77:435. doi: 10.1021/ac049214f. [DOI] [PubMed] [Google Scholar]
- 143.Tan W., Fan Z.H., Qiu C.X., Ricco A.J., Gibbons I. Electrophoresis. 2002;23:3638. doi: 10.1002/1522-2683(200210)23:20<3638::AID-ELPS3638>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 144.Stoyanov A.V., Das C., Fredrickson C.K., Fan Z.H. Electrophoresis. 2005;26:473. doi: 10.1002/elps.200406170. [DOI] [PubMed] [Google Scholar]
- 145.Das C., Fan Z.H. Electrophoresis. 2006;27:3619. doi: 10.1002/elps.200600013. [DOI] [PubMed] [Google Scholar]
- 146.Cui H.C., Horiuchi K., Dutta P., Ivory C.F. Anal. Chem. 2005;77:7878. doi: 10.1021/ac050781s. [DOI] [PubMed] [Google Scholar]
- 147.Li Y., DeVoe D.L., Lee C.S. Electrophoresis. 2003;24:193. doi: 10.1002/elps.200390013. [DOI] [PubMed] [Google Scholar]
- 148.Mao X.L., Wang K., Du Y.G., Lin B.C. Electrophoresis. 2003;24:3273. doi: 10.1002/elps.200305598. [DOI] [PubMed] [Google Scholar]
- 149.Mao X.L., Luo Y., Dai Z.P., Wang K.Y., Du Y.G., Lin B.C. Anal. Chem. 2004;76:6941. doi: 10.1021/ac049270g. [DOI] [PubMed] [Google Scholar]
- 150.Shen Z., Liu X.J., Zhou X.M., Liang A.Y., Wu D.P., Yu L.F., Dai Z.P., Qin J.H., Lin B.C. J. Sep. Sci. 2007;30:1544. doi: 10.1002/jssc.200600530. [DOI] [PubMed] [Google Scholar]
- 151.Liu X.J., Liu X., Liang A.Y., Shen Z., Zhang Y., Dai Z.P., Xiong B.H., Lin B.C. Electrophoresis. 2006;27:3125. doi: 10.1002/elps.200500890. [DOI] [PubMed] [Google Scholar]
- 152.Liu X.J., Liang A.Y., Shen Z., Liu X., Zhang Y., Dai Z.P., Xiong B.H., Lin B.C. Electrophoresis. 2006;27:5128. doi: 10.1002/elps.200600294. [DOI] [PubMed] [Google Scholar]
- 153.Zhang B.L., Foret F., Karger B.L. Anal. Chem. 2001;73:2675. doi: 10.1021/ac001432v. [DOI] [PubMed] [Google Scholar]
- 154.Lazar I.M., Ramsey R.S., Sundberg S., Ramsey J.M. Anal. Chem. 1999;71:3627. doi: 10.1021/ac990373m. [DOI] [PubMed] [Google Scholar]
- 155.Mao X.L., Chu I.K., Lin B.C. Electrophoresis. 2006;27:5059. doi: 10.1002/elps.200600349. [DOI] [PubMed] [Google Scholar]
- 156.Hoffmann P., Hausig U., Schulze P., Belder D. Angew. Chem., Int. Ed. 2007;46:4913. doi: 10.1002/anie.200605152. [DOI] [PubMed] [Google Scholar]
- 157.Herr A.E., Molho J.I., Drouvalakis K.A., Mikkelsen J.C., Utz P.J., Santiago J.G., Kenny T.W. Anal. Chem. 2003;75:1180. doi: 10.1021/ac026239a. [DOI] [PubMed] [Google Scholar]
- 158.Chen X.X., Wu H.K., Mao C.D., Whitesides G.M. Anal. Chem. 2002;74:1772. doi: 10.1021/ac0109422. [DOI] [PubMed] [Google Scholar]
- 159.Li Y., Buch J.S., Rosenberger F., DeVoe D.L., Lee C.S. Anal. Chem. 2004;76:742. doi: 10.1021/ac034765b. [DOI] [PubMed] [Google Scholar]
- 160.Wang Y.C., Choi M.N., Han J.Y. Anal. Chem. 2004;76:4426. doi: 10.1021/ac0497499. [DOI] [PubMed] [Google Scholar]
- 161.Rocklin R.D., Ramsey R.S., Ramsey J.M. Anal. Chem. 2000;72:5244. doi: 10.1021/ac000578r. [DOI] [PubMed] [Google Scholar]
- 162.Gottschlich N., Jacobson S.C., Culbertson C.T., Ramsey J.M. Anal. Chem. 2001;73:2669. doi: 10.1021/ac001019n. [DOI] [PubMed] [Google Scholar]
- 163.Ramsey J.D., Jacobson S.C., Culbertson C.T., Ramsey J.M. Anal. Chem. 2003;75:3758. doi: 10.1021/ac0264574. [DOI] [PubMed] [Google Scholar]
- 164.Shadpour H., Soper S.A. Anal. Chem. 2006;78:3519. doi: 10.1021/ac0600398. [DOI] [PubMed] [Google Scholar]
- 165.Foote R.S., Khandurina J., Jacobson S.C., Ramsey J.M. Anal. Chem. 2005;77:57. doi: 10.1021/ac049136w. [DOI] [PubMed] [Google Scholar]
- 166.Lee J.H., Chung S., Kim S.J., Han J.Y. Anal. Chem. 2007;79:6868. doi: 10.1021/ac071162h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Song S., Singh A.K., Kirby B.J. Anal. Chem. 2004;76:4589. doi: 10.1021/ac0497151. [DOI] [PubMed] [Google Scholar]
- 168.Hatch A.V., Herr A.E., Throckmorton D.J., Brennan J.S., Singh A.K. Anal. Chem. 2006;78:4976. doi: 10.1021/ac0600454. [DOI] [PubMed] [Google Scholar]
- 169.Herr A.E., Hatch A.V., Throckmorton D.J., Tran H.M., Brennan J.S., Giannobile W.V., Singh A.K. Proc. Natl. Acad. Sci. U.S.A. 2007;104:5268. doi: 10.1073/pnas.0607254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Long Z.C., Liu D.Y., Ye N.N., Qin J.H., Lin B.C. Electrophoresis. 2006;27:4927. doi: 10.1002/elps.200600252. [DOI] [PubMed] [Google Scholar]
- 171.Huang H.Q., Xu F., Dai Z.P., Lin B.C. Electrophoresis. 2005;26:2254. doi: 10.1002/elps.200410393. [DOI] [PubMed] [Google Scholar]
- 172.Mohamadi M.R., Kaji N., Tokeshi M., Baba Y. Anal. Chem. 2007;79:3667. doi: 10.1021/ac0623890. [DOI] [PubMed] [Google Scholar]
- 173.Kelly R.T., Li Y., Woolley A.T. Anal. Chem. 2006;78:2565. doi: 10.1021/ac0521394. [DOI] [PubMed] [Google Scholar]
- 174.Liu J.K., Sun X.F., Farnsworth P.B., Lee M.L. Anal. Chem. 2006;78:4654. doi: 10.1021/ac060204j. [DOI] [PubMed] [Google Scholar]
- 175.Wang Y.C., Stevens A.L., Han J.Y. Anal. Chem. 2005;77:4293. doi: 10.1021/ac050321z. [DOI] [PubMed] [Google Scholar]
- 176.Dishinger J.F., Kennedy R.T. Anal. Chem. 2007;79:947. doi: 10.1021/ac061425s. [DOI] [PubMed] [Google Scholar]