

Abstract
Oligonucleotides (ONs) are a new class of therapeutic compounds under investigation for the treatment of a variety of disease states, such as cancer and HIV, and for FDA approval of an anti‐CMV retinitis antisense molecule (Vitravene™, Isis Pharmaceuticals). However, these molecules are limited not only by poor cellular uptake, but also by a general lack of understanding regarding the mechanism(s) of ON cellular uptake. As a result, various delivery vehicles have been developed that circumvent the proposed mechanism of uptake, endocytosis, while improving target specific delivery and/or drug stability. This review describes various traditional and novel delivery mechanisms that have been employed to improve ON cellular delivery, cost effectiveness, and therapeutic efficacy. © 2003 Wiley‐Liss, Inc. and the American Pharmacists Association J Pharm Sci 92:1559–1573, 2003
Keywords: oligonucleotide, uptake, delivery, liposomes, carrier‐mediated delivery, novel delivery methods
INTRODUCTION
The approval of an antisense oligonucleotide (ON) for the treatment of HIV‐associated CMV retinitis (Vitravene™, Isis Pharmaceuticals) has signaled the birth of a new class of therapeutically useful compounds. Since their conceptual origins,1,2 ONs have been investigated as novel therapeutic agents, and they are currently under investigation for the treatment of a wide variety of diseases, such as cancer and viral diseases [human immunodeficiency virus (HIV), hepatitis, cytomegalovirus (CMV)].
ONs are short sequences of single stranded nucleic acid or nucleic acid analogs designed to bind intracellularly to target nucleic acids (RNA or DNA) or proteins in a sequence or conformation‐specific manner.3, 4, 5 Additionally, ONs containing a specific CpG motif can elicit an immune response.6, 7, 8, 9 Although the clinical relevance of ONs has been demonstrated, poor stability and inefficient cellular uptake, both in vitro and in vivo, limit the efficacy of ONs and have been a barrier to therapeutic development. Cellular uptake can be <2% of the dose,10,11 depending on ON chemistry and cell type, whereas further intracellular barriers hinder transport of the internalized ON to the active site. As a result, ON concentration at the active site, especially in terms of in vivo drug delivery, may be too low for an effective and sustained outcome. At the very least, the efficiency of ON delivery is rather poor in terms of cost‐effective drug development. Indeed, a majority of the current and past research focuses on the improvement of ON stability and delivery through modification of the ON structure itself5,12, 13, 14 or via the use of delivery vehicles, which can both protect the drug from degradation as well as improve ON delivery into cells. The purpose of this communication is to review the mechanism of ON cellular uptake as well as both some of the common and more novel ON cellular delivery methods.
OLIGONUCLEOTIDE CELLULAR UPTAKE
Mechanisms of Cellular Uptake
Advances in ON drug delivery by a variety of mechanisms have developed from a need to circumvent poor cellular uptake of “naked” ONs. However, a basic understanding of the proposed and potential actual mechanism(s) of ON uptake would be most useful for not only delivery of naked ONs but also development of optimal delivery systems. The most widely accepted mechanism of ON uptake is adsorptive endocytosis, whereby adsorption of the ON to the cell membrane results in transport of ON into the cell via endocytosis.15,16 However, intracellular localization of ON varies depending on cell type or experimental method. In some uptake studies, a significant proportion of internalized ON remains solely sequestered within the endosomal compartment, as evidenced by a punctate distribution of fluorescently labeled ON in the cell.15,17 If the ON is unable to escape the endosome, lysosomal degradation is likely. However, other research suggests that the ON escapes the endosome and is either rapidly transported to the nucleus or accumulates in perinuclear organelles.11,18,19 Currently, the mechanisms by which ONs escape lysosomal degradation and are transported into the cytosol and nucleus remain unclear. Other mechanisms of ON cellular uptake have been proposed such as pinocytosis20 and caveolar potocytosis.18
The multitude of proposed mechanisms indicates that ON uptake is not easily defined. Indeed, the mechanism can vary depending on ON chemical structure. A number of modifications to the native phosphodiester (PO) DNA backbone have been synthesized to improve the target affinity, intracellular delivery, or stability of ONs in the presence of abundant exo‐ and endonucleases. Currently, the most commonly investigated ONs are the phosphorothioate (PS), methylphosphonate (MP), and chimeric backbones containing mixed backbone chemistries, and peptide nucleic acids (PNA; Figure 1). Although the PS modification retains the aqueous solubility of the native PO backbone while increasing ON stability in the presence of nucleases, the anionic charge hinders uptake through the lipophilic membrane, resulting in poor cellular uptake (Table 1). However, ONs with an anionic backbone, like the PO and PS modifications, bind to cell surface proteins resulting in adsorptive endocytosis. In contrast to anionic ONs, neutral molecules like the MP and PNA have surprisingly low cellular uptake, resulting from low cell surface protein binding, and therefore exhibit decreased adsorptive endocytosis.
Figure 1.
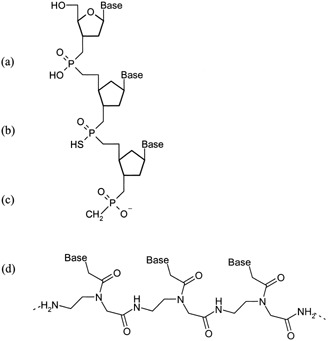
Backbone structures for commonly used oligonucleotides: (a) phosphodiester (PO); (b) phosphorothioate (PS); (c) methylphosphonate (MP); (d) peptide nucleic acid (PNA).
Table 1.
Oligonucleotide Stability and Cellular Uptakea
| Backbone Chemistry | Nuclease Stability | Cellular Uptake | Charge |
|---|---|---|---|
| PO | ↓ | ↓ | — |
| PS | ↑ | ↓ | — |
| MP | ↑ | ↓↓ | Ø |
| PNA | ↑ | ↓↓ | Ø |
Key: (↓) low; (↓↓) very low; (↑) high; (—) negative; (Ø) neutral.
Cellular uptake is also highly variable depending on cell type and cell cycle. For example, cellular uptake of a c‐myc ON into Rauscher erythroleukemia cells occurs via a nonspecific protein and energy‐dependent pathway immune to endocytic inhibition, which is inconsistent with endocytosis alone.21,22 Cellular uptake involves significant surface binding as well as both cation and cell cycle dependency;23 thus, multiple mechanisms of ON uptake in the Rauscher cell line may be occurring, some of which have yet to be completely characterized. Current evidence may also suggest a porin‐like transporter or pore may be involved in cellular uptake.21,24 Finally, some researchers have demonstrated nuclear or perinuclear fluorescence without a punctate fluorescence pattern, suggesting either a nonendocytic uptake mechanism or rapid endosomal escape.11,25, 26, 27 Therefore, alternate nonendocytic or multiple mechanisms of ON cellular uptake are plausible.
Barriers to Efficient Cellular Uptake
Despite the unknown mechanism of uptake for ONs or other hydrophilic macromolecules, intracellular transport and activity of “naked” ONs are observed, although uptake is quite inefficient. Indeed, several barriers to cellular uptake exist, which must be overcome improve ON cellular delivery. The first barrier is the lipophilic cell membrane, through which these large, anionic molecules must pass to reach the site of action. Additionally, many ONs appear to enter the cell by endocytosis, as already mentioned, resulting in sequestration of the molecules inside endosomes. Again, the ON must pass through the endosomal membrane or risk lysosomal degradation. Finally, although evidence exists that once inside the cytosol ONs are rapidly transported to the nucleus,28,29 nucleases within the intracellular milieu can degrade the ON, rendering it inactive. Ideally, optimal delivery of ONs to the site of action would be based on a thorough understanding of the mechanism of ON uptake. However, because the mechanism of ON transport into the cell remains to be fully elucidated, many researchers have focused on the empirical development of various novel delivery vehicles that circumvent the barriers to uptake to improve the efficacy and ultimately the activity of ONs.
GENERAL DELIVERY METHODS
Liposomes
The most widely used method for improving ON stability and intracellular delivery utilizes ON‐lipid conjugates or liposome formulations. Liposomes are composed from artificial cationic lipids such as N‐[1‐(2,3‐dioleoyloxy) propyl]‐N,N,N‐trimethylammonium chloride (DOTMA) and N‐[1‐(2,3‐dioleoyloxy) propyl]‐N,N,N‐trimethylammonium methyl sulfate (DOTAP), where the cationic head group associates with negatively charged ONs, neutralizing the anionic charge and facilitating delivery of the large anionic molecules through the lipophilic cell membrane.30, 31, 32, 33,35 Uptake occurs via endocytosis, in which the cationic liposomes induce membrane “flip–flop”, releasing the ON from the liposome and into the cytosol.34 Additionally, the addition of dioleyl phosphatidylethanolamine (DOPE) to liposome formulations aids in destabilization of endosomal membranes, facilitating the release of ON to the site of action.35,36 Encapsulation of ONs within liposomes also affords protection against abundant nuclease degradation in the intracellular milieu.37,38
However, cationic liposomes are cytotoxic, especially at high concentrations, which limits their usefulness both in vitro and in vivo.38,39 To combat some of the drawbacks associated with cationic liposomes, modifications to the basic liposome formulation, such as changing the lipid, composition of lipids, or lipid concentration, have been extensively investigated. For example, a novel nontoxic liposome formulation has been used to deliver a phosphorothioate ON antisense to neuropeptide corticotropin‐releasing factor receptor (CRF‐R) mRNA in CHO cells using the amphiphilic pyridinium‐based lipid SAINT‐2.40 ON–lipid complexes were formed using 20 μM SAINT‐2/DOPE (1:1) and 100 nM PS or PO anti‐CRF‐R ON. A 100–250‐fold increase in cell‐associated ON was observed following a 24‐h incubation of complex in CHO cells compared with free ON. In addition, in vitro antisense activity resulted in a 50% decrease in CRF‐R mRNA expression using complexes, whereas no effect was observed for free ON or lipid controls.
Alternatively, pH‐sensitive fusogenic liposomes are used to bypass endosomal sequestration. pH‐sensitive liposomes are composed of amphipathic lipids, such as choleseryl hemisuccinate (CHEMS) and DOPE. At physiologic pH, CHEMS stabilizes DOPE into lamellar phase liposomes. However, at acidic pH, such as that in the early endosome, CHEMS undergoes protonation, resulting in formation of a hexagonal HII phase and subsequent fusion or destabilization of the endosomal membrane and release of ON to the cytosol.41, 42, 43, 44, 45 By altering their lipid composition, pH‐sensitive liposomes can have increased stability at physiologic pH, which would improve circulation time in vivo. Additionally, pH‐sensitive liposomes also have fusogenic properties at low pH, resulting in release of the ON from the endosome to the cytosol.44 pH‐sensitive liposomes were used to deliver antisense ON against the Na+/myo‐inositol co‐transporter mRNA in astrocytes.43 Cellular uptake of ON was more efficient when pH‐sensitive liposomes were used, compared with free ON or non‐pH‐sensitive liposomes (lecithin), as determined using fluorescence microscopy. In addition, pH‐sensitive liposome delivery resulted in a 50‐fold decrease of target mRNA expression compared with free ON, suggesting that liposome encapsulation can have a significant impact of ON delivery to the site of action. However, evidence suggests that small (<200 nm) pH‐sensitive liposomes composed of DOPE/OA are stabilized in the presence of human plasma via insertion of hydrophobic plasma molecules into the liposome.46 Consequently, the liposomes lose their pH sensitivity and therefore may have decreased drug delivery capability in vivo. Therefore, the size, lipid composition, and interactions with plasma of pH‐sensitive liposomes need to be carefully characterized and adjusted to achieve optimal drug delivery.
Liposomes have been modified for targeted delivery by coupling receptor ligands47, 48, 49 or antibodies (immunoliposomes)50, 51, 52, 53, 54, 55, 56 to the liposome. For example, maleylated‐ON–BSA conjugates effectively delivered ON into Leishmania‐infected macrophages via the scavenger receptor system, resulting in nearly a 5‐fold decrease in parasite‐infected macrophages compared with the results with free ON.48 Pagnan et al.50 encapsulated c‐myb antisense ON into cationic liposomes coated with neutral lipids and covalently coupled to anti‐ganglioside GD2 monoclonal antibody (GD2‐CCL).50 Cellular uptake of ON into GD2 positive neuroblastoma cells was significantly higher for the immunoliposome‐encapsulated ON compared with free ON, resulting in a significant target‐specific decrease in c‐myb expression. Additionally, immunoliposome‐directed uptake was specific for GD2 receptors because no increase in uptake compared with free ON was observed in HeLa cell, a GD2 negative cell line, or when a nonspecific immunoliposome was used. The results suggest that non‐target GD2 negative cells may be largely unaffected if the GD2‐CCL delivery system is used in vivo in the antisense treatment of neuroblastoma.
Carrier Molecules
Receptor‐Mediated Endocytosis (RME)
RME‐directed uptake utilizes import mechanisms already present in the cell membrane for the uptake of biomolecules necessary for cell function. ONs can be linked directly to a carrier protein via a covalent bond or noncovalently via poly‐l‐lysine (PLL)–carrier conjugates. The choice of carrier is dependent on its known ability to bind to specific cell membrane receptors and accumulate in the cell via endocytosis. Not only can internalization of ON potentially be improved, but cell‐specific delivery can also be achieved by targeting receptors solely expressed or over‐expressed on certain target cells.
For example, although folate receptors are found on all dividing cells, they are overexpressed in epithelial cancer cells and therefore have been investigated for cell‐specific targeting of drugs. Ginobbi and co‐workers57 used folic acid–PLL–ON conjugates to target antisense phosphorothioate c‐myc ON to M‐14 human melanoma cells. An increase in intracellular delivery of ON was observed, resulting in a significant decrease in both c‐myc protein levels and cell growth. As a control, the investigators used a transferrin–PLL–ON conjugate, for which no RME should be observed due to the lack of transferrin receptors on M‐14 cells. Indeed, no improvement of ON uptake and activity were observed, suggesting that targeting specific receptors is a viable means to limit drug delivery to specific cell populations. However, effective drug‐targeting by RME depends not only on the affinity of the ligand for the receptor, but also on limitation of the selected receptor to target cells, especially in vivo. Additionally, the ligand itself must either be inactive or augment the therapeutic outcome to avoid potential toxicity associated with the delivery vehicle. Therefore, thorough characterization of the selected receptor and its in vivo ubiquity, or lack thereof, is a prerequisite to developing RME ON delivery.
Hepatocyte‐specific ON drug delivery for the treatment of hepatic diseases has been investigated by targeting asialoglycoprotein receptors.58, 59, 60, 61, 62, 63 Biessen and co‐workers59 delivered a random sequence ON to parenchymal liver cells by derivatizing ON with L3G4, a lysine‐based galactoside synthetic ligand with high affinity for asialoglycoprotein receptors. Although conjugation of ON to L3G4 slightly decreased the affinity of L3G4 for ASOR receptors (K d = 23 versus 6.5 nM), binding of ON to the target sequence in an in vitro assay was not significantly affected by the presence of L3G4, suggesting that the in vitro activity of the ON would not be affected by the ligand. The authors also suggested that uptake occurred via active transport because intracellular ON concentrations were 40 nM, which is 4‐fold higher than the extracellular concentration, whereas cellular uptake decreased by at least 70% on the addition of various uptake inhibitors. In addition, uptake of ON–L3G4 conjugate was significantly decreased by almost 35‐fold in the presence of the competitive ASOR receptor ligands GalNAc and ASOR. However, confocal analysis of ON‐ligand uptake using rhodamine‐labeled ON showed mainly a punctate fluorescence pattern with minor amounts of fluorescence in the cytosol, suggesting ON is sequestered within endosomal compartments.
In vivo intravenous administration of 32P‐ON–L3G4 conjugates to mice demonstrated a significant alteration of ON distribution. Plasma clearance for ON was rapid; that is, at 2 min after injection, only 14.3 and 11.5% of free and conjugated ON, respectively, remained in plasma. However, whereas only 19.1% of free ON was found in liver, L3G4 conjugation increased delivery to the liver 4‐fold to 77.6%. Not only did conjugation increase selective ON delivery to the liver, but also conjugates successfully bypassed reticuloendothelial clearance and scavenger‐receptor uptake within the liver. A 25‐fold increase of ON conjugate delivery to parenchymal liver rather than Kupffer or endothelial liver cells was observed compared with free ON hepatic distribution. Therefore, ON conjugates may potentially be employed to selectively deliver ONs using RME both in vitro and in vivo. However, further studies regarding the in vitro or in vivo activity of ON conjugates are necessary to determine the overall efficacy of ASOR RME delivery of ONs to the liver.
Many other cell‐specific receptors may prove useful for targeted ON delivery. Enhancement of ON delivery has been investigated using mannose delivery to alveolar macrophages64 and human tumor targeting using ErbB‐265 and transferrin66,67 or anti‐transferrin antibody.68 Anti‐transferrin antibody–ON conjugates have also been used to target ON delivery into the brain,69 CD14‐ON conjugates have been used to target monocytes,70 and epidermal growth factor (EGF) receptor has been targeted for delivery of EGF–ON conjugates to epithelial cancer cells.71 However, RME generally results in sequestration of ON in endosomal compartments, thereby limiting the utility of this method for delivering therapeutic levels of drug to intracellular targets. Additionally, ON–protein conjugates must be carefully designed such that the ON does not hinder receptor‐ligand binding by blocking receptor‐binding sites on the carrier protein.
Fusogenic Peptides
Fusogenic peptides have been used to promote peptide fusion of ON–peptide conjugates with either cell or lysosomal membranes.72, 73, 74 For example, Morris et al. developed a peptide vector containing hydrophobic HIV gp41 fusion peptide and hydrophilic SV40 T‐antigen to create ON–peptide conjugates via electrostatic interaction between ON and the hydrophilic peptide.74 Using confocal microscopy, cellular uptake of the conjugates was observed to occur via a non‐endocytic pathway because ONs were localized to the nucleus within 1 h in 90% of cells, whereas very little punctate fluorescence was observed. In contrast, following endocytosis of ON conjugates to influenza virus hemagglutinin (HA) envelope fusion peptide, acidification of the endosomal compartment resulted in a conformational change in HA, destabilization of the lysosomal membrane, and release of ON conjugate to the cytosol.75, 76, 77 Freulon and co‐workers investigated the uptake of the anti‐HIV‐1 antisense ON GEM 91 in A549 and HeLa cells by coincubating unconjugated ON and either monomeric or dimeric E5 C‐terminal subunit of HA2 with A549 and HeLa cells in slightly acidic media.77 Under acidic conditions, HA2 peptides destabilized endosomal membranes, allowing for ON delivery into the cytosol. Complete uptake of 0.1 μM fluorescently labeled ON occurred using 40 μM E5 monomer at pH 5.8 following a 30‐min incubation at 37°C. Uptake was similar using different fluorophores, cell lines, and ON backbones but was dependent on pH, supporting the role of endosome acidification in HA fusion peptide‐mediated delivery. Confocal analysis indicated that ON and E5 peptide were co‐localized in endosomal compartments prior to acidification. However, following acidification and endosomal membrane destabilization, ONs were localized to the nucleus, whereas E5 remained in the cytosol. Although in vitro activity studies were not done, the potential for increased activity at lower ON concentrations was suggested because ON remained free (uncomplexed to the peptide vector) and delivery to the nucleus was improved compared with ON alone.
Signal Peptides
Selective import of nuclear proteins from the cytosol into the nucleus is mediated by short peptide sequences called nuclear localization signals (NLS). NLS peptide derivatives of SV‐40,78, 79, 80 transcription nuclear factor κB,81 influenza viral nucleoprotein,82 and yeast α2 protein82 have been used to facilitate ON transport to the nucleus. In addition to NLS, signal import peptides (IP), which promote cellular uptake of IP or DNA, have been derived from Kaposi's fibroblast growth factor (K‐FGF)81,83 and DNA uptake stimulating factor (DUSF) of Neurospora crassa, 84 respectively.
The synthetic IP from K‐FGF promoted transient pore formation in cell membranes of P59, A549, SV40, and RAW264.7 cells.83 Uptake of ON–PLL–IP conjugates was dose dependent at 50–500 μM IP–PL and resulted in strong intracellular cytoplasmic and nuclear fluorescence via a nonendocytic pathway, as demonstrated by confocal microscopy and endocytosis inhibition studies. Competition studies also indicated that the uptake mechanism was not receptor mediated. However, similar studies using ON–K‐FGF disulfide conjugates in monkey kidney fibroblast CV1‐P, CHO, and human retinal pigmented epithelial D407 cells resulted in punctate distribution of ON, with faint nuclear and cytosolic fluorescence, suggesting that the mechanism of K‐FGF‐mediated ON delivery varied among cell lines or that the method of ON–peptide conjugation strongly affected the delivery mechanism.
Finally, multiple import mechanisms may be combined for optimal ON delivery. Peptides derived from HIV tat or Drosophila antennapedia proteins serve as both cell membrane translocation signals as well as nuclear transport signals.85 Fusion peptide–NLS conjugates are often used to deliver ONs and include delivery assemblages like K‐FGF–NLS,81 gp41–NLS,74,80 and protamine–NLS.79
NOVEL DELIVERY METHODS
Cyclodextrins
Cyclodextrins (CDs) are cyclic oligosaccharides that consist of a hydrophobic central cavity and multiple hydroxyl groups on their outer surface (Figure 2). Because of their unique ability to associate with other molecules, their water solubility, and the potential for derivatization of the hydroxyl groups in the central cavity, CDs have been investigated for the delivery of a variety of drug molecules.86,87 CD delivery systems have also been used to minimize drug toxicity88 or enhance bioavailability via multiple delivery routes.88,89 β‐CDs were used to deliver a 25‐mer phosphorothioate ON into a variety of human T‐cell leukemia cells lines.90 The authors reported that not only did cellular uptake of ON improve in the presence of CD (2‐ to 3‐fold over 24 h), but that uptake was dependent on both the concentration of CD and ON. Importantly, ONs remained active when CD delivery was used.91 The antiviral activity of an antisense ON targeted to coronavirus in adenocarcinoma cells increased almost 8‐fold when 7.5 μM ON was delivered using a CD derivative, 6‐deoxy‐6‐S‐β‐d‐galactopyranosyl‐6‐thio‐cyclomaltoheptaose, compared with free ON. No viral inhibition or cell toxicity was associated with the CD itself.
Figure 2.
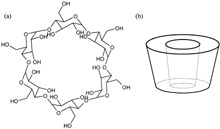
Chemical structure (a) and schematic diagram (b) of βcyclodextrin.
CDs have also been used to deliver ONs in vivo.92,93 Zhao and co‐workers used CDs to deliver ONs containing immunogenic CpG sequences to mice.93 At a dose of 50 mg/kg ON in varying concentrations of CD, the authors found that CD‐mediated delivery resulted in a decrease in the mitogenicity of the ON, a lack of splenomegaly, and increased IgM production compared with free ON. However, when coadministered with ON, the protective effects of the CD against ON‐associated toxicity decreased over time because the clearance of CD was much more rapid than that of the ON. Although the mechanism by which CDs inhibit immune stimulation by ONs was unclear, the authors suggested the possibility that inclusion of ON within the CD cavity blocks potential ON–protein binding with proteins involved in lymphocyte activation. Therefore, CD–ON formulations must be optimized to retain the improved delivery and stability of ONs while lengthening the time frame in which CD are also protective. Similarly, Barrett et al.92 found that cellular uptake of β‐CD–adamantyl–ON conjugates in PC12 cells was enhanced compared with free ON. Additionally, following a 2‐week systemic administration of the conjugate to mice, uptake to dorsal root ganglia, liver, and kidneys, but not brain, increased compared with free ON, suggesting CD may be used to target specific disease sites in vivo.
Dendrimers
Dendrimers are highly branched macromolecules synthesized by multiplication of a series of repetitive units, typically polyamides (Figure 3). The molecules have surface functional groups, such as primary amino groups, which interact with other molecules via electrostatic interaction. As a result, rapid and highly reproducible complex formation occurs, yielding complexes with weak cytotoxicity. An increase in research investigating the potential of dendrimers as ON and/or DNA delivery vehicles has recently occurred.94, 95, 96, 97
Figure 3.
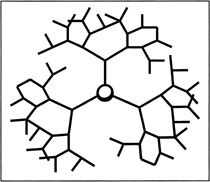
Schematic diagram of a generation 4 dendrimer.
Bielinska et al. investigated the cellular uptake and in vitro activity of a 27‐mer anti‐luciferase ON, either as free ON or complexed with generation 3 starburst PAMAM (polyamidoamine) dendrimers, using luciferase expressing U937, Jurkat, D5, and Rat 2 cells.98 Dendrimer–ON complexes at various charge ratios resulted in a 4‐ to 5‐fold increase in cellular uptake and 30–60% decreases in luciferase expression compared with free ON in all cell lines, suggesting increased ON cytosolic delivery. In addition, ONs delivered using dendrimers had significantly higher retention in cells, up to 4 days, compared with <24 h with free ON. Similarly, Yoo and Juliano investigated the mechanism of dendrimer‐mediated ON cellular uptake in HeLa cells using TAMRA‐labeled ON–Oregon Green 488‐labeled generation 5 PAMAM dendrimers.94 Dendrimer–ON complexes remained intact intracellularly and, although presumably bound to the plasma membrane and in endosomal compartments, also co‐localized in the cytoplasm and nucleus to a much higher extent than free ON. Similar to the results of Bielinska et al., the data suggested that dendrimers enhanced the extra‐endosomal delivery of ONs. Additionally, delivery of ON in the presence of 30% FCS was possible using dendrimers, whereas liposome formulations, such as lipofectamine, are typically inactive in the presence of serum. Therefore, the lack of cytotoxicity and enhanced delivery of ONs, even in the presence of serum, has possibly given dendrimers an advantage over traditional delivery vehicles like liposomes.
Nanoparticles and Microparticles
Cyanoacrylate nanoparticles have been studied as drug delivery tools since the late 1970s99,100 and as ON delivery tools since the early 1990s.101, 102, 103 Drug–nanoparticle association is achieved through hydrophobic van der Waals interactions. The anionic charge of the ON necessitates formation of ion pairs using various hydrophobic cations prior to loading nanoparticles. Typically, alkylcyanoacrylates, such as polyisohexyl‐ (PIHCA), polyisobutyl‐ (PIBCA), or polyhexycyanoacrylate (PHCA), are used to form the nanoparticles, although a variety of lipophilic cation–ON ion pairs have been tested.103, 104, 105 Adsorption of ON to nanoparticulate systems is dependent on not only the type of cyanoacrylate used and its respective hydrophobicity, but also ON length and the type of hydrophobic cation used. In addition, ON adsorption to nanoparticles is inversely related to ionic strength during formulation.103
Using ON–CTAB (hexadecylmethylammonium)–PIHCA nanoparticles, Helene and co‐workers investigated the cellular uptake and stability of ONs in U937 cells.101 In cell‐free systems, nanoparticle‐encapsulated ON was stable for >20 h in the presence of phosphodiesterase at 37°C, whereas free ON was degraded within 10 min. Similarly, in serum‐containing cell culture medium, nanoparticles were able to protect ONs from nuclease degradation for up to 3 h, whereas they were rapidly degraded when unencapsulated. Finally, uptake of ON nanoparticles was ∼6‐fold higher than unencapsulated ONs following a 24‐h incubation with U937 cells, with no observed cytotoxicity.
Little in vivo work was done using nanoparticle ON delivery systems.102,106,107 For example, tumor growth in nude mice was inhibited by intratumoral injection of anti‐Ha‐ras ON adsorbed to PIHCA nanoparticles and required significantly less ON than injection of free ON alone.102 Similarly, Lambert et al. found that Ewing sarcoma‐related tumor size in nude mice was ∼3‐fold smaller following intratumoral injection of anti‐EWS Fli‐1 ON adsorbed to PICA nanoparticles compared with free ON following ∼15–20 days of treatment.106 Finally, studies using 33P‐d(T)16‐CTAB‐PIBCA nanoparticles to deliver ON to mice indicated that ON stability was improved compared with free ONs in both plasma and liver.107 Additionally, while plasma half‐life (t½) was not significantly altered, distribution of ONs, when encapsulated by nanoparticles, was shifted to the liver and out of other organs.
Nanoparticle encapsulation can increase in vitro and in vivo cellular uptake of ONs with little cytotoxicity, while increasing in vitro ON nuclease stability. Although cell uptake and presumably activity are limited by endosomal sequestration, chemical modifications to nanoparticles to improve cytosolic and nuclear delivery of ONs in vivo may be possible. In addition, nanoparticles may be an advantageous delivery system for targeted delivery of ONs to the liver.
Recently, the use of microparticulate delivery systems has been investigated for sustained‐release delivery of ONs.108, 109, 110 Microparticles or microspheres are micron‐sized delivery systems typically composed of the biodegradable polymer poly(d,l‐lactide‐co‐glycolide) [P(LA‐GA)]. The advantage of microsphere formulation is slow release of encapsulated drug. For example, De Rosa et al. investigated the cellular uptake of poly dT(4–16) ONs encapsulated in 30–60 μM P(LA‐GA) micropheres in HeLa cells.108 Release of ON from microspheres was triphasic, indicating an initial burst phase, a slow‐release phase occurring over ∼15 days, and eventually bulk erosion of the microsphere. Additionally, both encapsulation efficiency and release rate of ON could be controlled by the addition of polyethylenimine (PEI). Interestingly, confocal microscopy of HeLa cells incubated with ON, ON/PEI, or microsphere‐encapsulated ON/PEI demonstrated increased cytosolic and nuclear delivery of rhodamine‐labeled ON. The authors proposed that the extracellular controlled release of ON from micropheres prevented saturation of pinocytotic uptake, resulting in increased cytoplasmic accumulation and nuclear delivery of ONs. Similar results have been demonstrated following encapsulation of c‐myc or tat PO‐ONs in 10–20 μm P(LA‐GA) microspheres.110 The authors found that ON release was triphasic, with an initial burst phase during the first 48 h and microsphere degradation after 25 days when microspheres were incubated in physiologic buffer. In addition, the stability of the ONs was significantly improved in the presence of serum, yet ON‐RNA hybridization was not hindered by microsphere encapsulation. Therefore, the development of microsphere delivery systems for ONs may not only improve cellular delivery of ONs, but will minimize the need for multiple dosing of ONs.
Physical Methods
In addition to molecular‐ and chemical‐based strategies for delivering ONs into cells, physical methods of delivery have also been reported. Such delivery methods utilize the physical forces of electricity, pressure, and sound.
Electroporation and Iontophoresis
Electroporation is a delivery technique that utilizes high intensity electric fields to destabilize lipid bilayers, thereby permeabilizing cell membranes and allowing for increased transport of drug or ionized drugs (iontophoresis) into target cells. Electroporation has been successfully used to enhance the transdermal delivery of ONs111, 112, 113, 114, 115, 116 and deliver ON in ex vivo and in vivo studies.114,117 Zewert and co‐workers successfully increased transdermal delivery of c‐myb and c‐myc antisense‐ONs through heat‐stripped stratum corneum using electroporation.115 Increases of 6‐ to 12‐fold in c‐myb‐ and c‐myc‐ON transport, respectively, were observed when a 1.1‐ms pulse of 80 V was applied every 5 s for 1 h compared with pre‐pulsed ON transport.115 Transport occurred as a result of electrophoresis through the skin, rather than through preexisting pores or hair follicles.
Electroporation has been applied successfully to the in vivo delivery of bcl‐2 antisense‐ONs in the rat.114 Application of 0.5 A through a 0.3–0.6‐cm section of rat liver in vivo resulted in selective transfer of liposome‐encapsulated ON to the liver within the target zone, yielding an increased apoptosis of precancerous cells. Therefore, delivery of anticancer ONs in conjunction with surgical removal and/or exposure of cancerous tissue may prove to be an effective combination treatment. In addition to in vivo experiments, ex vivo treatment of bone marrow with c‐myc antisense‐ON and electroporation has been investigated for the purging of bone marrow contaminated with cancer cells for autologous transplantation.117 Studies in mice have shown an immediate decrease in myc protein in ON‐treated samples, rapid and selective cell death of cancerous cells, and little observed effect on hematopoeitic reconstitution. The combined results from the literature suggest that electroporation will potentially be an effective method for delivery of ONs via the transdermal route and may facilitate selective ON delivery either in vivo or ex vivo.
Pressure‐Mediated Delivery
Application of pressure to “naked” ON delivery systems has been investigated both in vitro and ex vivo to inhibit bypass graft and cardiac allograft rejection.118,119 Studies in rat carotid arteries have demonstrated a pressure‐dependent uptake of FITC‐ON.119 Using an infusion pressure of 760 mmHg, ∼84, 64, and 92% neointimal, medial, and adventitial cell nuclei, respectively, were positive for FITC‐ON after 4 days. In an ex vivo experiment, excised rat hearts were submersed in an ON solution followed by injection of anti‐IL‐6– or ICAM‐1–ON solution into the coronary circulation under 0.5–2 atm of pressure above ambient pressure.118 The hearts were then transplanted and harvested 90 days postoperatively, and levels of IL‐6 or ICAM‐1 were measured. A maximal uptake efficiency of 53% was observed when ON was applied both to the myocardial surface as well as into the circulation for 30 min at 2 atm, as already described, whereas only 3.5% uptake occurred when the ON was applied under ambient pressure. No neutrophil infiltration, edema, or histological changes were observed, but microscopy of myocardial cells suggested that uptake bypasses endosomal sequestration, resulting in direct transport of ON to the nucleus. Although ON delivery into cells and/or tissues was improved using pressure‐mediated delivery, the need for a closed system under pressure may limit the technique to ex vivo procedures.
Shockwaves
The use of shockwaves, acoustic high‐energy pressure pulses, as a novel method of cytoplasmic ON delivery has recently been introduced.120 Shock waves are delivered through a fluid medium, focused on a targeted area of delivery. Sequential positive and negative pressure waves cause air bubbles and secondary shock wave emission resulting in transient pore formation in cell membranes (cavitation). A lithotripter (80 nF, 1 Hz, 25 kV) was used to deliver shockwaves to PBMC and HeLa cells in the presence of 50 μM anti‐TNFα–ON. Following the shockwave, cells were washed and placed under 3 bar of pressure. FITC‐ON uptake was measured by flow cytometry and fluorescence microscopy, and TNFα production was measured by ELISA. Cell viability remained >95%. Cell uptake was dependent on the number of shockwaves and ON concentration. ON distribution in the cell was initially homogenous, followed by accumulation in the nucleus. At 50 μM ON and using 250 shockwaves, up to 62% inhibition of TNFα was observed compared with cells treated with ON in the absence of shockwaves. Comparatively, electroporation resulted in spotty fluorescence on the cell membrane, with little nuclear accumulation and lower in vitro ON activity. Shockwaves therefore may be an advantageous delivery mechanism because non‐endosomal delivery, the level of which can be adjusted by altering the concentration of ON, is achieved. Additionally, delivery is reproducible and can be applied to various cell types. Although the high volume of ON solution required for the technique may somewhat limit its widespread applicability, improved in vivo gene transfer into mouse melanoma cells has been demonstrated by administration of 800 shockwaves following injection of air (10% tumor volume) to improve acoustic cavitation. Therefore, shockwave‐mediated delivery may potentially be a successful method for ON delivery.121
Ultrasound
Ultrasound is an acoustic technique similar to shockwaves but one that uses higher frequencies (MHz versus Hz) and shorter overall application times (seconds to minutes). Recently, the use of ultrasound as an enhancer of DNA transfection in gene therapy has been investigated.122, 123, 124 In in vitro experiments, ultrasound waves were transmitted through cell culture flask walls, permeabilizing cell membranes and allowing DNA to enter the cell. At 1‐MHz continuous wave frequency, primary rat fibroblasts or chondrocytes were transiently transfected with lacZ or neo gene‐containing plasmids at an efficiency of up to 2.4%.122 Additionally, if the duration of 1 MHz ultrasound treatment was <60 s, little decrease in cell viability was observed.123 Huber et al. compared shockwave and ultrasound in the transfection of β‐gal and luciferase expressing plasmids into HeLa cells.124 Luciferase expression was maximal following 100 Hz ultrasound for a duration of 2 min under 1.5 mPa pressure. This treatment resulted in 3% transfection of viable cells, which is about a 5‐fold increase over transfection in the absence of ultrasound. In comparison, although shock‐wave‐mediated transfection increased as the number of waves delivered increased, only a 0.08% transfection rate was observed, partially due to a reduction in cell viability. Therefore, ultrasound may prove to be a more efficient and less toxic means than shockwaves of delivering DNA into cells. Future studies may prove that ultrasound can also be used to effectively deliver shorter DNA strands, like ONs, into cells or tissues whether in vitro or in vivo.
CONCLUSIONS
In vitro uptake of naked ONs into cells is highly inefficient and necessitates novel delivery methods. In addition, ON endosomal sequestration is commonly observed, limiting ON concentration at the target site in the cytosol and/or nucleus. This review has described various standard and novel techniques currently in use for the enhancement of ON delivery into cell culture. Several of the techniques (electroporation, liposomes, nanoparticles, pressure, and dendrimers) have also been investigated in vivo as potential delivery systems for therapeutic application of ONs. In addition, delivery methods that bypass or destabilize the endosome may prove to be most useful because endosomal escape is a major limiting factor for drug accumulation at the target site.28,74,83 Significant progress has been made in the area of ON drug delivery and, although questions remain regarding toxicity, stability, and optimization of the various delivery systems, the therapeutic use of such systems may prove to be necessary for the effective use of ONs in clinical settings.
REFERENCES
- 1. Zamecnik PC, Stephenson ML. 1978. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA 75: 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stephenson ML, Zamecnik PC. 1978. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA 75: 285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crooke ST. 2000. Progress in antisense technology: the end of the beginning. Methods Enzymol 313: 3–45. [DOI] [PubMed] [Google Scholar]
- 4. Stull RA, Szoka FC Jr. 1995. Antigene, ribozyme and aptamer nucleic acid drugs: Progress and prospects. Pharm Res 12: 465–483. [DOI] [PubMed] [Google Scholar]
- 5. Helene C, Toulme JJ. 1990. Specific regulation of gene expression by antisense, sense and antigene nucleic acids. Biochim Biophys Acta 1049: 99–125. [DOI] [PubMed] [Google Scholar]
- 6. Klinman DM, Barnhart KM, Conover J. 1999. CpG motifs as immune adjuvants. Vaccine 17: 19–25. [DOI] [PubMed] [Google Scholar]
- 7. Krieg AM. 2000. CpG oligonucleotides as immune adjuvants. Ernst Schering Res Found Workshop 30: 105–118. [DOI] [PubMed] [Google Scholar]
- 8. Krieg AM. 2000. Immune effects and mechanisms of action of CpG motifs. Vaccine 19: 618–622. [DOI] [PubMed] [Google Scholar]
- 9. Weiner GJ, Liu HM, Wooldridge JE, Dahle CE, Krieg AM. 1997. Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc Natl Acad Sci USA 94: 10833–10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wickstrom EL, Bacon TA, Gonzalez A, Freeman DL, Lyman GH, Wickstrom E. 1988. Human promyelocytic leukemia HL‐60 cell proliferation and c‐myc protein expression are inhibited by an antisense pentadecadeoxynucleotide targeted against c‐myc mRNA. Proc Natl Acad Sci USA 85: 1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marti G, Egan W, Noguchi P, Zon G, Matsukura M, Broder S. 1992. Oligodeoxyribonucleotide phosphorothioate fluxes and localization in hematopoietic cells. Antisense Res Dev 2: 27–39. [DOI] [PubMed] [Google Scholar]
- 12. Peyman A, Uhlmann E. 1996. Minimally modified oligonucleotides‐combination of end‐capping and pyrimidine‐protection. Biol Chem Hoppe Seyler 377: 67–70. [DOI] [PubMed] [Google Scholar]
- 13. Uhlmann E, Ryte A, Peyman A. 1997. Studies on the mechanism of stabilization of partially phosphorothioated oligonucleotides against nucleolytic degradation. Antisense Nucleic Acid Drug Dev 7: 345–350. [DOI] [PubMed] [Google Scholar]
- 14. Gilar M, Belenky A, Budman Y, Smisek DL, Cohen AS. 1998. Impact of 3′‐exonuclease stereoselectivity on the kinetics of phosphorothioate oligonucleotide metabolism. Antisense Nucleic Acid Drug Dev 8: 35–42. [DOI] [PubMed] [Google Scholar]
- 15. Shoji Y, Akhtar S, Periasamy A, Herman B, Juliano RL. 1991. Mechanism of cellular uptake of modified oligodeoxynucleotides containing methylphosphonate linkages. Nucleic Acids Res 19: 5543–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yakubov LA, Deeva EA, Zarytova VF, Ivanova EM, Ryte AS, Yurchenko LV, Vlassov VV. 1989. Mechanism of oligonucleotide uptake by cells: Involvement of specific receptors? Proc Natl Acad Sci USA 86: 6454–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loke SL, Stein CA, Zhang XH, Mori K, Nakanishi M, Subasinghe C, Cohen JS, Neckers LM. 1989. Characterization of oligonucleotide transport into living cells. Proc Natl Acad Sci USA 86: 3474–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zamecnik P, Aghajanian J, Zamecnik M, Goodchild J, Witman G. 1994. Electron micrographic studies of transport of oligodeoxynucleotides across eukaryotic cell membranes. Proc Natl Acad Sci USA 91: 3156–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farrell CL, Bready JV, Kaufman SA, Qian YX, Burgess TL. 1995. The uptake and distribution of phosphorothioate oligonucleotides into vascular smooth muscle cells in vitro and in rabbit arteries. Antisense Res Dev 5: 175–183. [DOI] [PubMed] [Google Scholar]
- 20. Stein CA, Tonkinson JL, Zhang LM, Yakubov L, Gervasoni J, Taub R, Rotenberg SA. 1993. Dynamics of the internalization of phosphodiester oligodeoxynucleotides in HL60 cells. Biochemistry 32: 4855–4861. [DOI] [PubMed] [Google Scholar]
- 21. Wu‐Pong S, Weiss TL, Hunt CA. 1992. Antisense c‐myc oligodeoxyribonucleotide cellular uptake. Pharm Res 9: 1010–1017. [DOI] [PubMed] [Google Scholar]
- 22. Wu‐Pong S, Weiss TL, Hunt CA. 1994. Antisense c‐myc oligonucleotide cellular uptake and activity. Antisense Res Dev 4: 155–163. [DOI] [PubMed] [Google Scholar]
- 23. Wu‐Pong S, Bard J, Huffman J, Jimerson J. 1997. Oligonucleotide biological activity: Relationship to the cell cycle and nuclear transport. Biol Cell 89: 257–261. [PubMed] [Google Scholar]
- 24. Wu‐Pong S. 2000. Alternative interpretations of the oligonucleotide transport literature: Insights from nature. Adv Drug Deliv Rev 44: 59–70. [DOI] [PubMed] [Google Scholar]
- 25. Laktionov P, Dazard JE, Piette J, Vives E, Rykova E, Vlassov V, Lebleu B. 1999. Uptake of oligonucleotides by keratinocytes. Nucleosides Nucleotides 18: 1697–1699. [DOI] [PubMed] [Google Scholar]
- 26. Noonberg SB, Garovoy MR, Hunt CA. 1993. Characteristics of oligonucleotide uptake in human keratinocyte cultures. J Invest Dermatol 101: 727–731. [DOI] [PubMed] [Google Scholar]
- 27. Iversen PL, Zhu S, Meyer A, Zon G. 1992. Cellular uptake and subcellular distribution of phosphorothioate oligonucleotides into cultured cells. Antisense Res Dev 2: 211–222. [DOI] [PubMed] [Google Scholar]
- 28. Chin DJ, Green GA, Zon G, Szoka FC Jr, Straubinger RM. 1990. Rapid nuclear accumulation of injected oligodeoxyribonucleotides. New Biol 2: 1091–1100. [PubMed] [Google Scholar]
- 29. Leonetti JP, Mechti N, Degols G, Gagnor C, Lebleu B. 1991. Intracellular distribution of microinjected antisense oligonucleotides. Proc Natl Acad Sci USA 88: 2702–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fimmel S, Saborowski A, Orfanos CE, Zouboulis CC. 2000. Development of efficient transient transfection systems for introducing antisense oligonucleotides into human epithelial skin cells. Horm Res 54: 306–311. [DOI] [PubMed] [Google Scholar]
- 31. Stuart DD, Allen TM. 2000. A new liposomal formulation for antisense oligodeoxynucleotides with small size, high incorporation efficiency and good stability. Biochim Biophys Acta 1463: 219–229. [DOI] [PubMed] [Google Scholar]
- 32. Lappalainen K, Urtti A, Jaaskelainen I, Syrjanen K, Syrjanen S. 1994. Cationic liposomes mediated delivery of antisense oligonucleotides targeted to HPV 16 E7 mRNA in CaSki cells. Antiviral Res 23: 119–130. [DOI] [PubMed] [Google Scholar]
- 33. Lappalainen K, Urtti A, Soderling E, Jaaskelainen I, Syrjanen K, Syrjanen S. 1994. Cationic liposomes improve stability and intracellular delivery of antisense oligonucleotides into CaSki cells. Biochim Biophys Acta 1196: 201–208. [DOI] [PubMed] [Google Scholar]
- 34. Zelphati O, Szoka FC Jr. 1996. Mechanism of oligonucleotide release from cationic liposomes. Proc Natl Acad Sci USA 93: 11493–11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Farhood H, Gao X, Son K, Yang YY, Lazo JS, Huang L, Barsoum J, Bottega R, Epand RM. 1994. Cationic liposomes for direct gene transfer in therapy of cancer and other diseases. Ann NY Acad Sci 716: 2334; discussion 34–35. [DOI] [PubMed] [Google Scholar]
- 36. Jaaskelainen I, Monkkonen J, Urtti A. 1994. Oligonucleotide‐cationic liposome interactions. A physicochemical study. Biochim Biophys Acta 1195: 115–123. [DOI] [PubMed] [Google Scholar]
- 37. Thierry AR, Dritschilo A. 1992. Intracellular availability of unmodified, phosphorothioated and liposomally encapsulated oligodeoxynucleotides for antisense activity. Nucleic Acids Res 20: 5691–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Capaccioli S, Di Pasquale G, Mini E, Mazzei T, Quattrone A. 1993. Cationic lipids improve antisense oligonucleotide uptake and prevent degradation in cultured cells and in human serum. Biochem Biophys Res Commun 197: 818–825. [DOI] [PubMed] [Google Scholar]
- 39. Konopka K, Rossi JJ, Swiderski P, Slepushkin VA, Duzgunes N. 1998. Delivery of an anti‐HIV‐1 ribozyme into HIV‐infected cells via cationic liposomes. Biochim Biophys Acta 1372: 55–68. [DOI] [PubMed] [Google Scholar]
- 40. Shi F, Nomden A, Oberle V, Engberts JB, Hoekstra D. 2001. Efficient cationic lipid‐mediated delivery of antisense oligonucleotides into eukaryotic cells: Down‐regulation of the corticotropin‐releasing factor receptor. Nucleic Acids Res 29: 2079–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Legendre JY, Szoka FC Jr. 1992. Delivery of plasmid DNA into mammalian cell lines using pH‐sensitive liposomes: Comparison with cationic liposomes. Pharm Res 9: 1235–1242. [DOI] [PubMed] [Google Scholar]
- 42. Ropert C, Lavignon M, Dubernet C, Couvreur P, Malvy C. 1992. Oligonucleotides encapsulated in pH sensitive liposomes are efficient toward Friend retrovirus. Biochem Biophys Res Commun 183: 879–885. [DOI] [PubMed] [Google Scholar]
- 43. Lubrich B, van Calker D, Peschka‐Suss R. 2000. Inhibition of inositol uptake in astrocytes by antisense oligonucleotides delivered by pH‐sensitive liposomes. Eur J Biochem 267: 2432–2438. [DOI] [PubMed] [Google Scholar]
- 44. Ponnappa BC, Dey I, Tu GC, Zhou F, Aini M, Cao QN, Israel Y. 2001. In vivo delivery of antisense oligonucleotides in pH‐sensitive liposomes inhibits lipopolysaccharide‐induced production of tumor necrosis factor‐α in rats. J Pharmacol Exp Ther 297: 1129–1136. [PubMed] [Google Scholar]
- 45. Koltover I, Salditt T, Radler JO, Safinya CR. 1998. An inverted hexagonal phase of cationic liposome‐DNA complexes related to DNA release and delivery. Science 281: 78–81. [DOI] [PubMed] [Google Scholar]
- 46. Liu DX, Huang L. 1989. Small, but not large, unilamellar liposomes composed of dioleoylphosphatidylethanolamine and oleic acid can be stabilized by human plasma. Biochemistry 28: 7700–7707. [DOI] [PubMed] [Google Scholar]
- 47. Waelti ER, Gluck R. 1998. Delivery to cancer cells of antisense l‐myc oligonucleotides incorporated in fusogenic, cationic‐lipid‐reconstituted influenza‐virus envelopes (cationic virosomes). Int J Cancer 77: 728–733. [DOI] [PubMed] [Google Scholar]
- 48. Chaudhuri G. 1997. Scavenger receptor‐mediated delivery of antisense mini‐exon phosphorothioate oligonucleotide to Leishmania‐infected macrophages. Selective and efficient elimination of the parasite. Biochem Pharmacol 53: 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S, Lee RJ, Cauchon G, Gorenstein DG, Low PS. 1995. Delivery of antisense oligodeoxyribonucleotides against the human epidermal growth factor receptor into cultured KB cells with liposomes conjugated to folate via polyethylene glycol. Proc Natl Acad Sci USA 92: 3318–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pagnan G, Stuart DD, Pastorino F, Raffaghello L, Montaldo PG, Allen TM, Calabretta B, Ponzoni M. 2000. Delivery of c‐myb antisense oligodeoxynucleotides to human neuroblastoma cells via disialoganglioside GD(2)‐targeted immunoliposomes: Antitumor effects. J Natl Cancer Inst 92: 253–261. [DOI] [PubMed] [Google Scholar]
- 51. Selvam MP, Buck SM, Blay RA, Mayner RE, Mied PA, Epstein JS. 1996. Inhibition of HIV replication by immunoliposomal antisense oligonucleotide. Antiviral Res 33: 11–20. [DOI] [PubMed] [Google Scholar]
- 52. Ma DD, Wei AQ. 1996. Enhanced delivery of synthetic oligonucleotides to human leukaemic cells by liposomes and immunoliposomes. Leuk Res 20: 925–930. [DOI] [PubMed] [Google Scholar]
- 53. Zelphati O, Imbach JL, Signoret N, Zon G, Rayner B, Leserman L. 1994. Antisense oligonucleotides in solution or encapsulated in immunoliposomes inhibit replication of HIV‐1 by several different mechanisms. Nucleic Acids Res 22: 4307–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zelphati O, Wagner E, Leserman L. 1994. Synthesis and anti‐HIV activity of thiocholesteryl‐coupled phosphodiester antisense oligonucleotides incorporated into immunoliposomes. Antiviral Res 25: 13–25. [DOI] [PubMed] [Google Scholar]
- 55. Zelphati O, Zon G, Leserman L. 1993. Inhibition of HIV‐1 replication in cultured cells with antisense oligonucleotides encapsulated in immunoliposomes. Antisense Res Dev 3: 323–338. [DOI] [PubMed] [Google Scholar]
- 56. Leonetti JP, Machy P, Degols G, Lebleu B, Leserman L. 1990. Antibody‐targeted liposomes containing oligodeoxyribonucleotides complementary to viral RNA selectively inhibit viral replication. Proc Natl Acad Sci USA 87: 2448–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ginobbi P, Geiser TA, Ombres D, Citro G. 1997. Folic acid‐polylysine carrier improves efficacy of c‐myc antisense oligodeoxynucleotides on human melanoma (M14) cells. Anticancer Res 17: 29–35. [PubMed] [Google Scholar]
- 58. Biessen EA, Vietsch H, Rump ET, Fluiter K, Bijsterbosch MK, van Berkel TJ. 2000. Targeted delivery of antisense oligonucleotides to parenchymal liver cells in vivo. Methods Enzymol 314: 324–342. [DOI] [PubMed] [Google Scholar]
- 59. Biessen EA, Vietsch H, Rump ET, Fluiter K, Kuiper J, Bijsterbosch MK, van Berkel TJ. 1999. Targeted delivery of oligodeoxynucleotides to parenchymal liver cells in vivo. Biochem J 340: 783–792. [PMC free article] [PubMed] [Google Scholar]
- 60. Michael SI, Huang CH, Romer MU, Wagner E, Hu PC, Curiel DT. 1993. Binding‐incompetent adenovirus facilitates molecular conjugate‐mediated gene transfer by the receptor‐mediated endocytosis pathway. J Biol Chem 268: 6866–6869. [PubMed] [Google Scholar]
- 61. Wu GY, Walton CM, Wu CH. 2001. Targeted polynucleotides for inhibition of hepatitis B and C viruses. Croat Med J 42: 463–466. [PubMed] [Google Scholar]
- 62. Wu GY, Wu CH. 1992. Specific inhibition of hepatitis B viral gene expression in vitro by targeted antisense oligonucleotides. J Biol Chem 267: 12436–12439. [PubMed] [Google Scholar]
- 63. Chiou HC, Tangco MV, Levine SM, Robertson D, Kormis K, Wu CH, Wu GY. 1994. Enhanced resistance to nuclease degradation of nucleic acids complexed to asialoglycoprotein‐polylysine carriers. Nucleic Acids Res 22: 5439–5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liang WW, Shi X, Deshpande D, Malanga CJ, Rojanasakul Y. 1996. Oligonucleotide targeting to alveolar macrophages by mannose receptor‐ mediated endocytosis. Biochim Biophys Acta 1279: 227–234. [DOI] [PubMed] [Google Scholar]
- 65. Fominaya J, Wels W. 1996. Target cell‐specific DNA transfer mediated by a chimeric multidomain protein. Novel non‐viral gene delivery system. J Biol Chem 271: 10560–10568. [DOI] [PubMed] [Google Scholar]
- 66. Cotten M, Langle‐Rouault F, Kirlappos H, Wagner E, Mechtler K, Zenke M, Beug H, Birnstiel ML. 1990. Transferrin‐polycation‐mediated introduction of DNA into human leukemic cells: Stimulation by agents that affect the survival of transfected DNA or modulate transferrin receptor levels. Proc Natl Acad Sci USA 87: 4033–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wagner E, Zenke M, Cotten M, Beug H, Birnstiel ML. 1990. Transferrin‐polycation conjugates as carriers for DNA uptake into cells. Proc Natl Acad Sci USA 87: 3410–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu D, Boado RJ, Pardridge WM. 1996. Pharmacokinetics and blood‐brain barrier transport of [3H]‐biotinylated phosphorothioate oligodeoxynucleotide conjugated to a vector‐mediated drug delivery system. J Pharmacol Exp Ther 276: 206–211. [PubMed] [Google Scholar]
- 69. Penichet ML, Kang YS, Pardridge WM, Morrison SL, Shin SU. 1999. An antibody‐avidin fusion protein specific for the transferrin receptor serves as a delivery vehicle for effective brain targeting: Initial applications in anti‐HIV antisense drug delivery to the brain. J Immunol 163: 4421–4426. [PubMed] [Google Scholar]
- 70. Stephens AC, Rivers RP. 1997. Suppression of human monocyte tissue factor synthesis by antisense oligodeoxynucleotide. Thromb Res 85: 387–398. [DOI] [PubMed] [Google Scholar]
- 71. Deshpande D, Toledo‐Velasquez D, Thakkar D, Liang W, Rojanasakul Y. 1996. Enhanced cellular uptake of oligonucleotides by EGF receptor‐mediated endocytosis in A549 cells. Pharm Res 13: 57–61. [DOI] [PubMed] [Google Scholar]
- 72. Soukchareun S, Haralambidis J, Tregear G. 1998. Use of Nalpha‐Fmoc‐cysteine(S‐thiobutyl) derivatized oligodeoxynucleotides for the preparation of oligodeoxynucleotide‐ peptide hybrid molecules. Bioconjug Chem 9: 466–475. [DOI] [PubMed] [Google Scholar]
- 73. Soukchareun S, Tregear GW, Haralambidis J. 1995. Preparation and characterization of antisense oligonucleotide‐peptide hybrids containing viral fusion peptides. Bioconjug Chem 6: 43–53. [DOI] [PubMed] [Google Scholar]
- 74. Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. 1997. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res 25: 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bongartz JP, Aubertin AM, Milhaud PG, Lebleu B. 1994. Improved biological activity of antisense oligonucleotides conjugated to a fusogenic peptide. Nucleic Acids Res 22: 4681–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Smith CC, Kulka M, Aurelian L. 2000. Modified adenovirus penton base protein (UTARVE) as a non‐replicating vector for delivery of antisense oligonucleotides with antiviral and/or antineoplastic activity. Int J Oncol 17: 841–850. [DOI] [PubMed] [Google Scholar]
- 77. Freulon I, Roche AC, Monsigny M, Mayer R. 2001. Delivery of oligonucleotides into mammalian cells by anionic peptides: Comparison between monomeric and dimeric peptides. Biochem J 354: 671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. de La Torre BG, Albericio F, Saison‐Behmoaras E, Bachi A, Eritja R. 1999. Synthesis and binding properties of oligonucleotides carrying nuclear localization sequences. Bioconjug Chem 10: 1005–1012. [DOI] [PubMed] [Google Scholar]
- 79. Benimetskaya L, Guzzo‐Pernell N, Liu ST, Lai JC, Miller P, Stein CA. 2002. Protamine‐fragment peptides fused to an sv40 nuclear localization signal deliver oligonucleotides that produce antisense effects in prostate and bladder carcinoma cells. Bioconjug Chem 13: 177–187. [DOI] [PubMed] [Google Scholar]
- 80. Chaloin L, Vidal P, Lory P, Mery J, Lautredou N, Divita G, Heitz F. 1998. Design of carrier peptide‐oligonucleotide conjugates with rapid membrane translocation and nuclear localization properties. Biochem Biophys Res Commun 243: 601–608. [DOI] [PubMed] [Google Scholar]
- 81. Antopolsky M, Azhayeva E, Tengvall U, Auriola S, Jaaskelainen I, Ronkko S, Honkakoski P, Urtti A, Lonnberg H, Azhayev A. 1999. Peptide‐oligonucleotide phosphorothioate conjugates with membrane translocation and nuclear localization properties. Bioconjug Chem 10: 598–606. [DOI] [PubMed] [Google Scholar]
- 82. Reed MW, Fraga D, Schwartz DE, Scholler J, Hinrichsen RD. 1995. Synthesis and evaluation of nuclear targeting peptide‐antisense oligodeoxynucleotide conjugates. Bioconjug Chem 6: 101–108. [DOI] [PubMed] [Google Scholar]
- 83. Dokka S, Rojanasakul Y. 2000. Novel non‐endocytic delivery of antisense oligonucleotides. Adv Drug Deliv Rev 44: 35–49. [DOI] [PubMed] [Google Scholar]
- 84. Toth G, Schlammadinger J, Szabo G. 1994. DNA uptake stimulating protein from Neurospora crassa enhances DNA and oligonucleotide uptake also in mammalian cells. Biochim Biophys Acta 1219: 314–320. [DOI] [PubMed] [Google Scholar]
- 85. Astriab‐Fisher A, Sergueev DS, Fisher M, Shaw BR, Juliano RL. 2000. Antisense inhibition of P‐glycoprotein expression using peptide‐oligonucleotide conjugates. Biochem Pharmacol 60: 83–90. [DOI] [PubMed] [Google Scholar]
- 86. Albers E, Muller BW. 1995. Cyclodextrin derivatives in pharmaceutics. Crit Rev Ther Drug Carrier Syst 12: 311–337. [DOI] [PubMed] [Google Scholar]
- 87. Brewster ME, Simpkins JW, Hora MS, Stern WC, Bodor N. 1989. The potential use of cyclodextrins in parenteral formulations. J Parenter Sci Technol 43: 231–240. [PubMed] [Google Scholar]
- 88. Arima H, Kondo T, Irie T, Uekama K. 1992. Enhanced rectal absorption and reduced local irritation of the anti‐inflammatory drug ethyl 4‐biphenylylacetate in rats by complexation with water‐soluble β‐cyclodextrin derivatives and formulation as oleaginous suppository. J Pharm Sci 81: 1119–1125. [DOI] [PubMed] [Google Scholar]
- 89. Merkus FW, Verhoef JC, Romeijn SG, Schipper NG. 1991. Absorption enhancing effect of cyclodextrins on intranasally administered insulin in rats. Pharm Res 8: 588–592. [DOI] [PubMed] [Google Scholar]
- 90. Zhao Q, Temsamani J, Agrawal S. 1995. Use of cyclodextrin and its derivatives as carriers for oligonucleotide delivery. Antisense Res Dev 5: 185–192. [DOI] [PubMed] [Google Scholar]
- 91. Abdou S, Collomb J, Sallas F, Marsura A, Finance C. 1997. β‐Cyclodextrin derivatives as carriers to enhance the antiviral activity of an antisense oligonucleotide directed toward a coronavirus intergenic consensus sequence. Arch Virol 142: 1585–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Epa WR, Greferath U, Shafton A, Rong P, Delbridge LM, Bennie A, Barrett GL. 2000. Downregulation of the p75 neurotrophin receptor in tissue culture and in vivo, using β‐cyclodextrin‐adamantane‐oligonucleotide conjugates. Antisense Nucleic Acid Drug Dev 10: 469–478. [DOI] [PubMed] [Google Scholar]
- 93. Zhao Q, Temsamani J, Iadarola PL, Agrawal S. 1996. Modulation of oligonucleotide‐induced immune stimulation by cyclodextrin analogs. Biochem Pharmacol 52: 1537–1544. [DOI] [PubMed] [Google Scholar]
- 94. Yoo H, Juliano RL. 2000. Enhanced delivery of antisense oligonucleotides with fluorophore‐ conjugated PAMAM dendrimers. Nucleic Acids Res 28: 4225–4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sato N, Kobayashi H, Saga T, Nakamoto Y, Ishimori T, Togashi K, Fujibayashi Y, Konishi J, Brechbiel MW. 2001. Tumor targeting and imaging of intraperitoneal tumors by use of antisense oligo‐DNA complexed with dendrimers and/or avidin in mice. Clin Cancer Res 7: 3606–3612. [PubMed] [Google Scholar]
- 96. Helin V, Gottikh M, Mishal Z, Subra F, Malvy C, Lavignon M. 1999. Uptake and intracellular distribution of oligonucleotides vectorized by a PAMAM dendrimer. Nucleosides Nucleotides 18: 1721–1722. [DOI] [PubMed] [Google Scholar]
- 97. Helin V, Gottikh M, Mishal Z, Subra F, Malvy C, Lavignon M. 1999. Cell cycle‐dependent distribution and specific inhibitory effect of vectorized antisense oligonucleotides in cell culture. Biochem Pharmacol 58: 95–107. [DOI] [PubMed] [Google Scholar]
- 98. Bielinska A, Kukowska‐Latallo JF, Johnson J, Tomalia DA, Baker JR Jr. 1996. Regulation of in vitro gene expression using antisense oligonucleotides or antisense expression plasmids transfected using starburst PAMAM dendrimers. Nucleic Acids Res 24: 2176–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Couvreur P, Kante B, Roland M, Speiser P. 1979. Adsorption of antineoplastic drugs to polyalkylcyanoacrylate nanoparticles and their release in calf serum. J Pharm Sci 68: 1521–1524. [DOI] [PubMed] [Google Scholar]
- 100. Couvreur P, Kante B, Roland M, Guiot P, Bauduin P, Speiser P. 1979. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: Preparation, morphological and sorptive properties. J Pharm Pharmacol 31: 331–332. [DOI] [PubMed] [Google Scholar]
- 101. Chavany C, Saison‐Behmoaras T, Le Doan T, Puisieux F, Couvreur P, Helene C. 1994. Adsorption of oligonucleotides onto polyisohexylcyanoacrylate nanoparticles protects them against nucleases and increases their cellular uptake. Pharm Res 11: 1370–1378. [DOI] [PubMed] [Google Scholar]
- 102. Schwab G, Chavany C, Duroux I, Goubin G, Lebeau J, Helene C, Saison‐Behmoaras T. 1994. Antisense oligonucleotides adsorbed to polyalkylcyanoacrylate nanoparticles specifically inhibit mutated Ha‐ras‐mediated cell proliferation and tumorigenicity in nude mice. Proc Natl Acad Sci USA 91: 10460–10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chavany C, Le Doan T, Couvreur P, Puisieux F, Helene C. 1992. Polyalkylcyanoacrylate nanoparticles as polymeric carriers for antisense oligonucleotides. Pharm Res 9: 441–449. [DOI] [PubMed] [Google Scholar]
- 104. Zimmer A. 1999. Antisense oligonucleotide delivery with polyhexylcyanoacrylate nanoparticles as carriers. Methods 18: 286–295, 322. [DOI] [PubMed] [Google Scholar]
- 105. Lambert G, Fattal E, Pinto‐Alphandary H, Gulik A, Couvreur P. 2001. Polyisobutylcyanoacrylate nanocapsules containing an aqueous core for the delivery of oligonucleotides. Int J Pharm 214: 13–16. [DOI] [PubMed] [Google Scholar]
- 106. Lambert G, Bertrand JR, Fattal E, Subra F, Pinto‐Alphandary H, Malvy C, Auclair C, Couvreur P. 2000. EWS fli‐1 antisense nanocapsules inhibits ewing sarcoma‐related tumor in mice. Biochem Biophys Res Commun 279: 401–406. [DOI] [PubMed] [Google Scholar]
- 107. Nakada Y, Fattal E, Foulquier M, Couvreur P. 1996. Pharmacokinetics and biodistribution of oligonucleotide adsorbed onto poly(isobutylcyanoacrylate) nanoparticles after intravenous administration in mice. Pharm Res 13: 38–43. [DOI] [PubMed] [Google Scholar]
- 108. De Rosa G, Quaglia F, La Rotonda MI, Appel M, Alphandary H, Fattal E. 2002. Poly(lactide‐co‐glycolide) microspheres for the controlled release of oligonucleotide/polyethylenimine complexes. J Pharm Sci 91: 790–799. [DOI] [PubMed] [Google Scholar]
- 109. Freytag T, Dashevsky A, Tillman L, Hardee GE, Bodmeier R. 2000. Improvement of the encapsulation efficiency of oligonucleotide‐containing biodegradable microspheres. J Controlled Release 69: 197–207. [DOI] [PubMed] [Google Scholar]
- 110. Lewis KJ, Irwin WJ, Akhtar S. 1998. Development of a sustained‐release biodegradable polymer delivery system for site‐specific delivery of oligonucleotide: Characterization of P(LA‐GA) copolymer microspheres in vitro. J Drug Target 5: 291–302. [DOI] [PubMed] [Google Scholar]
- 111. Regnier V, De Morre N, Jadoul A, Preat V. 1999. Mechanisms of a phosphorothioate oligonucleotide delivery by skin electroporation. Int J Pharm 184: 147–156. [DOI] [PubMed] [Google Scholar]
- 112. Regnier V, Le Doan T, Preat V. 1998. Parameters controlling topical delivery of oligonucleotides by electroporation. J Drug Target 5: 275–289. [DOI] [PubMed] [Google Scholar]
- 113. Regnier V, Preat V. 1998. Localization of a FITC‐labeled phosphorothioate oligodeoxynucleotide in the skin after topical delivery by iontophoresis and electroporation. Pharm Res 15: 1596–1602. [DOI] [PubMed] [Google Scholar]
- 114. Baba M, Iishi H, Tatsuta M. 2000. In vivo electroporetic transfer of bcl‐2 antisense oligonucleotide inhibits the development of hepatocellular carcinoma in rats. Int J Cancer 85: 260–266. [PubMed] [Google Scholar]
- 115. Zewert TE, Pliquett UF, Langer R, Weaver JC. 1995. Transdermal transport of DNA antisense oligonucleotides by electroporation. Biochem Biophys Res Commun 212: 286–292. [DOI] [PubMed] [Google Scholar]
- 116. Oldenburg KR, Vo KT, Smith GA, Selick HE. 1995. Iontophoretic delivery of oligonucleotides across full thickness hairless mouse skin. J Pharm Sci 84: 915–921. [DOI] [PubMed] [Google Scholar]
- 117. Bergan R, Hakim F, Schwartz GN, Kyle E, Cepada R, Szabo JM, Fowler D, Gress R, Neckers L. 1996. Electroporation of synthetic oligodeoxynucleotides: A novel technique for ex vivo bone marrow purging. Blood 88: 731–741. [PubMed] [Google Scholar]
- 118. Mann MJ, Gibbons GH, Hutchinson H, Poston RS, Hoyt EG, Robbins RC, Dzau VJ. 1999. Pressure‐mediated oligonucleotide transfection of rat and human cardiovascular tissues. Proc Natl Acad Sci USA 96: 6411–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. von der Leyen HE, Braun‐Dullaeus R, Mann MJ, Zhang L, Niebauer J, Dzau VJ. 1999. A pressure‐mediated nonviral method for efficient arterial gene and oligonucleotide transfer. Hum Gene Ther 10: 2355–2364. [DOI] [PubMed] [Google Scholar]
- 120. Tschoep K, Hartmann G, Jox R, Thompson S, Eigler A, Krug A, Erhardt S, Adams G, Endres S, Delius M. 2001. Shock waves: A novel method for cytoplasmic delivery of antisense oligonucleotides. J Mol Med 79: 306–313. [DOI] [PubMed] [Google Scholar]
- 121. Bao S, Thrall BD, Gies RA, Miller DL. 1998. In vivo transfection of melanoma cells by lithotripter shock waves. Cancer Res 58: 219–221. [PubMed] [Google Scholar]
- 122. Kim HJ, Greenleaf JF, Kinnick RR, Bronk JT, Bolander ME. 1996. Ultrasound‐mediated transfection of mammalian cells. Hum Gene Ther 7: 1339–1346. [DOI] [PubMed] [Google Scholar]
- 123. Unger EC, McCreery TP, Sweitzer RH. 1997. Ultrasound enhances gene expression of liposomal transfection. Invest Radiol 32: 723–727. [DOI] [PubMed] [Google Scholar]
- 124. Huber PE, Jenne J, Debus J, Wannenmacher MF, Pfisterer P. 1999. A comparison of shock wave and sinusoidal‐focused ultrasound‐induced localized transfection of HeLa cells. Ultrasound Med Biol 25: 1451–1457. [DOI] [PubMed] [Google Scholar]