

Abstract
The severe acute respiratory syndrome coronavirus papain-like protease (SARS-CoV PLpro) carries out N-terminal processing of the viral replicase polyprotein, and also exhibits Lys48-linked polyubiquitin chain debranching and ISG15 precursor processing activities in vitro. Here, we used SDS–PAGE and fluorescence-based assays to demonstrate that ISG15 derivatives are the preferred substrates for the deubiquitinating activity of the PLpro. With kcat/KM of 602,000 M−1 s−1, PLpro hydrolyzes ISG15-AMC 30- and 60-fold more efficiently than Ub-AMC and Nedd8-AMC, respectively. Data obtained with truncated ISG15 and hybrid Ub/ISG15 substrates indicate that both the N- and C-terminal Ub-like domains of ISG15 contribute to this preference. The enzyme also displays a preference for debranching Lys48- over Lys63-linked polyubiquitin chains. Our results demonstrate that SARS-CoV PLpro can differentiate between ubiquitin-like modifiers sharing a common C-terminal sequence, and that the debranching activity of the PLpro is linkage type selective. The potential structural basis for the demonstrated specificity of SARS-CoV PLpro is discussed.
Keywords: Severe acute respiratory syndrome, Coronavirus, Papain-like protease, Deubiquitination, Ubiquitin, ISG15, Specificity
The severe acute respiratory syndrome coronavirus (SARS-CoV) 1 is the cause of an atypical form of pneumonia which emerged in China in late 2002 [1], [2]. SARS-CoV is a single-stranded positive-sense RNA virus that replicates in the cytoplasm of infected cells. Replication is mediated by the replicase polyprotein, which is translated directly from the viral genome, and is processed by two coronaviral proteases present in the replicase polyprotein itself [3], [4]. One of these enzymes, the papain-like protease (PLpro), is responsible for N-terminal processing of the replicase polyprotein at three sites, releasing nsp1 to nsp3 in this order [5]. In addition to its function in viral replicase processing, the SARS-CoV PLpro has been shown recently to possess deubiquitinating activity [6], [7]. As predicted based on the similarity of its catalytic core domain to the corresponding domain of ubiquitin-specific protease 7 (USP7) [8], the enzyme debranches Lys48-linked polyubiquitin chains and hydrolyzes the general deubiquitinating enzyme (DUB) substrate Ub-7-amino-4-methylcoumarin (Ub-AMC) [6], [7]. The SARS-CoV PLpro was also found to process fusion proteins of the ubiquitin-like modifier (Ubl) ISG15 (15 kDa protein encoded by an interferon-stimulated gene) [7]. Ubl family members are 8–17 kDa proteins which are defined by a common β-grasp three-dimensional fold [9], [10]. Amongst the Ubls, ISG15 and FAT10 are unique in that they comprise two tandem β-grasp folds [9], [10]. Prior to our findings, only two enzymes had been shown to possess deISGylating activity: USP18 as well as the adenovirus protease adenain [11], [12].
The recent determination of the three-dimensional structure of the enzyme has confirmed that the SARS-CoV PLpro is a member of the USP family of DUBs [13]. The enzyme consists of a circularly permuted Zn-ribbon domain inserted between the two subdomains of a papain-like cysteine protease fold, preceded by an N-terminal ubiquitin-like domain. USPs recognize the Ubl moieties of their substrates mainly through two regions: intimate binding of the Ubl C-terminal tail at the enzyme active site, and interaction of the corresponding β-grasp fold of the substrate with the Zn-ribbon of the USP [14], [15], [16]. Both ISG15 and Ub share a common C-terminal sequence, consisting of LRLRGG. The last four residues of this sequence match the narrow LXGG sequence specificity at SARS-CoV PLpro processing sites [3], [5], [17]. Detailed examination of the PLpro structure, modeled in complex with ubiquitin, indeed shows that the active site is highly complementary to the LRGG sequence and establishes extensive hydrogen bonding with the substrate mainchain residues [8], [13], [18].
Polyprotein processing by SARS-CoV PLpro plays an important role in viral replication, and has been well characterized. In particular, the specificity determinants of polyprotein processing have been outlined and are well understood at the molecular level [5], [6], [8], [18]. Specificity of the deubiquitinating activity of the SARS-CoV PLpro, on the other hand, has yet to be characterized in detail. Substrate specificity of DUBs can be expressed through recognition of the Ubl moiety, the target protein to which the moiety is conjugated, and, in the case of polyubiquitin chains, the ubiquitin lysine residue of isopeptide bond linkage, and the chain length. Presently, it is not clear to what extent the PLpro can be selective for these various substrate features. This is also true of cellular USPs which, for the most part, remain largely uncharacterized at the molecular and functional levels [19]. In order to characterize further the specificity of the PLpro for the Ubl moiety, and to initiate the probing of its structural determinants, Ubl derivatives have been designed and produced as substrates. The results presented in this paper demonstrate that SARS-CoV PLpro is able to differentiate between ubiquitin-like modifiers sharing a common C-terminal sequence, and that the debranching activity of the PLpro is linkage type selective.
Materials and methods
DNA cloning and recombinant protein expression
For the production of recombinant SARS-CoV PLpro, P-PLpro(C2)-His6 was expressed in Escherichia coli and purified as previously described [7]. P refers to the eight amino acid C-terminal pro-sequence (GTEPGGRS) of the ISG15 precursor protein, and PLpro(C2) corresponds to residues Gly1507 to Thr1858 of the SARS-CoV polyprotein. The purified protein was digested with 1 μg of trypsin (Roche Diagnostics, Laval, QC) per 100 μg of protein for 45 min at 22 °C. The reaction was quenched by 1 mM benzamidine (Eastman Kodak, NY), and was subjected to chromatography on a MonoQ HR 5/5 column (Amersham Biosciences, Inc., Piscataway, NJ), showing the same elution behavior as P-PLpro(C2)-His6 [cf. 7]. The purified PLpro migrated at an approximate molecular weight of 37 kDa in reducing SDS–PAGE compared to 41 kDa for undigested purified P-PLpro(C2)-His6. N-terminal sequencing of two independent PLpro preparations showed that they consisted of two PLpro species each present in the protein band in close to equal amounts, starting with residues Ser1538 and Glu1541 of the SARS-CoV polyprotein, indicating the loss of the first 41 and 44 residues of P-PLpro(C2)-His6, respectively. Analytical gel filtration was consistent with the absence of these N-terminal sequences in the purified protein (not shown). The PLpro band was further detected by anti-His6 Western blotting (not shown), demonstrating the presence of an intact C-terminus. The two protein species in the PLpro preparation differ in length by only three amino acids at the N-terminus. They essentially comprise the catalytic core domain plus the N-terminal ubiquitin-like domain of SARS-CoV PLpro, and this represents the complete PLpro [13]. After addition of 2 mM dithiothreitol (DTT), purified PLpro was stored at −80 °C retaining its enzymatic activity for at least 6 months.
The cDNA of human Ub52 (GenBank Accession No. BM706733) and the ISG15 precursor gene (IMAGE clone 3545944) were purchased from Open Biosystems (Huntsville, AL). ISG15 was modified to carry the stabilizing C78S mutation [9] using the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). GFP refers to a variant of Aequorea victoria green fluorescent protein with the F64L/S65C/I167T/K238N mutations (Qbiogene, Montreal, QC), and the sequence of the E. coli maltose binding protein (MBP), including a C-terminal polylinker, was taken from the pMAL vector (New England Biolabs LTD, Pickering, ON). In order to generate fusion proteins that could serve as polyprotein substrates for the PLpro, sequences derived from MBP, Ub, ISG15, and GFP were PCR-amplified and cloned into the BamHI and XhoI sites of pET-21a(+) (Novagen, Madison, WI) with the T7 and the His6 tags at the N- and C-terminus, respectively (see supplementary material). In brief, in T7-MBP(6)-P-GFP-His6 and T7-MBP(12)-P-GFP-His6, the MBP is followed by the C-terminal six and twelve residues of mature ISG15, respectively (Fig. 1 ). The latter linker was included because results of a previous study using peptidyl vinyl sulfone inhibitors suggested that the 12 C-terminal residues of a Ubl sequence alone may contribute significantly to enzyme recognition [20]. T7-ISG15(C)-P-GFP-His6, where ISG15(C) refers to the C-terminal portion of ISG15 starting at Leu82 and ending at Gly157, was derived from T7-ISG15-P-GFP-His6 by deletion of the N-terminal Ub-like domain of ISG15. T7-ISG15(N)-Ub-P-GFP-His6, where ISG15(N) refers to the N-terminal portion of ISG15 ending at residue Pro81, resulted from T7-Ub-P-GFP-His6 by insertion of this domain upstream of the Ub sequence. Protein expression in E. coli BL21(DE3) cells (Novagen) was induced with 0.4 mM isopropyl-1-thio-b-d-galactopyranoside for 3 h at 22 °C. Proteins were purified by Ni2+ affinity chromatography, followed by buffer exchange to 20 mM Tris–HCl (pH 7.8), and further purification on a MonoQ HR 5/5 column (Amersham Biosciences, Inc.) as described for PLpro. Samples were stable at 4 °C for at least 1 month. Protein concentrations for the PLpro and polyprotein substrates were determined using the Bio-Rad protein assay (Hercules, CA) with bovine serum albumin (Sigma, Saint Louis, MO) as the standard.
Fig. 1.
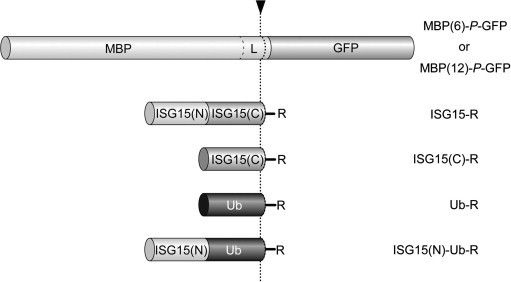
Schematic representation of the various substrates used in this work. ISG15(N) and ISG15(C) represent the N- and C-terminal domains of ISG15. R refers to P-GFP or AMC. L is a linker consisting of the C-terminal polylinker from the pMAL vector, followed by the sequence LRLRGG-P (for MBP(6)-P-GFP) or STVFMNLRLRGG-P (for MBP(12)-P-GFP), as described in Materials and methods. The arrowhead and dashed line indicate the site of proteolytic cleavage. In addition, the P-GFP fusion proteins include T7- and His6 tags at their N- and C-terminus, respectively.
For the generation of AMC substrates, the sequences corresponding to ISG15, ISG15(C), Ub, and ISG15(N)-Ub, excluding the T7 epitope and the C-terminal glycine residue in each case (Fig. 1), were subcloned from the respective P-GFP fusion constructs (described in the supplementary material) into the NdeI and SapI sites of the pTYB1 vector (New England Biolabs LTD) placing them directly upstream of the intein–chitin binding domain (CBD).
Production of ISG15/Ub-based fluorogenic substrates for PLpro
ISG15-AMC (full-length), ISG15(C)-AMC, Ub-AMC and ISG15(N)-Ub-AMC (Fig. 1) were obtained according to the method outlined by Wilkinson et al. [21] for the synthesis of Nedd8-AMC. The Ub- and ISG15-based constructs were expressed in E. coli BL21(DE3) and their thioester derivatives were purified on Chitin–Sepharose (New England Biolabs LTD). For the synthesis of ISG15-AMC, Ub-AMC, and ISG15(N)-Ub-AMC, the respective protein thioesters were allowed to react with Gly-AMC (Chem-Impex Int., Wood Dale, IL) in the presence of 20 mM N-hydroxysuccinamide (Sigma), while the coupling reaction to generate ISG15(C)-AMC was carried out in urea [cf. 21]. The Ubl-AMC substrates were purified by gel filtration on two Superose 12 columns connected in series and equilibrated with 10% glycerol in 20 mM Tris–HCl pH 7.4 in 150 mM NaCl.
Products were analyzed by electrospray ionisation mass spectrometry (Q-TOF Ultima Global, Waters, Manchester, UK), which revealed that the E. coli expression host had processed the N-terminal Met for ISG15 and ISG15(N)-Ub, but not for Ub and ISG15(C). It must be noted that for both ISG15(N)-Ub-AMC and ISG15(C)-AMC, minor peaks could be observed by MS at molecular mass higher than the predicted values by 16 and 32 kDa, indicating oxidation. Purification and storage of the substrates in presence of 1 mM of the reducing agent Tris(2-carboxyethyl)phosphine hydrochloride prevented the appearance of the minor peaks. Substrate oxidation did not, however, affect the rates of hydrolysis by the PLpro (data not shown). HPLC analysis of purified AMC substrates detected approximately 2–10% of non-labeled protein, and 50% of Gly-AMC. Control experiments, however, showed that the presence of the non-labeled protein and of Gly-AMC did not interfere with the activity measurements. Approximately 0.4 mg of Ub-AMC and ISG15-AMC were obtained after processing of 1L of E. coli culture, while yields for the other constructs were somewhat lower (0.05–0.1 mg).
Enzyme activity assays
Lys48- and Lys63-linked polyubiquitin chains with lengths of two to seven (Ub2–7) or four (Ub4) units, AMC substrates Ub-AMC, Nedd8-AMC and SUMO-AMC, as well as the enzymes ubiquitin C-terminal hydrolase 3 (UCH-L3) and USP5, were obtained from Boston Biochem (Cambridge, MA). Enzymatic hydrolysis of polyubiquitin chains was tested by incubating 5 μg of Ub2-7 or Ub4 from 1 mg/mL stock solutions in 50 mM Tris–HCl (pH 7.9) and 200 ng of either SARS-CoV PLpro or USP5, in a final volume of 25 μl of 50 mM Tris–HCl (pH 7.9), 200 mM NaCl, and 2 mM DTT for 2 h at 37 °C. For enzymatic hydrolysis of linear P-GFP fusion proteins and Lys48-linked Ub4, substrate proteins (200 nM) and PLpro (20 nM) were incubated at 37 °C in a buffer containing 20 mM Tris–HCl, 290 mM NaCl, 2 mM DTT (pH 7.8). Controls were incubated without enzyme. At indicated times, aliquots of reaction mixtures were quenched by the addition of Laemmli buffer and kept on ice until analysis. For analysis, samples were boiled and proteins were separated by reducing SDS–PAGE. PLpro remained fully active under these assay conditions for up to 180 min as measured by its ability to hydrolyze Ub-AMC (see below). Enzymatic hydrolysis of linear P-GFP fusion proteins was quantified by densitometric analysis of the gels, using the image analysis software ImageQuant TL (Amersham Biosciences Inc.).
The activity of PLpro and, for comparison, UCH-L3 and USP5 against the fluorogenic AMC derivatives of Ubl and variants was assessed as previously described for Ub-AMC [7]. For PLpro, the reaction conditions consisted of 50 mM Tris–HCl (pH 7.8), 2 mM dithiothreitol (DTT), 200 mM NaCl, 1 mM benzamidine and 2% (vol/vol) dimethyl sulfoxide. UCH-L3 and USP5 were assayed in 50 mM Hepes (pH 7.8), 0.5 mM EDTA, 2 mM DTT and 2% (vol/vol) dimethyl sulfoxide. For USP5 enzyme assays, 100 nM of ubiquitin was also included in the reaction mixture [22]. Temperature was set at 25 °C for all assays involving AMC substrates. Reactions were initiated by the addition of enzyme to the cuvette, yielding final concentrations of enzyme of 3.3 nM for the assay with the substrate ISG15-AMC and 100 nM with the other AMC substrates. For UCH-L3 and USP5, enzyme concentrations were in the range of 0.05–1.0 nM and 2.0–7.5 nM, respectively, depending on the substrate used. The kinetic parameters for substrate hydrolysis were determined by monitoring full progress curves (i.e., by allowing complete hydrolysis of the substrate) at [S] << K M. Under these conditions, the progress curves follow first-order kinetics and values of k cat/K M were obtained by dividing the first-order rate constant by enzyme concentration. Controls were performed to insure that the enzyme did not loose activity during the course of the reaction, and that no product inhibition interfered with progress curve analysis. The reported k cat/K M values were within ±14% of the mean for at least three independent measurements. For poor substrates (i.e., k cat/K M values below ca 10,000 M−1 s−1), k cat/K M could only be estimated using initial rate measurements.
Results and discussion
Hydrolytic activity of PLpro against Ubl fusion proteins
SARS-CoV PLpro has been shown to exhibit activity against Ub-based substrates, as well as ISG15 precursor processing activity [7]. To investigate the influence of the ubiquitin-like domain sequences of ISG15 and Ub on the efficiency of hydrolysis by SARS-CoV PLpro in a precursor processing assay, we incubated the PLpro with a series of artificial precursor molecules illustrated in Fig. 1. Fig. 2 shows the analyses of the reaction mixtures by SDS–PAGE. The extent of substrate hydrolysis by the PLpro was quantified by densitometric analysis of the gels (see supplementary material). For T7-ISG15-P-GFP-His6, 50% hydrolysis was reached after 10 min of incubation (Fig. 2a). By comparison, hydrolysis of T7-Ub-P-GFP-His6 had reached only about 30% after 180 min (Fig. 2b). No hydrolysis was detectable for T7-MBP(6)-P-GFP-His6 and T7-MBP(12)-P-GFP-His6, where the non-Ubl protein MBP is fused to P-GFP by a linker containing the six and twelve C-terminal residues, respectively, of mature ISG15 (Fig. 2c). Therefore, hydrolysis of Ubl fusion proteins and variants by PLpro do not reflect solely the recognition of the LRLRGG sequence by the enzyme. In order to gain insight into the nature of the molecular determinants of substrate specificity, we have generated ISG15/Ub variants. A fusion protein containing only the C-terminal ubiquitin-like domain of ISG15 (T7-ISG15(C)-P-GFP-His6) was hydrolyzed more slowly than T7-ISG15-P-GFP-His6, reaching 50% hydrolysis after 60 min (Fig. 2d). We have also generated the fusion protein T7-ISG15(N)-Ub-P-GFP-His6, which contains a hybrid Ubl protein including the N-terminal residues of ISG15 followed by Ub. However, the purified protein was found to be unstable under assay conditions, and hydrolysis by PLpro could not be assessed. Overall, the results clearly show that the ISG15 fusion was hydrolyzed at least 15 times more efficiently than the Ub fusion, with the truncation of ISG15 missing the N-terminal Ub-like domain, i.e., ISG15(C), adopting an intermediate position.
Fig. 2.
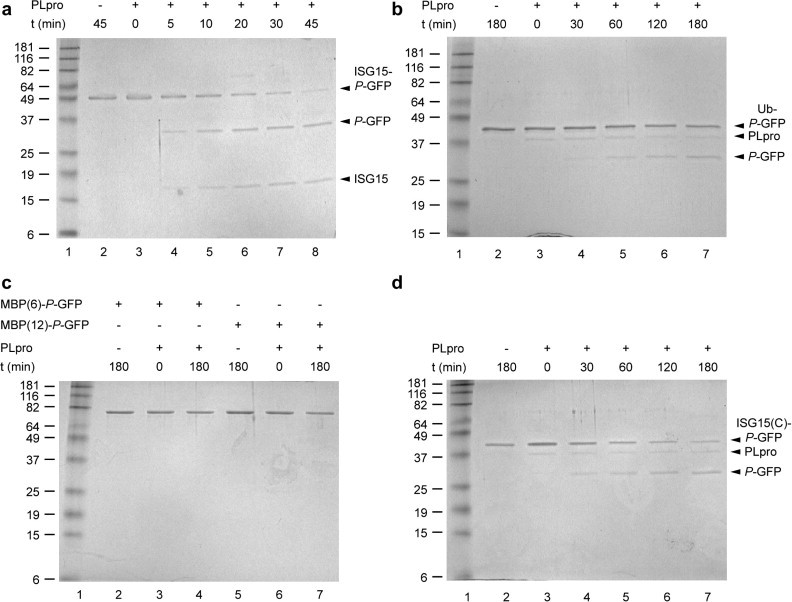
Precursor protein processing by SARS-CoV PLpro. (a) T7-ISG15-P-GFP-His6, (b) T7-Ub-P-GFP-His6, (c) T7-MBP(6)-P-GFP-His6 and T7-MBP(12)-P-GFP-His6, (d) T7-ISG(C)-P-GFP-His6. The purified proteins were incubated with PLpro at 37 °C for the indicated times. In each case, enzyme and substrate were present at 20 nM and 200 nM, respectively. Equivalents of 100 ng of input substrate protein per lane were resolved by 12% SDS–PAGE and revealed by Coomassie blue staining. The molecular weights of the marker proteins are indicated on the left of each gel (in kDa). Visible protein species are indicated on the right of the gel as identified by their apparent molecular weights. For MBP(6)-P-GFP and MBP(12)-P-GFP, the intact fusion proteins are the only species visible before and after incubation. In addition to the full-length substrates and hydrolysis products, (b) and (d) also show a band corresponding to PLpro due to shorter gel destaining times that were required in order to achieve similarly intense coloration of the substrate proteins in all gels.
Specificity of PLpro against fluorogenic Ubl-AMC substrates
Dynamic range and sensitivity for the detection of hydrolysis products by Coomassie staining after SDS–PAGE are limited. Hence, we synthesized a series of substrates representing fluorogenic AMC derivatives of the Ubl variants (Fig. 1) and used them to quantitatively determine catalytic efficiencies. The substrates were also evaluated against two well characterized DUBs, namely USP5 and UCH-L3. The results, illustrated in Fig. 3 , are in agreement with the activity profile obtained by SDS–PAGE analysis of fusion protein hydrolysis by PLpro. ISG15-AMC is a good substrate for PLpro, with k cat/K M of 602,000 M−1 s−1. By comparison, the activity against Ub-AMC was 30-fold lower, with k cat/K M = 19,800 M−1 s−1. PLpro is also able to hydrolyze Nedd8-AMC at an appreciable rate, with k cat/K M = 10,500 M−1 s−1, a value 60-fold lower than that obtained with ISG15-AMC. Among the Ubls, Nedd8 displays the highest level of sequence identity to Ub (∼60%), and features a C-terminal sequence (LALRGG) that is similar to the one of Ub and ISG15 (LRLRGG). Activity against SUMO1-AMC, which has a different C-terminal sequence (QEQTGG), was another three orders of magnitude lower (k cat/K M = 14 M−1 s−1), which agrees with the strict requirement of SARS-CoV PLpro for a LXGG sequence at the N-terminal side of the scissile bond [18]. In contrast to PLpro, USP5 and UCH-L3 are very specific for Ub-AMC, displaying more than three (USP5) and four (UCH-L3) orders of magnitude preference for Ub-AMC over ISG15-AMC (Fig. 3). UCH-L3 is also able to efficiently hydrolyze Nedd8-AMC, in agreement with its reported dual specificity for Ub- and Nedd8-conjugated proteins [23].
Fig. 3.
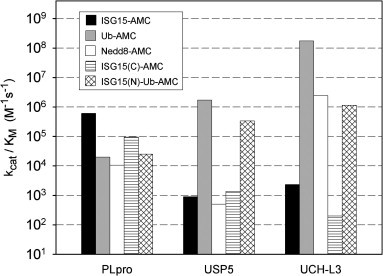
Specificity profiles of SARS-CoV PLpro, USP5 and UCH-L3 against fluorogenic AMC derivatives of Ubl and variants. The catalytic efficiency (kcat/KM) against the AMC substrates is plotted on a log scale. All reactions were carried out at pH 7.8, in presence of 2 mM DTT, 2% DMSO, and at 25 °C. For USP5 enzyme assays, 100 nM of free ubiquitin was also included. The substrates used are ISG15-AMC (black), Ub-AMC (gray), Nedd8-AMC (white), ISG15(C)-AMC (horizontal lines) and ISG15(N)-Ub-AMC (crosshair).
Contribution of Ubl domains to specificity
Given the identical C-terminus of ISG15 and Ub, the observed specificity of SARS-CoV PLpro most likely arises from interactions outside of the catalytic cleft of the enzyme. Removal of the N-terminal ubiquitin-like domain of ISG15-AMC, to generate ISG15(C)-AMC, reduces the activity of PLpro 6-fold (from a k cat/K M of 602,000 M−1 s−1 to 94,000 M−1 s−1), while it remains still 5-fold higher than against Ub-AMC (k cat/K M = 19,800 M−1 s−1). This indicates that part of the selectivity for ISG15 resides in the C-terminal Ub fold of ISG15, and that the N-terminus contributes to this property as well. This could be due to specific interactions between the ISG15(N) domain and the enzyme, and/or reflect interactions between the two ISG15 domains stabilizing a particular conformation of ISG15, as exemplified by the crystal structure of ISG15 [9]. The latter alternative is consistent with the observation that SARS-CoV PLpro activity against the hybrid molecule ISG15(N)-Ub-AMC (k cat/K M = 24,800 M−1 s−1) and Ub-AMC are very similar.
USP5 and UCH-L3 exhibit very strong preferences (three to five orders of magnitude) for Ub-AMC over ISG15-AMC and ISG15(C)-AMC (Fig. 3). Addition of the ISG15(N) domain to the N-terminus of Ub in ISG15(N)-Ub-AMC moderately decreased k cat/K M for USP5 (5-fold) in comparison to Ub-AMC, whereas a more dramatic drop was observed for UCH-L3 (150-fold). However, for both enzymes the activity against the hybrid molecule was still over two orders of magnitude higher than against both ISG15-AMC and its truncation, ISG15(C)-AMC. This indicates that the presence of the Ub domain, both in the context of Ub-AMC and ISG15(N)-Ub-AMC, is a major specificity determinant for USP5 and UCH-L3. The molecular basis for the inhibitory effect of the addition of the ISG15(N) domain to Ub-AMC in the hybrid on UCH-L3 catalytic activity remains unclear.
Specificity of PLpro against polyubiquitin chains
SARS-CoV PLpro has been shown to debranch Lys48-linked polyubiquitin chains [6], [7]. We next tested whether PLpro was able to also disassemble Lys63-linked polyubiquitin chains. USP5, which acts on both linkage types [24], [25], was used as a positive control. As expected, SDS–PAGE analysis (Fig. 4 ) shows that USP5 debranches both types of Ub2–7 chains, only leaving, besides monoubiquitin, detectable amounts of Ub2 and Ub3 species (lanes 4 and 7). PLpro, however, did not significantly process Lys63-linked polyubiquitin chains (lane 6). Nevertheless, a small amount of monoubiquitin could be detected, but it is not clear from the gel from which of the higher Ub-polymers it had been liberated. As previously observed with P-PLpro(C2)-His6 [7], Lys48-linked polyubiquitin chains were hydrolyzed by PLpro, generating mainly Ub2 as well as smaller amounts of monoubiquitin (lane 3). Incubation of PLpro with Lys48-linked Ub4 at the same concentrations of substrate (200 nM) and enzyme (20 nM) as in the Ubl fusion protein cleavage assay described above, led to the rapid appearance of Ub2 with 50% conversion after 5 min (data not shown), suggesting that this debranching reaction occurred at rates superior even to ISG15 precursor processing.
Fig. 4.
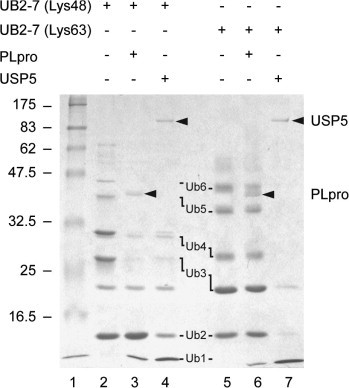
Hydrolysis of differently branched polyubiquitin chains by SARS-CoV PLpro and USP5. Lys48-linked Ub2-7 (lanes 2–4) and Lys63-linked Ub2-7 (lanes 5–7) were incubated for 2 h at 37 °C with PLpro (lanes 3 and 6) or USP5 (lanes 4 and 7). Enzymes and substrates were present at 8 μg/mL and 200 μg/mL, respectively. Proteins were resolved by a 10% SDS–PAGE and revealed by Coomassie blue staining. The molecular weights of the marker proteins are indicated on the left of the gel (in kDa). Arrowheads indicate the positions of PLpro and USP5.
Conclusion
With the SARS-CoV PLpro, we have for the first time assessed deISGylating specificity for a DUB in a quantitative manner. The enzyme’s specificity profile, outlined in our study, shows marked differences with the corresponding profiles for USP5 and UCH-L3. Overall, PLpro displays a broader specificity profile than the two other DUBs, with a preference for ISG15. This preference originates both from interaction with the C-terminal ubiquitin-like domain of ISG15, and from recognition of an additional, likely conformational feature in the context of the complete two-domain ISG15. The absence of a crystal structure or a relevant structural model of an ISG15-PLpro complex makes it difficult to delineate the molecular basis of the Ubl specificity of the PLpro. Available crystal structures for a number of Ub-USP adducts [14], [15], [16] reveal that the β-grasp fold of Ub interacts with the Zn-ribbon domain characteristic of the USP family of DUBs. A similar interaction has been postulated for PLpro [8], [13], [18]. We are presently evaluating the possible contribution of interactions in this and other regions to substrate specificity for the SARS-CoV PLpro and other USPs.
We have also established a strong preference for the isopeptidase activity of SARS-CoV PLpro to debranch Lys48- versus Lys63-linked polyubiquitin chains setting forth evidence for dual polyubiquitin debranching/deISGylating specificity. While viral strategies to take advantage of protein ubiquitination are well documented [26], viral interactions with deubiquitination are also increasingly reported. Several new enzymes from viruses and bacteria, able to deubiquitinated substrates in vitro, have been identified in the last few years [27], [28]. As discussed recently by Catic et al. [28], it remains to be shown if these enzymes truly act as deubiquitinating enzymes in vivo. The involvement of SARS-CoV PLpro in viral polyprotein processing is well established. Deubiquitination of host substrate(s) in the context of a SARS-CoV infection, however, remains to be demonstrated. The observed in vitro activity against Ubl derivatives suggests that the SARS coronavirus could benefit from the deubiquitinating activity of PLpro through deconjugation of ISGylated proteins, or through polyubiquitin deconjugation, leading to protection of host and/or viral proteins from degradation [6], [7]. In the present study, we have shown that the SARS-CoV PLpro displays a broader specificity profile than USP5 and UCH-L3, indicating that the enzyme is more promiscuous than these host (human) DUBs. Activity of the PLpro against Ubl derivatives in vitro may therefore be a reflection of this broader specificity. In vivo, however, the SARS-CoV PLpro is part of nsp3 localized to the membrane-bound viral replicase complex [29]. Although we previously reported PLpro mediated cleavage of an ISG15-nsp2 fusion protein in a cell-based assay [7], we could not detect any clear decrease in ubiquitinated or ISGylated proteins using anti-Ub and anti-ISG15 Western blotting of cell lysates incubated with purified PLpro (data not shown). Therefore, as an isopeptidase, PLpro may exhibit more restricted substrate specificity. PLpro may also act on modified viral proteins that require its confined membrane localization as part of the replication complex. Identification of putative PLpro deubiquitination substrates during virus infection will require a more extensive, proteome-wide, approach.
Acknowledgments
We thank Nasser Fotouhi-Ardakani, Alain Alary and Eunice Ferreira Ajamian for technical assistance, and Traian Sulea and Allan Matte for discussion. This is NRCC Publication No. 49516.
Footnotes
Abbreviations used: SARS-CoV, severe acute respiratory syndrome coronavirus; PLpro, papain-like protease; USP7, ubiquitin-specific protease 7; Ub-AMC, Ub-7-amino-4-methylcoumarin; DTT, dithiothreitol; MBP, maltose binding protein; UCH-L3, ubiquitin C-terminal hydrolase 3.
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.abb.2007.07.006.
Appendix A. Supplementary data
References
- 1.Peiris J.S., Yuen K.Y., Osterhaus A.D., Stohr K. N. Engl. J. Med. 2003;349:2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 2.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziebuhr J. Curr. Top. Microbiol. Immunol. 2005;287:57–94. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. J. Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindner H.A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Menard R. J. Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sulea T., Lindner H.A., Purisima E.O., Menard R. J. Virol. 2005;79:4550–4551. doi: 10.1128/JVI.79.7.4550-4551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narasimhan J., Wang M., Fu Z., Klein J.M., Haas A.A., Kim J.J. J. Biol. Chem. 2005;280:27356–27365. doi: 10.1074/jbc.M502814200. [DOI] [PubMed] [Google Scholar]
- 10.Kerscher O., Felberbaum R., Hochstrasser M. Annu. Rev. Cell Dev. Biol. 2006;22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 11.Malakhov M.P., Malakhova O.A., Kim K.I., Ritchie K.J., Zhang D.E. J. Biol. Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 12.Balakirev M.Y., Jaquinod M., Haas A.L., Chroboczek J. J. Virol. 2002;76:6323–6331. doi: 10.1128/JVI.76.12.6323-6331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Proc. Natl. Acad. Sci. USA. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu M., Li P., Li M., Li W., Yao T., Wu J.W., Gu W., Cohen R.E., Shi Y. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 15.Hu M., Li P., Sing L., Jeffrey P., Chenova T., Wilkinson K.D., Cohen R.E., Shi Y. EMBO J. 2005;24:3747–3756. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renatus M., Parrado S.G., D’Arcy A., Eidhoff U., Gerhartz B., Hassiepen U., Pierrat B., Riedl R., Vinzenz D., Worpenberg S., Kroemer M. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 18.Sulea T., Lindner H.A., Purisima E.O., Menard R. Proteins. 2006;62:760–775. doi: 10.1002/prot.20802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nijman S.M., Luna-Vargas M.P., Velds A., Brummelkamp T.R., Dirac A.M., Sixma T.K., Bernards R. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Borodovsky A., Ovaa H., Meester W.J., Venanzi E.S., Bogyo M.S., Hekking B.G., Ploegh H.L., Kessler B.M., Overkleeft H.S. Chembiochem. 2005;6:287–291. doi: 10.1002/cbic.200400236. [DOI] [PubMed] [Google Scholar]
- 21.Wilkinson K.D., Gan-Erdene T., Kolli N. Methods Enzymol. 2005;399:37–51. doi: 10.1016/S0076-6879(05)99003-4. [DOI] [PubMed] [Google Scholar]
- 22.Reyes-Turcu F.E., Horton J.R., Mullally J.E., Heroux A., Cheng X., Wilkinson K.D. Cell. 2006;124:1197–1208. doi: 10.1016/j.cell.2006.02.038. [DOI] [PubMed] [Google Scholar]
- 23.Gong L., Kamitani T., Millas S., Yeh E.T.H. J. Biol. Chem. 2000;275:14212–14216. doi: 10.1074/jbc.275.19.14212. [DOI] [PubMed] [Google Scholar]
- 24.Amerik A.Y., Hochstrasser M. Biochim. Biophys. Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 25.Hadari T., Warms J.V., Rose I.A., Hershko A. J. Biol. Chem. 1992;267:719–727. [PubMed] [Google Scholar]
- 26.Shackelford J., Pagano J.S. Essays Biochem. 2005;41:139–156. doi: 10.1042/EB0410139. [DOI] [PubMed] [Google Scholar]
- 27.Lindner H.A. Virology. 2007;362:245–256. doi: 10.1016/j.virol.2006.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catic A., Misaghi S., Korbel G.A., Ploegh H.L. PloS ONE. 2007;2:e381. doi: 10.1371/journal.pone.0000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziebuhr J., Schelle B., Karl N., Minskaia E., Bayer S., Siddell S.G., Gorbalenya A.E., Thiel V. J. Virol. 2007;81:3922–3932. doi: 10.1128/JVI.02091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.