

Abstract
The central importance of cell polarity control is emphasized by the frequency with which it is targeted by many diverse viruses. It is clear that in targeting key polarity control proteins, viruses affect not only host cell polarity, but also influence many cellular processes, including transcription, replication, and innate and acquired immunity. Examination of the interactions of different virus proteins with the cell and its polarity controls during the virus life cycles, and in virally‐induced cell transformation shows ever more clearly how intimately all cellular processes are linked to the control of cell polarity.
Keywords: Cell polarity, viruses
Abbreviations: ABP, apico-basal polarity; PCP, planar cell polarity; MCP, migrating cell polarity; ICP, immune cell polarity; Scrib, Scribble; Dlg1, Discs Large 1; Par, Partitioning defective; TJs, tight junctions; AJs, adherens junctions; HPV, human papillomavirus; EBV, Epstein–Barr virus; KSHV, Kaposi's sarcoma herpesvirus; PBM, PDZ-binding motif; EMT, epithelial-to-mesenchymal transition; HSV, herpes simplex virus; VZV, varicella zoster virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HCC, hepatocellular carcinoma; HIV-1, human immunodeficiency virus-1; HTLV-1, human T lymphotrophic virus-1; AdV, adenovirus; CAR, coxsackie and adenovirus receptor; TBEV, tick-borne encephalitis virus; DENV, Dengue virus; SARS, severe acute respiratory syndrome; MV, measles virus; RaV, rabies virus; CMV, cytomegalovirus; MCPyV, Merkel cell polyoma virus; IAV, Influenza A virus; WNV, West Nile virus; MuV, mumps virus
Graphical Abstract
Highlights
-
•
Multiple viruses target cell polarity.
-
•
Viral targeting of polarity frequently occurs through PDZ recognition.
-
•
Biological effects include immune-avoidance, cell proliferation and apoptosis inhibition.
Introduction
The control of cell polarity is a fundamental characteristic of multicellular organisms. Correct cell polarity leads to the ordered orientation of cells, both internally and with respect to one another, that is essential for the development of ordered systems, from intracellular organelles such as the endoplasmic reticulum, to the highly complicated networks of the brain. This being so, it is inevitable that the mechanisms and controls of cellular polarity will both affect and be affected by the presence of intracellular parasites such as viruses.
The control of normal cell polarity and tissue structure essentially defines most multicellular organisms, and its loss or misregulation essentially defines most tumors [1]. Cell polarity can be divided into four main types (shown in Fig. 1 ) that are highly conserved throughout evolution, with many vital regulatory elements being conserved from Drosophila melanogaster and Caenorhabditis elegans through to mice and humans [2]: apico-basal polarity (ABP), which describes the orientation of cells perpendicular to the basement membrane; planar cell polarity (PCP), describing their orientation parallel to it; migrating cell polarity (MCP), defining the leading and lagging edges of a migrating cell; and cell division polarity, defined by the difference between symmetric and asymmetric cell division in tissue differentiation. Additional, more tissue-specific, forms of polarity control include neuronal cell polarity, defining the direction of neuronal signal transmission, and immune cell polarity (ICP) induced in activated cells of the immune system, such as T lymphocytes, macrophages or dendritic cells [3].
Fig. 1.

Some types of cellular polarity control.
PCP is controlled by factors in the non-canonical Wnt/PCP pathway and by the interplay of a large number of signaling pathways in response to mechanical and biochemical forces [4], and PCP is established through the asymmetric and mutually exclusive localization of a highly conserved core set of proteins. These include the Scribble (Scrib) complex proteins, Scribble (Scrib) and Discs Large 1 (Dlg1), which are also critical regulators of ABP (see below), and which may represent the intersection of PCP and ABP controls [5]. PCP is essential for oriented cell division, collective cell migration and the development and function of many tissues [6].
ABP is a defining characteristic of epithelial cells and is also controlled by the intersection of several pathways, but at its core is a multi-macromolecular interaction network (Fig. 2 ) of three distinct protein complexes: the Scrib, Partitioning defective (Par) and Crumbs (Crbs) complexes [2], [7], [8]. These connect cell polarity with the control of cell–cell contacts via multiple scaffolding proteins at tight junctions (TJs) [9] and cadherins at adherens junctions (AJs) [10], [11]. They also connect ABP with cell proliferation control through mitogen-activated signaling pathways [12], [13], the control of cell size through the Hippo pathway [14], [15], and directional cell migration and apoptosis through small GTPase signaling [16], [17], [18]. The function of this network is critically dependent on a number of factors, including precise regulation of the expression level and post-translational modifications of the network components [2]. ABP regulation is fundamentally dependent on the mutually exclusive localization of different network components, which depends, at least in part, upon differential phosphorylation of the Scrib and Par complex proteins, some of which is regulated through the atypical protein kinase C [19], [20]. This differential localization of components divides the cell into different zones, each defined by the presence of one of the three complexes of the pathway (Fig. 2 and other chapters in this volume); perturbation of the correct spatio-temporal localization of the individual components has profoundly deleterious effects on the functioning of the whole complex.
Fig. 2.

Cell polarity pathways are targeted by diverse human viruses.
Interplay between the Scribble (SCRIB), PAR, and Crumbs (CRB) complexes is central to the control of cell polarity. These link to the TJ and AJs that control cell–cell contacts, and to the Wnt signaling pathway, through the SCRIB complex and cadherins. The CRB complex links to cell growth and proliferation through the Hippo signaling pathway and YAP/TAZ. The SCRIB complex also links to apoptotic pathways through β-PIX, and to growth factor signaling-induced proliferation through ERK, and to interferon-activated innate immune responses through STAT activation. The virus proteins are indicated by hexagons: red for cancer-associated virus proteins or blue for non-cancer causing.
Studies in lower eukaryotes, such as Drosophila and C. elegans, show that perturbing any one of the proteins that function in this network can have very adverse effects on the overall functioning of the ABP pathway and have defined many of these proteins as potential tumor suppressors [21]. However, in higher eukaryotes, the picture is more confused; although expression of many polarity regulators is lost in tumors [22], [23], it is often unclear whether this is a cause or a consequence of the tumor development. However, in recent years, studies have shown that many human viruses, both oncogenic and not, target certain key polarity regulators, demonstrating the fundamental involvement of the ABP control network not only in different viral replication strategies, but also in in tumor suppression and tumorigenesis. Targeting of many of these cell polarity regulators by oncogenic viruses, such as human papillomaviruses (HPV), has focused attention on the cell proliferative regulating activities of these proteins, and how this is connected to the control of polarity. However, many non-oncogenic viruses target many of the same pathways as a means of avoiding immune detection, both through innate and acquired mechanisms. This therefore highlights the incredible multifunctional nature of these cell polarity regulators and emphasizes why viruses targeting these pathways can achieve multiple aims, all of which contribute to a successful virus infection: the generation of a replication-permissive environment, and the avoidance of immune detection. In rare cases, the combination of such perturbation of cellular homeostasis with a long-term persistent infection can give rise to events that ultimately result in malignancy.
Tumor Viruses and Polarity Control
Tumor viruses have been estimated to cause up to 12% of all human cancers [24], [25]. These viruses, as stated above, are from widely differing evolutionary backgrounds and cause many different malignancies. However, they commonly attack the same pathways, targeting, either directly or indirectly, some of the central elements of polarity control [26], [27] (see Fig. 2). The purpose of this targeting, from the point of view of the virus, is to render the cellular environment more conducive to viral replication, and any resulting malignancy is a rare event and often prevents an effective viral life-cycle. This emphasizes the importance of host cell polarity regulation to the replication strategy of the virus.
The majority of tumor viruses exhibit long-term persistent infections in the host, infecting precursor cells and using the cellular differentiation program for their replication. Examples include B-cell precursors infected by Epstein–Barr virus (EBV), endothelial precursors infected by Kaposi's sarcoma herpesvirus (KSHV), and epithelial precursors infected by the HPVs [28], [29], [30]. Long-term or persistent infections may continue for years and in some cases are lifelong, allowing the viral oncoproteins, or the oncogenic by-products of the viral life cycles, to interact with the cellular environment over long periods and to initiate the processes that lead eventually to malignant transformation and cancer.
HPV
HPVs are major carcinogens, being responsible for around 5% of all human cancers worldwide [31]. HPV-related malignancies are characterized by their addiction to the two viral oncoproteins, E6 and E7, which contribute directly to the development and maintenance of the tumor and of cells derived therefrom [32], [33], even after many years in tissue culture [34]. They act together to immortalize keratinocytes [35], which are the natural target cells of the virus, and have been shown to cause tumors in transgenic mouse models [36], [37], [38]. Thus, E6 and E7 have the potential to be excellent therapeutic targets [39].
HPV replication occurs in terminally differentiating keratinocytes and is completely dependent upon the differentiation program of the epithelium [40]. The E6 and E7 proteins cooperate to reactivate the cell cycle in these cells; E7 induces cell cycle re-entry, while E6 abrogates the resulting pro-apoptotic responses [39], [41], [42]. While usually resulting in a productive infection of a few months' duration, this perturbation of cellular controls can initiate the steps that lead eventually to malignant transformation.
Polarity targeting by HPV E6
Of the nearly 200 known papillomavirus types, only a few high-risk HPV types are defined as cancer causing and, of these, HPV types 16 and 18 are the most clinically important. These viruses all have a PDZ (PSD-95/Dlg1/ZO1)-binding motif (PBM) at the C-terminus of their E6 proteins, which is largely absent from those of low-risk viruses, which cause only benign lesions. The high-risk E6 proteins are thus able to bind to a number of polarity control proteins that contain PDZ domains. These include core components of ABP cell polarity control—hDlg1, hScrib, and PATJ—and the TJ protein MAGI-1, which are targeted by E6 for ubiquitin-mediated degradation at the proteasome, thus disrupting the stoichiometric equilibrium of these complexes [43], [44], [45], [46]. However, it is becoming very clear that all HPV E6 PBMs are not equal, and that subtle differences in the exact amino acid sequence of the PBM can markedly alter the substrate selection, as can be seen from Table 1 , the HPV E6 PBMs, whilst all having a core consensus PBM, have significant variation in the non-canonical amino acid residues. Indeed these differences can profoundly affect substrate selection, and the molecular basis for these differences has been extensively explored [47], [48], [49], showing the effect that even minor changes can have; for example, HPV-16 E6 was shown to preferentially interact with hScrib, and HPV-18 E6 with Dlg, and swapping the final amino acid also swaps these preferences [50]. More recently, the greater functional flexibility of the E6 PBM (i.e., the greater the number of different PDZ domain-containing proteins that it can bind) has been found to correlate with the degree of association of that HPV type with cervical cancer [51]. Interestingly, all of the PBMs tested in that study, including those from HPV types rarely (HPV-26, HPV-66), or never (HPV-40), found in cancer, were able to interact with hDlg, suggesting that this is a fundamental requirement for the replication of this group of HPV types. Interaction with hScrib and the TJ protein ZO2, however, was limited to the PBMs of virus types with a strong cancer association (HPV types 16, 18, 31, 33, 35, 51), suggesting that the greater disruption of the Scrib complex caused by the combined loss of hScrib and hDlg1, particularly in the context of a long-term persistent infection, has a greater risk of disrupting cellular polarity in a manner that favors malignant transformation. Indeed, this has been recently confirmed in cellular transformation assays [52]. Human cervical keratinocytes exogenously expressing activated Ras, plus HPV-16 wild-type E6 and E7, are capable of anchorage-independent growth and are tumorigenic in nude mice. Expression in this system of an E6 defective in PDZ-binding prevents this, but the tumorigenic phenotype can be slightly rescued by the individual ablation of DLG1 or DLG4 (also known as PSD-95) and partially rescued by ablation of either hScrib, MAGI-1 or PAR3. However, ablation of hScrib plus DLG1 and DLG4, or hScrib plus MAGI1 and PAR3 in this system actually enhances the tumorigenic phenotype above that of cells expressing the wild-type HPV-16 E6 [52], demonstrating the pivotal importance of hScrib targeting in HPV-induced malignancy.
Table 1.
The PBMs of viral proteins targeting cell polarity and their potential phospho-control. Many of the viral proteins that target cell polarity control do so by binding the PDZ domains of polarity control proteins through a PBM. The table shows the sequences of such viral PBMs, their cellular targets, and the potential of the PBM to be modified by phosphorylation. The T/S amino acid residues in green have been shown to be phosphorylated by the indicated kinase [76], [263]; those in blue have consensus kinase recognition sites, but phosphorylation has not been formally demonstrated.

It is interesting to note that in the rhesus papillomavirus (MmPV1), which causes cervical cancer in rhesus macacques (Macaca mulata), E6 has no PBM but instead the E7 oncoprotein has a PBM [53]. This has a very different sequence from that of the HPV E6 PBMs and structurally would appear to be quite diverse [47], [49]. Not surprisingly, proteomic and biochemical analyses showed that it binds to a different polarity regulator, the PAR3 protein [53], part of the PAR complex. Thus, MmPV1 targets the same overall polarity regulatory pathway as HPVs, and through the same mechanism, but using a different viral protein and with different cellular partners, demonstrating a marked evolutionarily conserved requirement for these viruses to target this pathway for viral replication, and most likely for their ability to cause cancer.
Evolutionary conservation of the polarity proteins is also illustrated in a new animal model for HPV E6 activity, where HPV E6 can bind to the Drosophila MAGI protein in a PBM-dependent fashion, and, if exogenously supplied with a human UBE3A (E6AP) ubiquitin-protein ligase, can induce its degradation [54]. Intriguingly, this model shows that a key factor in HPV-induced malignancy is the E6AP, which is used by E6 for substrate degradation, but is also required for E6 stability [55]. The Drosophila E6AP lacks the E6 interaction motif, and hence, human E6AP is required for E6 to exert a phenotype in flies. Notably, although both Dlg and MAGI would appear to be perturbed in this model by E6, it is MAGI that appears to play the critical role in the tumor phenotype [54].
Cell polarity and the HPV life cycle
The life cycle of HPVs is completely dependent on the differentiation program of the infected epithelium, and studies of infection in organotypic raft cultures show that viruses defective in binding the polarity proteins produce fewer progeny virus and their genomes are more unstable and prone to integration in the host DNA [56], [57], [58].
The reason for loss of correct virus genome segregation in the absence of the E6 PBM is as yet unknown, although it may be related to the levels of cell proliferation in the lesion. The E6 PBM plays a key role in expanding the number of proliferating cells (i.e., those capable of replicating viral DNA), by targeting the polarity proteins to uncouple the link between cell polarity control and cell proliferation control. The orderly asymmetric cell division seen in the normal differentiating epithelium is maintained by strict control of mitotic spindle orientation, maintenance of ABP, and correct formation of the cell–cell junctions [59], [60]. The HPV E7 protein inappropriately stimulates cell cycle progression in the epithelial mid-layer, while E6 perturbs the Scrib and Par complexes to expand the population of infected cells capable of replicating the viral DNA. Expression of E7 alone has been shown to induce the formation of aberrant spindle poles [61], [62], while disruption of Dlg and the Par complex also perturbs mitotic spindle orientation [63], [64], all of which contributes to enhanced symmetrical cell division, thus expanding the population of replication-competent cells and accounting for the disordered epithelium observed in viral lesions.
As noted above, cell–cell communication through cell junctions is also affected in HPV-infected cells. MAGI-1 is targeted for degradation by high-risk HPV E6 proteins [65], possibly to counteract the signaling role of non-junctional MAGI-1 in the induction of apoptosis [66]. Indeed, when a mutant MAGI-1, which was no longer susceptible to E6-induced degradation, was re-expressed in HeLa cells, it was found to induce cell growth arrest and apoptosis [67]. On the other hand, E6 induces the stabilization of the TJ protein ZO-2, which appears to increase cell proliferation and enhance the wound healing ability of HeLa cells [68], [51]. In addition, both E6 and E7 target AJs by downregulation of E-cadherin, at least partly through the induction of Cdc6 [69], [70], [71], [72], thus increasing proliferative signaling. Potentially, this disruption of cell junction control, combined with enhanced proliferation and decreased apoptotic signaling, could also contribute to the disordered epithelial structure characteristic of HPV lesions and, further, increase the risk of pro-oncogenic mutations arising in those lesions.
Regulation of PBM/PDZ binding
It is becoming very clear that PBM/PDZ binding, while allowing proteins a wide flexibility in choice of binding partners, is subject to a greater level of specificity and control than was originally thought [73]. The steric and electrostatic characteristics of the respective PBM and PDZ sequences give an irreducible level of specificity to the interactions of the papillomavirus PBMs [47], [48], [49]. However, the HPV PBMs are also bi-functional: there is a phospho-acceptor site embedded within the PBM, which, when phosphorylated, prevents PDZ binding [74] and instead confers affinity for proteins of the 14-3-3 family [75]. Examples of phospho-acceptor sites on the PBMs of diverse viral proteins are shown in Table 1. These proteins link to a variety of different pathways, including Hippo and p53 regulation, while kinases such as AKT and PKA have been implicated in regulating E6 interaction with the cell polarity regulators [76]. Recent data also suggest that the E6 PBM may be phosphorylated by DNA damage response kinases, allowing E6 to modulate p53's transcriptional transactivation activity [77]. It seems likely that this would reduce p53's apoptotic response, while allowing the cellular DNA damage repair enzymes to enhance viral genome replication [78], [79]. Taken together with studies that show phospho-regulation of Dlg for E6 regulation [80], [81], these findings highlight a complex pattern of phospho-regulation controlling the E6 PBM interactions. Perturbation of these controls is likely to have profound consequences for successful viral life cycles, and for the potential progression to malignancy.
HPV-induced malignancy and cell polarity
As mentioned above, the productive HPV life cycle occurs in the differentiating squamous epithelium: the virus infects cells in the basal layer of the stratified epithelium and progeny virions are shed as the surface layer of cells sloughs off. There is considerable debate surrounding the identity of the cells that are subject to malignant transformation: are they squamous cells in which a normally productive infection has stalled and become persistent? or are they cells that do not stratify, such as the columnar cells of the simple epithelium of the endocervix? It is clear that the majority of cervical (> 99%) and anal (> 90%) tumors occur at the transformation zone between the stratified and columnar epithelia [82], and many cervical tumor cells have an embryonic stem cell expression signature [83]. It has been suggested that many cervical tumors may have their origin in the cuboidal cells of the squamocolumnar junction: these cells are more accessible than the endocervical cells; they are highly susceptible to infection by HPV, but are unable to stratify and permit a productive infection; and they are located in an anatomical site that appears to be somewhat immune-privileged. Thus, they appear to be convincing candidates for malignant transformation [84], [85], and interestingly, a similar population of cells has been described at the anorectal junction [84], where HPV is also a potent carcinogen. However, other studies suggest that they arise in a population of stem cells, known as reserve cells, that maintain both stratified and columnar epithelia, depending upon tissue context [86], [87], [30]. Interestingly, while the majority of HPV-16-induced cervical cancers are derived from the squamous cells of the ectocervix, HPV-18 is found in a very high proportion of adenocarcinomas [88], which would be consistent with infection of the columnar epithelium of the endocervix. These differences may reflect the finding that E6 PBMs from different HPV types have distinct binding profiles with respect to their cellular PDZ-containing targets, many of which are polarity control proteins [51], and it would be interesting to further determine whether the E6 PBM has any role in the frequency with which different HPV types are found associated with adenocarcinomas.
Studies in transgenic mice have shown that the E6–PDZ interaction is required for the ability of E6 and E7 to induce hyperplasia and tumors in the skin and in the cervix [89], [90], suggesting that deregulation of polarity control contributes to malignant transformation. This is further confirmed by the finding that E6 proteins mutated in the PBM lose the ability to induce an epithelial-to-mesenchymal transition (EMT) in keratinocytes and fail to cooperate with E7 in inducing cell transformation in certain in vitro settings [91]. E7 downregulates E-cadherin expression by epigenetic modification [70] and E6 also has a downregulatory effect, probably through the degradation of PDZ domain-containing polarity complex proteins leading to the perturbation of cell–cell adhesion complexes [91], [92].
Up to 20 different PDZ domain-containing targets of high-risk E6 proteins have been described [93], many of them targeted by E6 for ubiquitin-mediated degradation, and it is not clear which of these losses is/are associated with the induction of malignancy. Dlg1 was the first PDZ-containing target of HPV E6s to be identified [43] and seemed for some years to be a likely candidate, but recent work suggests that E6 PBMs from many HPV types can bind to Dlg1, regardless of the degree of association with cancer [51]. As shown in Fig. 3 , this may point more to this association being an adaptation for the viral life cycle and replication at mucocutaneous anatomical sites, rather than to an association with malignancy [94], and suggests that the ability to target hDlg1 may be a necessary but not sufficient feature of cancer-causing HPV types. Other E6 PBM degradation targets that have also been validated in vivo [44], [95], such as MAGI-1 and hScrib, are also associated with polarity control, and the ability to bind to hScrib appears to be confined to those HPV types with highest cancer risk [49]. The key element here appears to be the degree of flexibility of the E6 PBM, with the ability to bind multiple PDZ-containing cell polarity regulators conferring the ability to target multiple signaling pathways.
Fig. 3.

The contribution of HPV E6 PBM target selection to HPV-induced carcinogenesis. The evolutionary acquisition of a PBM on HPV E6 proteins is probably associated with the colonizing of a new niche—and is associated with Dlg1 binding, but not with oncogenicity. Further adaptations of the PBM allow binding to more diverse polarity proteins, thus targeting more pathways to enhance the viral life cycle, but potentially also perturbing controls of epithelial differentiation, seen clinically as CIN I (cervical intraepithelial neoplasia, grade 1). Further adaptation of the PBM leads to a more flexible target binding profile, greater polarity perturbation, mislocalization or deletion of PDZ polarity proteins, and the acquisition of pro-oncogenic properties by Dlg1 and hScrib, manifest as CIN III (cervical intraepithelial neoplasia, grade 3) and its potential progression to cancer. Restoration of functional polarity proteins can lead to apoptosis of cervical cancer cells and may indicate a possible direction for therapeutic strategies.
Interestingly, it has recently been shown that hScrib expression in HeLa cells is required to maintain E6 expression levels, through hScrib regulation of mTORC signaling [96]. In the development of HPV-induced tumors, disruption of polarity control appears to drive malignancy, with both hDlg1 and hScrib being mislocalized, and often overexpressed, in medium- to high-grade lesions [97], [98]. This suggests that in certain cellular locations, both hDlg1 and hScrib might have pro-oncogenic properties: studies that ablated each protein individually would seem to support this [96], [99], and this effect has also been seen in non-HPV related tumor development [100], [101], [102]. However, in late-stage cancers, hDlg1, hScrib, and E-cadherin are often lost, which is consistent with data from various experimental settings showing that under certain conditions, they can act as tumor suppressors [97], [100], [103].
Human Herpesviruses
The human herpesviruses are large, enveloped, double-stranded DNA viruses that establish lifelong latent infections, which can be periodically reactivated by various stimuli to cause a productive lytic infection.
Tumor-associated herpesviruses
There are two human tumor viruses in the herpesvirus family: KSHV, which causes cancers only infrequently, and mainly in immunosuppressed patients; and EBV, which causes a large number of diverse tumor types. A number of the viral proteins that are expressed to maintain the latent phase of infection have oncogenic potential [24].
KSHV can infect a wide range of cells, including lymphoid cells, endothelial cells, and keratinocytes [24]; it also expresses a number of potentially oncogenic proteins [29]. These include LANA, which is essential for the establishment and maintenance of KSHV latency [24]; LANA expression upregulates Par3 and SNAIL, leading to the downregulation of E-cadherin and the promotion of EMT [104], [105] In addition, the latency protein K15 upregulates miR-31 to inhibit the FAT4 cadherin [106], which has been implicated in the control of both apico-basal and planar polarity [107], [108], and is a component of the Hippo pathway [109]; thus, expression of the K15 protein is likely to have wide-ranging effects on ABP and PCP cellular polarity.
EBV infection is generally found in B-lymphocytes, but the virus is also thought to infect polarized epithelium through the formation of direct adhesions between the B cells and the basolateral surface of epithelial cells [110], [111], utilizing the endocytic recycling pathways of both donor and recipient cell to facilitate direct cell–cell transmission of the virus [112], [113]. It has also been suggested that the virus exploits the endosomal machinery to cross the epithelial cells of the oral mucosa, without initiating an infection, to directly infect the B lymphocyes in the underlying dermis [112], [114].
EBV, like all herpesviruses, persists latently for the life of the host in memory B cells and different latency states, characterized by specific gene-expression profiles, are associated with the development of specific tumor types [24]. None of the transforming proteins of EBV—EBNA1, LMP1, and LMP2a—has been detected in direct interaction with cell polarity regulators, but through upregulation of the TWIST, SNAIL, and SLUG transcription repressors, they severally downregulate expression of E-cadherin [115], [116], [117], [118], [119] and upregulate β-catenin [116], [120], [121]. In addition LMP1 stimulates CDC42 activation [122], leading to increased cell motility, and potentially to invasion, through CDC42's RhoGTPase activity, which is required for Scrib's ability to control polarized cell migration through actin remodeling [123], [124], [125]. Thus, although there is no direct interaction between KSHV or EBV proteins and the core cell polarity control proteins, they perturb many peripheral aspects of the pathway, leading to increased cell proliferation and an increased risk of malignant transformation.
Non-oncogenic herpesviruses
Herpes simplex viruses (HSV1 and HSV2), cytomegalovirus, and the varicella zoster virus (VZV) infect epithelial cells where the lytic, or productive, virus life cycle occurs and it is not clear whether the polarity of these cells is involved in the virus replication cycle. However, in plaque assays, HSV infection has been shown to induce polarized cell migration (MCP) in uninfected keratinocytes, which migrate toward the site of infection, and become infected in their turn, [126], while VZV glycoprotein E expression colocalizes with the ZO1 TJ protein and can increase cell–cell contacts and enhance TJ formation in low calcium, presumably to allow cell-to-cell spread of the virus particles [127]. The cytomegalovirus gpUS9 protein has similarly been reported to colocalize with cell junction components such as ZO1 and E-cadherin in polarized epithelial cells [128], although this finding has been disputed [129]. In addition to their lytic cycle, HSV and VZV form latent infections in the dorsal ganglia, forming reactivated lesions at the original site under certain stimuli, which argues a very sophisticated interaction with the neuronal cell polarity determinants of the neuronal cell. The glycosylated US9 and gE/gI proteins of both HSV and VZV, and also the rodent pseudorabies virus (PsRV) have been implicated in the microtubule-mediated ferrying of the virus capsid within the axon [130], [131], [132].
Thus, it appears that effects on the core regulators of cell polarity are seen unequivocally only with the tumorigenic herpesviruses, confirming again that perturbation of the correct functioning of polarity control is one of the primary features of malignancy.
Hepatitis Viruses
Hepatitis viruses are the major cause of hepatitis worldwide (World Health Organization) and chronic infections of hepatocytes with hepatitis B virus (HBV; a double-stranded DNA hepadnavirus) and hepatitis C virus (HCV; a single-stranded RNA flavivirus) are associated with over 80% of hepatocellular carcinomas (HCCs) [24]. Hepatocytes are polygonal multipolar cells, with multiple apical and basal surfaces and hepatic function is dependent upon the maintenance of hepatocyte PCP and the correct functioning of the hepatocyte's numerous TJs [133]. The high rates of cell division and double-strand DNA breaks characteristic of chronic hepatitis are thought to be the means by which HBV DNA integrates into the cellular genome, where it is found in more than 85% of HBV-induced HCCs [134]. Studies suggest that the integration site is random, but may promote the inappropriate activation of neighboring cellular genes, with potentially pro-oncogenic sequelae.
HBV expresses a protein, X (Hbx), that contributes to tumorigenesis through its effects on many cellular mechanisms, including polarity control pathways. The virus uses the various activities of Hbx to optimize viral replication through epigenetic modification and transcriptional reprogramming of the cell. It suppresses E-cadherin expression though epigenetic modification [135], as well as upregulating SNAIL expression and downregulating miR-373 expression [136], while also perturbing the β-catenin interaction with the APC tumor suppressor, thus increasing β-catenin levels [137], which, in turn, further downregulates E-cadherin. Hbx also upregulates YAP expression, thus stimulating cell proliferation [138]. The resulting perturbation of PCP promotes the EMT, which, in combination with the increased cell division, thus contributes to the development of HCC.
HCV is a very different virus from HBV, but given the similarity of their replicative environments and clinical outcomes, it is not surprising that they target a number of similar cellular pathways [139]. It is clear that HCV targets certain proteins of the TJ, including claudin, occludin, and the interferon-induced transmembrane protein 1 [140], [141], [142], during virus entry into the basolateral membrane of the hepatocyte [143]. In contrast, intact TJs have also been shown to be instrumental in defending the cell against HCV entry [144], [145]; however, virus envelope components have been shown to relocalize TJ components, possibly to prevent superinfection of the cell, but potentially leading to the compromise of junctional integrity [146], [147].
During infection, the HCV core protein has been shown to inhibit the SHIP2 phosphatase, leading to downregulation of Dlg1 and Scrib at the basolateral membrane. This disrupts both apico-basal and PCP and morphology and causes the formation of multi-lumen cysts, which compromise hepatocyte function [148], [149]. The core protein also induces methylation of the E-cadherin promoter, leading to its down-regulation [150], while the viral NS5A protein activates PI3 kinase activity, leading to increased β-catenin stability and upregulating c-Myc expression [151], [152], these activities together tending to promote EMT-associated changes in cell polarity. Such changes are further promoted by the synergy between NS5A activation of TWIST2 [153], while the viral core and Env proteins induce increased TGF-β signaling [154]. It has recently been reported that the NS4B protein binds to Scrib, through at least three PDZ domains, and induces its proteasome-mediated degradation [155]. This appears to protect the infected cell from Scrib-mediated apoptosis, a result that also appears to contribute directly to an increased likelihood of cell transformation. Thus, the virus uses multiple ways to manipulate host cell polarity control, which, in combination with chronic hepatic inflammation and hence accelerated cell division, can contribute to drive malignant transformation.
Merkel Cell Polyomavirus
MCPyV is the first polyomavirus found to be associated with a human cancer [156], [157], and over 80% of Merkel cell carcinomas have been found to harbor viral DNA sequences [156]. Although MCPyV infection is widespread in normal skin, a number of characteristics define it as a probable causative agent: clonal integration of the viral genome into the host cell DNA appears to precede tumor development [158], [159]; the tumor cells require persistent expression of the viral small T antigen (sT) to survive [160], [161]; and the association with immunesuppression strongly supports a viral etiology for the cancer [158].
Interactome analysis has shown that the MCPyV sT binds to a wide range of cellular proteins, most relevantly including CD44 and emerin [162]. CD44 is a widely expressed protein, involved in stabilizing the TJ, thus regulating epithelial barrier function and cell–cell communication [163]. Knockout of CD44 has been shown to result in mislocalization of Par3 to the cell membrane and disruption of the TJ barrier function in mouse skin. Emerin is a component of the LINC complex, which connects the nuclear lamina to the actin cytoskeleton [164]; it binds β-catenin, restricting its nuclear entry [165] and, at least in certain cell types, constraining the Wnt/β-catenin signaling [166] that is required for the MCPyV life cycle in human dermal fibroblasts [167]; and it therefore seems probable that the sT interaction with emerin would relieve this inhibition.
T Lymphotrophic Tumor Viruses
Two retroviruses that infect T lymphocytes, HIV-1 (human immunodeficiency virus-1) and HTLV-1 (human T lymphotrophic virus-1) are known to be cancer-causing.
HIV-1 is defined as tumorigenic owing to the tumors that develop as a direct result of its immunosuppressive impact on the host, but although tumors are common in HIV patients, HIV-1 is not a tumor virus in the classic sense. The cancers most commonly associated with HIV-1-infection are induced by other viruses: Kaposi's sarcoma, induced by KSHV; anogenital carcinomas induced by HPV; and lymphomas, induced mainly by EBV.
To further its life cycle, HIV-1 impinges upon several aspects of the cellular polarity control machinery. The virus envelope protein gp120 has been reported to decrease the ZO-1 levels at the TJ, and this may contribute to efficient virus entry [168]. Normal T-cell activation depends on cell polarity pathways, with Rac1, cdc42 and the Scrib/β-Pix complex signaling downstream to PAK at the immune synapse [169]. However, in HIV-1-infected T cells, the viral Nef protein activates PAK independently [170], inducing a pseudo-activated state that is favorable for viral replication [171], but which also induces anti-apoptotic signals [172], leading to increased survival of the infected cell and enhanced viral pathogenesis [173]. Late in infection, the viral Vpu protein has been shown to sequester β-TrcP in the cytoplasm [174], preventing its E3 ligase activity and thereby stabilizing Snail and β-catenin [175], destabilizing E-cadherin, and enhancing the release of progeny virus particles [176]. β-TrcP is similarly targeted by the Vaccinia virus A49 protein [177] and by the Rotavirus NSP1 protein [178]; these conserved virus strategies indicating a key point at which viral activity can redirect host cell functions to serve the virus's end. Fresh cycles of virus infection may also be enhanced by secreted HIV-1 Tat protein from already-infected cells, which can also result in decreased ZO-1 expression at the TJ in uninfected cells [179], [180].
HIV-1 thus perturbs ICP cellular polarity to enhance the efficiency of virus infection, leading to host immunosuppression and increased susceptibility to cancers induced by other viruses.
Unlike HIV-1, HTLV-1 is a true tumor virus, being a direct cause of adult T-cell leukemia, and maintenance of the malignancy, as with HPV-induced tumors, depends on the continued expression of a viral oncogene, in this case the viral HBZ gene. The Tax oncoprotein, although lost in many later-stage T-cell leukemia cells, is required for viral replication and is implicated in many of the steps that lead to tumor development. It has a C-terminal PBM, through which, like HPV E6, it binds a number of proteins involved in cell polarity control, including Dlg, hScrib, MAGI-1, MAGI-3, and Lin7 [181], [182], [183], [184], [185], [186]. Tax binding to these proteins results, at least in part, in their aggregation or mislocalization within the cell [184], [187], [188]. Since Tax competes with PTEN for binding to Dlg [189], this attenuates PTEN-mediated restriction of the PI3K/Akt pathway [190], increasing Akt activation and enhancing cell proliferation. As with HPV E6, the Tax PBM has been shown to be required for persistent viral infection [191], for IL2-independent growth of HTLV-1-infected T cells [192], and also for its transforming potential in cooperation with NF-κB [193], [194]. The importance of the PBM to the carcinogenic phenotype of HTLV-1 is underlined by its absence from the Tax protein of non-oncogenic HTLV-2 [182].
The viral envelope glycoprotein Env also has a PBM, through which it binds hDlg1 [195] and recruits it to the virological synapse that forms between neighboring T cells, and which is thought to be a means of transmitting virus directly from cell to cell. It is thought that the recruitment of hDlg1 may stabilize these synapses to enhance viral transmission [196]. This process is also enhanced by Tax, which is closely associated with the microtubule organizing center (MTOC) on the cell surface, to which it recruits intercellular adhesion molecule 1 (ICAM-1) [197], [198], thus activating MEK/ERK signaling and inducing the reorganization of MTOC polarity to form the virological synapse [199]. The involvement of ERK signaling may also suggest a role for hScrib in this process [13]. Thus, the virus exploits the normal cell polarity machinery to enhance its ability to enter cells directly, while evading any risk of immune detection [200].
Non-tumorigenic Viruses
Adenoviruses
Adenoviruses (AdVs) are large DNA viruses with linear double-stranded genomes; they usually cause self-limiting, lytic respiratory or gastrointestinal infections in humans. AdV infection occurs through the virion binding to the coxsackie and adenovirus receptor (CAR), which is located on the basolateral cell surface and is essential for TJ integrity [201]. CAR has a C-terminal PBM that mutational studies suggest is not involved in CAR's basolateral targeting, but is required for interactions with other PDZ domain-containing TJ proteins: MAGI-1, ZO-1, and DLG4, amongst others [202]. Thus, the initial steps of AdV infection involve interaction with proteins essential in maintaining correct cell–cell interactions and polarity.
AdVs are not usually associated with any tumorigenic activity in humans; however, the human adenovirus 9 (AdV9) has, uniquely, been long been known to have transformation potential in animal cells and can cause tumors in laboratory rodents [203]. In human cells, it has no such effect, but it has been used as a model for virus-induced tumorigenesis [204].
The main oncoprotein of AdV 9 is the early-expressed E4-ORF1 protein, which has at its C-terminus the first virus PBM to be discovered [181], [205]. Through this, E4-ORF1, like high-risk HPV E6, binds to Dlg, MUPP, MAGI-1, ZO-2, and PATJ [45], [181], [206], [207], [208]. Unlike E6, E4-ORF1 does not induce the degradation of its PDZ-containing targets, but sequesters or relocates them. The E4-ORF1 protein exists in either monomeric or trimeric states, with different binding specificities. Monomeric E4-ORF1 binds to its PDZ-containing targets associated with the TJ (MUPP1, MAGI-1, and ZO-2), and with the Crumbs cell polarity complex (MUPP1 and PATJ), sequestering them in insoluble complexes within the cytoplasm, thus disrupting ABP control and compromising the TJ barrier function [209]. In contrast, the trimeric form of E4-ORF1 binds only to Dlg, relocating it to the plasma membrane and, as with the HTLV-1 Tax/Dlg interaction [189], resulting in the constitutive activation of the PI3K signaling pathway [210], [211]. Thus, under certain circumstances, perturbation of Dlg by E4-ORF1 turns Dlg into a pro-oncogenic factor [212], [213], resulting in increased cell cycle turnover, and thus probably an enhanced environment for AdV9 replication; however, it is likely to also increase the potential for EMT and the risk of malignant transformation. Thus, in enhancing the virus life cycle, the AdV9 E4-ORF1 can compromise both cell junction integrity and cell polarity control. There is a clear parallel here with the early stages of HPV-induced cervical cancer [98], where mislocalization and upregulation of Dlg may also play a pro-oncogenic role. Both of these cases thus underline the multifunctional nature of the polarity control proteins, and the contribution that the complex interplay of their function and location makes to the determination of cell fate.
Orthomyxoviruses
These are negative-strand RNA viruses, of which the best-studied is Influenza A virus, which infects the respiratory epithelia of many birds and mammals [214]. Indeed, the frequent transmission of different influenza virus serotypes between host species renders it a major global health risk for many species of economic importance, such as chickens and pigs, as well as humans.
The influenza A virus non-structural protein NS1 is a multifunctional virulence factor that has a number of protein interaction domains, including an SH3 domain, through which it binds and activates PI3K [215], [216], [217], leading to Akt activation and enhanced cell growth, and also inhibiting pro-apoptotic factors, thus extending the life-span of the infected cell to allow enhanced viral replication [218], [219], [220].
The vast majority of NS1 proteins analyzed also have a C-terminal PBM, the presence and sequence of which correlate with viral virulence, since it is often mutated or lost in attenuated viruses [221], [222], [223]. Interestingly, the avian and human subtypes of influenza A virus have quite different and characteristic PBM sequences (ESEV and RSKV, respectively), which differ in their target specificities, and with their virulence in mammalian cells [224], [225]. The avian-type PBM binds strongly to the cellular polarity proteins Scrib, Dlg, MAGI-1, MAGI-2, MAGI-3, PDLIM2, and PSD-95 (Dlg4) [226], [227], [228], [229], while the human-type PBM binds more weakly or not at all. The interaction with Scrib relocalizes Scrib from the plasma membrane to cytoplasmic puncta, protecting infected cells from Scrib's pro-apoptotic functions [226], and reducing interferon-induced STAT activation [227], an effect also seen with the Tick-Borne Encephalitis virus (TBEV) NS5 protein [230]. Dlg has also been shown to localize with Scrib in these cytoplasmic puncta, disrupting cell–cell junctions and ABP and PCP polarity control [231]. The NS1 PBM binding also colocalizes MAGI-1 in these puncta, potentially perturbing its regulation of interferon-β signaling [232]. The Dlg/Scrib interaction has also recently been suggested to be involved in dendritic cell maturation, and to be targeted there by NS1, thus potentially leading to evasion of both innate and acquired immune systems [219], [233].
Flaviviruses
These are single-stranded RNA viruses that are mainly zoonotic and transmitted to mammals by insect vectors. TBEV causes a severe encephalitis in humans, with a high mortality rate (20%–30%). The TBEV NS5 protein, and that of the closely related West Nile Virus (WNV), is an RNA-dependent RNA polymerase that is essential for replication of the virus genome, but it also has a rare internal PBM, through which it binds to Scrib and relocates to the plasma membrane. Binding Scrib blocks interferon-mediated JAK/STAT signaling, presumably to evade the host's innate immune response [230]. TBEV and WMV NS5 also bind to ZO-1, apparently stabilizing it at the TJ, and also binds to the RIMS2 protein [234], [235], [236], a PDZ-containing neurone-specific protein, [237] reflecting the viruses' neuronal etiology. However, it is not yet clear what role this NS5/ZO-1/RIMS complex may play in the virus infectious cycle.
Dengue virus (DENV), which causes the severe febrile illness Dengue fever, is also insect-borne and has an NS5 protein very similar to the TBEV NS5, and which also binds ZO-1 through its internal PBM [234], but in the case of DENV, ZO-1 appears to be sequestered in the nuclear or perinuclear regions of the infected endothelial cell. DENV infection also increases the expression of macrophage inhibitory factor, probably to evade the host immune response and this may also displace ZO-1 from the TJ [238]. The resulting TJ impairment is probably responsible for the vascular leakage that is a hallmark of severe Dengue infection [239], [240].
These studies highlight unexpected roles for ABP cell polarity regulation in the control of immune signaling, and it seems likely that some of the well-studied human oncogenic viruses which target these cell polarity regulators are also taking advantage of the immune avoidance opportunities that these interactions provide, and not just promoting an increase in cell proliferation.
Coronaviruses
Coronaviruses are large single- and positive-stranded enveloped RNA viruses which infect the gastrointestinal and respiratory tracts of birds and mammals. The most studied human coronavirus, SARS-CoV, causes severe acute respiratory syndrome (SARS) and its original major outbreak is thought to have originated as a zoonosis from a bat coronavirus. Like most coronaviruses, SARS-CoV infects polarized epithelium specifically through the apical cell membrane [241], [242], [243]. The virus envelope protein (E) has a C-terminal PBM, through which it binds to the cellular PALS protein [244], a component of the Crumbs polarity complex that defines the apical domain, and controls the establishment of apicobasal polarity and the formation of TJs. In infected cells, PALS relocalizes specifically to the perinuclear regions of the Golgi, where virus assembly occurs. Although the location of other TJ proteins, such as ZO-1, does not appear to be affected, the formation of TJs was shown to be aberrant in SAR-CoV-infected cells, and apicobasal polarity is defective in cyst formation of MDCK cells ectopically expressing E, but not EΔPBM. It is not clear whether SARS-CoV requires the E/PALS interaction for virus entry, but the interaction may contribute to virus budding. The importance of the Crumbs complex [245], [246] in the control of polarity is shown clearly here; the mislocalization of just one Crm component completely disrupting the epithelial integrity, which may, in turn, be one cause of the particularly severe pathology of SARS.
The MERS (Middle East respiratory syndrome)-CoV, in contrast, can enter and exit cells through both apical and basolateral membranes [247], and viral release occurs through inducing apoptosis in the infected cell, but no specific interactions with the polarity proteins have been reported.
Paramyxoviruses
These are enveloped, negative-, and single-strand RNA viruses, which include the highly contagious measles, mumps and (now-eradicated) Rinderpest viruses. The measles virus (MV) is spread by aerosols, but curiously does not directly infect the respiratory epithelium. Instead, in the alveoli it infects activated, and hence polarized, myeloid cells, through binding of the virus H glycoprotein to the signaling lymphocyte activation molecule on the cell surface [248]. The virus then replicates extensively in myeloid cells in the lymphatic organs, and ultimately the circulating infected myeloid cells deliver the viral genomes directly to the columnar epithelial cells of the upper airway. Making use of the ABP of these cells, the virus enters via their basolateral surfaces, through binding to the AJ protein Nectin 4 [249], [250]. The Nectin 4 connection, via Afadin, to the actin cytoskeleton is then required for MV infection to spread directly from cell to cell through the formation of pores in the lateral surfaces of adjacent epithelial cells [251]. This results in the formation of large infectious centers in the tracheal epithelium, which are thought to eventually detach and be expelled in aerosols, facilitating transmission of the virus. Thus, the life cycle of MV uses both the ICP of activated myeloid cells in the lungs to infect the host and the ABP of tracheal epithelium as a means of egress and further cycles of contagion.
The mumps virus (MuV) differs from MV, as its receptor, sialic acid, is expressed on both apical and basolateral membranes, allowing its initial infection via the apical surface of airway epithelia, and its secondary infection in the viremic phase through the basolateral surfaces of the epithelia of the various target organs of MuV. Virus release, however, occurs solely from the apical surface of epithelial cells and requires the Rab11 protein, a protein found in recycling endosomes and characteristic of differentiated epithelium [252], [253], again showing virus exploitation of normal cellular mechanisms.
Rhabdoviruses
Rabies virus (RaV) is a small single- and negative-strand RNA virus that infects the neuronal tissue of a very wide host range, causing an acute, and usually fatal, encephalitis. The virus infects the peripheral nervous tissue from wounds or bites, and travels along the axon to the central nervous system. The RV glycoprotein (G) has a C-terminal PBM [254] that appears to be a virulence factor, depending upon its amino acid sequence, similar to that of the Influenza A virus NS1. The PBM of virulent RaV strains (QTRL) has only one known binding partner: it binds with very high specificity to the MAST2 serine–threonine kinase. MAST2 is a normal binding partner of PTEN, which it phosphorylates, allowing phospho-PTEN to enter the nucleus and inhibit the PI3kinase/Akt pathway, limiting neurosurvival. The virulent RaV G protein binds to MAST2 preventing PTEN phosphorylation and leading to Akt activation, thus promoting neuronal cell survival [255]. The PBM of attenuated RaV (ETRL) is more promiscuous in its binding: it does not compete with PTEN for MAST2 binding, instead it binds to a number of PDZ-containing proteins, including Dlg2 (PSD-93) and MUPP1. It also binds to the tyrosine phosphatase PTPN4, which binding promotes apoptosis and neuronal cell death [256], [257]. Interestingly, in the absence of any binding ligand, the PDZ domain of PTPN4 inhibits the catalytic activity of the PTP domain, while ligand binding activates the PTP activity [258]. This lends further weight to the notion that certain PDZ domains may not be simply molecular interactors, but might also act in the dynamic regulation of signaling pathways, suggesting yet another means by which viruses may exert control over specific cellular processes.
The identification of the differences in virus targets between virulent and non-virulent strains of RaV has practical applications: studies are ongoing to identify peptides that could be used therapeutically, either binding MAST2 to promote neuro-regeneration or binding PTPN4 as oncolytic therapies in neuronal tumors [259], [260]. The apoptotic bodies induced by non-virulent RaV are strong potentiators of the antiviral immune response [261], which may potentially contribute to vaccine design [256]. In addition, they are being examined as predictors of neurovirulence in the safety testing of live viral vaccines, such as those against yellow fever, polio, or measles [262].
Conclusions
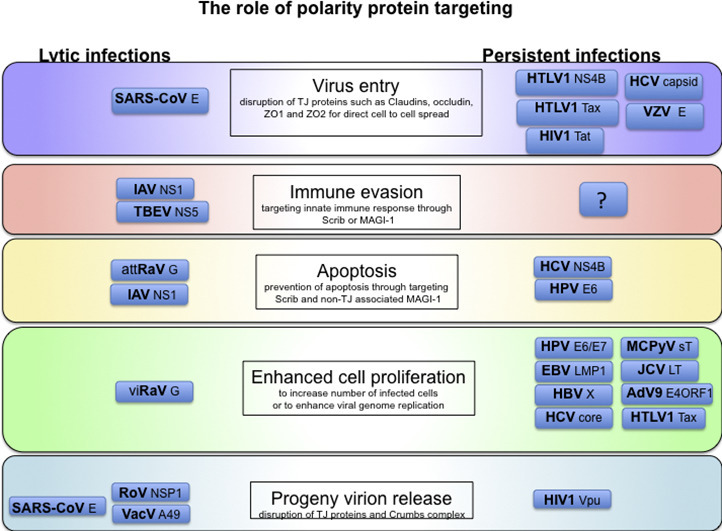
The central importance of cell polarity control is emphasized by the frequency with which it is targeted by many diverse viruses. Figure 4 shows an overview of the role of polarity protein targeting in persistent and lytic virus infections. It is clear that in targeting key polarity control proteins, viruses not only affect host cell polarity, but also influence many cellular processes, including transcription, replication, and innate and acquired immunity. Examination of the interactions of different virus proteins with the cell and its polarity controls during the virus life cycles, and in virally‐induced cell transformation, shows ever more clearly how intimately all cellular processes are linked to the control of cell polarity. Viruses act as signposts, indicating the key cellular pathways and controls that maintain cellular and tissue homeostasis.
Fig. 4.

The role of polarity targeting in virus life cycles. Cellular polarity control is targeted by many diverse virus types to enhance their productive life-cycle. However, the effects of the targeting are somewhat different. Lytic viruses, such as influenza A virus (IAV), tend to have a short replication cycle within each infected cell, and the polarity targeting contributes to immune evasion and virus release. Persistent viruses, such as HPVs, have a long replicative cycle, and the polarity targeting tends more towards activating the life-span and proliferation of the infected cell to increase the pool of cells harboring viral genomes and thus to increase the productivity of infection.
Acknowledgments
We are most grateful to all members of the Banks Lab and other colleagues for many fruitful discussions. L.B. acknowledges research support provided by the Associazione Italiana per la Ricerca sul Cancro (Grant No. 18578).
Declarations of Interest: None.
Edited by Marc Kvansakul
References
- 1.McCaffrey L.M., Macara I.G. Epithelial organization, cell polarity and tumorigenesis. Trends Cell Biol. 2011;21:727–735. doi: 10.1016/j.tcb.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Belmonte F., Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer. 2010;12:23–38. doi: 10.1038/nrc3169. [DOI] [PubMed] [Google Scholar]
- 3.Stephens R., Lim K., Portela M., Kvansakul M., Humbert P.O., Richardson H.E. The Scribble cell polarity module in the regulation of cell signalling in tissue development and morphogenesis. J. Mol. Biol. 2018 Feb 7 doi: 10.1016/j.jmb.2018.01.011. (pii: S0022-2836(18)30037-8) [DOI] [PubMed] [Google Scholar]
- 4.Devenport D. The cell biology of planar cell polarity. J. Cell Biol. 2014;207:171–179. doi: 10.1083/jcb.201408039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milgrom-Hoffman M., Humbert P.O. Regulation of cellular and PCP signalling by the Scribble polarity module. Semin. Cell Dev. Biol. 2017 Nov 22 doi: 10.1016/j.semcdb.2017.11.021. (pii: S1084-9521(16)30335-4) [DOI] [PubMed] [Google Scholar]
- 6.Devenport D. Tissue morphodynamics: translating planar polarity cues into polarised cell behaviours. Semin. Cell Dev. Biol. 2016;55:99–110. doi: 10.1016/j.semcdb.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assémat E., Bazellières E., Pallesi-Pocachard E., Le Bivic A., Massey-Harroche D. Polarity complex proteins. Biochim. Biophys. Acta. 2008;1778:614–630. doi: 10.1016/j.bbamem.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Ellenbroek S.I.J., Iden S., Collard J.G. Cell polarity proteins and cancer. Semin. Cancer Biol. 2012;22:208–215. doi: 10.1016/j.semcancer.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 9.Tsukita S., Yamazaki Y., Katsuno T., Tamura A., Tsukita S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene. 2008;27:6930–6938. doi: 10.1038/onc.2008.344. [DOI] [PubMed] [Google Scholar]
- 10.Qin Y., Capaldo C., Gumbiner B.M., Macara I.G. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J. Cell Biol. 2005;171:1061–1071. doi: 10.1083/jcb.200506094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georgiou M., Marinari E., Burden J., Baum B. Cdc42, Par6, and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr. Biol. 2008;18:1631–1638. doi: 10.1016/j.cub.2008.09.029. [DOI] [PubMed] [Google Scholar]
- 12.Dow L.E., Elsum I.A., King C.L., Kinross K.M., Richardson H.E., Humbert P.O. Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene. 2008;27:5988–6001. doi: 10.1038/onc.2008.219. [DOI] [PubMed] [Google Scholar]
- 13.Nagasaka K., Pim D., Massimi P., Thomas M., Tomaić V., Subbaiah V.K., Kranjec C., Nakagawa S., Yano T., Taketani Y., Myers M., Banks L. The cell polarity regulator hScrib controls ERK activation through a KIM site-dependent interaction. Oncogene. 2010;29:5311–5321. doi: 10.1038/onc.2010.265. [DOI] [PubMed] [Google Scholar]
- 14.Cordenonsi J., Zanconato F., Azzolin L., Forcato M., Rosato A., Frasson C., Inui M., Montagner M., Parenti A.R., Poletti A., Daidone M.G., Dupont S., Basso G., Bicciato S., Piccolo S. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- 15.Varelas X., Samavarchi-Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B.G., Rossant J., Wrana J.L. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell. 2006:2395–2405. doi: 10.1016/j.devcel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Osmani N., Vitale N., Borg J.P., Etienne-Manneville S. Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr. Biol. 2006;16:2395–2405. doi: 10.1016/j.cub.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 17.Dow L.E., Kauffman J.S., Caddy J., Zarbalis K., Peterson A.S., Jane S.M., Russell S.M., Humbert P.O. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene. 2007;26:2272–2282. doi: 10.1038/sj.onc.1210016. [DOI] [PubMed] [Google Scholar]
- 18.Zhan L., Rosenberg A., Bergami K.C., Yu M., Xuan Z., Jaffe A.B., Allred C., Muthuswamy S.K. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for polarity in carcinoma. Cell. 2008;135:865–878. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagai-Tamai Y., Mizuno K., Hirose T., Suzuki A., Ohno S. Regulated protein–protein interaction between aPKC and PAR-3 plays an essential role in the polarization of epithelial cells. Genes Cells. 2002;7:1161–1171. doi: 10.1046/j.1365-2443.2002.00590.x. [DOI] [PubMed] [Google Scholar]
- 20.Betschinger J., Mechtler K., Knoblich J.A. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. 2003;422:326–330. doi: 10.1038/nature01486. [DOI] [PubMed] [Google Scholar]
- 21.Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev. 2004;18:1909–1925. doi: 10.1101/gad.1211604. [DOI] [PubMed] [Google Scholar]
- 22.Gardiol D., Zacchi A., Petrera F., Stanta G., Banks L. Human discs large and scribble are localized at the same regions in colon mucosa and changes in their expression patterns are correlated with loss of tissue architecture during malignant progression. Int. J. Cancer. 2006;119:1285–1290. doi: 10.1002/ijc.21982. [DOI] [PubMed] [Google Scholar]
- 23.Pearson H.B., Perez-Mancera P.A., Dow L.E., Ryan A., Tennstedt P., Bogani D., Elsum I., Greenfield A., Tuveson D.A., Simon R., Humbert P.O. SCRIB expression is deregulated in human prostate cancer, and its deficiency in mice promotes prostate neoplasia. J. Clin. Invest. 2011;121:4257–4267. doi: 10.1172/JCI58509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.vol. 100B. International Agency for Research on Cancer; Lyon: 2012. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Biological Agents. (ISBN 978 92 832 1319 2) [Google Scholar]
- 25.Bouvard V., Baan R., Straif K., Grosse Y., Secretan B., El Ghissassi F., Benbrahim-Tallaa L., Guha N., Freeman C., Galichet L., Cogliano V., WHO International Agency for Research on Cancer Monograph Working Group A review of human carcinogens—Part B: biological agents. Lancet Oncol. 2009;10:321–322. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- 26.Moore P.S., Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer. 2010;10:878–889. doi: 10.1038/nrc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banks L., Pim D., Thomas M. Human tumour viruses and the deregulation of cell polarity in cancer. Nat. Rev. Cancer. 2012;12:877–886. doi: 10.1038/nrc3400. [DOI] [PubMed] [Google Scholar]
- 28.Katamine S., Otsu M., Tada K., Tsuchiya S., Sato T., Ishida N., Honjo T., Ono Y. Epstein–Barr virus transforms precursor B cells even before immunoglobulin gene rearrangements. Nature. 1984;309:369–372. doi: 10.1038/309369a0. [DOI] [PubMed] [Google Scholar]
- 29.Mesri E.A., Cesarman E., Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer. 2010;10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egawa N., Egawa K., Griffin H., Doorbar J. Human papillomaviruses; epithelial tropisms, and the development of neoplasia. Viruses. 2015;16:3863–3890. doi: 10.3390/v7072802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Martel C., Plummer M., Vignat J., Franceschi S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer. 2017;141:664–670. doi: 10.1002/ijc.30716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshinouchi M., Yamada T., Kizaki M., Fen J., Koseki T., Ikeda Y., Nishihara T., Yamato K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. 2003;8:762–768. doi: 10.1016/j.ymthe.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Magaldi T.G., Almstead L.L., Bellone S., Prevatt E.G., Santin A.D., DiMaio D. Primary human cervical carcinoma cells require human papillomavirus E6 and E7 expression for ongoing proliferation. Virology. 2012;422:J114–124. doi: 10.1016/j.virol.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butz K., Ristriani T., Hengstermann A., Denk C., Scheffner M., Hoppe-Seyler F. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene. 2003;22:5938–5945. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- 35.Barbosa M., Schlegel R. The E6 and E7 genes of HPV-18 are sufficient for inducing two-stage in vitro transformation of human keratinocytes. Oncogene. 1989;4:1529–1532. [PubMed] [Google Scholar]
- 36.Lambert P.F., Pan H., Pitot H.C., Liem A., Jackson M., Griep A.E. Epidermal cancer associated with expression of human papillomavirus type 16 E6 and E7 oncogenes in the skin of transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 1993;90:5583–5587. doi: 10.1073/pnas.90.12.5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song S., Liem A., Miller J.A., Lambert P.F. Human papillomavirus type 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267:141–150. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- 38.Riley R.R., Duensing S., Brake T., Münger K., Lambert P.F., Arbeit J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–4871. [PubMed] [Google Scholar]
- 39.Thomas M., Narayan N., Pim D., Tomaić V., Massimi P., Nagasaka K., Kranjec C., Gammoh N., Banks L. Human papillomaviruses, cervical cancer and cell polarity. Oncogene. 2008;27:7018–7030. doi: 10.1038/onc.2008.351. [DOI] [PubMed] [Google Scholar]
- 40.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond.) 2006;110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 41.McLaughlin-Drubin M.E., Münger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143:195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moody C., Laimins L.A. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 43.Gardiol D., Kuhne C., Glaunsinger B., Lee S.S., Javier R., Banks L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene. 1999;18:5487–5496. doi: 10.1038/sj.onc.1202920. [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa S., Huibregtse J. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6-AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000;20:8244–8253. doi: 10.1128/mcb.20.21.8244-8253.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glaunsinger B., Lee S.S., Thomas M., Banks L., Javier R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene. 2000;19:1093–1098. doi: 10.1038/sj.onc.1203906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storrs C., Silverstein S. PATJ, a tight junction-associated PDZ protein, is a novel degradation target of high-risk human papillomavirus E6 and the alternatively spliced isoform 18E6*. J. Virol. 2007;8:4080–4090. doi: 10.1128/JVI.02545-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y., Dasgupta J., Ma R.Z., Banks L., Thomas M., Chen X.S. Structures of a human papillomavirus (HPV) E6 polypeptide bound to MAGUK proteins: mechanisms of targeting tumor suppressors by a high-risk HPV oncoprotein. J. Virol. 2007;81:3618–3626. doi: 10.1128/JVI.02044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas M., Dasgupta J., Zhang Y., Chen X., Banks L. Analysis of specificity determinants in the interactions of different HPV E6 proteins with their PDZ domain-containing substrates. Virology. 2008;376:371–378. doi: 10.1016/j.virol.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 49.Charbonnier S., Stier G., Orfanoudakis G., Kieffer B., Atkinson R.A., Travé G. Defining the minimal interacting regions of the tight junction protein MAGI-1 and HPV16 E6 oncoprotein for solution structure studies. Protein Expr. Purif. 2008;60:64–73. doi: 10.1016/j.pep.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 50.Thomas M., Massimi P., Navarro C., Borg J.-P., Banks L. The hScrib/Dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene. 2005;24:6222–6230. doi: 10.1038/sj.onc.1208757. [DOI] [PubMed] [Google Scholar]
- 51.Thomas M., Myers M.P., Massimi P., Guarnaccia C., Banks L. Analysis of multiple HPV E6 PDZ interactions defines type-specific PDZ fingerprints that predict oncogenic potential. PLoS Pathog. 2016;12(8) doi: 10.1371/journal.ppat.1005766. (doi: 10.1371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshimatsu Y., Nakahara T., Tanaka K., Inagawa Y., Narisawa-Saito M., Yugawa T., Ohno S.-I., Fujita M., Nakagama H., Kiyono T. Roles of the PDZ-binding motif of HPV 16 E6 protein in oncogenic transformation of human cervical keratinocytes. Cancer Sci. 2017;108:1303–1309. doi: 10.1111/cas.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tomaić V., Gardiol D., Massimi P., Ozbun M., Myers M., Banks L. Human and primate tumour viruses use PDZ binding as an evolutionarily conserved mechanism of targeting cell polarity regulators. Oncogene. 2009;28:1–8. doi: 10.1038/onc.2008.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Padash Barmchi M., Gilbert M., Thomas M., Banks L., Zhang B., Auld V.J. A Drosophila model of HPV E6-induced malignancy reveals essential roles for Magi and the insulin receptor. PLoS Pathog. Aug 18 2016;12(8) doi: 10.1371/journal.ppat.1005789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomaić V., Pim D., Banks L. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology. 2009;393:7–10. doi: 10.1016/j.virol.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 56.Lee C., Laimins L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004;78:12366–12377. doi: 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicolaides L., Davy C., Raj K., Kranjec C., Banks L., Doorbar J. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology. 2011;414:137–145. doi: 10.1016/j.virol.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 58.Marsh E.K., Delury C.P., Davies N.J., Weston C.J., Miah M.A.L., Banks L., Parish J.L., Higgs M.R., Roberts S. Mitotic control of human papillomavirus genome-containing cells is regulated by the function of the PDZ-binding motif of the E6 oncoprotein. Oncotarget. 2017;8:19491–19506. doi: 10.18632/oncotarget.14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lechler T., Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275–280. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poulson N.D., Lechler T. Robust control of mitotic spindle orientation in the developing epidermis. J. Cell Biol. 2010;191:915–922. doi: 10.1083/jcb.201008001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu Y., Munger K. Human papillomavirus type 16 E7 oncoprotein engages but does not abrogate the mitotic spindle assembly checkpoint. Virology. 2012;432:120–126. doi: 10.1016/j.virol.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Y., Munger K. Human papillomavirus type 16 E7 oncoprotein inhibits the anaphase promoting complex/cyclosome activity by dysregulating EMI1 expression in mitosis. Virology. 2013;446:251–259. doi: 10.1016/j.virol.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hao Y. Par3 controls epithelial spindle orientation by aPKC-mediated phosphorylation of apical Pins. Curr. Biol. 2010;20:1809–1818. doi: 10.1016/j.cub.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson C.A., Hirono K., Pehoda K.E., Do C.Q. Identification of an Aurora-A/Pinslinker/Dlg spindle orientation pathway using induced cell polarity in S2 cells. Cell. 2009;138:1150–1163. doi: 10.1016/j.cell.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thomas M., Laura R., Hepner K., Guccione E., Sawyers C., Lasky L., Banks L. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene. 2002;21:5088–5096. doi: 10.1038/sj.onc.1205668. [DOI] [PubMed] [Google Scholar]
- 66.Gregorc U., Ivanova S., Thomas M., Guccione E., Glaunsinger B., Javier R., Turk V., Banks L., Turk B. Cleavage of MAGI-1, a tight junction PDZ protein, by caspases is an important step for cell–cell detachment in apoptosis. Apoptosis. 2007;12:343–354. doi: 10.1007/s10495-006-0579-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kranjec C., Massimi P., Banks L. Restoration of MAGI-1 expression in human papillomavirus-positive tumor cells induces cell growth arrest and apoptosis. J. Virol. 2014;88:7155–7169. doi: 10.1128/JVI.03247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hernández-Monge J., Garay E., Raya-Sandino A., Vargas-Sierra O., Díaz-Chávez J., Popoca-Cuaya M., Lambert P.F., González-Mariscal L., Gariglio P. Papillomavirus E6 oncoprotein up-regulates occludin and ZO-2 expression in ovariectomized mice epidermis. Exp. Cell Res. 2013;319:2588–2603. doi: 10.1016/j.yexcr.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 69.Matthews K., Leong C.M., Baxter L., Inglis E., Yun K., Bäckström B.T., Doorbar J., Hibma M. Depletion of Langerhans cells in human papillomavirus type 16-infected skin is associated with E6-mediated downregulation of E-cadherin. J. Virol. 2003;77:8378–8385. doi: 10.1128/JVI.77.15.8378-8385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laurson J., Khan S., Chung R., Cross K., Raj K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis. 2010;31:918–926. doi: 10.1093/carcin/bgq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caberg J.H., Hubert P.M., Begon D.Y., Herfs M.F., Roncarati P.J., Boniver J.J., Delvenne P.O. Silencing of E7 oncogene restores functional E-cadherin expression in human papillomavirus 16-transformed keratinocytes. Carcinogenesis. 2008;29:1441–1447. doi: 10.1093/carcin/bgn145. [DOI] [PubMed] [Google Scholar]
- 72.Faghihloo E., Sadeghizadeh M., Shahmahmoodi S., Mokhtari-Azad T. Cdc6 expression is induced by HPV16 E6 and E7 oncogenes and represses E-cadherin expression. Cancer Gene Ther. 2016 doi: 10.1038/cgt.2016.51. [DOI] [PubMed] [Google Scholar]
- 73.Nourry C., Grant S.G.N., Borg J.-P. Sci. STKE. 2003;179 doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- 74.Kühne C., Gardiol D., Guarnaccia C., Amenitsch H., Banks L. Differential regulation of human papillomavirus E6 by protein kinase A: conditional degradation of human discs large protein by oncogenic E6. Oncogene. 2000;19:5884–5891. doi: 10.1038/sj.onc.1203988. [DOI] [PubMed] [Google Scholar]
- 75.Boon S.S., Banks L. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3ζ in a PDZ binding motif-dependent manner. J. Virol. 2013;87:1586–1595. [Google Scholar]
- 76.Boon S.S., Tomaić V., Thomas M., Roberts S., Banks L. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J. Virol. 2015;89:1579–1586. doi: 10.1128/JVI.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thatte J, Massimi P, Boon SS, Banks L. The HPV E6 PDZ binding motif links DNA damage response signalling to E6 inhibition of p53 transcriptional activity. PLoS Pathog. (submitted for publication) [DOI] [PMC free article] [PubMed]
- 78.Bristol M.L., Das D., Morgan I.M. Why human papillomaviruses activate the DNA damage response (DDR) and how cellular and viral replication persists in the presence of DDR signaling. Viruses. 2017;9 doi: 10.3390/v9100268. (pii E268) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bordignon V., Di Domenico E.G., Trento E., D'Agosto G., Cavallo I., Pontone M., Pimpinelli F., Mariani L., Ensoli F. How human papillomavirus replication and immune evasion strategies take advantage of the host DNA damage repair machinery. Viruses. 2017;9:E390. doi: 10.3390/v9120390. (doi: 10.3390) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Massimi P., Narayan N., Cuenda A., Banks L. Phosphorylation of the discs large tumour suppressor protein controls its membrane localisation and enhances its susceptibility to HPV E6-induced degradation. Oncogene. 2006;25:4276–4285. doi: 10.1038/sj.onc.1209457. [DOI] [PubMed] [Google Scholar]
- 81.Narayan N., Massimi P., Banks L. CDK phosphorylation of the discs large tumour suppressor controls its localisation and stability. J. Cell Sci. 2009;122:65–74. doi: 10.1242/jcs.024554. [DOI] [PubMed] [Google Scholar]
- 82.Mirkovic J., Howitt B.E., Roncarati P., Demoulin S., Suarez-Carmona M., Hubert P., McKeon F.D., Xian W., Li A., Delvenne P., Crum C.P., Herfs M. Carcinogenic HPV infection in the cervical squamo-columnar junction. J. Pathol. 2015;236:265–271. doi: 10.1002/path.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Organista-Nava J., Gómez-Gómez Y., Gariglio P. Embryonic stem cell-specific signature in cervical cancer. Tumour Biol. 2014;35:1727–1738. doi: 10.1007/s13277-013-1321-y. [DOI] [PubMed] [Google Scholar]
- 84.Herfs M., Yamamoto Y., Laury A., Wang X., Nucci M.R., McLaughlin-Drubin M.E., Münger K., Feldman S., McKeon F.D., Xian W., Crum C.P. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. U. S. A. 2012;109:10516–10521. doi: 10.1073/pnas.1202684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Herfs M., Soong T.R., Delvenne P., Crum C.P. Deciphering the multifactorial susceptibility of mucosal junction cells to HPV infection and related carcinogenesis. Viruses. 2017;9 doi: 10.3390/v9040085. (pii: E85) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Griffin H., Soneji Y., Van Baars R., Arora R., Jenkins D., van de Sandt M., Wu Z., Quint W., Jach R., Okon K., Huras H., Singer A., Doorbar J. Stratification of HPV-induced cervical pathology using the virally encoded molecular marker E4 in combination with p16 or MCM. Mod. Pathol. 2015;28:977–993. doi: 10.1038/modpathol.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Isaacson Wechsler E., Wang Q., Roberts I., Pagliarulo E., Jackson D., Untersperger C., Coleman N., Griffin H., Doorbar J. Reconstruction of human papillomavirus type 16-mediated early-stage neoplasia implicates E6/E7 deregulation and the loss of contact inhibition in neoplastic progression. J. Virol. 2012;86:6358–6364. doi: 10.1128/JVI.07069-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith J.S., Lindsay L., Hoots B., Keys J., Franceschi S., Winer R., Clifford G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int. J. Cancer. 2007;121:621–632. doi: 10.1002/ijc.22527. [DOI] [PubMed] [Google Scholar]
- 89.Nguyen M.L., Nguyen M.M., Lee D., Griep A.E., Lambert P.F. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J. Virol. 2003;77:6957–6964. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shai A., Brake T., Somoza C., Lambert P.F. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 2007;67:1626–1635. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watson R.A., Thomas M., Banks L., Roberts S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 2003;116:4925–4934. doi: 10.1242/jcs.00809. [DOI] [PubMed] [Google Scholar]
- 92.D'Costa Z.J., Jolly C., Androphy E.J., Mercer A., Matthews C.M., Hibma M.H. Transcriptional repression of E-cadherin by human papillomavirus type 16 E6. PLoS One. 2012;7(11) doi: 10.1371/journal.pone.0048954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.James C.D., Roberts S. Viral Interactions with PDZ domain-containing proteins-an oncogenic trait? Pathogens. 2016;5 doi: 10.3390/pathogens5010008. (pii: E8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Doorslaer K., DeSalle R., Einstein M.H., Burk R.D. Degradation of human PDZ-proteins by human alphapapillomaviruses represents an evolutionary adaptation to a novel cellular niche. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1004980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kranjec C., Banks L. A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J. Virol. 2011;85:1757–1764. doi: 10.1128/JVI.01756-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kranjec C., Tomaić V., Massimi P., Nicolaides L., Doorbar J., Banks L. The high-risk HPV E6 target scribble (hScrib) is required for HPV E6 expression in cervical tumour-derived cell lines. Papillomavirus Res. 2016;2:70–77. doi: 10.1016/j.pvr.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Watson R.A., Rollason T.P., Reynolds G.M., Murray P.G., Banks L., Roberts S. Changes in expression of the human homologue of the Drosophila discs large tumour suppressor in high-grade premalignant cervical neoplasias. Carcinogenesis. 2002;23:1791–1796. doi: 10.1093/carcin/23.11.1791. [DOI] [PubMed] [Google Scholar]
- 98.Cavatorta A.L., Fumero G., Chouhy D., Aguirre R., Nocito A.L., Giri A.A., Banks L., Gardiol D. Differential expression of the human homologue of Drosophila discs large oncosuppressor in histologic samples from human papillomavirus-associated lesions as a marker for progression to malignancy. Int. J. Cancer. 2004;111:373–380. doi: 10.1002/ijc.20275. [DOI] [PubMed] [Google Scholar]
- 99.Massimi P., Zorzi P., Roberts S., Banks L. Differential regulation of cell–cell contact, invasion and anoikis by hScrib and hDlg in keratinocytes. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhan L., Rosenberg A., Bergami K., Yu M., Xuan Z., Jaffe A., Allred C., Muthuswamy S. Deregulation of Scribble promotes mammary tumourigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865–878. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elsum I., Martin C., Humbert P. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J. Cell Sci. 2013;126:3990–3999. doi: 10.1242/jcs.129387. [DOI] [PubMed] [Google Scholar]
- 102.Feigin M., Akshinthala D., Araki K., Rosenberg A., Muthuswamy L., Martin B., Lehmann B., Berman H., Pietenpol J., Cardiff R., Muthuswamy S. Mislocalization of the cell polarity protein Scribble promotes mammary tumourigensis and is associated with basal breast cancer. Cancer Res. 2014;74:3180–3194. doi: 10.1158/0008-5472.CAN-13-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jeanes A., Gottardi C.J., Yap A.S. Cadherins and cancer: how does cadherin dysfunction promote tumour progression. Oncogene. 2008;27:6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fujimoro M. A novel viral mechanism for dysregulation of β-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 2003;9:300–306. doi: 10.1038/nm829. [DOI] [PubMed] [Google Scholar]
- 105.Jha H.C., Sun Z., Upadhyay S.K., El-Naccache D.W., Singh R.K., Sahu S.K., Robertson E.S. KSHV-mediated regulation of Par3 and SNAIL contributes to B-cell proliferation. PLoS Pathog. 2016 doi: 10.1371/journalppat.1005801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu Y.H. The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial motility. Blood. 2011;111:2896–2905. doi: 10.1182/blood-2011-01-330589. [DOI] [PubMed] [Google Scholar]
- 107.Saburi S., Hester I., Fischer E., Pontoglio M., Eremina V., Gessler M., Quaggin S.E., Harrison R., Mount R., McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- 108.Ishiuchi T., Misaki K., Yonemura S., Takeichi M., Tanoue T. Mammalian fat and dachsous cadherins regulate apical membrane organization in the embryonic cerebral cortex. J. Cell Biol. 2009;185:959–967. doi: 10.1083/jcb.200811030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Matakatsu H., Blair S.S. Separating planar cell polarity and Hippo pathway activities of the protocadherins fat and dachsous. Development. 2012;139:1498–1508. doi: 10.1242/dev.070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shannon-Lowe C., Rowe M. Epstein–Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen J., Jardetzky T.S., Longnecker R. The cytoplasmic tail domain of Epstein–Barr virus gH regulates membrane fusion activity through altering gH binding to gp42 and epithelial cell attachment. MBio. 2016;7:e01871–16. doi: 10.1128/mBio.01871-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tugizov S.M., Herrera R., Palefsky J.M. Epstein–Barr virus transcytosis through polarized oral epithelial cells. J. Virol. 2013;87:8179–8194. doi: 10.1128/JVI.00443-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nanbo A., Kachi K., Yoshiyama H., Ohba Y. Epstein–Barr virus exploits host endocytic machinery for cell-to-cell viral transmission rather than a virological synapse. J. Gen. Virol. 2016;97:2989–3006. doi: 10.1099/jgv.0.000605. [DOI] [PubMed] [Google Scholar]
- 114.Hutt-Fletcher L.M. The long and complicated relationship between Epstein–Barr virus and epithelial cells. J. Virol. 2016;91:e01677–16. doi: 10.1128/JVI.01677-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsai C.N., Tsai C.L., Tse K.P., Chang H.Y., Chang Y.S. The Epstein–Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10084–10089. doi: 10.1073/pnas.152059399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scholle F., Bendt K.M., Raab-Traub N. Epstein–Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000;74:10681–10689. doi: 10.1128/jvi.74.22.10681-10689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Horikawa T., Yang J., Kondo S., Yoshizaki T., Joab I., Furukawa M., Pagano J.S. Twist and epithelial–mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007;67:1970–1978. doi: 10.1158/0008-5472.CAN-06-3933. [DOI] [PubMed] [Google Scholar]
- 118.Horikawa T., Yoshizaki T., Kondo S., Furukawa M., Kaizaki Y., Pagano J.S. Epstein–Barr Virus latent membrane protein 1 induces Snail and epithelial–mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer. 2011;104:1160–1167. doi: 10.1038/bjc.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gaur N., Gandhi J., Robertson E.S., Verma S.C., Kaul R. Epstein–Barr virus latent antigens EBNA3C and EBNA1 modulate epithelial to mesenchymal transition of cancer cells associated with tumor metastasis. Tumour Biol. 2015;36:3051–3060. doi: 10.1007/s13277-014-2941-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shackelford J., Maier C., Pagano J.S. Epstein–Barr virus activates beta-catenin in type III latently infected B lymphocyte lines: association with deubiquitinating enzymes. Proc. Natl. Acad. Sci. U. S. A. 2003;100:15572–15576. doi: 10.1073/pnas.2636947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Morrison J.A., Raab-Traub N. Roles of the ITAM and PY motifs of Epstein Barr virus latent membrane protein 2A in the inhibition of epithelial cell differentiation and activation of β-catenin signaling. J. Virol. 2005;79:2375–2382. doi: 10.1128/JVI.79.4.2375-2382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu H.P., Chen C.C., Wu C.C., Huang Y.C., Liu S.C., Liang Y., Chang K.P., Chang Y.S. Epstein–Barr virus-encoded LMP1 interacts with FGD4 to activate Cdc42 and thereby promote migration of nasopharyngeal carcinoma cells. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- 124.Mack N.A., Georgiou M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases. 2014;5:10. doi: 10.4161/21541248.2014.973768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gandalovičová A., Vomastek T., Rosel D., Brábek J. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget. 2016;7:25022–25049. doi: 10.18632/oncotarget.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abaitua F., Zia F.R., Hollinshead M., O'Hare P. Polarized cell migration during cell-to-cell transmission of herpes simplex virus in human skin keratinocytes. J. Virol. 2013;87:7921–7932. doi: 10.1128/JVI.01172-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mo C., Schneeberger E.E., Arvin A.M. Glycoprotein E of varicella-zoster virus enhances cell–cell contact in polarized epithelial cells. J. Virol. 2000;74:11377–11387. doi: 10.1128/jvi.74.23.11377-11387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maidji E., Tugizov S., Abenes G., Jones T., Pereira L. A novel human cytomegalovirus glycoprotein, gpUS9, which promotes cell-to-cell spread in polarized epithelial cells, colocalizes with the cytoskeletal proteins E-cadherin and F-actin. J. Virol. 1998;72:5717–5727. doi: 10.1128/jvi.72.7.5717-5727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huber M.T., Tomazin R., Wisner T., Boname J., Johnson D.C. Human cytomegalovirus US7, US8, US9, and US10 are cytoplasmic glycoproteins, not found at cell surfaces, and US9 does not mediate cell-to-cell spread. J. Virol. 2002;76:5748–5758. doi: 10.1128/JVI.76.11.5748-5758.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Snyder A., Polcicova K., Johnson D.C. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 2008;82:10613–10624. doi: 10.1128/JVI.01241-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Howard P.W., Howard T.L., Johnson D.C. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J. Virol. 2013;87:403–414. doi: 10.1128/JVI.02465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tomishima M.J., Enquist L.W. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 2001;154:741–752. doi: 10.1083/jcb.200011146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lara-Pezzi E., Roche S., Andrisani O.M., Sánchez-Madrid F., López-Cabrera M. The hepatitis B virus HBx protein induces adherens junction disruption in a src-dependent manner. Oncogene. 2001;20:3323–3331. doi: 10.1038/sj.onc.1204451. [DOI] [PubMed] [Google Scholar]
- 134.Bonilla Guerrero R., Roberts L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005;42:760–777. doi: 10.1016/j.jhep.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 135.Lee J.O., Kwun H.J., Jung J.K., Choi K.H., Min D.S., Jang K.L. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24:6617–6625. doi: 10.1038/sj.onc.1208827. [DOI] [PubMed] [Google Scholar]
- 136.Arzumanyan A., Friedman T., Kotei E., Ng I.O., Lian Z., Feitelson M.A. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. 2012;31:563–572. doi: 10.1038/onc.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hsieh A., Kim H.S., Lim S.O., Yu D.Y., Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett. 2011;300:162–172. doi: 10.1016/j.canlet.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 138.Zhang T., Zhang J., You X., Liu Q., Du Y., Gao Y., Shan C., Kong G., Wang Y., Yang X., Ye L., Zhang X. Hepatitis B virus X protein modulates oncogene Yes-associated protein by CREB to promote growth of hepatoma cells. Hepatology. 2012;56:2051–2059. doi: 10.1002/hep.25899. [DOI] [PubMed] [Google Scholar]
- 139.Tsai W.L., Chung R.T. Viral hepatocarcinogenesis. Oncogene. 2010;29:2309–2324. doi: 10.1038/onc.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Evans M.J., von Hahn T., Tscherne D.M., Syder A.J., Panis M., Wölk B., Hatziioannou T., McKeating J.A., Bieniasz P.D., Rice C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 141.Ploss A., Evans M.J., Gaysinskaya V.A., Panis M., You H., de Jong Y.P., Rice C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilkins C., Woodward J., Lau D.T., Barnes A., Joyce M., McFarlane N., McKeating J.A., Tyrrell D.L., Gale M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013;57:461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Belouzard S., Danneels A., Fénéant L., Séron K., Rouillé Y., Dubuisson J. Entry and release of Hepatitis C virus in polarized human hepatocytes. J. Virol. 2017;91:e00478–17. doi: 10.1128/JVI.00478-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mee C.J., Grove J., Harris H.J., Hu K., Balfe P., McKeating J.A. Effect of cell polarization on hepatitis C virus entry. J. Virol. 2008;82:461–470. doi: 10.1128/JVI.01894-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu S., Yang W., Shen L., Turner J.R., Coyne C.B., Wang T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 2009;83:2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Benedicto I., Molina-Jiménez F., Barreiro O., Maldonado-Rodríguez A., Prieto J., Moreno-Otero R., Aldabe R., López-Cabrera M., Majano P.L. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology. 2008;48:1044–1053. doi: 10.1002/hep.22465. [DOI] [PubMed] [Google Scholar]
- 147.Benedicto I., Molina-Jiménez F., Bartosch B., Cosset F.L., Lavillette D., Prieto J., Moreno-Otero R., Valenzuela-Fernández A., Aldabe R., López-Cabrera M., Majano P.L. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009;83:8012–8020. doi: 10.1128/JVI.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Awad A., Sar S., Barré R., Cariven C., Marin M., Salles J.P., Erneux C., Samuel D., Gassama-Diagne A. SHIP2 regulates epithelial cell polarity through its lipid product, which binds to Dlg1, a pathway subverted by hepatitis C virus core protein. Mol. Biol. Cell. 2013;24:2171–2185. doi: 10.1091/mbc.E12-08-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Awad A., Gassama-Diagne A. PI3K/SHIP2/PTEN pathway in cell polarity and hepatitis C virus pathogenesis. World J. Hepatol. 2017;8:18–29. doi: 10.4254/wjh.v9.i1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Arora P., Kim E.O., Jung J.K., Jang K.L. Hepatitis C virus core protein downregulates E-cadherin expression via activation of DNA methyltransferase 1 and 3b. Cancer Lett. 2008;261:244–252. doi: 10.1016/j.canlet.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 151.Street A., Macdonald A., McCormick C., Harris M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005;79:5006–5016. doi: 10.1128/JVI.79.8.5006-5016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Higgs M.R., Lerat H., Pawlotsky J.M. Hepatitis C virus-induced activation of β-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene. 2013;32:4683–4693. doi: 10.1038/onc.2012.484. [DOI] [PubMed] [Google Scholar]
- 153.Akkari L., Grégoire D., Floc'h N., Moreau M., Hernandez C., Simonin Y., Rosenberg A.R., Lassus P., Hibner U. Hepatitis C viral protein NS5A induces EMT and participates in oncogenic transformation of primary hepatocyte precursors. J. Hepatol. 2012;57:1021–1028. doi: 10.1016/j.jhep.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 154.Battaglia S., Benzoubir N., Nobilet S., Charneau P., Samuel D., Zignego A.L., Atfi A., Bréchot C., Bourgeade M.F. Liver cancer-derived hepatitis C virus core proteins shift TGF-beta responses from tumor suppression to epithelial–mesenchymal transition. PLoS One. 2009;4 doi: 10.1371/journal.pone.0004355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hu B., Li S., Zhang Z., Xie S., Hi Y., Huang X., Zheng Y. HCV NS4B targets Scribble for proteasome-mediated degradation to facilitate cell transformation. Tumor Biol. 2016;37:12387–12396. doi: 10.1007/s13277-016-5100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu W., MacDonald M., You J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol. 2016;20:20–27. doi: 10.1016/j.coviro.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.DeCaprio J.A. Merkel cell polyomavirus and Merkel cell carcinoma. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017;372 doi: 10.1098/rstb.2016.0276. (20160276) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Laude H.C., Jonchère B., Maubec E., Carlotti A., Marinho E., Couturaud B., Peter M., Sastre-Garau X., Avril M.F., Dupin N., Rozenberg F. Distinct merkel cell polyomavirus molecular features in tumour and non-tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shuda M., Feng H., Kwun H.J., Rosen S.T., Gjoerup O., Moore P.S., Chang Y. T antigen mutations are a human tumour-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Houben R., Shuda M., Weinkam R., Schrama D., Feng H., Chang Y., Moore P.S., Becker J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010;84:7064–7072. doi: 10.1128/JVI.02400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rozenblatt-Rosen O., Deo R.C., Padi M., Adelmant G., Calderwood M.A., Rolland T., Grace M., Dricot A., Askenazi M., Tavares M., Pevzner S.J., Abderazzaq F., Byrdsong D., Carvunis A.R., Chen A.A., Cheng J., Correll M., Duarte M., Fan C., Feltkamp M.C., Ficarro S.B., Franchi R., Garg B.K., Gulbahce N., Hao T., Holthaus A.M., James R., Korkhin A., Litovchick L., Mar J.C., Pak T.R., Rabello S., Rubio R., Shen Y., Singh S., Spangle J.M., Tasan M., Wanamaker S., Webber J.T., Roecklein-Canfield J., Johannsen E., Barabási A.L., Beroukhim R., Kieff E., Cusick M.E., Hill D.E., Münger K., Marto J.A., Quackenbush J., Roth F.P., DeCaprio J.A., Vidal M. Interpreting cancer genomes using systematic host network perturbations by tumor virus proteins. Nature. 2012;487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kirschner N., Haftek M., Niessen C.M., Behne M.J., Furuse M., Moll I., Brandner J.M. CD44 regulates tight-junction assembly and barrier function. J. Investig. Dermatol. 2011;131:932–943. doi: 10.1038/jid.2010.390. [DOI] [PubMed] [Google Scholar]
- 164.Meinke P., Nguyen T.D., Wehnert M.S. The LINC complex and human disease. Biochem. Soc. Trans. 2011;39:1693–1697. doi: 10.1042/BST20110658. [DOI] [PubMed] [Google Scholar]
- 165.Markiewicz E., Tilgner K., Barker N., van de Wetering M., Clevers H., Dorobek M., Hausmanowa-Petrusewicz I., Ramaekers F.C., Broers J.L., Blankesteijn W.M., Salpingidou G., Wilson R.G., Ellis J.A., Hutchison C.J. The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J. 2006;25:3275–3285. doi: 10.1038/sj.emboj.7601230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stubenvoll A., Rice M., Wietelmann A., Wheeler M., Braun T. Attenuation of Wnt/β-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum. Mol. Genet. 2015;24:802–813. doi: 10.1093/hmg/ddu498. [DOI] [PubMed] [Google Scholar]
- 167.Liu W., Yang R., Payne A.S., Schowalter R.M., Spurgeon M.E., Lambert P.F., Xu X., Buck C.B., You Y. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe. 2016;19:775–787. doi: 10.1016/j.chom.2016.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nazli A., Chan O., Dobson-Belaire W.N., Ouellet M., Tremblay M.J., Gray-Owen S.D., Arsenault A.L., Kaushic C. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog. 2010;6(4) doi: 10.1371/journal.ppat.1000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hawkins E.D., Russell S.M. Upsides and downsides to polarity and asymmetric cell division in leukemia. Oncogene. 2008;27:7003–7017. doi: 10.1038/onc.2008.350. [DOI] [PubMed] [Google Scholar]
- 170.Renkema G.H., Manninen A., Saksela K. Human immunodeficiency virus type 1 Nef selectively associates with a catalytically active subpopulation of p21-actvated kinase (PAK2) independently of PAK2 binding to Nck or β-PIX. J. Virol. 2001;75:2154–2160. doi: 10.1128/JVI.75.5.2154-2160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stevenson M. HIV-1 pathogenesis. Nat. Med. 2003;9:853–860. doi: 10.1038/nm0703-853. [DOI] [PubMed] [Google Scholar]
- 172.Wolf D., Witte V., Laffert B., Blume K., Stromer E., Trapp S., d'Aloja P., Schürmann A., Baur A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001;7:1217–1224. doi: 10.1038/nm1101-1217. [DOI] [PubMed] [Google Scholar]
- 173.Stoddart C.A., Geleziunas R., Ferrell S., Linquist-Stepps V., Moreno M.E., Bare C., Xu W., Yonemoto W., Bresnahan P.A., McCune J.M., Greene W.C. Human immunodeficiency virus type 1 Nef-mediated downregulation of CD4 correlates with Nef enhancement of viral pathogenesis. J. Virol. 2003;77(3):2124–2133. doi: 10.1128/JVI.77.3.2124-2133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Margottin F., Bour S.P., Durand H., Selig L., Benichou S., Richard V., Thomas D., Strebel K., Benarous R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell. 1998;1(4):565–574. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 175.Besnard-Guerin C., Belaïdouni N., Lassot I., Segeral E., Jobart A., Marchal C., Benarous R. HIV-1 Vpu sequesters beta-transducin repeat-containing protein (betaTrCP) in the cytoplasm and provokes the accumulation of beta-catenin and other SCFbetaTrCP substrates. J. Biol. Chem. 2004;279(1):788–795. doi: 10.1074/jbc.M308068200. [DOI] [PubMed] [Google Scholar]
- 176.Salim A., Ratner L. Modulation of beta-catenin and E-cadherin interaction by Vpu increases human immunodeficiency virus type 1 particle release. J. Virol. 2008;82(8):3932–3938. doi: 10.1128/JVI.00430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mansur D.S., Maluquer de Motes C., Unterholzner L., Sumner R.P., Ferguson B.J., Ren H., Strnadova P., Bowie A.G., Smith G.L. Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 2013;9(2) doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Morelli M., Dennis A.F., Patton J.T. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-κB activation by using molecular mimicry to target β-TrCP. MBio. 2015;6(1):e02490–14. doi: 10.1128/mBio.02490-14. (pii) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pu H., Tian J., Andras I.E., Hayashi K., Flora G., Hennig B., Toborek M. HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK 1/2 activation. J. Cereb. Blood Flow Metab. 2005;25(10):1325–1335. doi: 10.1038/sj.jcbfm.9600125. [DOI] [PubMed] [Google Scholar]
- 180.Zhong Y., Zhang B., Eum S.Y., Toborek M. HIV-1 Tat triggers nuclear localization of ZO-1 via Rho signaling and cAMP response element-binding protein activation. J. Neurosci. 2012;32(1):143–150. doi: 10.1523/JNEUROSCI.4266-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lee S.S., Weiss R.S., Javier R.T. Binding of human virus oncoproteins tohDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. U. S. A. 1997;94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hirata A., Higuchi M., Niinuma A., Ohashi M., Fukushi M., Oie M., Akiyama T., Tanaka Y., Gejyo F., Fujii M. PDZ domain-binding motif of human T-cell leukemia virus type 1 Tax oncoprotein augments the transforming activity in a rat fibroblast cell line. Virology. 2004;318:327–336. doi: 10.1016/j.virol.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 183.Arpin-André C., Mesnard J.M. The PDZ domain-binding motif of the human T cell leukemia virus type 1 tax protein induces mislocalisation of the tumor suppressor hScrib in T cells. J. Biol. Chem. 2007;282:33132–33141. doi: 10.1074/jbc.M702279200. [DOI] [PubMed] [Google Scholar]
- 184.Makokha G.N., Takahashi M., Higuchi M., Saito S., Tanaka Y., Fujii M. Human T-cell leukemia virus type 1 Tax protein interacts with and mislocalizes the PDZ domain protein MAGI-1. Cancer Sci. 2013;104:313–320. doi: 10.1111/cas.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ohashi M., Sakurai M., Higuchi M., Mori N., Fukushi M., Oie M., Coffey R.J., Yoshiura K., Tanaka Y., Uchiyama M., Hatanaka M., Fujii M. Human T-cell leukemia virus type 1 Tax oncoprotein induces and interacts with a multi-PDZ domain protein, MAGI-3. Virology. 2004;320:52–62. doi: 10.1016/j.virol.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 186.Rousset R., Fabre S., Desbois C., Bantignies F., Jalinot P. The C-terminus of the HTLV-1 Ttax oncoprotein mediates interaction with the PDZ domain of cellular proteins. Oncogene. 1998;16:643–654. doi: 10.1038/sj.onc.1201567. [DOI] [PubMed] [Google Scholar]
- 187.Okajima M., Takahashi M., Higuchi M., Ohsawa T., Yoshida S., Yoshida Y., Oie M., Tanaka Y., Gejyo F., Fujii M. Human T-cell leukemia virus type 1 Tax induces an errant clustering of the tumor suppressor Scribble through the PDZ domain-binding motif dependent and independent interaction. Virus Genes. 2008;37:231–240. doi: 10.1007/s11262-008-0259-4. [DOI] [PubMed] [Google Scholar]
- 188.Marziali F., Bugnon Valdano M., Brunet Avalos C., Moriena L., Cavatorta A.L., Gardiol D. Interfernce of HTLV-1 Tax protein with cell polarity regulators: defining the subcellular localization of the Tax–DLG1 Interaction. Viruses. 2017;9:355. doi: 10.3390/v9120355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cherian M.A., Bydoun H.H., Al-Saleem J., Shkribai N., Kvaratskhelia M., Green P., Ratner L. Akt activation by human T-cell leukemia virus type 1 Tax oncoprotein. J. Biol. Chem. 2015;290:26270–26281. doi: 10.1074/jbc.M115.684746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sotelo N.S., Valiente M., Gil A., Pulido R. A functional network of tumour suppressors APC, hDlg, and PTEN that relies on recognition of specific PDZ domains. J. Cell. Biochem. 2012;113:2661–2670. doi: 10.1002/jcb.24141. [DOI] [PubMed] [Google Scholar]
- 191.Xie L., Yamamoto B., Haoudi A., Semmes O.J., Green P.L. PDZ binding motif of HTLV-1 Tax promotes virus-mediated T-cell proliferation in vitro and persistence in vivo. Blood. 2006;107:1980–1988. doi: 10.1182/blood-2005-03-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tsubata C., Higuchi M., Takahashi M., Oie M., Tanaka Y., Gejyo F., Fujii M. PDZ domain-binding motif of human T-cell leukemia virus type 1 Tax oncoprotein is essential for the interleukin 2 independent growth induction of a T-cell line. Retrovirology. 2005;2:46. doi: 10.1186/1742-4690-2-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.James M.A., Lee J.H., Klingelhutz A.J. Human papillomavirus type 16 E6 activates NF-κB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J. Virol. 2006;80:5301–5307. doi: 10.1128/JVI.01942-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Higuchi M., Tsubata C., Kondo R., Yoshida S., Takahashi M., Oie M., Tanaka Y., Mahieux R., Matsuoka M., Fijii M. Cooperation of NK-kappaB2/p100 activation and the PDZ domain binding motif signal in human T-cell Leukemia virus type 1 (HTLV-1) Tax1 but not HTLV-2 Tax2 is crucial for interleukin-2-independent growth transformationof a T-cell line. J. Virol. 2007;81:11900–11907. doi: 10.1128/JVI.00532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Blot V., Delamarre L., Perugi F., Pham D., Bénichou, Benarous R., Hanada T., Chishti A.H., Dokhélar M.-C., Pique C. Human Dlg protein binds to the envelope glycoproteins of human T-cell leukemia virus type 1 and regulates envelope mediated cell–cell fusion in T lymphocytes. J. Cell Sci. 2004;117:3983–3993. doi: 10.1242/jcs.01266. [DOI] [PubMed] [Google Scholar]
- 196.Yoshida S., Higuchi M., Shoji T., Yoshita M., Ishioka K., Takahashi M., Oie M., Tanaka Y., Uchiyama M., Fujii M. Knockdown of synapse-associated protein Dlg1 reduces syncytium formation induced by human T-cell leukemia virus type 1. Virus Genes. 2008;37(1):9–15. doi: 10.1007/s11262-008-0234-0. [DOI] [PubMed] [Google Scholar]
- 197.Tanaka Y., Fukudome K., Hayashi M., Takagi S., Yoshie O. Induction of ICAM-1 and LFA-3 by Tax1 of human T-cell leukemia virus type 1 and mechanism of down-regulation of ICAM-1 or LFA-1 in adult-T-cell-leukemia cell lines. Int. J. Cancer. 1995;60:554–561. doi: 10.1002/ijc.2910600421. [DOI] [PubMed] [Google Scholar]
- 198.Barnard A., Igakura T., Tanaka Y., Taylor G.P., Bangham C.R.M. Engagement of specific T-cell surface molecules regulates cytoskeletal polasization in HTLV-1-infected lymphocytes. Blood. 2005;106:988–995. doi: 10.1182/blood-2004-07-2850. [DOI] [PubMed] [Google Scholar]
- 199.Nejmeddine M., Neghi V.S., Mukherjee S., Tanaka T., Orth K., Taylor G.P., Bangham C.R.M. HTLV-1-tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood. 2009;114:1016–1025. doi: 10.1182/blood-2008-03-136770. [DOI] [PubMed] [Google Scholar]
- 200.Igakura T., Stinchcombe J.C., Goon P.K.C., Taylor G.P., Weber J.N., Griffiths G.M., Tanaka Y., Osame M., Bangham C.R.M. Spread of HTLV-1 between lymphocytes by virus-induced polarization of the cytoskeleton. Science. 2003;299:1713–1716. doi: 10.1126/science.1080115. [DOI] [PubMed] [Google Scholar]
- 201.Cohen C.J., Gaetz J., Ohman T., Bergelson J.M. Multiple regions within the coxsackievirus and adenovirus receptor cytoplasmic domain are required for basolateral sorting. J. Biol. Chem. 2001;276:25392–25398. doi: 10.1074/jbc.M009531200. [DOI] [PubMed] [Google Scholar]
- 202.Cohen C.J., Shieh J.T., Pickles R.J., Okegawa T., Hsieh J.T., Bergelson J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. U. S. A. 2001;98:15191–15196. doi: 10.1073/pnas.261452898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.McAllister R.M., Nicolson M.O., Reed G., Kern J., Gilden R.V., Huebner R.J. Transformation of rodent cells by adenovirus 19 and other group D adenoviruses. J. Natl. Cancer Inst. 1969;43:917–923. [PubMed] [Google Scholar]
- 204.Javier R.T., Rice A.P. Emerging theme: cellular PDZ proteins as common targets of pathogenic viruses. J. Virol. 2011;85:11544–11556. doi: 10.1128/JVI.05410-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Weiss R.S., Javier R.T. A carboxy-terminal region required by the adenovirustype 9 E4-ORF1 oncoprotein for transformation mediates direct binding to cellular polypeptides. J. Virol. 1997;71:7873–7880. doi: 10.1128/jvi.71.10.7873-7880.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lee S.S., Glaunsinger B., Mantovani F., Banks L., Javier R.T. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 oncoproteins. J. Virol. 2000;74:9680–9693. doi: 10.1128/jvi.74.20.9680-9693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Glaunsinger B.A., Weiss R.S., Lee S.S., Javier R.T. Link of the unique oncogenic properties of adenovirus type 9 E4-ORF1 to a select interaction with the candidate tumour suppressor protein ZO-2. EMBO J. 2001;20:5578–5586. doi: 10.1093/emboj/20.20.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Latorre I.J., Roh M.H., Frese K.K., Weiss R.S., Margolis B., Javier R.T. Viral oncoprotein-induced mislocalisation of select PDZ proteins disrupts tight junctions and causes polarity defects in epithelial cells. J. Cell Sci. 2005;118:4283–4293. doi: 10.1242/jcs.02560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chung S.H., Weiss R.S., Frese K.K., Prasad B.V., Javier R.T. Functionally distinct monomers and trimers produced by a viral oncoprotein. Oncogene. 2008;27:1412–1420. doi: 10.1038/sj.onc.1210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Frese K.K., Lee S.S., Thomas D.L., Latorre I.J., Weiss R.S., Glaunsinger B.A., Javier R.T. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene. 2003;22:710–721. doi: 10.1038/sj.onc.1206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Frese K.K., Latorre I.J., Chung S.H., Caruana G., Bernstein A., Jones S.N., Donehower L.A., Justice M.J., Garner C.C., Javier R.T. Oncogenic function for the Dlg1 mammalian homolog of the Drosophila discs-large tumor suppressor. EMBO J. 2006;25:1406–1417. doi: 10.1038/sj.emboj.7601030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Kumar M., Kong K., Javier R. Hijacking Dlg1 for oncogenic phosphatidylinositol 3-kinase activation in human epithelial cells is a conserved mechanism of human adenovirus E4-ORF1 proteins. J. Virol. 2014;88:14268–14277. doi: 10.1128/JVI.02324-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kong K., Kumar M., Taruishi M., Javier R. The human adenovirus E4-ORF1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wang Q., Tao Y.J., editors. Influenza: Current Research. Caister Academic Press; 2016. [Google Scholar]
- 215.Erhardt C., Wolff T., Pleschka S., Planz O., Beerman W., Bode J.G. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signalling responses. J. Virol. 2007;81:3058–3067. doi: 10.1128/JVI.02082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shin Y.K., Li Y., Liu Q., Anderson D.H., Babiuk L.A., Zhou Y. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 2007;81:12730–12739. doi: 10.1128/JVI.01427-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hale B.G., Randall R.E. PI3K signalling during influenza A virus infections. Biochem. Soc. Trans. 2007;35:186–187. doi: 10.1042/BST0350186. [DOI] [PubMed] [Google Scholar]
- 218.Zhirnov O.P., Konakova T.E., Wolff T., Klenk T.D. NS1 protein of Influenza A virus down-regulates apoptosis. J. Virol. 2002;76:1617–1625. doi: 10.1128/JVI.76.4.1617-1625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Herold S., Ludwig S., Pleschka S., Wolff T. Apoptosis signalling in influenza virus propagation, innate host defense, and lung injury. J. Leukoc. Biol. 2012;92:75–82. doi: 10.1189/jlb.1011530. [DOI] [PubMed] [Google Scholar]
- 220.Hirata N., Suizu F., Matsuda-Lennikov M., Edamura T., Bala J., Noguchi M. Inhibition of Akt kinase activity suppresses entry and replication of influenza virus. Biochem. Biophys. Res. Commun. 2014;450:891–898. doi: 10.1016/j.bbrc.2014.06.077. [DOI] [PubMed] [Google Scholar]
- 221.Li Z., Jiang Y., Jiao P., Wang A., Zhao F., Tian G., Wang X., Yu K., Bu Z., Chen H. The NS1 gene contributes to the virulence of H5N1 avian inguenza viruses. J. Virol. 2006;80:11115–11123. doi: 10.1128/JVI.00993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cauthen A.N., Swayne D.E., Sekellick M.J., Marcus P.I., Suarez D.L. Amelioration of influenza virus pathogenesis in chickens attributed to the enhanced interferon-inducing capacity of a virus with a truncated NS1 gene. J. Virol. 2007;81:1838–1847. doi: 10.1128/JVI.01667-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Soubies S.M., Volmer C., Croville G., Loupias J., Peralta B., Costes P., Lacroux C., Guérin J.L., Volmer R. Species-specific contribution of the four C-terminal amino acids of influenza virus NS1 protein to virulence. J. Virol. 2010;84:6733–6747. doi: 10.1128/JVI.02427-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jackson D., Hossain M.J., Hickman D., Perez D.R., Lamb R.A. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4381–4386. doi: 10.1073/pnas.0800482105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fan S., Macken C.A., Li C., Ozawa M., Goto H., Iswahyudi N.F., Nidom C.A., Chen H., Neumann G., Kawaoka Y. Synergistic effect of the PDZ and p85β-binding domains of the NS1 protein on virulence of an avian H5N1 influenza A virus. J. Virol. 2013;87:4861–4871. doi: 10.1128/JVI.02608-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Liu H., Golebiewski L., Dow E.C., Krug R.M., Javier R.T., Rice A.P. The ESEV PDZ-binding motif of the avian influenza A virus NS1 protein protects infected cells from apoptosis by directly targeting Scribble. J. Virol. 2010;84:11164–11174. doi: 10.1128/JVI.01278-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thomas M., Kranjec C., Nagasaka K., Matlashewski G., Banks L. Analysis of the PDZ binding specificities of Influenza A virus NS1 proteins. Virol. J. 2011;8(25) doi: 10.1186/1743-422X-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Yu J., Li X., Wang Y., Li B., Li H., Li Y., Zhou W., Zhang C., Wang Y., Rao Z., Bartlam M., Cao Y. PDlim2 selectively interacts with the PDZ binding motif of highly pathogenic avian H5N1 influenza A virus NS1. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019511. (doi: 10.1371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zhang H., Li W., Wang G., Su Y., Zhang C., Chen X., Xu Y., Li K. The distinct binding properties between avian/human influenza A virus NS1 and Postsynaptic density protein-95 (PSD-95), and inhibition of nitric oxide production. Virol. J. 2011;8:298. doi: 10.1186/1743-422X-8-298. (doi: 10.1186) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Werme K., Wigerius K.M., Johansson M. Tick-borne encephalitis virus NS5 associates with membrane protein scribble and impairs interferon-stimulated JAK-STAT signalling. Cell. Microbiol. 2008;10:696–712. doi: 10.1111/j.1462-5822.2007.01076.x. [DOI] [PubMed] [Google Scholar]
- 231.Golobiewski L., Liu H., Javier R.T., Rice A.P. The avian influenza virus NS1 ESEV PDZ binding motif associates with Dlg1 and Scribble to disrupt cellular tight junctions. J. Virol. 2011;85:10639–10648. doi: 10.1128/JVI.05070-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kumar M., Liu H., Rice A.P. Regulation of interferon-β by MAGI-1 and its interaction with influenza A virus NS1 protein with ESEV PBM. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Barreda D., Sánchez-Galindo M., López-Flores J., Nava-Castro K.E., Bobadilla K., Salgado-Aguayo A., Santos-Mendoza T. PDZ proteins are expressed and regulated in antigen-presenting cells and are targets of influenza A virus. J. Leukoc. Biol. 2018 Apr;103(4):731–738. doi: 10.1002/JLB.4AB0517-184R. [DOI] [PubMed] [Google Scholar]
- 234.Ellencrona K., Syed A., Johansson M. Flavivirus NS5 associates with host-cell proteins zonula occludens-1 (ZO-1) and regulating synaptic membrane exocytosis-2 (RIMS2) via an internal PDZ binding mechanism. Biol. Chem. 2009;390:319–323. doi: 10.1515/BC.2009.041. [DOI] [PubMed] [Google Scholar]
- 235.Wigerius M., Melik W., Elväng A., Johansson M. Rac1 and Scribble are targets for the arrest of neurite outgrowth by TBE virus NS5. Mol. Cell. Neurosci. 2010;44:260–271. doi: 10.1016/j.mcn.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 236.Melik W., Ellencrona K., Wigerius M., Hedström C., Elväng A., Johansson M. Two PDZ binding motifs within NS5 have roles in tick-borne encephalitis virus replication. Virus Res. 2012;169:54–62. doi: 10.1016/j.virusres.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 237.Kaeser P.S., Deng L., Fan M., Südhof T.C. RIM genes differentially contribute to organizing presynaptic release sites. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11830–11835. doi: 10.1073/pnas.1209318109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Chuang Y.C., Lei H.Y., Liu H.S., Lin Y.S., Fu T.F., Yeh T.M. Macrophage migration inhibitory factor induced by dengue virus infection increases vascular permeability. Cytokine. 2011;54:222–231. doi: 10.1016/j.cyto.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 239.Kanlaya R., Pattanakitsakul S.N., Sinchaikul S., Chen S.T., Thongboonkerd V. Alterations in actin cytoskeletal assembly and junctional protein complexes in human endothelial cells induced by dengue virus infection and mimicry of leukocyte transendothelial migration. J. Proteome Res. 2009;8:2551–2562. doi: 10.1021/pr900060g. [DOI] [PubMed] [Google Scholar]
- 240.Soe H.J., Khan A.M., Manikam R., Samudi Raju C., Vanhoutte P., Sekaran S.D. High dengue virus load differentially modulates human microvascular endothelial barrier function during early infection. J. Gen. Virol. 2017;98:2993–3007. doi: 10.1099/jgv.0.000981. [DOI] [PubMed] [Google Scholar]
- 241.Schultze B., Zimmer G., Herrler G. Virus entry into a polarized epithelial cell line (MDCK): similarities and dissimilarities between influenza C virus and bovine coronavirus. J. Gen. Virol. 1996;77:2507–2514. doi: 10.1099/0022-1317-77-10-2507. [DOI] [PubMed] [Google Scholar]
- 242.Tseng C.T., Tseng J., Perrone L., Worthy M., Popov V., Peters C.J. Apical entry and release of severe acute respiratory syndrome-associated coronavirus in polarized Calu-3 lung epithelial cells. J. Virol. 2005;79:9470–9479. doi: 10.1128/JVI.79.15.9470-9479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Cong Y., Ren X. Coronavirus entry and release in polarized epithelial cells: a review. Rev. Med. Virol. 2014;24:308–315. doi: 10.1002/rmv.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Teoh K.T., Siu Y.L., Chan W.L., Schlüter M.A., Liu C.J., Peiris J.S.M., Bruzzone R., Margolis B., Nal B. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell. 2010;21:3838–3852. doi: 10.1091/mbc.E10-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Román-Fernández A., Bryant D.M. Complex polarity: building multicellular tissues through apical membrane traffic. Traffic. 2016;17:1244–1261. doi: 10.1111/tra.12417. [DOI] [PubMed] [Google Scholar]
- 246.Campanale J.P., Sun T.Y., Montell D.J. Development and dynamics of cell polarity at a glance. J. Cell Sci. 2017;130:1201–1207. doi: 10.1242/jcs.188599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Tao X., Hill T.E., Morimoto C., Peters C.J., Ksiazek T.J., Tseng C.T.K. Bilateral entry and release of Middle East respiratory syndrome coronavirus induces profound apoptosis of human bronchial epithelial cells. J. Virol. 2013;87:9953–9958. doi: 10.1128/JVI.01562-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Tatsuo H., Ono N., Tanaka K., Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406(6798):893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 249.Mühlebach M.D., Mateo M., Sinn P.L., Prüfer S., Uhlig K.M., Leonard V.H.J., Navaratnarajah C.K., Frenzke M., Wong X.X., Sawatsky B., Ramachandran S., McCray P.B., Cichutek K., von Messling V., Lopez M., Cattaneo R. Adherens junction protein Nectin-4 is the epithelial receptor for measles virus. Nature. 2011;480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Noyce R., Richardson C.D. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol. 2012;20:429–439. doi: 10.1016/j.tim.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 251.Singh B.K., Hornick A.L., Krishnamurthy S., Locke A.C., Mendoza C.A., Mateo M., Miller-Hunt C.L., Cattaneo R., Sinn P.L. The Nectin-4/Afadin protein complex and intercellular membrane pores contribute to rapid spread of measles virus in primary human airway epithelia. J. Virol. 2015;89:7089–7096. doi: 10.1128/JVI.00821-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Katoh H., Nakatsu Y., Kubota T., Sakata M., Takeda M., Kidokoro M. Mumps virus is released from the apical surface of polarized epithelial cells and the release is facilitated by a Rab11-mediated transport system. J. Virol. 2015;89:12026–12034. doi: 10.1128/JVI.02048-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Nandy N., Roy J.K. Rab11, a potential determinant of epithelial morphogenesis and gastrulation, in Drosophila development. Mech. Dev. 2017;145:S68. [Google Scholar]
- 254.Terrien E., Simenel C., Prehaud C., Buc H., Delepierre M., Lafon M., Wolff N. 1H, 13C and 15N resonance assignments of the PDZ of microtubule-associated serine/threonine kinase 205 (MAST205) in complex with the C-terminal motif from the rabies virus glycoprotein. Biomol. NMR Assign. 2009;3:45–48. doi: 10.1007/s12104-008-9138-0. [DOI] [PubMed] [Google Scholar]
- 255.Terrien E., Chaffotte A., Lafage M., Khan Z., Préhaud C., Cordier F., Simenel C., Delepierre M., Buc H., Lafon M., Wolff N. Interference with the PTEN–MAST2 interaction by a viral protein leads to cellular relocalization of PTEN. Sci. Signal. 2012;5(237):ra58. doi: 10.1126/scisignal.2002941. [DOI] [PubMed] [Google Scholar]
- 256.Préhaud C., Wolff N., Terrien E., Lafage M., Mégret F., Babault N., Cordier F., Tan G.S., Maitrepierre E., Ménager P., Chopy D., Hoos S., England P., Delepierre M., Schnell M.J., Buc H., Lafon M. Attenuation of rabies virulence: takeover by the cytoplasmic domain of its envelope protein. Sci. Signal. 2010;3(105):ra5. doi: 10.1126/scisignal.2000510. (doi: 10.1126) [DOI] [PubMed] [Google Scholar]
- 257.Babault N., Cordier F., Lafage M., Cockburn J., Haouz A., Prehaud C., Rey F.A., Delepierre M., Buc H., Lafon M., Wolff N. Peptides targeting the PDZ domain of PTPN4 are efficient inducers of glioblastoma cell death. Structure. 2011;19:1518–1524. doi: 10.1016/j.str.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 258.Maisonneuve P., Caillet-Saguy C., Raynal B., Gilquin B., Chaffotte A., Pérez J., Zinn-Justin S., Delepierre M., Buc H., Cordier F., Wolff N. Regulation of the catalytic activity of the human phosphatase PTPN4 by its PDZ domain. FEBS J. 2014;281:4852–4865. doi: 10.1111/febs.13024. [DOI] [PubMed] [Google Scholar]
- 259.Khan Z., Lafon M. PDZ domain-mediated protein interactions: therapeutic targets in neurological disorders. Curr. Med. Chem. 2014;21(23):2632–2641. doi: 10.2174/0929867321666140303145312. (Review) [DOI] [PubMed] [Google Scholar]
- 260.Caillet-Saguy C., Maisonneuve P., Delhommel F., Terrien E., Babault N., Lafon M., Cordier F., Wolff N. Strategies to interfere with PDZ-mediated interactions in neurons: what we can learn from the rabies virus. Prog. Biophys. Mol. Biol. 2015;119:53–59. doi: 10.1016/j.pbiomolbio.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 261.Mégret F., Préhaud C., Lafage M., Batejat C., Escriou N., Lay S., Thoulouze M.I., Lafon M. Immunopotentiation of the antibody response against influenza HA with apoptotic bodies generated by rabies virus G-ERA protein-driven apoptosis. Vaccine. 2005;23:5342–5350. doi: 10.1016/j.vaccine.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 262.Seo W., Prehaud C., Khan Z., Sabeta C., Lafon M. Investigation of rabies virus glycoprotein carboxyl terminus as an in vitro predictive tool of neurovirulence. A 3R approach. Microbes Infect. 2017;19:476–484. doi: 10.1016/j.micinf.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 263.Bidoia C., Mazzorana M., Pagano M.A., Arrigoni G., Meggio F., Pinna L.A., Bertazzoni U. The pleiotropic protein kinase CK2 phosphorylates HTLV-1 Tax protein in vitro, targeting its PDZ-binding motif. Virus Genes. 2010;41:149–157. doi: 10.1007/s11262-010-0494-3. [DOI] [PubMed] [Google Scholar]